

Abstract
It has been demonstrated that dexmedetomidine (Dex) can protect patients with acute kidney injury from experiencing further tissue damage, however its mechanism of action remains unclear. The present study investigated the immune modulatory functions of Dex in rats with acute kidney injury (AKI) induced via injection of lipopolysaccharide into the tail vein. ELISA analysis showed that Dex reduced the levels of inflammatory cytokines in rats with AKI in a dose dependent manner. Furthermore, the regulatory effects of Dex on cytokine production disappeared when the α-2 adrenergic receptor antagonist Yohimbine (YOH) was added. For a detailed investigation on how Dex regulates the immune response in rats with AKI, the impact of Dex on the viability of splenocytes and lymphocytes was determined and it was determined that Dex did not influence splenocyte and lymphocyte viability. In addition, ELISA tests showed that Dex regulated the production of the T-helper (Th) 17 cytokines interleukin (IL)-17 and IL-23, but not the Th1 cytokine tumor necrosis factor α, in splenocytes and lymphocytes. To confirm whether Dex functioned as an α-2-adrenergic receptor in these immune regulations, YOH was administered together with Dex. When Dex and YOH were administered together, the regulatory functions of Dex were reduced, confirming that Dex acted as an agonist on the α-2-adrenergic receptor. Thus the results of the current study may provide novel insights regarding how Dex modulates immune functions in AKI.
Keywords: dexmedetomidine, acute kidney damage, cytokines, lymphocytes, splenocytes, inflammation
Introduction
Complex interactions occur between the immune and nervous systems of mammals and humans. When an animal experiences injury or pain, its immune system reacts to the injury via immediate nociceptor-induced sympathetic activation, where immune cells from lymphoid organs, including the spleen and lymph nodes, stimulate adrenoceptors (1). These immune cells respond to injury either by increasing the cytotoxic activity of natural killer cells or by producing cytokines through other immune cells (2,3). Through these immune responses, pain and injury are modulated (4).
Acute kidney injury (AKI) is a disease that usually requires a long period of hospitalization and tends to have high morbidity and mortality rates (5). AKI occurs in both transplanted and native kidneys, often occurring due to ischemic reperfusion injury. In such cases, tubulointerstitial inflammation, as well as strong inflammatory responses, usually occur (6). AKI generally begins with anoxic and hypoxic cell injury, and is then followed by inflammation. Inflammatory components, including pro-inflammatory cytokines and chemokines, usually flow within the blood and are delivered to the injured sites. Subsequently, leukocytes are recruited to these sites. A complete immune response following AKI usually begins with the passive release of factors from injured cells and is followed by the production of active factors from ischemic cells. The third and fourth steps usually involve the recognition of injured cells and reduced secretion of anti-inflammatory factors from injured cells following recognition of the injury (7). Based on these processes and interactions, reno-protective therapies targeting components relevant to the immune response, including chemokines, complements and adhesion molecules, have been employed to support the functions of innate immune responses in the pathogenesis of AKI (8,9).
Dexmedetomidine (Dex) is an agonist that acts selectively on the α-2-adrenergic receptor. It is widely used to sedate patients that go through mechanical ventilation within 24 h of AKI occurrence (10). By targeting the α-2-adrenergic receptor in the nervous system, Dex is able to immediately inhibit the production of catecholamines in patients. Dex is also used as an adjuvant in the perioperative period, since it has sedative and hypotensive functions (11) and by receiving Dex, patients with sepsis experience an improvement in brain function and require reduced mechanical ventilation. In particular, α-2 agonists can inhibit the deterioration of renal functions associated with AKI, as they exert potential renal protective functions, including inhibiting the release of renin and increasing glomerular filtration by activating the α-2-adrenoceptor. Importantly, Dex can protect renal functions associated with AKI by stabilizing the sympathetic systems and inducing anti-inflammation factors, such as cytokines (12). Therefore the aim of the present study was to investigate the role of Dex in regulating immune functions.
Materials and methods
Animal model
A total of 60 male Sprague Dawley rats weighing 280–300 g and 8–10 weeks of age were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Rats were housed in an environment at 25°C and 40% humidity with a 12 h light/dark cycle and free access to food and water. Rat AKI models were established by injecting lipopolysaccharide (LPS) via rat tail vein (5 mg/kg; Sigma Aldrich; Merck kGaA, Darmstadt, Germany) following a previously established protocol (13–15). Yohimbine (YOH) was used as an α-2-adrenergic receptor antagonist (1 mg/kg; Sigma-Aldrich; Merck kGaA). Rats involved in the present study were divided into 5 groups: CTRL, LPS, Dex, Dex + LPS, Dex + LPS + YOH (n=8). For the LPS + Dex group, Dex (3–20 µg/kg, Sigma-Aldrich; Merck KGaA) was injected 10 min prior to LPS injection. For the Dex + LPS + YOH group, YOH was administered to rats 30 min prior to Dex injection. The treatments were performed daily. For in vivo inflammation study, a Dex dose study was performed using 3, 5, 10 or 20 µg/kg Dex plus 5 mg/kg LPS. To test the impact of YOH, 1 mg/kg YOH was employed, followed by treating rats with 20 µg/kg Dex and 5 mg/kg LPS. The rats that were treated with 1 mg/kg YOH plus 5 mg/kg LPS was used as control. Rats were sacrificed for experiments 4–6 h following treatment was completed. All experiments were conducted under the guidance of Animal Care and Use Committee in Tianjin First Center Hospital (Tianjin, China). All experiments followed the guidelines of the National Institutes of Health (NIH) for the Care and Use of Laboratory Animals. The experimental performance also followed the Ethical Guidelines for Investigations of Experimental Pain in Conscious Animals. The present study was approved by the Animal Ethics Committee at Tianjin First Center Hospital.
Assays
Blood was drawn from the abdominal artery to assess levels of cytokines or drugs. The levels of interleukin (IL)-6, IL-18 and tumor necrosis factor (TNF)-α were assessed as follows: Blood was collected via the abdominal artery in heparinized tubes, followed by centrifugation (2,500 × g, 15 min at 4°C). Serum was collected and stored at −80°C prior to further use. ELISA kits for IL-6 (cat. no. 550319), IL-8 (cat. no. 555,244) and TNF-α (cat. no. 558535) in the serum were used (BD Biosciences, Franklin Lakes, NJ, USA) following the manufacturer's protocol. The concentration of Dex in the rat plasma was assessed using gas chromatography (GC)-mass spectrometry (M; Agilent Technologies Model 6890; Agilent Technologies, Inc., Santa Clara, CA, USA), with the lowest detectable limit set at 0.01 ng/ml. Capillary GC with a 6 m HP-1 column (internal diameter, 0.25 mm; film thickness, 0.25 µm) was employed. A splitless injection mode was used. The temperature of the initial column was 50°C for 1 min. Temperature increased at a rate of 60°C per min. The temperature was then programmed to 260°C at a rate of 10°C per min. The temperatures for injector, transfer line and ion source were set at 160, 245 and 100°C, respectively. Mass spectra and selected ion monitoring measurements were obtained with a negative ion mode. Methane was used as a reagent gas. For sample preparation, a C-18 Bakerbond spe was washed with methanol and water. A total of 0.1 ml plasma sample was added with 75 pmol d4-cortisol added in a mixture of methanol and phosphoric acid (50% methanol added with 1 ml phosphoric acid, pH 1). The samples were maintained at room temperature for 30 min and then moved to the column. The column was washed with 2 ml water and 2 ml methanol (30% v/v), then eluted with 2 ml methanol (100% v/v) into a conical reaction vial. The eluate was then processed under nitrogen into dry powder. The samples were then moved to 20 µl acetonitrile, followed by reacting with 20 µl pentafluoroporpionic anhydride for 2 h. An ~1 µl sample from the mixture was injected into the GC/MS for analysis.
Isolation of splenocytes and lymphocytes
Rats receiving different treatments were sacrificed via cervical dislocation. Subsequently, the spleen was dissected and rat lymph nodes were drained. The lymph nodes were smashed through 40 µm cell strainers by mechanical force with a syringe plunger to obtain isolated cells. Spleens were minced into small pieces (<1 mm3), followed by treatment with 20 U/ml collagenase (Sigma-Aldrich; Merck kGaA) and 20 U/ml DNase I (Sigma-Aldrich; Merck KGaA) in cell culture medium (RPMI culture medium with 2% fetal calf serum; Sigma-Aldrich, Merck KGaA) for 30 min at room temperature. EDTA (10 mM) was then added to the suspension to dissociate cell aggregates into single cells. Following collection and washing of cells with PBS, cells were cultured in RPMI culture medium for 48 h prior to ELISA analysis or staining.
ELISA analysis of cell culture medium of splenocytes and lymphocytes
The levels of cytokines in the splenocyte or lymphocyte culture were analyzed using ELISA. Production of TNF-α (cat. no. 560478; BD Biosciences), IL-17 (cat. no. 560666; BD Biosciences) and IL-23 (cat. no. ab119545; Abcam, Cambridge, UK) were analyzed using ELISA reagents (BD Biosciences) following the manufacturer's protocol. The cells in culture medium were centrifuged at 1,000 × g at 4°C for 5 min to remove the cells, followed by centrifugation at 12,000 × g at 4°C for 5 min to obtain supernatant at 4°C. A volume of 20 µl supernatant was employed in each analysis. A standard curve was set for quantification.
Whole splenocyte and lymphocyte isolation and staining
Spleens and lymph nodes were isolated from rats and isolated into single cell suspensions between glass slides. Following two washes with PBS, RBC-lysed single cell suspensions from mice receiving different treatments (CTRL, LPS, Dex, Dex + LPS, Dex + LPS + YOH) were cultured in round bottom 96-well plates with different treatments. For flow cytometry analysis, cells were treated with 80 µl 2 mM EDTA to produce a single cell suspension and were washed twice with PBS. Cells were centrifuged at 1,000 × g at 4°C for 5 min to remove PBS and then re-suspended in staining buffer (PBS with 10% fetal calf serum and 0.5 M EDTA). Fc-block (clone 93; Biolegend, 1:25 dilution at room temperature for 10 min) was used to block non-specific binding. Following washing with PBS twice, DC (DEC 205; 138205; 1:200; Biolegend, Inc., San Diego, CA, USA) and B220 [Fluorescein isothiocyanate (FITC) labeled anit-B220; 553087; 1:200; BD Biosciences] were then used to stain the cells for 25 min at room temperature. After washing with PBS, cells were either analyzed directly or fixed with 4% paraformaldehyde (PFA) solution for 30 min at room temperature and stored in a 1% PFA at 4°C solution until analysis. To determine the ratio of Th1 and Th17, cells were stained with a Th1/Th17 Phenotyping kit (560758; BD Pharmingen, San Diego, CA, USA). The kit was a Th1/Th17 Phenotyping Cocktail, including cluster of differentiation (CD) 4 (PerCP-Cy5.5 labeled) and IL-17A PE (TNF-α, FITC labeled), as well as fixation buffer and washing buffer. The staining process was performed following the manufacturer's protocol (cat. no. 560758; BD Biosciences). After staining, the cells were analyzed on BD LSRII (BD Biosciences, USA) Flow cytometer.
Statistical analysis
All data is presented as the mean ± standard error of the mean. Unpaired t-tests were employed to study the differences between different groups. One-way analysis of variance was used to analyze multiple comparisons. P<0.05 was considered to indicate a statistically significant difference. GraphPad Prism (version 6.02) was used to perform the analysis (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Dex concentrations in rats decrease over time
The concentration of Dex in rats was measured and the association between plasma Dex concentration and time is presented in Fig. 1. The two groups of rats were studied in this test: Rats treated with Dex and rats treated with Dex + LPS. For both groups, in vivo concentrations of Dex decreased over time. Dex concentration was high for the first 100 min following injection in all rats (Fig. 1). Rats treated with Dex + LPS had slightly lower concentrations of Dex compared with the control group (rats treated with Dex alone; Fig. 1, red). In both groups, the concentration of Dex was not detectable D min following initial injection (Fig. 1).
Figure 1.
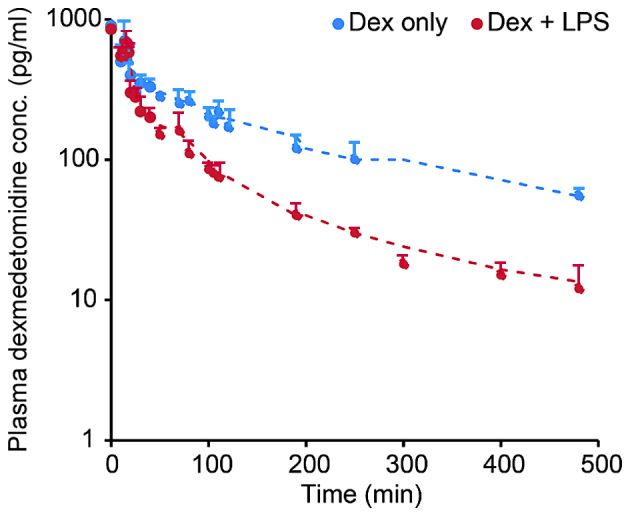
Concentration of Dex in rats over time (0–500 min). Rats were treated with Dex (red line) or Dex+LPS (blue line). The dots represent the concentration of Dex measured at different times. Dex concentrations in both groups were barely detectable after 8 h. Dex, dexmedetomidine; LPS, lipopolysaccharide.
Dex regulates levels of inflammatory cytokines in plasma
Following assessment of Dex levels in plasma, levels of the inflammatory cytokines IL-6 and TNF-α were measured using ELISA. There was an increase in IL-6 production in rats treated with LPS, indicating that potent inflammation was induced by LPS (Fig. 2A). The production of IL-6 was reduced when Dex was used to treat rats (Fig. 2A). Dex decreased IL-6 production in a dose dependent manner and an enhanced reduction of IL-6. Rats that received 20 µg/kg Dex had a higher level of cytokines than those that received 3, 5, or 10 µg/kg Dex (Fig. 2). To further investigate the role of Dex, another group of rats was treated with YOH + Dex + LPS. YOH is an α-2-adrenergic receptor antagonist and the α-2-adrenergic receptor is the target for DEX. Following the addition of YOH, production of IL-6 increased to levels similar to that in untreated LPS mice (Fig. 2A), indicating that YOH inhibited the activity of DEX. A similar trend was observed in the production of TNF-α. LPS treatment increased TNF-α levels and administration of Dex reduced levels of TNF-α in the serum. However, when YOH was added, TNF-α levels increased to levels similar to those in the LPS group (Fig. 2B).
Figure 2.

Concentration of IL-6 and TNF-α in plasma of rats undergoing different treatments. Rats were treated with different concentrations of Dex (3, 5, 10 or 20 µg/kg) or YOH (1.0 mg/kg) for 6 h after operation. ELISA was employed to analyze the concentrations of (A) IL-6 and (B) TNF-α. CTRL, control; LPS, lipopolysaccharide; Dex, dexmedetomidine; IL, interleukin; TNF, tumor necrosis factor; YOH, yohimbine; Dex 3; 3 µg/kg Dex; Dex 5, 5 µg/kg Dex; Dex 10, 10 µg/kg Dex; Dex 20, 20 µg/kg Dex. *P<0.05 and **P<0.01.
DEX does not affect viability of splenocytes and lymphocytes
It was determined that addition of Dex did not affect the viability of splenocytes (Fig. 3A). Viability of splenocytes was not significantly affected by the other treatments (LPS, Dex + LPS, or YOH + Dex + LPS) administered (Fig. 3A). Similar results were observed regarding the viability of lymphocytes; none of the treatments significantly affected lymphocyte viability (Fig. 3B).
Figure 3.

Viability of (A) splenocytes and (B) lymphocytes in rats undergoing different treatments. The rats were treated with Dex 10, LPS, Dex + LPS, YOH + Dex10 + LPS or YOH + LPS. There were no differences in the viability of splenocytes and lymphocytes in rats that had received different treatments. CTRL, control; LPS, lipopolysaccharide; Dex, dexmedetomidine; YOH, yohimbine; Dex 10, 10 µg/kg Dex.
DEX inhibits production of inflammatory cytokines in splenocytes and lymphocytes
Levels of inflammatory cytokine production were assessed (Fig. 4). Briefly, rats that underwent different treatments were sacrificed for the collection of spleen and lymph nodes. This was followed by the processing of organs into cell suspension. Cells were cultured in cell culture medium for 48 h and levels of cytokines in the cell culture medium were assessed with ELISA. LPS treatment of rats induced higher production of IL-6s in splenocytes (Fig. 4A) and lymphocytes (Fig. 4C) compared with mice that underwent no treatment. The use of Dex + LPS to treat rats reduced production of IL-6 compared with LPS alone, indicating that Dex inhibited IL-6 production (Fig. 4A and C). The impact of Dex + LPS treatment on IL-6 production occurred in a dose dependent manner; a higher dose of Dex yielded a more potent reduction of IL-6 levels (Fig. 4A and C). In addition, the effect of Dex on IL-6 production was attenuated when YOH was administered alongside Dex (Fig. 4A and C). Similar results were observed regarding the production of TNF-α and it was demonstrated that Dex inhibited TNF-α production in splenocytes and lymphocytes in a dose dependent manner, compared with LPS alone (Fig. 4B and D). When the antagonist for Dex, YOH, was added, the effect of Dex with regards to inhibiting TNF-α production was weakened (Fig. 4B and D).
Figure 4.

Expression of IL-6 and TNF-α in splenocytes and lymphocytes from rats undergoing different treatments. Expression of (A) IL-6 and (B) TNF-α in splenocytes; expression of (C) IL-6 and (D) TNF-α in lymphocytes. Rats were treated with different concentrations of Dex (3, 5, 10 or 20 µg/kg) or YOH (1.0 mg/kg) for 6 h after operation. ELISA was employed to analyze the concentration of IL-6 and TNF-α. CTRL, control; LPS, lipopolysaccharide; Dex, dexmedetomidine; IL, interleukin; TNF, tumor necrosis factor; YOH, yohimbine; Dex 3; 3 µg/kg Dex; Dex 5, 5 µg/kg Dex; Dex 10, 10 µg/kg Dex; Dex 20, 20 µg/kg Dex. *P<0.05 and **P<0.01.
DEX affects ratio of immune cells in the spleen and lymphocytes
The ratio of major immune cells in the splenocytes and lymphocytes was subsequently assessed. LPS injection increased the ratio of B cells, Th1 and Th17 cells and cluster of differentiation 11 c+ cells (DCs) in the spleen and lymph nodes (Fig. 5). Dex alone did not affect the ratios of immune cells in both lymphatic organs-levels were similar to those observed in control cells (Fig. 5). However, the use of Dex prior to LPS injection reduced the relative ratio of B cells, Th17 cells and DCs, compared to rats injected with LPS; when Dex and LPS (i.e., Dex + LPS) were administered together, the role of Dex in regulating the ratio of B cells, Th17 cells and DCs was significantly inhibited, as illustrated by a lower level of this cell in the group (Fig. 5A, C and D; P<0.05 in LPS group vs. LPS + Dex group). In Th1 cells, the use of Dex prior to LPS injected slightly, but not significantly, reduced the ratio of Th1 cells in the lymphocytes compared with the use of LPS alone. (Fig. 5B).
Figure 5.

Relative ratio of major immune cells in rat spleen (black bar) and lymph nodes (white bar) with different treatments. Rats underwent no treatments, or were treated with LPS, Dex 10, Dex 10+LPS or YOH+Dex 10+LPS. The ratio of (A) B cells, (B) Th1 cells, (C) Th17 cells and (D) DC cells were analyzed. Number of cells in rats was expressed as a relative amount, with the rats that received no treatment used as a control (*P<0.05). No treat, no treatment; LPS, lipopolysaccharide; Dex, dexmedetomidine; Th, T helper cells; YOH, yohimbine; Dex 10, 10 µg/kg Dex; DC, Cluster of differentiation 11 c+ cells. *P<0.05.
Dex regulates secretion of Th17 cytokines (IL-23) in splenocytes and lymphocytes
The production of Th1 (TNFα) and Th17 (IL-17 and IL-23) cytokines in splenocytes and lymphocytes were assessed (Fig. 6). There were no significant changes in the production of Th1 cytokines in rats treated with Dex + LPS compared with rats injected with LPS, although both groups exhibited increased secretion of INFγ compared to those injected with LPS (Fig. 6A and D). There were no significant alterations in cytokine levels following the use of YOH, indicating that YOH does not affect the production of the cytokines assessed (Fig. 6A-D). Similar results were also observed regarding the production of IL-17; neither Dex nor YOH had any effect on IL-17 production (Fig. 6B and E). Dex inhibited the secretion of IL-17 stimulated by LPS injection (Fig. 6B and E) and inhibited the secretion of IL-23 (Fig. 6C and F). Production of these two cytokines in splenocytes and lymphocytes from rats treated with Dex + LPS was reduced, compared with those injected with LPS (Fig. 6B-F). Notably, the inhibitory effects of Dex on the production of Th17 cytokines were significantly reduced when YOH was involved in the treatment (P<0.05; samples treated with YOH + DEX + LPS; Fig. 6B-F).
Figure 6.

Relative expression levels of Th1 (TNF-α), Th17 (IL-17 and IL-23) cytokines in rat splenocytes and lymphocytes. Relative expression of (A) TNF-α, (B) IL-17 and (C) IL-23 in the supernatant of splenocytes. Relative expression of (D) TNF-α, (E) IL-17 and (F) IL-23 in the supernatant of lymphocytes. The rats either received no treatment or were treated with CTRL, LPS, Dex, Dex + LPS or YOH + Dex + LPS. Concentrations of Dex and YOH used in this experiment were 10 µg/kg and 1 mg/kg, respectively. Splenocytes and lymphocytes were isolated from rat spleen or lymph nodes, respectively, followed by culturing cell culture plates for 48 h. Cell culture supernatant was used for ELISA analysis of Th1 or Th17 cytokines (*P<0.05). CTRL, control; LPS, lipopolysaccharide; Dex, dexmedetomidine; IL, interleukin; TNF, tumor necrosis factor; Th, T helper cells; YOH, yohimbine. *P<0.05.
Discussion
In clinical practice, AKI is a common occurrence in patients with sepsis (5). The mechanism of acute kidney injury is very complex and numerous explanations have been posited so far. Previous studies have associated AKI with renal ischemia reperfusion injury, nitric oxide release and oxidative stress-related injury (16,17). It has been suggested that sepsis-induced AKI is directly associated with inflammatory responses; however, this mechanism of action remains unclear due to the multiple mechanisms involved (10). The induction of rat sepsis model has been widely used to study the protective functions of Dex in AKI (4). Therefore, the current study used this model to study the role of DEX in AKI and observed that Dex had a protective role in AKI, by acting as an α-2 adrenergic receptor. To examine the role of Dex in our study, a well-known α-2 adrenergic receptor antagonist, YOH, was used to reveal the role of Dex in regulating immune functions in AKI (10,13).
The present study began by assessing the biodistribution of Dex in rats following LPS injection and it was found that there was a higher concentration of Dex detectable in the rat serum (Fig. 1). The concentration of Dex decreased slowly, possibly due to the fact that Dex was excreted in multiple ways, including through the liver and urinary system. Rats treated with Dex had a higher concentration of Dex than those treated with Dex + LPS. This was probably because LPS induced a strong immune response in rats (Fig. 1), which Dex is involved in. Rats with induced AKI had a slightly lower concentration of Dex in the serum than controls. This is consistent with previously recorded results (18,19). Additionally, this is consistent with other previous studies investigating the change in concentration of Dex over time in rats (20).
In the present study, the α-2-adrenergic receptor antagonist, YOH, was adopted, where α-2-adrenergic receptor is the target for DEX. This aids in investigating whether DEX inhibits the inflammatory responses via acting on the α-2-adrenergic receptor. Following the detection of Dex in blood serum, the level of inflammatory cytokines in rat blood was assessed using ELISA (Fig. 2A and B). LPS induced strong production of inflammatory cytokines in rat blood, possibly due to the injury it induced (Fig. 2A and B). These cytokines were also associated with the inflammatory responses that commonly occur in AKI. Regarding the role of Dex, the present study determined that Dex reduced the level of inflammatory cytokines in a dose-dependent manner. Although the level of cytokines in Dex-treated rats did not return to the base level (the level in control rats that received no treatment; Fig. 2A and B), even such a small reduction in the levels of inflammatory cytokines is considered desirable, since it can protect organs from damage.
As well as measuring the levels of inflammatory cytokines in rat blood and the effect of Dex on these levels, the current study aimed to investigate the role of Dex in regulating the immune response in two major immune organs: The spleen and lymph nodes. To assess the role of Dex in splenocytes and lymphocytes, cells were isolated from the spleen and lymph nodes and treated with Dex. There was no significant effect on cell viability following treatment with Dex and no other treatments employed in the current study affected cell viability (Fig. 3). Subsequently, the production of cytokines and the ratio of immune cells in the spleen and lymph nodes were investigated. The present study found that Dex treatment attenuated the increase in the ratio of B cells induced by LPS injection (Fig. 5A). Similar effects were also observed in other major immune cells (B cells, Th17 cells and DCs) that reside in the spleen and lymph nodes (Fig. 5A, C and D). To further investigate the inflammatory response in splenocytes and lymphocytes, the expression of Th1 and Th17 cytokines in these cells were assessed (Fig. 6). The results determined that Dex had no significant effects on Th1 cytokine production in the spleen and lymph nodes, but decreased levels of the Th17 regulatory cytokines IL-17 and IL-23 compared with rats that received LPS injection without Dex treatment (Fig. 6B, C, E and F). Further studies investigating the role of Dex on the production of other cytokines in the spleen and lymph nodes are therefore required.
In conclusion, the present study investigated the role of Dex in regulating the immune response in a rat AKI model. The results indicated that Dex is able to regulate the inflammatory response in rat blood serum. The study also revealed that while Dex did not affect the viability of the cells in the two major immune organs, it regulated the ratio of the immune cells ratio in the spleen and lymph node of rats induced with AKI, particularly the ratio of Th17 cells. Dex also affected the levels of cytokines secreted by these cells. Furthermore, assessment of cytokine production demonstrated that these ratio variations were associated with the production of cytokines that regulate immune inflammatory responses. Therefore the results of the present study may aid understanding regarding the role of Dex in regulating immune functions in AKI diseases.
References
- 1.Chapman CR, Tuckett RP, Song CW. Pain and stress in a systems perspective: Reciprocal neural, endocrine, and immune interactions. J Pain. 2008;9:122–145. doi: 10.1016/j.jpain.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharify A, Mahmoudi M, Izad MH, Hosseini MJ, Sharify M. Effect of acute pain on splenic NK cell activity, lymphocyte proliferation and cytokine production activities. Immunopharmacol Immunotoxicol. 2007;29:465–476. doi: 10.1080/08923970701619877. [DOI] [PubMed] [Google Scholar]
- 3.Greisen J, Hokland M, Grøfte T, Hansen PO, Jensen TS, Vilstrup H, Tønnesen E. Acute pain induces an instant increase in natural killer cell cytotoxicity in humans and this response is abolished by local anaesthesia. Br J Anaesth. 1999;83:235–240. doi: 10.1093/bja/83.2.235. [DOI] [PubMed] [Google Scholar]
- 4.Flores LR, Dretchen KL, Bayer BM. Potential role of the autonomic nervous system in the immunosuppressive effects of acute morphine administration. Eur J Pharmacol. 1996;318:437–446. doi: 10.1016/S0014-2999(96)00788-1. [DOI] [PubMed] [Google Scholar]
- 5.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 6.Bonventre JV, Zuk A. Ischemic acute renal failure: An inflammatory disease? Kidney Int. 2004;66:480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 7.Thurman JM. Triggers of inflammation after renal ischemia/reperfusion. Clin Immunol. 2007;123:7–13. doi: 10.1016/j.clim.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takada M, Nadeau KC, Shaw GD, Marquette KA, Tilney NL. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. Inhibition by a soluble P-selectin ligand. J Clin Invest. 1997;99:2682–2690. doi: 10.1172/JCI119457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singbartl K, Ley K. Protection from ischemia-reperfusion induced severe acute renal failure by blocking E-selectin. Crit Care Med. 2000;28:2507–2514. doi: 10.1097/00003246-200007000-00053. [DOI] [PubMed] [Google Scholar]
- 10.Martin E, Ramsay G, Mantz J, Sum-Ping ST. The role of the alpha2-adrenoceptor agonist dexmedetomidine in postsurgical sedation in the intensive care unit. J Intensive Care Med. 2003;18:29–41. doi: 10.1177/0885066602239122. [DOI] [PubMed] [Google Scholar]
- 11.Mantz J, Josserand J, Hamada S. Dexmedetomidine: New insights. Eur J Anaesthesiol. 2011;28:3–6. doi: 10.1097/EJA.0b013e32833e266d. [DOI] [PubMed] [Google Scholar]
- 12.Tasdogan M, Memis D, Sut N, Yuksel M. Results of a pilot study on the effects of propofol and dexmedetomidine on inflammatory responses and intraabdominal pressure in severe sepsis. J Clin Anesth. 2009;21:394–400. doi: 10.1016/j.jclinane.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Yu X, Wang F, Wang Y, Wang Y, Li H, Lv X, Lu D, Wang H. Yohimbine promotes cardiac NE release and prevents LPS-induced cardiac dysfunction via blockade of presynaptic α2A-adrenergic receptor. PLoS One. 2013;8:e63622. doi: 10.1371/journal.pone.0063622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Restitutti F, Laitinen MR, Raekallio MR, Vainionpää M, O'Brien RT, Kuusela E, Vainio OM. Effect of MK-467 on organ blood flow parameters detected by contrast-enhanced ultrasound in dogs treated with dexmedetomidine. Vet Anaesth Analg. 2013;40:E48–E56. doi: 10.1111/vaa.12058. [DOI] [PubMed] [Google Scholar]
- 15.Bayraktar O, Tekin N, Aydin O, Akyuz F, Musmul A, Burukoglu D. Effects of S-allyl cysteine on lung and liver tissue in a rat model of lipopolysaccharide-induced sepsis. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:327–335. doi: 10.1007/s00210-014-1076-z. [DOI] [PubMed] [Google Scholar]
- 16.Venkatachalam MA, Weinberg JM. The conundrum of protection from AKI by adenosine in rodent clamp ischemia models. Kidney Int. 2013;84:16–19. doi: 10.1038/ki.2013.101. [DOI] [PubMed] [Google Scholar]
- 17.Anjaneyulu M, Chopra K. Effect of irbesartan on the antioxidant defence system and nitric oxide release in diabetic rat kidney. Am J Nephrol. 2004;24:488–496. doi: 10.1159/000080722. [DOI] [PubMed] [Google Scholar]
- 18.Dyck JB, Maze M, Haack C, Vuorilehto L, Shafer SL. The pharmacokinetics and hemodynamic effects of intravenous and intramuscular dexmedetomidine hydrochloride in adult human volunteers. Anesthesiology. 1993;78:813–820. doi: 10.1097/00000542-199305000-00002. [DOI] [PubMed] [Google Scholar]
- 19.De Wolf AM, Fragen RJ, Avram MJ, Fitzgerald PC, Rahimi-Danesh F. The pharmacokinetics of dexmedetomidine in volunteers with severe renal impairment. Anesth Analg. 2001;93:1205–1209. doi: 10.1097/00000539-200111000-00031. [DOI] [PubMed] [Google Scholar]
- 20.Tan F, Chen Y, Yuan D, Gong C, Li X, Zhou S. Dexmedetomidine protects against acute kidney injury through downregulating inflammatory reactions in endotoxemia rats. Biomed Rep. 2015;3:365–370. doi: 10.3892/br.2015.427. [DOI] [PMC free article] [PubMed] [Google Scholar]