

Summary
Embryonic stem cells (ESCs), with their dual capacity to self-renew and differentiate, are commonly used to study differentiation, epigenetic regulation, lineage choices, and more. Using non-directed retroviral integration of a YFP/Cherry exon into mouse ESCs, we generated a library of over 200 endogenously tagged fluorescent fusion proteins and present several proof-of-concept applications of this library. We show the utility of this library to track proteins in living cells; screen for pluripotency-related factors; identify heterogeneously expressing proteins; measure the dynamics of endogenously labeled proteins; track proteins recruited to sites of DNA damage; pull down tagged fluorescent fusion proteins using anti-Cherry antibodies; and test for interaction partners. Thus, this library can be used in a variety of different directions, either exploiting the fluorescent tag for imaging-based techniques or utilizing the fluorescent fusion protein for biochemical pull-down assays, including immunoprecipitation, co-immunoprecipitation, chromatin immunoprecipitation, and more.
Keywords: embryonic stem cells, imaging, live imaging, fluorescence, differentiation, pluripotency, GFP, microscopy, DNA damage, protein dynamics
Graphical Abstract
Highlights
-
•
We generated a library of endogenously tagged fluorescent fusion proteins in ESCs
-
•
We show its potential use for a variety of screens, including live imaging
-
•
The library is useful for dynamic studies, e.g., photobleaching-based experiments
-
•
The YFP/Cherry tag can be used to pull down proteins for biochemical assays
Using a gene-tagging approach, Meshorer and colleagues describe in this article the generation of an endogenously tagged fluorescent fusion library in mouse ESCs, providing the community with over 200 YFP/Cherry-tagged clones highly expressed in ESCs. The paper describes the generation of the library as well as several potential uses and applications.
Introduction
The study of protein expression and localization in living cells has been made possible by the development of fluorescent reporters, especially by the use of a cDNA encoding GFP or its numerous variants fused to the gene of interest. The protein product of such a fusion construct renders the selected protein of interest fluorescent and allows monitoring of its localization, as well as its dynamic turnover using laser-based photobleaching methods, in living cells (Meshorer, 2008). However, such fusion proteins are usually expressed in cells under non-endogenous promoters and, therefore, their expression level does not correlate with endogenous levels. For expression of GFP-fusion proteins under their own endogenous promoters, enhancers, and control elements, GFP can be knocked in using genomic technologies, including homologous recombination, zinc-finger nucleases (ZFNs), TALE nucleases, and, more recently, CRISPR technology (Hockemeyer et al., 2011, Miller et al., 2007, Miller et al., 2011, Wang et al., 2013). Bacterial artificial chromosomes (BACs) have also been engineered to include GFP as part of a reading frame of a gene of choice, allowing the introduction of an entire BAC into living cells, thus expressing the protein of choice under its own control elements (Hutchins et al., 2010). Despite these advances, the aforementioned methods are time consuming and require engineering steps that limit the visualization to a few GFP-fusion proteins at best.
A library of endogenously labeled GFP-fusion proteins was previously generated in human lung carcinoma cells, H1299, based on non-directed integration of a retrovirus carrying the YFP (yellow fluorescent protein) coding region flanked by splice signals (Sigal et al., 2006). This library allowed analysis of the cell-cycle dependence of the proteins' expression (Sigal et al., 2007) to measure the proteins' expression response to a drug (Cohen et al., 2008) and drug combination (Geva-Zatorsky et al., 2010), to correlate RNA and protein levels in single cells (Cohen et al., 2009), and to measure the protein's half-life using “bleach-chase” (Eden et al., 2011). Here we used a similar strategy to endogenously tag over 200 proteins in mouse ESCs. We first generated a smaller library of YFP-tagged genes, then used one of the YFP-tagged clones (topoisomerase I [TopoI]) for the generation of a second library of Cherry-tagged TopoI-YFP double-labeled clones. Over 90% of the proteins in our library show correct localization. We show several proof-of-concept experiments demonstrating the usefulness of the library. We demonstrate that our libraries can be used to track the expression of individual proteins in living cells; to screen for general pluripotency markers that are reduced during ESC differentiation; to analyze cell-to-cell heterogeneity and identify heterogeneously expressed proteins; to analyze the dynamics of endogenously labeled proteins using fluorescence recovery after photobleaching (FRAP); to track proteins that are recruited to sites of DNA damage; to immunoprecipitate tagged proteins using YFP/Cherry pull-down; and to generate knockin mice. We further discuss additional potential uses of our libraries.
Results
Generation of an Endogenously Tagged Fluorescent Fusion Protein Library in Mouse ESCs
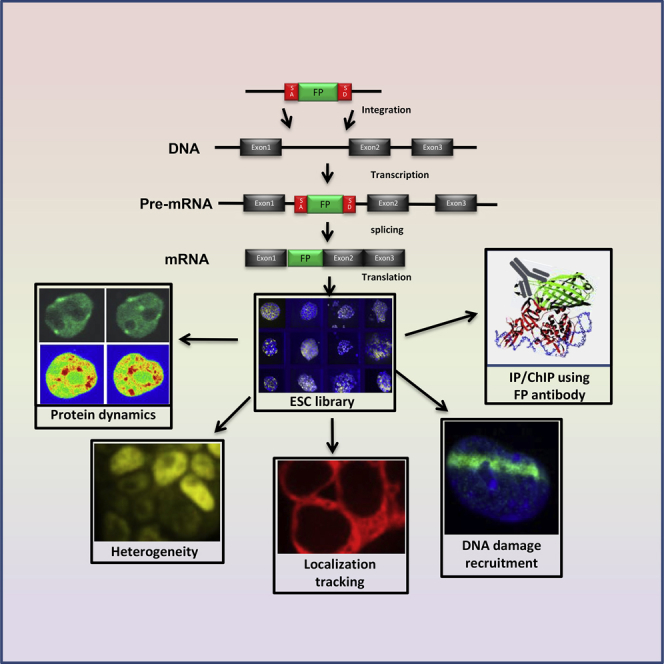
To generate an ESC clone library of fluorescently labeled proteins under their endogenous control elements, we used the central dogma (CD) tagging approach (Jarvik et al., 1996, Jarvik et al., 2002). This tagging approach, which utilizes a non-directed retroviral integration of an exon encoding a fluorescent protein (YFP or Cherry), allowed us to endogenously label genes, which are expressed at relatively high levels in ESCs. This approach has a strong resemblance to gene-trap technologies (Brennan and Skarnes, 1999), although the CD approach does not create a truncated protein, and in most cases the resulting fusion protein is expected to be functional. Importantly, the labeled protein is expressed at endogenous levels, and recapitulates the endogenous levels of the labeled protein at both the RNA and the protein level. To create the library of endogenously labeled fluorescent clones in ESCs, we used non-directed retroviral integration of a YFP exon (Sigal et al., 2006) which, when integrated inside a gene (usually within the first intron), is spliced together with that gene into the open reading frame, generating a YFP-fusion protein (Figure 1A). To create the library we infected low-passage R1 ESCs with the retrovirus, and the resulting fluorescent cells were single-cell sorted into 96-well plates. The clonal populations arising from the sorted single cells were expanded and the tagged genes were identified using the rapid amplification of cDNA ends (3′-RACE) system (Figure S1A). A list of all clones in the library is provided in Table S1. Our Cherry library was built on top of one of our YFP clones, namely TOPOI-YFP, making it a double-color (YFP/Cherry) library. TOPOI-YFP clone was selected because it displayed stable nuclear expression during ESC differentiation (Movie S1). This allowed efficient automated tracking of cells using nuclear TOPOI fluorescence in the green channel and different tagged proteins in the red channel (e.g., Cherry-tagged C1QBP, Movie S2). Most of the tagged proteins showed correct localization, in agreement with their previously reported localization (Figures 1B, 1C, and S2A) and as previously reported (Sigal et al., 2007).
Figure 1.

Clone Library Generation
(A) A schematic representation of the gene-tag clone library preparation. The pBABEpuro CD (Central Dogma)-tagging retroviral vector was used. Fluorescent-tag (FL) labeled ESCs were single sorted using fluorescence-activated cell sorting (FACS) into single wells of 96-well plates. The gene-tagging approach was achieved by the integration of the YFP or Cherry exon (which include strong splice sites but no start/termination codons) inside protein-coding genes, resulting in YFP or Cherry fusion proteins (inset). SA, splice acceptor; SD, splice donor; LTR, long terminal repeat.
(B) Examples, showing different localizations, of endogenously labeled fluorescent fusion proteins: cytoplasmic (NACA); nuclear (TOPOI); whole cell (HN1); and nucleolar (RRP15). Cherry clones (bottom): cytoplasmic (ACTN4); nuclear (HP1β); whole cell (UBE2n); and plasma membrane (CTNNA1). Scale bar, 200 μm.
(C) Endogenously tagged YFP and Cherry fusion proteins are properly localized. Clones expressing YFP-tagged (green, top and middle left) or Cherry-tagged (red, bottom left) proteins were fixed and labeled with the corresponding antibodies (right, Alexa 561-red for the YFP clones and Alexa 488-green for the Cherry clones). From top to bottom: NASP, SET, and HP1β. In all cases perfect co-localizations were observed. Scale bars, 20 μm.
(D) Integration site statistics. The majority of the integration events occurred inside intron 1 and exon 1.
We next tested whether our tagged proteins respond correctly to external stimuli. G3BP2 and CAPRIN1 are known stress granule proteins, which are involved in the assembly of stress granules (Kedersha et al., 2016). During normal growth these proteins are mainly cytoplasmic. Under stress conditions these proteins assemble into ribonucleoprotein granules, which are thought to protect mRNA (Kedersha et al., 2016). To induce stress granule assembly, we treated our cells with arsenite for 30 min. Reassuringly, G3BP2 and CAPRIN1 both behaved similarly to endogenous wild-type proteins, relocalizing to stress foci, following arsenite treatment (Figures S2B and S2C). Assembly of tagged G3BP2 and CAPRIN1 into stress granules clearly shows that the YFP did not alter the localization of the tagged protein (Figures S2B and S2C).
Analyzing the integration sites of all our clones, we found that the integration event was most frequent in intron 1, followed by exon 1 (Figure 1D), thus generating, in most of the cases, N-terminal fusion proteins or nearly N-terminal fusions. Interestingly, viral integration has a preference for certain chromosomes over others (Figure S1B). Not surprisingly, our clone library lacks integration events inside the small and gene-poor Y chromosome. Gene ontology (GO)-term analysis of genes tagged in the library revealed enrichment of genes belonging to housekeeping processes such as translation, transcription, and other pathways (Figure S1C). With increasing library size we will be able to tag most, if not all of the genes in individual GO terms, enabling studies involving whole pathways. Furthermore, this growing clone library encompasses a wide variety of labeled YFP- and Cherry-tagged proteins (Table S1) with different cellular localizations, allowing us to monitor numerous cellular processes for the first time in a non-invasive manner in ESCs.
Screening for Downregulated Proteins during ESC Differentiation
Our endogenously labeled fluorescent fusion libraries consist of ∼60 YFP-tagged and ∼150 Cherry-tagged clones. Since our labeling approach mostly targets the highly expressing genes in ESCs, this platform is perfect for imaging studies and allows the identification of proteins that are decreased during ESC differentiation. To screen for proteins downregulated during ESC differentiation, we followed the fluorescence intensity of individual clones following retinoic acid (RA) treatment and leukemia inhibitor factor (LIF) withdrawal for 4 days (Figure 2A). Most clones displayed little change in expression during this time frame. However, we were able to identify several different clones that were significantly decreased during the early course of ESC differentiation. These included CAPRIN1, NASP, ANP32A, and NPM1 (Figure 2B). Decrease in protein levels was verified using western blots with anti-GFP antibodies (Figure 2C), as well as real-time RT-PCR (Figure 2D). Interestingly, while Caprin1, Nasp, and Anp32a showed a concomitant decrease in their RNA levels, Npm1 RNA expression remained unaltered during the first 48 hr of differentiation, demonstrating selective regulation of NPM1 at the protein level. Another protein, SET, which showed decreased expression of one of its isoforms (SETα), was analyzed in depth (Edupuganti et al., 2017 [this issue of Stem Cell Reports]). SET could not have been identified by monitoring RNA levels during differentiation nor by mass spectrometry-based methods, because of an SETα/SETβ isoform switch: the downregulation of SETα is masked by the upregulation of SETβ. These two transcripts share more than 90% of their RNA and protein sequence, and our serendipitous labeling of only one of these two isoforms (SETα) allowed us to visualize its disappearance during early ESC differentiation (Edupuganti et al., 2017). These results demonstrate that our fluorescently labeled ESC libraries are a powerful resource enabling the search for pluripotency-related factors.
Figure 2.

Screening for Pluripotency-Related Candidates
(A) Schematic depicting the screening procedure.
(B) Fluorescent (top) and phase-contrast (bottom) images showing changes in the YFP-tagged protein levels before (left) and after (right) 48 hr of RA-induced differentiation. Scale bar, 100 μm. LIF, leukemia inhibitory factor.
(C and D) Western blot analyses using anti-GFP antibodies (C) and qPCR (D) of the selected clones shown in (B) before and after 48 hr of RA-induced differentiation. Protein and mRNA levels were normalized to GAPDH protein and mRNA, respectively. Data were obtained from 3 independent experiments; error bars represent ±SD.
Screening for Heterogeneously Expressed Proteins in ESCs
Next, we wished to screen for heterogeneously expressed proteins. Heterogeneous expression was demonstrated for several different proteins in ESCs, including NANOG (Kalmar et al., 2009, Singh et al., 2007), REX1 (Toyooka et al., 2008), and ZSCAN4 (Zalzman et al., 2010), to name a few, and was suggested to be functionally important for the stem cell state (Cahan and Daley, 2013, Hayashi et al., 2008). Therefore, heterogeneous expression is expected to have functional implications and warrants the identification of additional heterogeneously expressed clones in ESCs. Because the YFP and Cherry fluorescent proteins are integrated in our libraries as part of the reading frame, the resulting fluorescence mirrors the endogenous protein level. Also, since our libraries are clonal, heterogeneous expression reflects endogenous variation rather than differences in the cell origin. Thus, using fluorescence intensity quantification of confocal images, we calculated the coefficient of variation of several randomly selected clones, directly reflecting expression heterogeneity at the protein level (Figures 3A and S2D). We were able to identify several proteins (i.e., MDK, SRGAP11, PPP1CA1, CBX1, HNRNPC1/C2, ANP32A), which showed above average expression variation between cells within the same colony (Figure 3A). Single cells isolated from such heterogeneous colonies generated heterogeneous colonies, suggesting that heterogeneity in these clones is inherent and is not due to genetic heterogeneity. To verify that heterogeneity is an inherent state and to measure the kinetics by which heterogeneity is achieved, we selected two variable clones (HNRNPC1/C2 and MDK), sorted the 10% most highly expressing cells (“High”) and 10% most lowly expressing cells (“Low”), and plated each cell population in a separate plate. Remarkably, after 5 days of culture, both High cells and Low cells reverted back to the original distribution at the population level (Figures 3B and 3C). To ask whether these variations in protein levels are also reflected at the RNA level, we used previously published data from single-cell RNA sequencing (RNA-seq) experiments (Hashimshony et al., 2012, Sasagawa et al., 2013, Tang et al., 2010). We found that while Mdk1 is variable also at the RNA level, hnRNPc1/c2 showed homogeneous RNA expression among the different single cells (Figure 3D), suggesting, once again, selective regulation at the protein level. These results demonstrate the usefulness of the ESC clone libraries in identifying heterogeneously expressing proteins, and demonstrate that heterogeneity is an inherent state, at least in the case of hnRNPc1/c2 and Mdk, while heterogeneity at the protein level is not necessarily manifested at the RNA level.
Figure 3.

Screening for Heterogeneously Expressed Proteins in ESCs and Measuring Protein Dynamics
(A) Coefficient of variation (CV) of fluorescence of 10 randomly selected clones.
(B) Inter-convertible distribution of HNRNPC1/C2 in ESCs. The HNRNPC1/C2 cells were sorted into low and high subpopulations (top) and subcultured individually for 5 days. The subcultured cells were reanalyzed using FACS to assess the fluorescence distribution (bottom). Sorted populations reverted back to the parental population. R1 ESCs were used as negative controls.
(C) MDK showing characteristic distribution similar to HNRNPC1/C2.
(D) CV of the RNA level for hnRNPc1/c2 and Mdk. Data were obtained from 3 independent publications reporting single-cell RNA-seq experiments, including (left to right) Tang et al., 2010, Hashimshony et al., 2012, and Sasagawa et al. (2013).
(E) Representative cell images depicting fluorescence bleaching and recovery of TOPOI-YFP in ESC and differentiated cells. Scale bar, 5 μm.
(F) FRAP recovery curves of TOPOI-YFP in ESCs (blue) and in RA-differentiated ESCs (red) (n = 3 independent experiments; ∗p < 0.05, 2-tailed t test).
(G) Kinetic parameters of the FRAP curves depicted in (F).
Measuring Protein Dynamics in Living Cells Expressed at Endogenous Levels
Next, we wished to utilize our fluorescent proteins expressed at endogenous levels to measure their dynamics. We selected TOPOI, which showed no changes in fluorescence intensity during differentiation (Movie S1), and studied its dynamic association with chromatin using FRAP (Cheutin et al., 2003, Festenstein et al., 2003, Melcer et al., 2012, Meshorer et al., 2006). This is in fact the first time that such studies can be performed on proteins expressed at endogenous levels. Most FRAP studies, including our own (Melcer et al., 2012), relied on transient transfections of fusion proteins expressed under viral promoters. Here, we compared the dynamic association of TOPOI with chromatin before and after RA-induced differentiation (Figure 3E). We found comparable kinetics but an increased bleach depth following differentiation, reflecting a reduced highly mobile fraction of TOPOI in the differentiating cells (Figures 3F and 3G). These results may reflect the increased dynamics we previously demonstrated for structural chromatin proteins in ESCs (Melcer et al., 2012, Meshorer et al., 2006), or simply a change in the highly mobile fraction of TOPOI itself. Regardless, our libraries now enable, for the first time in systematic fashion, analysis of the dynamics of proteins expressed at endogenous levels in ESCs and during ESC differentiation.
Identifying Proteins Recruited to Sites of DNA Damage
Next, we tested several proteins for their propensity to relocalize to sites of DNA damage following UV irradiation. UV laser irradiation induces localized DNA damage. The histone variant H2A.X is selectively phosphorylated in the damaged site, followed by localized and selective recruitment of DNA repair proteins. Our libraries offer an easy and convenient screening platform for proteins potentially involved in the DNA damage/repair pathways. To this end, we irradiated several of our clones using a 355-nm UVA laser, inducing localized DNA damage in a specified region. We first verified, using immortalized mouse embryonic fibroblasts (MEFs), that our UVA is inducing cyclobutane pyrimidine dimers (CPDs), and that CPDs induce the expected recruitment (Figure S3A). We tested HP1β-GFP, which was previously shown to be recruited to sites of DNA damage (Ayoub et al., 2008). As expected, HP1β-GFP showed significant accumulation at the UV-irradiated region, marked by phosphorylation of H2A.X or 53BP1 (Figure 4A). We then screened for additional proteins that are recruited to sites of UVA-induced damage. We first tested SETα, the protein we identified to be downregulated during ESC differentiation and which was previously shown to be involved in the DNA damage response (Kalousi et al., 2015). We found a slight accumulation of SETα at sites of damage (Figure S3B). An additional protein, which showed accumulation at sites of damage, was TOPOI (Figure S3C), while other proteins, such as NPM1 (Figure 4B) and CCAP1, did not respond to UV irradiation (Figure S3D).
Figure 4.

Functional Validation of Clone Library
(A) Endogenously tagged HP1β fluorescent ESC clones were microirradiated using a 355-nm UVA laser. Cherry accumulation was monitored at the sites of damage. Cells were monitored from 10 s up to 15 min after UVA irradiation. A colony is shown on top (left: fluorescence; middle: bright-field; right: merge), and 1–2 selected cells are shown below. Green boxes depict area irradiated by UV-A. White arrows point at accumulation of HP1β at the DNA damage site. Scale bars (from top to bottom), 50 μm, 10 μm, 4 μm.
(B) Nucleophosmin (NPM1) did not show localization to DNA damage sites. In each panel, a colony is shown on top (left: fluorescence; middle: bright-field; right: merge), and 1–2 selected cells are shown below. Green boxes depict the area irradiated by UV-A. Scale bars (from top to bottom), 4 μm, 2 μm.
(C) Scheme representing the approach used to identify interaction partners combining the clone library with Nano-Trap technology.
(D) Western blots using anti-RFP (top) and anti-DNMT3B (bottom) antibodies showing CoIP of HP1α-Cherry and DNMT3B. RFP trap (lanes 1–3) and Control trap (lanes 4–6) are shown. Arrow indicates the presence of HP1α partner DNMT3B in the bound fraction.
Biochemical Pull-Down Applications
Next, we turned to utilizing our libraries in biochemical assays using the YFP/Cherry as bait for pull-down experiments. Isolation of proteins and accompanying complexes are notoriously difficult due to variability of the antibodies targeting the native proteins. We set out to pull down selected proteins using anti-Cherry antibodies (RFP trap) and examine potential interaction partners (Figure 4C). To this end, we examined whether we were able to detect a known association partner using co-immunoprecipitation (coIP) with the RFP-trap system, and another antibody against a known interaction partner. We selected our HP1α-Cherry clone for the pull-down experiment and labeled the blot with an anti-DNMT3B antibody. A weak interaction between these two proteins was previously reported (Lehnertz et al., 2003), and we wished to test whether we could recapitulate this weak interaction using our system. Reassuringly, we observed an interaction between HP1α-Cherry and DNMT3B (Figure 4D), suggesting that we can detect known interactions, and that the Cherry fusion did not alter the interaction of HP1α with DNMT3B. This approach, though not utilized at the single-cell level, is extremely powerful since it allows performance of biochemical assays such as chromatin immunoprecipitation (ChIP)-PCR and ChIP followed by high-throughput sequencing, and identification of interaction partners of many different proteins using a single antibody. Importantly, this obviously also applies to proteins for which antibodies are not available or which are inferior to the anti-GFP or anti-Cherry antibodies.
Chimeric Contribution
Finally, we tested the potential in vivo differentiation of some of our library clones by chimeric contribution. We injected tagged ESCs into mouse blastocysts and returned the injected blastocysts into pseudopregnant recipient female mice. We found that injected cells were able to generate chimeric mice and significantly contributed to the tissues of the resulting chimeric mice (Figure S3E). This indicates that despite extensive manipulation of the cells during the generation of the library, the clonal library cells can potentially be used in vivo in transgenic mouse production.
Discussion
Using non-directed retroviral integration of YFP and Cherry exons, we generated an endogenously labeled fluorescent protein library in mouse ESCs. Here, we demonstrated that this resource allows one to do the following: monitor protein expression levels in living cells; follow potential changes during ESC differentiation; identify heterogeneously expressing proteins; measure the dynamics of endogenously labeled proteins with photobleaching methods; pull down essentially all tagged proteins using a single (anti-YFP or anti-Cherry) antibody; and generate transgenic mice. Apart from these selected proof-of-concept experiments, these endogenously labeled cells can be used for additional screening and basic biological purposes (Figure 5). For example, drugs affecting protein expression and/or localization can be screened alone or in combinations. Additionally, a major effort in the field of DNA damage and repair is to identify proteins that are recruited to the site of damage. Such a screen can be easily performed using our endogenously labeled fluorescent libraries by irradiating (using a UV laser) a small portion of the nucleus and tracking the fluorescence intensity in the irradiated site (Figure 4A). Finally, the proteins' half-life can be measured using “bleach-chase” approaches as previously demonstrated in a human cancer cell line (Eden et al., 2011).
Figure 5.

Multiple Applications for the Endogenously Tagged Fluorescent Library in ESCs
Several potential applications for the clone library are indicated.
Unlike in differentiated cells, where repressive chromatin structure may preclude viral integration in several regions of the genome, ESCs have a more open chromatin conformation, and viral integration can be expected to be more widespread. Importantly, we did not observe any silencing of the tagged genes due to viral backbone integration. However, the clone library is not without limitations. Tagging all proteins expressed in ESCs may be extremely difficult to achieve, due to biological and technical reasons. In addition, the CD identification approach requires polyadenylation for 3′-RACE to work, and although 5′-RACE or linker-end amplification are also possible, these are notoriously cumbersome and time-consuming processes, which are difficult to automate. Hence, our libraries, as well as the previously generated ones (Sigal et al., 2006), contain no non-polyadenylated transcripts (i.e., histones). Another challenging feature is that several genes are repeatedly labeled, indicating that these genes are hotspots for retroviral integration. We also observed tagged ESC colonies with barely discernible fluorescence, which might reflect spurious transcription (Efroni et al., 2008). Finally, despite the titration of the retrovirus to achieve single integrations, we nonetheless observed instances where double tagging occurred.
Despite these difficulties, we were able to generate a library of over 200 (and growing in number) endogenously fluorescently tagged proteins expressed in ESCs, which we trust will be useful for the scientific community as a whole. In addition to the various approaches described and discussed herein, the fluorescent ESCs can also be used to generate transgenic animals and to study the fluorescent proteins in a developmental context, opening the door to a myriad of additional possibilities.
Experimental Procedures
All mice were obtained from the Jackson Laboratory and maintained in the Whitehead Institute animal facility. All experiments were approved by the Committee on Animal Care (CAC) at the Massachusetts Institute of Technology, and animal procedures were performed following the NIH guidelines.
Cell Culture, Media, and Reagents
R1 ESCs were grown on gelatinized dishes in regular DMEM containing 15% ES-grade fetal bovine serum (FBS), 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 0.1 mM β-mercaptoethanol, 1× penicillin/streptomycin antibiotics, and 1,000 U/mL LIF. For high cell-stress events such as single-cell sorting, ESCs were grown on DR4 MEF feeder layers. Differentiation was induced by growing cells in ESC medium with 10% FBS, without LIF and in the presence of RA (1 μM).
Vectors, Retroviral Preparation, and Infection
The CD-tagging vectors pBABEpuro-YFP (V1, V2, and V3 variants encoding YFP in three reading frames) and pBABEpuro-Cherry (V1, V2, and V3 variants) were previously reported (Sigal et al., 2007). Retrovirus for infection was prepared by transfection of V1, V2, and V3 vectors in equal amounts into Phoenix cells using Lipofectamine 2000 (Invitrogen). One day after transfection, the medium was changed to ESC medium and the cells were incubated for 24 hr. Virus-enriched ESC medium was harvested and used immediately for infection. A day before infection ESCs were plated on DR4 MEF-coated 6-well plates (105 cells per well). To prevent multiple integrations, we diluted viral stock to less than one viral particle per cell. Polybrene (4 μg/mL) was used to increase the viral particle attachment to cells. On day 0, ESCs were infected by spinfection to increase the efficiency of viral attachment and infection. In brief, 6-well ESC plates with viral solution were sealed and spun at 2,000 rpm for 45 min at room temperature. Immediately after the spin, plates were incubated at 37°C with 5% CO2 for 12–16 hr. On day 1, medium containing viral particles was replaced with fresh medium and on day 2, puromycin (1.5 μg/mL) was added for selection. This step enriches for the infected cells and reduces the number of cells to be sorted. The cells were grown for an additional day or two depending on the confluence.
Single-Cell FACS Sorting
Cells were trypsinized to prepare single-cell suspension. Trypsin was neutralized by diluting the cells in Ca2+- and Mg2+-free PBS and washed with PBS once. For FACS sorting, cell number was adjusted by diluting the cells in 1 mL of PBS with 0.5% FBS per 10% confluence. To remove any cell clumps, we passed cells through a 70-μm cell strainer (BD Falcon). Cells were placed on ice until sorting. Feeder MEF plates were incubated with ESC medium to condition the medium prior to sorting. Conditioned medium was diluted 1:1 with regular ESC medium. Sorting was performed on a BD FACSAria III cell sorter at 4°C. Uninfected ESCs were used as negative controls. GFP- or Cherry-expressing ESCs were used as positive controls to set the sorting gate parameters. Immediately after sorting, plates were incubated with conditioned medium at 37°C with 5% CO2. Medium was changed on day 2 after sorting and cells were grown until they formed colonies big enough for trypsinization, expansion, and RNA extraction.
Identification of FL-Tagged Clones by 3′-RACE
RNA was extracted using either an RNAeasy kit (Qiagen) or a ZR-96 Quick RNA plate (Zymo Research). 3′-RACE reaction was performed to identify the clones as described in Sigal et al. (2007), with a few modifications. Extracted RNA was used for cDNA preparation using a high-capacity cDNA reverse transcription kit (Applied Biosystems). For the GFP library, AP first-strand primer (5′-GGC CAC GCG TCG ACT AGT AC(T)17-3′) was used for cDNA reactions. The cDNA was used in a nested PCR reaction (DreamTaq PCR master mix, Thermo Scientific). YFP-90 (5′-GCA GAA GAA CGG CAT CAA GG-3′) and AP-92 (5′-GGC CAC GCG TCG ACT AGT AC-3′) primers were used to amplify the first PCR reaction, and the product was used as a template in a second nested PCR reaction with YFP-85 (5′-CGC GAT CAC ATG GTC CTG CTG-3′) and AP-92 primers. The product was run on an agarose gel to purify the amplified DNA product. First PCR and second PCR products from each clone were run side by side on an agarose gel and correct-size bands were excised. DNA was purified using a Hi Yield Gel/PCR purification kit or a 96-well PCR clean-up kit (RBC Biosciences). Purified nested DNA was directly sequenced with YFP-85 primer. For the Cherry library, AP first strand (cDNA), Cherry 45 (5′-GTG GTG ACC GTG ACC CAG GA-3′) and AP-92 (first PCR), Cherry 46 (5′-GCG GAT GTA CCC CGA GGA CG-3′) and AP-92 (Second PCR), and Cherry 56 (5′-GAC TAC ACC ATC GTG GAA CA-3′) (sequencing) were used.
Author Contributions
A.H., R.R.E., and E.M. designed the study; M.S. performed heterogeneity studies and analyses; G.K.A. performed validation experiments; S.M. and R.J. performed chimera experiments; P.S., S.L., and E.B. performed DNA damage experiments; S.F.-A. and U.A. helped with initial experiments and contributed clones to the library; R.R.E., A.H., and E.M. wrote the manuscript.
Acknowledgments
This work was supported by the Israel Science Foundation (ISF 1140/2017 to E.M. and ISF 1349/15 to U.A.); the European Research Council (to E.M.); the European Union's Horizon 2020 research and innovation program FET-OPEN (CellViewer, no. 686637); and NIH grants HD045022, R37-CA084198, and R01 NS088538-01 (to R.J.). E.M. is the incumbent of the Arthur Gutterman Professor Chair. U.A. is the incumbent of the Abisch-Frenkel Professor Chair. R.R.E. and A.H. were supported by the Marie Curie Nucleosome4D network; R.R.E. was supported by a post-doctoral fellowship from the Lady Davis Foundation. The authors thank Dan Lehman for help with single-cell sorting, as well as Bill Breuer, Tamar Danon, Mayan Sheiba, David Gokhman, Naveh Evantal, and Lilya Verchovsky for technical assistance. We dedicate this paper to the memory of Lilya Verchovsky.
Published: September 28, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, three figures, one table, and two movies and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.08.022.
Supplemental Information
GO terms are presented in an additional sheet.
TopoI clone undergoing differentiation for 48 hr in the presence of RA. Note that fluorescence levels are constant throughout the differentiation process. Imaging was performed with a 60× oil objective.
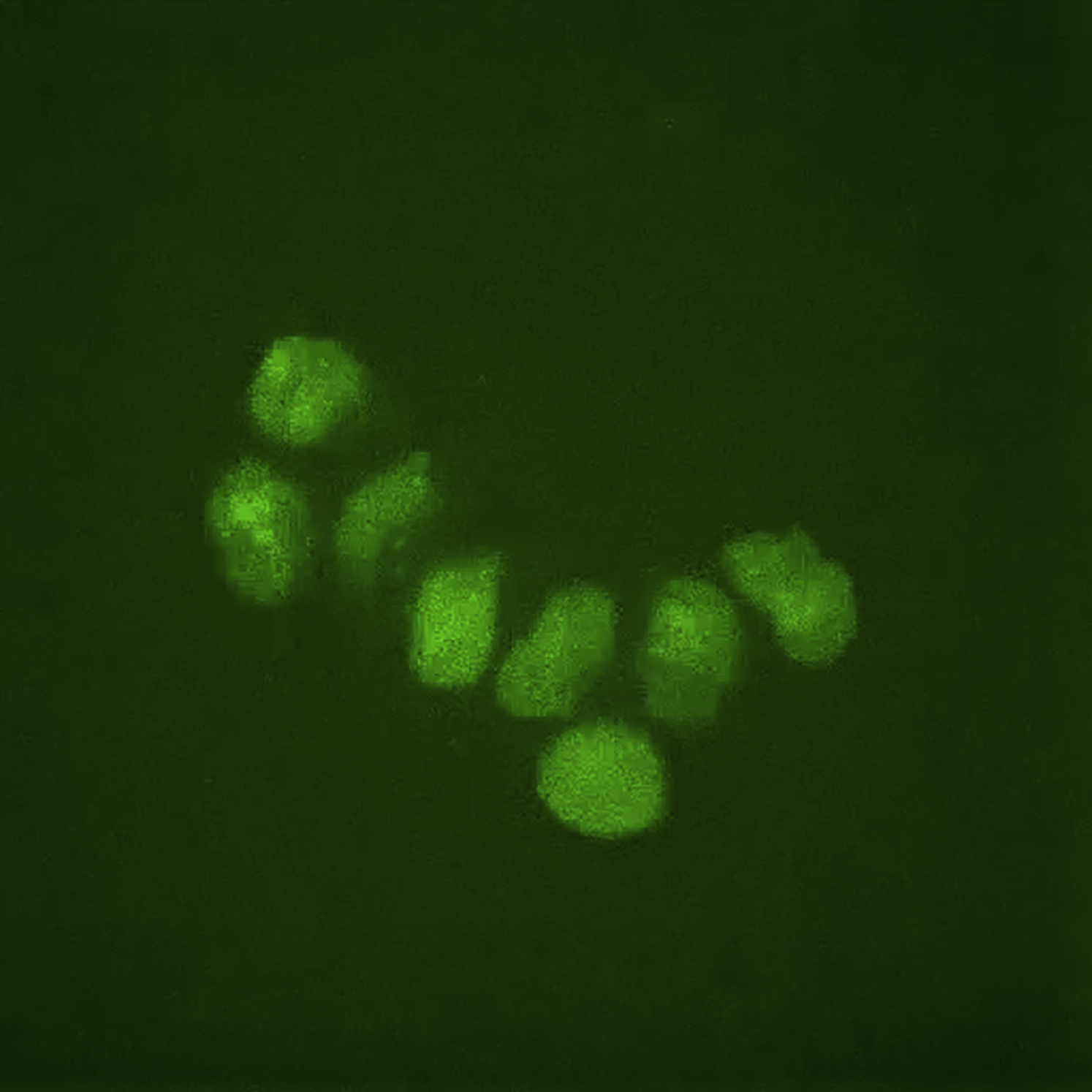{kind=link}
TOPOI used as a background clone (nuclear, green) to monitor the dynamics of a mitochondrial localized Cherry-tagged C1qbp protein (red). Imaging was performed with a 60× oil objective.
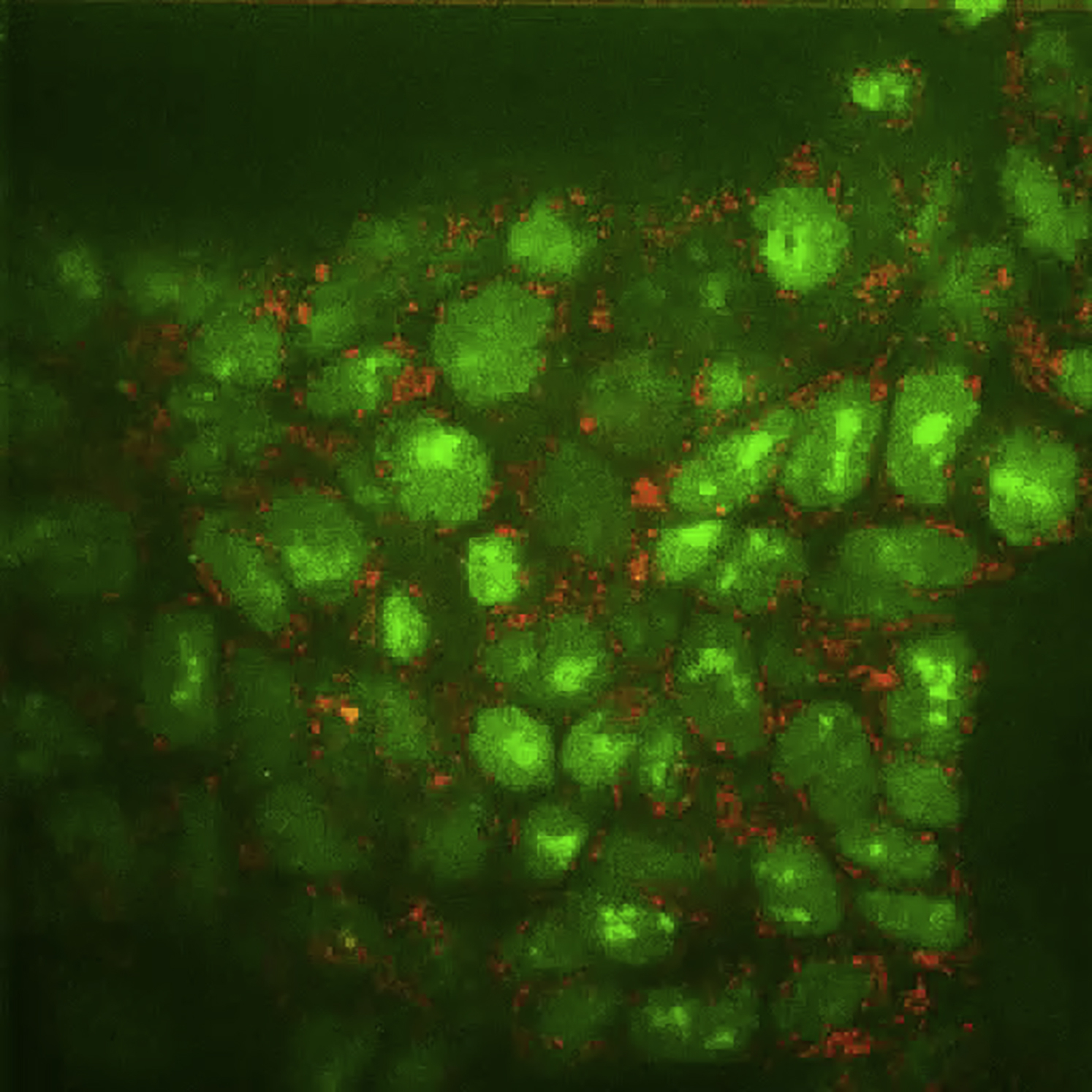{kind=link}
References
- Ayoub N., Jeyasekharan A.D., Bernal J.A., Venkitaraman A.R. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- Brennan J., Skarnes W.C. Gene trapping in mouse embryonic stem cells. Methods Mol. Biol. 1999;97:123–138. doi: 10.1385/1-59259-270-8:123. [DOI] [PubMed] [Google Scholar]
- Cahan P., Daley G.Q. Origins and implications of pluripotent stem cell variability and heterogeneity. Nat. Rev. Mol. Cell Biol. 2013;14:357–368. doi: 10.1038/nrm3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheutin T., McNairn A.J., Jenuwein T., Gilbert D.M., Singh P.B., Misteli T. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 2003;299:721–725. doi: 10.1126/science.1078572. [DOI] [PubMed] [Google Scholar]
- Cohen A.A., Geva-Zatorsky N., Eden E., Frenkel-Morgenstern M., Issaeva I., Sigal A., Milo R., Cohen-Saidon C., Liron Y., Kam Z. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–1516. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- Cohen A.A., Kalisky T., Mayo A., Geva-Zatorsky N., Danon T., Issaeva I., Kopito R.B., Perzov N., Milo R., Sigal A. Protein dynamics in individual human cells: experiment and theory. PLoS One. 2009;4:e4901. doi: 10.1371/journal.pone.0004901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E., Geva-Zatorsky N., Issaeva I., Cohen A., Dekel E., Danon T., Cohen L., Mayo A., Alon U. Proteome half-life dynamics in living human cells. Science. 2011;331:764–768. doi: 10.1126/science.1199784. [DOI] [PubMed] [Google Scholar]
- Edupuganti R.R., Harikumar A., Aaronson Y., Biran A., Sailaja B.S., Nissim-Rafinia M., Azad G.K., Cohen M.M., Park J.E., Shivalila C.S. Alternative SET/TAFI promoters regulate embryonic stem cell differentiation. Stem Cell Reports. 2017;9:1291–1303. doi: 10.1016/j.stemcr.2017.08.021. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni S., Duttagupta R., Cheng J., Dehghani H., Hoeppner D.J., Dash C., Bazett-Jones D.P., Le Grice S., McKay R.D., Buetow K.H. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2:437–447. doi: 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festenstein R., Pagakis S.N., Hiragami K., Lyon D., Verreault A., Sekkali B., Kioussis D. Modulation of heterochromatin protein 1 dynamics in primary mammalian cells. Science. 2003;299:719–721. doi: 10.1126/science.1078694. [DOI] [PubMed] [Google Scholar]
- Geva-Zatorsky N., Dekel E., Cohen A.A., Danon T., Cohen L., Alon U. Protein dynamics in drug combinations: a linear superposition of individual-drug responses. Cell. 2010;140:643–651. doi: 10.1016/j.cell.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Hashimshony T., Wagner F., Sher N., Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Hayashi K., Lopes S.M., Tang F., Surani M.A. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell. 2008;3:391–401. doi: 10.1016/j.stem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Wang H., Kiani S., Lai C.S., Gao Q., Cassady J.P., Cost G.J., Zhang L., Santiago Y., Miller J.C. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins J.R., Toyoda Y., Hegemann B., Poser I., Heriche J.K., Sykora M.M., Augsburg M., Hudecz O., Buschhorn B.A., Bulkescher J. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science. 2010;328:593–599. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvik J.W., Adler S.A., Telmer C.A., Subramaniam V., Lopez A.J. CD-tagging: a new approach to gene and protein discovery and analysis. Biotechniques. 1996;20:896–904. doi: 10.2144/96205rr03. [DOI] [PubMed] [Google Scholar]
- Jarvik J.W., Fisher G.W., Shi C., Hennen L., Hauser C., Adler S., Berget P.B. In vivo functional proteomics: mammalian genome annotation using CD-tagging. Biotechniques. 2002;33:852–854. doi: 10.2144/02334rr02. 856, 858-860 passim. [DOI] [PubMed] [Google Scholar]
- Kalmar T., Lim C., Hayward P., Munoz-Descalzo S., Nichols J., Garcia-Ojalvo J., Martinez Arias A. Regulated fluctuations in nanog expression mediate cell fate decisions in embryonic stem cells. PLoS Biol. 2009;7:e1000149. doi: 10.1371/journal.pbio.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalousi A., Hoffbeck A.S., Selemenakis P.N., Pinder J., Savage K.I., Khanna K.K., Brino L., Dellaire G., Gorgoulis V.G., Soutoglou E. The nuclear oncogene SET controls DNA repair by KAP1 and HP1 retention to chromatin. Cell Rep. 2015;11:149–163. doi: 10.1016/j.celrep.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Kedersha N., Panas M.D., Achorn C.A., Lyons S., Tisdale S., Hickman T., Thomas M., Lieberman J., McInerney G.M., Ivanov P. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 2016;212:845–860. doi: 10.1083/jcb.201508028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnertz B., Ueda Y., Derijck A.A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A.H. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- Melcer S., Hezroni H., Rand E., Nissim-Rafinia M., Skoultchi A., Stewart C.L., Bustin M., Meshorer E. Histone modifications and lamin A regulate chromatin protein dynamics in early embryonic stem cell differentiation. Nat. Commun. 2012;3:910. doi: 10.1038/ncomms1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E. Harvard Stem Cell Institute; Cambridge, MA: 2008. Imaging chromatin in embryonic stem cells. In StemBook. [PubMed] [Google Scholar]
- Meshorer E., Yellajoshula D., George E., Scambler P.J., Brown D.T., Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.C., Holmes M.C., Wang J., Guschin D.Y., Lee Y.L., Rupniewski I., Beausejour C.M., Waite A.J., Wang N.S., Kim K.A. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Sasagawa Y., Nikaido I., Hayashi T., Danno H., Uno K.D., Imai T., Ueda H.R. Quartz-Seq: a highly reproducible and sensitive single-cell RNA-Seq reveals non-genetic gene expression heterogeneity. Genome Biol. 2013;14:R31. doi: 10.1186/gb-2013-14-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal A., Danon T., Cohen A., Milo R., Geva-Zatorsky N., Lustig G., Liron Y., Alon U., Perzov N. Generation of a fluorescently labeled endogenous protein library in living human cells. Nat. Protoc. 2007;2:1515–1527. doi: 10.1038/nprot.2007.197. [DOI] [PubMed] [Google Scholar]
- Sigal A., Milo R., Cohen A., Geva-Zatorsky N., Klein Y., Alaluf I., Swerdlin N., Perzov N., Danon T., Liron Y. Dynamic proteomics in individual human cells uncovers widespread cell-cycle dependence of nuclear proteins. Nat. Methods. 2006;3:525–531. doi: 10.1038/nmeth892. [DOI] [PubMed] [Google Scholar]
- Singh A.M., Hamazaki T., Hankowski K.E., Terada N. A heterogeneous expression pattern for Nanog in embryonic stem cells. Stem Cells. 2007;25:2534–2542. doi: 10.1634/stemcells.2007-0126. [DOI] [PubMed] [Google Scholar]
- Tang F., Barbacioru C., Bao S., Lee C., Nordman E., Wang X., Lao K., Surani M.A. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell. 2010;6:468–478. doi: 10.1016/j.stem.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka Y., Shimosato D., Murakami K., Takahashi K., Niwa H. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development. 2008;135:909–918. doi: 10.1242/dev.017400. [DOI] [PubMed] [Google Scholar]
- Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalzman M., Falco G., Sharova L.V., Nishiyama A., Thomas M., Lee S.L., Stagg C.A., Hoang H.G., Yang H.T., Indig F.E. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature. 2010;464:858–863. doi: 10.1038/nature08882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GO terms are presented in an additional sheet.
TopoI clone undergoing differentiation for 48 hr in the presence of RA. Note that fluorescence levels are constant throughout the differentiation process. Imaging was performed with a 60× oil objective.
TOPOI used as a background clone (nuclear, green) to monitor the dynamics of a mitochondrial localized Cherry-tagged C1qbp protein (red). Imaging was performed with a 60× oil objective.