

Abstract

Nuclear resonance vibrational spectroscopy (NRVS; also known as nuclear inelastic scattering, NIS) is a synchrotron-based method that reveals the full spectrum of vibrational dynamics for Mössbauer nuclei. Another major advantage, in addition to its completeness (no arbitrary optical selection rules), is the unique selectivity of NRVS. The basics of this recently developed technique are first introduced with descriptions of the experimental requirements and data analysis including the details of mode assignments. We discuss the use of NRVS to probe 57Fe at the center of heme and heme protein derivatives yielding the vibrational density of states for the iron. The application to derivatives with diatomic ligands (O2, NO, CO, CN–) shows the strong capabilities of identifying mode character. The availability of the complete vibrational spectrum of iron allows the identification of modes not available by other techniques. This permits the correlation of frequency with other physical properties. A significant example is the correlation we find between the Fe–Im stretch in six-coordinate Fe(XO) hemes and the trans Fe–N(Im) bond distance, not possible previously. NRVS also provides uniquely quantitative insight into the dynamics of the iron. For example, it provides a model-independent means of characterizing the strength of iron coordination. Prediction of the temperature-dependent mean-squared displacement from NRVS measurements yields a vibrational “baseline” for Fe dynamics that can be compared with results from techniques that probe longer time scales to yield quantitative insights into additional dynamical processes.
1. Introduction
Vibrational spectroscopy methods have had a long tradition in the study of heme proteins and metalloporphyrins that range from the simple detection of a ligand vibration to the use of femtosecond spectroscopy for the elucidation of reaction processes. However, selection rules and/or a lack of selectivity can limit the utility of vibrational spectroscopy for the study of hemes and heme proteins, preventing the observation of many important vibrations. In this review we discuss how the application of the vibrational technique nuclear resonance vibrational spectroscopy (NRVS) circumvents these problems and moreover provides substantial additional information useful for understanding the molecular dynamics of systems containing an iron porphyrin or heme derivative. The studies detailed here, although restricted to systems containing a heme, provide a rather complete overview of the capability of NRVS applications that can be readily applied to other iron-containing materials. The technique is also sometimes called NRIXS (nuclear resonance inelastic X-ray scattering) or NIS (nuclear inelastic scattering).
NRVS is a synchrotron-based vibrational spectroscopy technique with unique selectivity for vibrations involving displacement of Mössbauer-active nuclei; these include 83Kr, 119Sn, 157Eu, 161Dy, 121Sb, 125Te, and 57Fe.1−14 To date, successful measurements utilize nuclear resonances with transition energies below 70 keV, because source intensity and detector efficiency both decrease with increasing photon energy and with excited state lifetimes longer than 0.6 ns, because of limited detector time resolution. The left panel of Figure 1 displays isotopes with nuclear resonances for which experimental capabilities currently exist, and NRVS measurements have been reported for those indicated by filled symbols. Because it meets these criteria as well as having a large absorption cross section, the 14.4125 keV resonance of 57Fe is especially favorable for NRVS, a fortunate circumstance given the widespread occurrence of iron compounds in both earth science and biological systems. There is a high incidence of iron-containing proteins, both with known and with unknown structures and functions. Figure 1 (right) displays relative abundances of metal ions that were assimilated by the proteome of the microbe Pyrococcus furiosus.15
Figure 1.

NRVS measurements on hemes exploit a fortuitous coincidence: Iron plays a prominent role in cellular metalloproteins, and the nuclear properties of the 57Fe isotope are particularly favorable for NRVS. Experimental facilities have been developed at one or more synchrotron light sources to allow measurements of the nuclear resonances plotted on the left as a function of photon energy and excited state lifetime, with areas proportional to the absorption cross section. Circles filled in red (for 57Fe) or cyan indicate resonances for which NRVS measurements have been reported. Data obtained from refs (12) and (14). Relative abundance of metalloproteins containing metals other than iron and zinc is less than 3% in P. furiosus, one of a few microorganisms for which the metalloproteome is well characterized (data reported in ref (15)).
The NRVS phenomenon results from a change in vibrational state coincident with excitation of the nucleus by an X-ray photon. This contrasts with the classical Mössbauer effect, which results from the absorption of a photon to excite the ground state nucleus to an excited nuclear state without a change in the vibrational state, thus achieving a recoil-free absorption of a photon with energy exactly equal to the energy difference between the nuclear ground state and the excited state. When the nucleus is in a bound state, the vibrational energy can only change by discrete amounts determined by normal mode frequencies. Under these circumstances, the absorption spectrum includes peaks displaced from the recoilless Mössbauer resonance by energies equal to the vibrational quanta. Although this phenomenon was predicted16,17 shortly after the original Mössbauer report,18 the practical observation of vibrationally shifted nuclear absorption required intense X-ray beams as described below. Materials containing the isotope 57Fe have yielded the majority of NRVS results to date, and of course, iron is at the center of hemes and heme proteins as well as many other biologically important centers. See, for example, the report by Cramer and co-workers on the hydride bridge in [NiFe]-hydrogenase for a significant nonheme NRVS result.19
The iron NRVS experiment selectively yields the complete set of modes that involves motion of the iron atom. The method has a selectivity that is reminiscent of that of resonance Raman spectroscopy, but again note the significant advantage that NRVS is not subject to the electromagnetic selection rules of Raman or infrared spectroscopy. Indeed, NRVS provides the ultimate limit in selectivity because only the vibrational dynamics of the probe nucleus contribute to the observed signal. Importantly, the NRVS intensity is directly related to the magnitude and direction of the motion; hence, the method has a unique quantitative component in the measured vibrational spectrum.
2. NRVS Basics
In this section we outline the basics of NRVS. We describe the instrumentation required, sample essentials and data collection, analysis, and a brief outline of the vibrational dynamics of iron in the porphyrin plane.
2.1. Experimental Issues—Instrumentation
Specialized synchrotron beamlines and end station instrumentation are required to carry out NRVS studies, with a tunable ultra-high-resolution monochromator providing an extremely narrow X-ray beam with energy resolution on the order of 1 meV or 7–8 cm–1. There are now four sites available for use located at APS (Argonne National Lab, Chicago, IL, USA), Petra III (Hamburg, Germany), SPring8 (Riken, Japan), and ESRF (Grenoble, France). All are user facilities available for use by the general scientific community. A nuclear resonance program including NRVS is also proposed for the new synchrotron facility to be constructed near Beijing.
In addition to the ultra-high-resolution monochromator, other operational requirements include appropriate timing electronics to discriminate the prompt elastic scattering signal (∼107 times larger than the NRVS signals) from the desired nuclear resonance signals, which arrive at delays distributed throughout the lifetime of the nuclear excited state (141 ns for 57Fe). The basic experimental setup is depicted schematically in Figure 2. In that diagram, the X-ray pulses are separated by 150 ns with a peak width of less than 150 ps that are characteristic of the standard timing mode at the APS ring. Further details on instrumentation can be found in two earlier reviews.20,21
Figure 2.

Schematic diagram illustrating the experimental setup for the NRVS experiment including the high-resolution monochromator. Synchrotron beam is run in pulse mode, and avalanche photodiode, APD, is disabled during the X-ray pulse (shown bottom left) in order to gate out elastically scattered 14.4 keV photons, so that only delayed photons corresponding to nuclear absorption are detected. Lateral APD detectors measure nuclear absorption, including the vibrational signal, by detecting 6.4 and 14.4 keV fluorescence emitted by excited 57Fe nuclei, while the coherent forward-scattered 14.4 keV photons reaching the second APD provide a real-time measure of the experimental resolution function. Photo at the left is the Advanced Photon Source at Argonne National Lab. Photo courtesy of Argonne National Laboratory.
2.2. Experimental Issues—Samples and Data Collection
Experimental samples can be either oriented (crystalline) or unoriented (powders or solution). The NRVS signal selectively derives from the Mössbauer active nucleus 57Fe, and samples are generally enriched to about 95% 57Fe (2.14% natural abundance) to provide an adequate signal. The 57Fe labeling necessity conveniently allows for the possibility of site selection in a multi-iron sample. The maximum utilized sample size is constrained by the size of the incident beam arriving from the monochromator and will be somewhat dependent on the measurement site; however, smaller sample sizes at all lines have become the norm. For optimal signal, randomly oriented samples are measured at grazing incidence in a channel wider than the beam width and long enough to utilize the full detector area. Typical sample volumes are currently 20 μL at the APS. The crystal size for single-crystal measurements is now at the size required for normal X-ray diffraction studies, ideally about 0.3 mm on a side.
Since the NRVS measurement is a bulk measurement, it is highly desirable that the sample be pure, because unknown molecular populations confound the quantitative extraction of atomic displacements, which is a unique feature of NRVS.22 Typically, the measurement is from −20 to −40 meV below the resonance energy to from 80 to 100 meV above the resonant energy and is dependent on the particular sample being analyzed. The energy sampling interval, typically 0.25 meV, needs to be smaller than the resolution of the monochromator, 1.0 meV. With a measuring time of 5 s per step, a full energy scan requires a bit less than 1 h. Several scans are typically measured and added, with the measurement time for a small molecule taking as little as 2–3 h, whereas a protein sample requires 1 day or more. Subtraction of the resolution function, as measured from the resonant forward scattering, yields the pure vibrational component of the signal.
As our experimental and spectral analysis experience developed, the importance of oriented single-crystal measurements became more evident. Whereas a powder measurement does include a signal for every Fe vibrational mode, this richness of information will likely include many overlapped peaks. Improved resolution and identification of the peaks can be obtained through the use of oriented single-crystal measurements that require specific crystallographic conditions described below. The power of oriented single-crystal measurements comes from the fact that NRVS intensity is proportional to the mean square displacement of iron motion along the direction of the exciting incident X-ray beam. Thus, if a crystal of a porphyrin derivative can be oriented so that all porphyrin planes are perpendicular to the beam, only modes in which there is nonzero iron motion projected on the heme normal will have intensity. Consequently, the out-of-plane modes will be clearly defined. A second measurement with the heme normal perpendicular to the X-ray beam will capture in-plane modes. However, since the exciting beam is typically at some arbitrary position in the porphyrin plane(s) not all peaks will be at their maximum intensity. Indeed, there is the possibility that a peak might be entirely missing. This problem will be minimized by comparison with a powder spectrum. We have termed this in-plane measurement as the generalized in-plane measurement.
Maximum information can be obtained in the case of solid-state derivatives crystallizing in the triclinic crystal system. In that crystal system two properly chosen orthogonal in-plane measurements will capture all in-plane intensity. We have termed this in-plane measurement as the special in-plane measurement. When such measurements have been made, the beam directions chosen are usually parallel and perpendicular to an axial ligand plane and thus provide additional information on the bonding effects of the axial ligand.
2.3. Experimental Issues—Data Analysis
Complete details of the analysis of NRVS data within the harmonic approximation have been given in refs (21) and (23). This ultimately provides a partial vibrational density of states (VDOS) specific for the probe nucleus. Briefly, the measured signal can be normalized using the sum rules according to Lipkin.24 The program PHOENIX25 is used to calculate of the VDOS from the resulting excitation probability. The program requires the summed sample data and the experimental resolution function appropriate to the measured spectrum and estimates of the sample temperature and background. The resolution function must be subtracted from summed sample data. The sample temperature is usually iterated to obtain a Boltzmann fit to the intensity of a feature observed at both positive and negative frequencies and should be more accurate than the probe temperature.
The peak areas in the output VDOS quantitatively provides the mode composition factor eFe2. The physical significance of eFe for any mode is that it gives the fraction of the kinetic energy associated with the motion of the iron in the mode and that it is proportional to the contribution of the mode to the mean square displacement of the iron atom. The experiment thus provides quantitative information in both frequency and amplitudes of the Fe, which can be compared with predicted values, particularly from DFT calculations.
2.4. General Experimental Results
The NRVS experiment provides a signal for every mode in which there is motion of the iron; all other modes are silent. Thus, the very selective NRVS signal is richer in details about the iron target center than that obtained from either IR or resonance Raman spectroscopy. Accompanying this obvious signal enhancement is a substantial challenge. The challenge is the necessity of making detailed vibrational assignments, the difficulty of which is exacerbated by significant mode mixing. Unlike the vibrations typically used for defining ligand binding, such as a carbonyl stretch, the modes involving iron are found at much lower frequencies and consequently are mixed with porphyrin ligand modes.
Spectra from heme samples typically encompass three main categories with all frequencies usually occurring below ∼600 cm–1. The highest frequency modes are typically the vibrational modes involving iron and the axial ligands (above about 400 cm–1), the midrange modes are primarily in plane, and the lowest frequency modes (below about 200 cm–1) are principally out-of-plane modes including the difficult to observe doming mode. The lowest frequency modes, typically broad and below about 50 cm–1, are phonon modes resulting from heme translation in the solid. Although this division is typically found (especially for those systems with the diatomic ligands), spectral overlaps are frequent possibilities that must be resolved in order to provide detailed assignments. Additional weak multiquantum features contribute to the unprocessed experimental data26 but are removed in the process of extracting the VDOS.
2.5. Spectral Analysis Procedures—Mode Assignment
A crucial challenge is the assignment of the iron mode for each observed frequency in the iron partial VDOS. Although the use of isotope labeling has real value, the utility of labeling is usually limited to a subset of the spectral features and that does not necessarily include all biologically important modes, especially low-frequency modes. Is there a global approach to the assignment of all observed modes in the NRVS spectrum?
In principle, a complete set of calculated mode assignments can be obtained either by an empirical approach (normal mode or coordinate analysis) or by de novo calculations (density functional theory). Empirical normal mode calculations are intrinsically underdetermined; however, two features of NRVS data do provide constraints in the fitting of the spectra and provide some confidence in the reliability of the final results. An important feature of NRVS data is that not only are all frequencies of the iron modes observed but also that the iron amplitudes of each mode are known quantitatively. Single-crystal measurements allow the unambiguous identification of many modes as having either in-plane or out-of-plane character. The approach is the application of normal mode calculations with adjustment of the force field to achieve better fits of the observed VDOS from the NRVS measurement. Normal mode calculations for [Fe(TPP)(NO)]27 were obtained by refining a starting set of Wilson-type force fields28 based on earlier work of Li et al.29 and Rush et al.30 The procedure has been described in more detail in ref (27).
This study clearly demonstrated that the modes observed are mixed and, perhaps surprisingly, that the peripheral substituents have substantial effects on the spectra particularly in the midrange (in-plane) modes. However, this calculation suggested a mode assignment with major contributions from the FeNO bend that later work demonstrated was mostly a porphyrin mode. Additional analyses were obtained for a five-coordinate high-spin iron(II) complex, [Fe(TPP)(2-MeHIm)], which gave an assignment for the Fe–N(Im) stretch.31 Assignments were also made for the six-coordinate derivative [Fe(TPP)(1-MeIm)(CO)], particularly for the Fe–N(Im) stretch that is not observable in resonance Raman spectroscopy.32 Normal mode analysis of the powder NRVS data of four-coordinate [Fe(OEP)] stressed the importance of utilizing the true point group symmetry rather than an idealized higher symmetry as the iron normal modes showed sensitivity to structural details.33
Nonetheless, the method is intrinsically underdetermined even when intermolecular interactions are considered,34 and the possibility of misassignments remains a concern. The complexes described above have been followed up with additional studies with DFT predictions and are considered in the appropriate subsequent sections.
We believe that the better answer to this assignment challenge has been met by a combination of theoretical and experimental approaches. The theoretical aspect of the analysis is the use of DFT calculations to predict both the nature and the intensity of the observed spectral features. However, the experience with DFT predictions shows that caution must be used in its application. The complete molecule must be used in the calculations, many features of the vibrational spectrum are strongly dependent on the peripheral groups of the porphyrin. The quality of the predictions are often found to depend crucially on the functional chosen for the calculation.35 Unlike predictions for the energy-minimized structure, which seem much less dependent on the computational details for satisfactory predictions of the experimental results, spectroscopic predictions are much more dependent on the computational details. The most successful approach for a complete assignment of the vibrational spectrum obtained from a NRVS experiment appears to a semiempirical one in which the choice of DFT functional is guided by the experimental results. Experimental single-crystal measurements are of particular utility since these provide information on both the intensity and the character, unlike powder measurements which provide information on intensity alone. The single-crystal measurement is especially powerful because the intensity of the NRVS signal is proportional to the projection of iron motion on the exciting X-ray beam. Thus, careful mounting and orientation using an (offline) diffractometer allows the in-plane and out-of-plane modes to be readily distinguished by a 90° goniometer rotation and provides substantial constraints on the theoretical predictions. This combined approach of calculations and experimental single-crystal measurements are emphasized in many of the results described below. Predicted frequencies for the out-of-plane modes involving the diatomic axial ligand(s) sometimes exhibit considerable variation with computational details such as the functional chosen for a DFT calculation; less difficulty is experienced for the in-plane modes.
2.6. Vibrational Dynamics of Fe in the Porphyrin Core
Porphyrin vibrations are commonly described using the nomenclature originally established36 for the vibrational normal modes of nickel porphine [Ni(P)] and later adapted for [Fe(P)].37 The porphine molecule has D4h symmetry, and Fe motion contributes only to a few modes of [Fe(P)] having either Eu or A2u symmetry. Figures 3 and 4 illustrate these normal modes.
Figure 3.
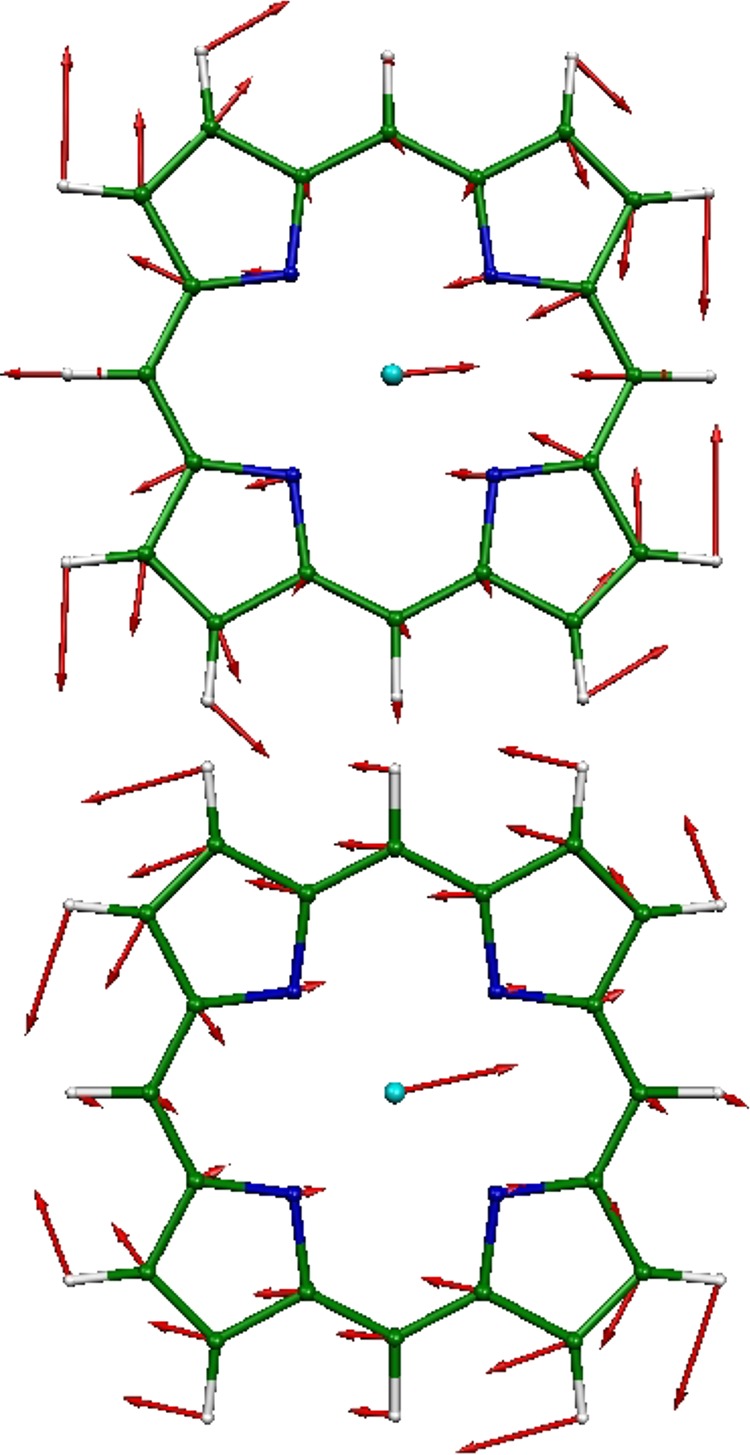
ν50 (top) and ν53 (bottom) modes of Eu symmetry comprise the predicted vibrational dynamics of iron parallel to the porphyrin plane for the ground state of [Fe(P)]. In this and subsequent figures, each arrow is 100(mj/mFe)1/2 times longer than the zero-point vibrational amplitude of atom j. This and all subsequent illustrations of vibrational modes were rendered with the program MOLEKEL.39
Figure 4.

From top to bottom, respectively, γ9, γ6, and γ7 modes predicted for the ground state of [Fe(P)].
In particular, two degenerate pairs of Eu modes, ν50 and ν53 (Figure 3), dominate Fe dynamics parallel to the porphine plane. The ν53 modes involve concerted stretching/compression of the Fe–N(pyrrole) bonds, coupled to rotation of the pyrrole rings in the plane to follow the motion of the iron. The same motions contribute to the ν50 modes, except that the relative phase has reversed. Notice that the Fe does not move in the ν8 mode (of A1g symmetry), which involves in-phase stretching of all four Fe–N(pyrrole) bonds, and thus, this mode does not contribute to the NRVS signal, although it is often observed in resonance Raman.
Three A2u modes (Figure 4) involve significant Fe motion perpendicular to the plane of the heme. γ9 is a “doming”' mode, where the four pyrrole rings swivel about the methine bridges to track the displacement of the Fe from the mean plane of the molecule. Reversal of the relative phase of the Fe and ligand motions characterizes the higher frequency γ6 mode, which has sometimes been described as “inverse doming”.37 Out-of-plane displacement of the methine hydrogens characterizes a third A2u mode, γ7, which involves minimal Fe motion for [Fe(P)], but can become relevant in more complex porphyrins.
To what extent do these porphine core vibrations remain useful descriptions for the vibrational dynamics of more complex porphyrins, where the addition of axial ligands and side chains reduces molecular symmetry and introduces additional vibrational degrees of freedom? We explored this question using NRVS measurements on a series of porphyrin halides, supported by quantitative projection of calculated normal modes onto the vibrational basis of the simpler [Fe(P)] molecule.38
Vibrational measurements on a series of porphyrin halides, supported by vibrational projection of calculations onto the simpler porphine molecule, explored the contribution of these porphine core vibrations to Fe dynamics. The results reveal that the character of these vibrational modes persists in the less symmetric molecules.
Binding of a monatomic halide ligand (L = Cl, Br) reduces the molecular symmetry to C4v. Inspection of the vibrations predicted by DFT calculations shows that the additional degree of freedom corresponding to halide translation blends with the γ6 and γ7 modes to produce the Fe–L stretching mode, a pattern that recurs in more complex porphyrins. A large observed frequency shift clearly identifies the Fe–Cl and Fe–Br stretching vibrations in the experimental VDOS of [Fe(P)(Cl)] and [Fe(P)(Br)] (See Figure 5). The doming mode (γ9) is very broad, perhaps because of vibrational coupling with other molecules in the crystal. This effect as well as low-frequency acoustic lattice vibrations are not captured by the calculations, which are performed on an isolated molecule. The dominant in-plane contribution is ν53, observed at 260 cm–1 in Figure 5. Secondary contributions to the in-plane vibrational dynamics of the Fe result not only from ν50 but also from γ23, which couples with the ν53 modes, because of the reduced C4v symmetry of the halide complexes.
Figure 5.

Experimental vibrational densities of states for [Fe(P)(Cl)] and [Fe(P)(Br)].
The presence of side chains in more complex porphyrins further reduces molecular symmetry and introduces additional vibrational degrees of freedom.38 As might be expected, the frequency of γ9 is sensitive to the nature of the side chains, while other effects turn out to be difficult to anticipate. On one hand, [Fe(OEP)(Cl)] exhibits a broad feature extending from 220 to 300 cm–1, and correlation analysis of the vibrational calculations indicates multiple unresolved vibrations in this region that overlap with the ν53 vibration of [Fe(P)(Cl)]. In fortuitous contrast, a relatively sharp feature attributable to ν53 dominates the in-plane contribution to the VDOS of [Fe(PPIX)(Cl)], the porphyrin which holds the greatest biological relevance, and the ν50 contribution is unexpectedly suppressed. The experimental Fe–Cl stretching band varies little among [Fe(P)(Cl)], [Fe(OEP)(Cl)], and [Fe(PPIX)(Cl)], although calculations again suggest unresolved complexity for [Fe(OEP)(Cl)].
In spite of structural variations in complex porphyrins, fundamental vibrations of the porphyrin core recur repeatedly in all of the porphyrins studied to date. The Fe–Np vibrations ν53 and ν50 form the main contribution to the in-plane vibrational dynamics of the Fe, while some mixture of γ6 and γ7 contributes to the stretching of the bond to the axial ligand, and lower frequency Fe vibrations have doming (γ9) character. The frequencies of these vibrations probe structural variations as well among the porphyrins investigated as well as the effects of axial ligands.
3. Diatomic Ligand Systems
A substantial portion of the heme NRVS effort has been the characterization of hemes or heme proteins with the three biologically significant gases O2, NO, and CO. The cyanide anion, CN–, is also included in this group of diatomic ligands. The biological function of O2 in aerobic life is evident, but other diatomic molecules are also substantial actors in biological sensing. Although there are substantial differences in the affinity of diatomics for free heme, most notably the 2 × 104 greater affinity of CO compared to O2 for free heme, heme proteins have evolved mechanisms for selective binding of a specific diatomic in the presence of another, an essential component of sensing. These NRVS studies have both clarified and provided new information concerning the nature of their vibrational dynamics.
3.1. Carbonyl (CO) Species
The coordination of carbon monoxide (CO) and an analysis of the vibrational spectra has often been used as a probe for heme proteins.40−45 It has long been recognized that the FeCO vibrational signals are a sensitive probe of ligands trans to CO and the electrostatic landscape of the distal pocket of heme proteins. The HOMOs of heme carbonyls are π bonding for the Fe–C bond and π antibonding for the C–O bond. Thus, any effect including environmental effects that change the π electron density will strengthen the Fe–C bond and weaken the C–O bond or vice versa.46 This bonding feature leads to the well-exploited monitoring of the bond strengths via changes in the ν(Fe–C) and ν(C–O) stretches. Plots of νFe–C vs νC–O show a linear relationship for all species with the same or similar ligand trans ligand, which is often called the backbonding correlation. There are distinct differences in these ν(Fe–C) vs ν(C–O) plots, so that imidazole (histidine) trans ligation is readily distinguished from trans thiolate (cysteinate) and imidazolate, tyrosinates, and also hemes with a solvent molecule trans to the CO. Hydrogen bonding to the distal carbonyl effects are apparent in these plots with proteins having strong hydrogen bonding to CO in the distal pocket being placed at the left (highest values of ν(Fe–C) and lowest value of ν(C–O)). Vibrational frequencies thus probe local electrostatic properties, which are crucial for stabilizing O2 binding.
The quantum yield for photodissociation of CO from heme proteins is higher than that for other diatomic ligands,48 and flash photolysis studies of carbonyls have been essential tools in the study of heme protein dynamics including the investigation of ligand dissociation49−51 and the migration of CO through the protein interior.52−55
Mammalian heme oxygenase,56 which oxidizes an iron porphyrin to biliverdin and releases iron and CO, is important not only in heme catabolism but also in iron homeostasis, cellular signaling,57 and O2 sensing.58 The CO released also competes with the O2 binding site of myoglobin and hemoglobin, and mechanisms for the selective binding of O2 have evolved to avoid CO poisoning.59 The sensing activity of CO in activating soluble guanylate cyclase,60,61 control of circadian rhythms,62,63 and bacterial signaling64−66 have further raised interest in the characterization of heme carbonyls.
These varied activities of heme carbonyls and vibrational spectroscopy demand that the vibrational spectra of their complexes be understood in detail, and we have applied NRVS to the task.
3.1.1. NRVS of Six-Coordinate CO Species
NRVS spectra have been measured for four distinct six-coordinate carbonyl derivatives of the form [Fe(Porph)(R-Im)(CO)].67 Powder data have been recorded for [Fe(TPP)(1-MeIm)(CO)], [Fe(TPP)(1-PhIm)(CO)], [Fe(TPP)(1,2-DiMeIm)(CO)], and [Fe(OEP)(1-MeIm)(CO)]. In addition, an oriented crystal, general in-plane measurement was made for [Fe(TPP)(1,2-DiMeIm)(CO)], and both a general in-plane and an out-of-plane measurement was made for [Fe(TPP)(1-MeIm)(CO)]. The powder data quickly allowed the assignment of the Fe–CO stretch with values of 507 (1-MeIm), 506 (1,2-DiMeIm), 502 (1-PhIm), and 499/513 cm–1 (OEP, 1-MeIm) with no significant imidazole dependence. The out-of-plane measurements for [Fe(TPP)(1-MeIm)(CO)] showed no signals at observed frequencies of 561 and 587 cm–1, which are the two Fe–C–O bending modes. However, the large splitting in the two peaks is likely not the result of lifting of the degeneracy by the trans, planar 1-methylimidazole. Rather the DFT predictions suggest the splitting is the result of porphyrin ligand modes mixing with the two bands. Figure 6 illustrates the predicted character of the two bands. The mixing of the FeCO bend and porphyrin modes and the importance of peripheral substituents in [Fe(TPP)(1-MeIm)(CO)] is further supported by the observation that only a single unresolved pair of bands at 580 cm–1 is observed in [Fe(OEP)(1-MeIm)(CO)]. The NRVS spectrum for MbCO has also been measured,67 and the Fe–CO stretch is found at 502 cm–1and the Fe–C–O bend at 572 cm–1 similar to those above.
Figure 6.

Predicted vibrational character of the in-plane modes for the FeCO fragment in [Fe(TPP)(1-MeIm)(CO)] with frequencies 560 (left) and 580 cm–1 (right) and observed at 569 and 586 cm–1. Relative phase of the FeCO bending and porphyrin motions reverses between the 560 and 580 the cm–1 modes. Illustration prepared using calculations originally reported in ref (67).
Ohta et al.69 also made NRVS measurements for an imidazole tailed, tetraaryl carbonyl derivative where the measured frequency for ν(Fe–C) was 470 cm–1 and the FeCO bending mode was found at 570 cm–1. The likely doming mode is at 110 cm–1. The in-plane motion pattern is similar to the other six-coordinate species. These NRVS measurements were made on a frozen solution, so no directional information was directly available.
The DFT calculations for the in-plane region of the spectrum (300–350 cm–1) for [Fe(TPP)(1-MeIm)(CO)] predicts the character of the Fe–Np stretches and predicts four bands at 318, 321, 323, and 329 cm–1, Figure 7. In addition, DFT predicts the appearance of an out-of-plane mode at 320 cm–1, which is overlapped with the in-plane modes. This peak is in fact observed at 331 cm–1 in the experimental out-of-plane spectrum of [Fe(TPP)(1-MeIm)(CO)]. The confirmation of this peak is only possible because of the measurement of oriented single-crystal spectra. The predicted character of this mode is the perpendicular movement of the FeCO group against the rest of the molecule. As will be further commented on the NO section, there are effects on the observed peaks in this region that are dependent on the peripheral substituents. There are also modest differences depending on the imidazole. Additional in-plane peaks are observed at 242 and 251, 413, and 466 cm–1.
Figure 7.

Predicted in-plane vibrational modes of [Fe(TPP)(1-MeIm) (CO)] with frequencies 318 (left) and 329 cm–1 (right), as viewed looking from the CO to the imidazole. Note the direction of iron motion toward a porphyrin nitrogen atom. Porphyrin component of motion corresponds to γ6. Illustration prepared using DFT calculations originally reported in ref (67).
The NRVS capability of observing all vibrations of iron with iron motion means that the spectra should provide information concerning the Fe–N(Im) stretch in six-coordinate heme derivatives, which are unavailable from resonance Raman. The predicted frequencies with substantial simultaneous motion of iron and imidazole do not conform to a simple two-body problem. Experimental data are nonetheless available. The out-of-plane bands observed in [Fe(TPP)(1-MeIm)(CO)] at 172, 225, and 330 cm–1 are found at slightly lower frequencies in the powder spectra for [Fe(TPP)(1,2-DiMeIm)(CO)].67 These data are consistent with the small differences (∼0.05 Å) in the Fe–N(Im) bond distances in the two derivatives.47 Accordingly, the peak at 225 cm–1 is most likely the Fe–N(Im) stretch and the 172 cm–1 band the Fe–N(Im) bend. These important observations will also be significant for other six-coordinate diatomic species to be discussed subsequently.
Low-frequency vibrational modes (below ∼200 cm–1) are observationally challenging, but NRVS is the most satisfactory experimental method. Low-frequency modes of particular interest in heme complexes includes observation of the doming mode. The doming mode can be described as the perpendicular motion of iron relative to the heme plane with the four pyrrole groups swiveling to follow the iron. Such doming motion is an important component of the association/dissociation of diatomic ligands including Perutz’s early model for hemoglobin cooperativity70 and in models for protein control of reactions of heme proteins.71−73 For [Fe(TPP)(1-MeIm)(CO)], two out-of-plane features at 64 and 127 cm–1 were considered likely candidates for doming mode motion, although the DFT calculations had predicted a single frequency at 96 cm–1.
Finally, we return to the issue of the Fe–C–O bending modes. A long-standing discussion in heme carbonyl structures has been the issue of the FeCO group not being linear but somehow “bent” from the normal to the heme plane, whereas small molecule species had linear Fe–C–O groups. This arises from the apparent deformability of the FeCO unit seen in several protein matrices. An interesting calculation by Ghosh and Bocian74 suggested a way out of this impasse. Their theoretical calculations suggested that the Fe–C–O bend and the Fe–CO tilt, which is a very low energy mode, are strongly coupled and allow for energetically accessible distortions. A later calculation by Kozlowski et al.75 detailed some possible limits to these geometrical distortions and the energies needed for the distortions.
The resulting mixing should lead to an out-of-phase combination observed at high frequency and predicted to be at somewhat higher frequencies than that expected without the tilt/bend mixing, i.e., near 580 cm–1 and an in-phase mode at very low frequency at about 80 cm–1. The high-frequency mode as observed at 561 and 587 cm–1 is in agreement with the prediction. Unfortunately, the low-frequency modes are predicted by DFT to involve negligible iron motion and indeed cannot be conclusively observed in the NRVS experimental spectrum.67 As described in ref (67), simple three-body predictions would predict higher intensity than observed for the in-phase mode and suggest the need for an improved description of the system and the energetics of the FeCO distortion.
3.1.2. NRVS of a Five-Coordinate CO Species
The synthesis of five-coordinate heme carbonyl derivatives (i.e., no trans ligand) in order to obtain vibrational spectra of a bona fide five-coordinate CO complex is more challenging than many believe. The reaction of CO with a four-coordinate iron(II) porphyrin leads, in solution, to a mixture of the CO complexes [Fe(Porph)(CO)] and [Fe(Porph)(CO)2].76−78 Rougee and Brault76 also showed that a number of weak ligands including DMF, H2O, and ethyl acetate would coordinate trans to the primary CO. Although the binding constants for these ligands are small, they are not negligible, especially in dilute solution.
The NRVS spectra obtained for crystalline [Fe(OEP)(CO)]79 (Figure 8, top) are the first vibrational data obtained for a crystallographically verified five-coordinate heme carbonyl.80 Oriented single-crystal data, an out-of-plane and a generalized in-plane measurement, uniquely identified the Fe–CO stretch at 573 cm–1 and the Fe–C–O bend at 505 cm–1. DFT calculations for [Fe(OEP)(CO)] also agree with these assignments. The 573 cm–1 band establishes a new upper limit for the Fe–CO stretch. Most significantly, that the Fe–CO stretch is well above the frequency for the Fe–C–O bend provides a unique vibrational signature for five-coordinate heme carbonyls. This reversal of stretch and bend, with the stretch at a significantly higher frequency than any previously observed, is to be contrasted with all data for six-coordinate carbonyl species where the frequency of the stretch is significantly below that of the bending frequency. The six-coordinate carbonyl pattern is well established by the NRVS data cited above as well as many other vibrational studies. A comparison of the spectra and predictions of five-coordinate [Fe(OEP)(CO)] and six-coordinate [Fe(OEP)(1-MeIm)(CO)] is shown in Figure 8.
Figure 8.

Experimental and calculated NRVS vibrational density of states (VDOS) for the Fe atom in [Fe(OEP)(CO)] and [Fe(OEP)(1-MeIm)(CO)] versus wavenumber shift. Top two panels are for [Fe(OEP)(CO)], with the first panel showing the experimental NRVS measurements on an oriented single crystal. Measurements yield the directional contributions to the Fe VDOS to a polycrystalline powder. Calculated VDOS for parallel (red), perpendicular (blue), and powder (black) are shown in the second panel. Experimentally derived VDOS for powdered [Fe(OEP)(1-MeIm)(CO)] is shown in the third panel with the predicted oriented VDOS from DFT calculations, revealing the ν(Fe–C) frequency below that of δ(FeCO). Reprinted with permission from ref (79). Copyright 2014 American Chemical Society.
The substantially higher stretch frequency compared to all heme carbonyls known to us is consistent with the substantially shorter Fe–C bond distance in five-coordinate [Fe(OEP)(CO)].80 The Fe–C(CO) values found (two derivatives) are 1.714 and 1.708 Å, compared to values at ∼1.76 Å for various six-coordinate heme carbonyls.81 The C–O stretching frequencies observed by FTIR is 1644 or 1648 cm–1, midway in the range of values observed in six-coordinate species, and the C–O bond distances are in the normal range. Plotting νFe–C vs νC–O for [Fe(OEP(CO)] on the traditional backbonding plot would clearly lead to points that are well above the points for any of the known species reported thus far.40 This clearly is shown in Figure 9 depicting the standard backbonding correlation with several sets of ligands trans to CO. The data for [Fe(OEP)(CO)] are shown as the solid red star.
Figure 9.
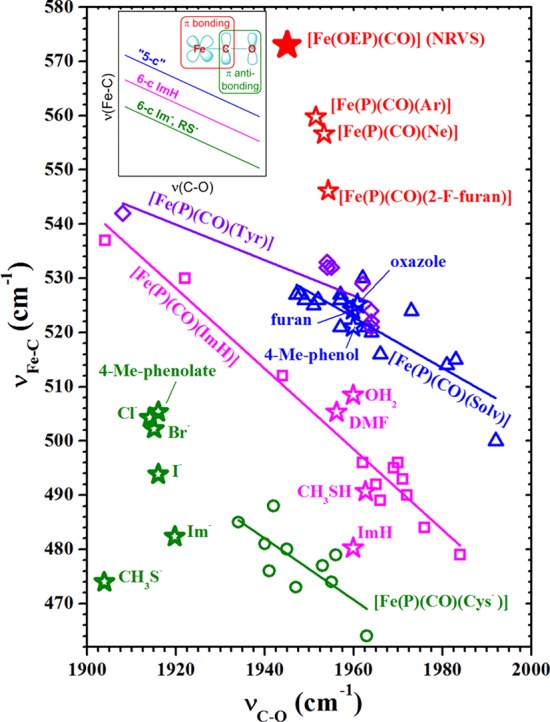
Backbonding correlation plot showing the vibrational signature of [Fe(OEP)(CO)] (solid red star), with its uniquely high experimental νFe–C frequency. Open round (green, anionic), square (magenta, neutral), and triangular (blue, trans-O-bound) points were taken from ref (82) and represent various porphyrins and proteins. Diamond (violet, trans-Tyr) points are Tyr-liganded proteins.45,85 Inset in the upper left shows the correlation lines based on experimental data. “5-c” line is expected for six-coordinate species with trans H2O ligands (see ref (79)). Open star-shaped points give predicted (DFT) νFe–C and νC–O frequencies for the indicated trans ligand.79 Frequencies were scaled to those of [Fe(OEP)(CO)]. Reprinted with permission from ref (79). Copyright 2014 American Chemical Society.
How have attempts to form spectroscopic samples of five-coordinate CO complexes been carried out previously? Synthetic procedures used involved aqueous dithionite to provide an iron(II) species followed by reaction with CO; hence, the possible presence of a water ligand trans to the CO cannot be eliminated. The success of such a synthetic strategy to provide a complex with no trans ligand is likely to be porphyrin ligand dependent.
Experiments by Rougee and Brault have shown that the six-coordinate species with trans water is a viable species.76 Resonance Raman spectra of a series of tetraarylporphyrin derivatives prepared in this fashion gave Fe–C stretching frequencies in the 500–530 cm–1 region with most between 520 and 530 cm–1.82 These values along with the observed C–O stretches, put these species on a distinct correlation line above those observed for imidazole derivatives and those for thiolate-ligated species. These are shown as the blue open triangles on Figure 9. Other carbonyl species on this new correlation line includes cytochrome c oxidase,83 heme catalases,84 and a number of bacterial heme trafficking proteins with tyrosine as the proximal ligand.45,85 Also along this line is an effector-activated CO form of soluble guanylyl cyclase; binding the effect leads to a +33 cm–1 increase in νFe–C and a 12 cm–1 decrease in νC–O relative to the native protein. Positions on this line have been associated as diagnostic of five coordination or a distally compressed heme carbonyl.40,86 The effector-induced activity of soluble guanylyl cyclase has been concluded to be five coordinate. Observe, however, that this correlation line falls between νFe–C of a known five-coordinate carbonyl and the lower two correlation lines (for imidazole- and thiolate-ligated heme carbonyls). We believe that the species along this new correlation line are more probably six coordinate with a weak-field trans ligand consistent with our observations and calculations described below.87
The NRVS spectrum of an [Fe(TMP)] carbonyl complex prepared by the aqueous dithionite reduction procedure69 displayed four peaks above 500 cm–1: peaks at 505, 520, 550, and 580 cm–1. Since the measurement was made on a frozen solution, no directional information is available. Ohta et al.69 assigned the 550 and 520 cm–1 peaks as the Fe–C stretch and bend, respectively. However, the presence of four significant peaks in this region suggests that two distinct species are in the frozen solution. Unfortunately, the frequencies of the C–O stretch were not reported, which could verify two CO species. We think that all four frequencies need to be assigned. Although any assignment will be ambiguous, we think that these frequencies match those expected for six-coordinate [Fe(TMP)(H2O)(CO)] (520 and 550 cm–1, stretch and bend) and five-coordinate [Fe(TMP)(CO)] (580 and 505 cm–1, stretch and bend). Any other possible combination of frequency pairs seems less likely.
We further explored the relationship between the trans ligand field and the frequency with a series of DFT calculations for model [Fe(OP)(L)(CO)] complexes, where P is porphine and L is one of a series of ligands of varying ligand field strength. Vibrational predictions for five-coordinate CO complexes, including [Fe(OEP)(CO)], shows that the reversal of the frequencies of the stretching and bending frequencies vis-a-vis six-coordinate species is expected.79 Calculations by Ohta et al. also found that this reversal is to be expected.69 These calculations showed that only extremely weak-field ligands (all much weaker than H2O) displayed the stretch and bend reversal.79 The reversal is also correlated with the displacement of iron from the porphyrin mean plane toward CO. Displacements of iron greater than ∼0.18 Å, which depends on the trans ligand (and field strength), lead to the reversal. Results from theses calculations are shown in Figure 9 as the open stars.
Other modes observed for [Fe(OEP)(CO)] include an out-of-plane peak at ∼157 cm–1, which is the likely doming mode. This is observed at higher frequency than that for the six-coordinate species as might be expected. The in-plane vibrational region for the six- and five-coordinate species is similar, consistent with a low-spin state for all. See Figure 8.
In conclusion, NRVS spectroscopy has shown that there are thus two distinct features for any five-coordinate CO derivative that must be seen to confirm any five-coordinate derivative. First is the reversal of the frequencies of the stretch and bend compared to six-coordinate species. Second will be the high value of the Fe–C stretch with the position on the backbonding correlation plot higher than those of the various six-coordinate species. As we stated elsewhere,79 we believe that all heme carbonyls reported prior to our report79 have been six-coordinate species with a weak-field ligand trans to CO.
3.2. Nitrosyl (NO) Species
The NO ligand coordinates to heme iron in both of its common oxidation states; both five- and six-coordinate complexes in both oxidation states have been studied with NRVS. The lower oxidation state, which is nominally iron(II), is often denoted as {FeNO}7. In the commonly used notation, {FeNO}n, first suggested by Enemark and Feltham,88n denotes the sum of the number of d electrons of the metal and the number of π* electrons in NO. This notation emphasizes the strongly covalent nature of the group. A celebrated system in heme-based NO sensing, that of a {FeNO}7 complex, is soluble guanylate cyclase activation where the sensing of NO leads to a cascade that controls the relaxation of endothelial cells of blood vessels and control of blood pressure. Genetic analysis has now demonstrated that there is a large class of related proteins that sense either O2 or NO; the binding domain is often called H-NOX.90 A notable biologically important example of an {FeNO}6 complex is that of the nitrophorins found in two classes of blood-sucking insects that allow the organism to obtain a blood meal from its victim.91 {FeNO}6 complexes are also important in denitrification processes.92,93
3.2.1. NRVS of Five-Coordinate {FeNO}7 Species
The earliest NRVS studies involving five-coordinate {FeNO}7 complexes were made on [Fe(TPP)(NO)].23,94,95 Both powder and a crystalline array were measured for this complex. The crystal array, a forerunner to more detailed single-crystal measurements made subsequently, allowed conclusive assignments of frequencies resulting from out-of-plane modes. These included the Fe–NO stretch at 540 cm–1 and the doming modes at 74 and 128 cm–1. The DFT calculations in support of the complete vibrational assignments revealed important limitations of the calculations but also affirmed the general utility of DFT predictions for assignments. An important observation was that predicting Fe–NO frequencies was especially challenging and dependent on the DFT functional chosen for the computation. Both under- and overestimation of the axial Fe–NO stretching frequency was noted. Difficulties in predicting axial ligand frequencies appear to be true across all diatomic species. However, other modes, specifically the in-plane modes, were well predicted both in frequency and in intensity, reinforcing the idea that, with care, DFT calculations are important components of achieving complete vibrational assignments.
Importantly, NRVS powder measurements made on four additional five-coordinate nitrosyl complexes ([FeOEP)(NO)] and [Fe(PPIX DME)(NO)] and two related species) demonstrated the importance of the peripheral substituents on the details of the in-plane motions of iron. Even changing only two of the eight β peripheral substituents leads to significant changes in the in-plane spectra. The out-of-plane ligand modes were, however, little affected.94 Subsequently, computational studies demonstrated that the varying possible conformation of the peripheral ethyl groups in [Fe(OEP)(NO)] have significant effects on the in-plane vibrations but not the iron NO vibrations.96 Isotope labeling (15NO for 14NO) led to assignments for the Fe–NO stretch and bend, consistent with our earlier assignments, but the lower frequency in-plane and out-plane vibrations of [Fe(OEP)(NO)] were not assigned.96
These earlier NRVS studies clearly demonstrated that the nonlinear Fe–N–O group (angle 140–150°) reduces the symmetry of the porphyrin species and consequently enriches the vibrational dynamics by removing in-plane degeneracy. In-plane asymmetry was recognized and preceded by the structures of several five-coordinate nitrosyl species, all unencumbered by crystallographic disorder.97,98 All species showed an off-axis tilt of the FeNO group with the in-plane Fe–Np bonds in the direction of the off-axis tilt shorter than those opposite to the tilt, i.e., substantial geometric in-plane asymmetry.
The in-plane anisotropy was explored by the first of a series of very detailed single-crystal NRVS spectral measurements on [Fe(OEP)(NO)] with interesting results.99 In this experiment, two orthogonal spectra were taken in the porphyrin plane. One direction was parallel to the projection of the bent FeNO group onto the porphyrin plane (x direction) and the second perpendicular (y direction) to this direction. The projection of the Fe–N–O plane is about halfway between a pair of Fe–Np bond directions (i.e., approximately bisecting the in-plane Np–Fe–Np angle). The two in-plane spectra display significant differences in both frequencies and intensities.
The two orthogonal in-plane spectra are illustrated in Figure 10, where the red or green lines show the observed NRVS signal with measurements taken in the directions shown by the insets. This is presented as a “mirror” plot with two directions sharing a common frequency axis. The Fe–N–O bending mode, as expected, is only found in the x in-plane and z out-of-plane directions at 394 cm–1. However, other differences in the two in-plane spectra are more unexpected.
Figure 10.
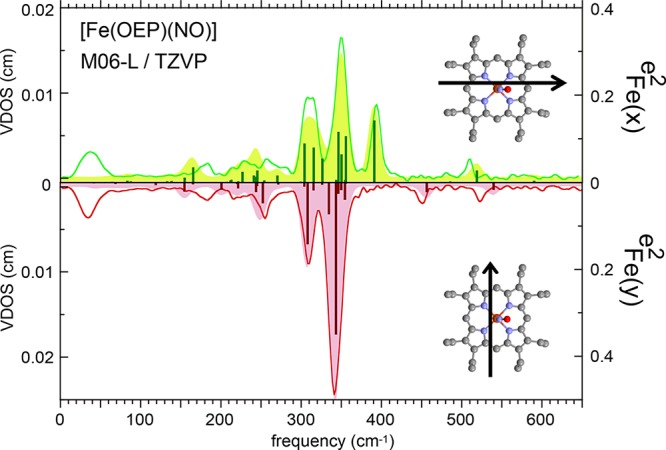
Measured (lines, ref (99)) and predicted (shaded and based on an MO6L functional35) contributions to the VDOS from iron motion. efFe2 values (bars) over the 125–480 cm–1 range are also presented. Adapted wth permission from ref (100). Copyright 2014 American Chemical Society.
The combination of the experimental data and theoretical calculations led to the surprising conclusion that the in-plane motion of the iron is along the x and y directions and not along the Fe–Np bond directions99 as shown in Figure 11. This strong preference for the in-plane iron motion parallel and perpendicular to the FeNO plane must represent asymmetry from the relatively strong Fe–N(NO) bond. The interaction between the iron dπ orbitals and the NO π* orbitals are parallel and perpendicular to the FeNO plane.
Figure 11.

Plots of several predicted in-plane modes for [Fe(OEP)(NO)] are shown. Directions of iron motion are either parallel and perpendicular to the FeNO plane and not along the Fe–Np bonds. Illustration prepared using DFT results reported in ref (99).
Subsequently, an equivalent oriented crystal study for [Fe(DPIX DME)(NO)] showed similar patterns in the motions of iron in the parallel and perpendicular directions to the FeNO plane.100 The pattern is not quite so strongly developed, which may result from the asymmetric peripheral substitution pattern. Observed out-of-plane modes in [Fe(DPIX DME)(NO)] include the bending mode at 399 cm–1, the Fe–NO stretch at 528 cm–1, and the doming mode at 183 cm–1.100 The corresponding modes in [Fe(OEP)(NO)] are the Fe–NO stretch at 517 cm–1 and the doming mode at 158 cm–1.
3.2.2. NRVS of Six-Coordinate {FeNO}7 Species
It has long been recognized101−104 that the NO group has a strong structural trans effect that leads to extremely long and weak Fe–L bonds on the other side of the porphyrin plane. Even though the trans bond is very weak, the interactions does lead to an ∼0.03 increase in the Fe–N(NO) bond distance relative to the five-coordinate complexes. In recognition of this we sought to understand how the Fe–NO stretch could be assigned at 552 cm–1 in six-coordinate MbNO when the stretch was assigned at 521 cm–1 in five-coordinate MbNO. The 30 cm–1 increase is inconsistent with the 0.03 Å longer and thus weaker Fe–NO bond found in the small molecule analogues. The 552 cm–1 assignment was based on a 15 cm–1 shift in this frequency when 14NO was replaced by 15NO (in resonance Raman studies). However, see further discussion in section 3.5. Oriented single-crystal NRVS on [Fe(TPP)(1-MeIm)(NO)] provided a solution to this conundrum.105 The single-crystal data clearly identify the out-of-plane modes. The iron contribution to the high-frequency peak is very modest, with most of the kinetic energy contribution resulting from motion of the central nitrogen of the FeNO group and not out-of-plane iron motion. This also provides understanding for the substantial lack of iron dependence in the difference Raman spectra between [54FeMbNO] and [57FeMbNO] in the high-frequency region.105 With the large contribution of the kinetic energy from the central nitrogen atom, this observed frequency at 556 cm–1 is best considered as the in-plane Fe–N–O bend. Rather the Fe–NO stretch is found at 443 and 473 cm–1, almost 100 cm–1 lower than that found for the five-coordinate species and quite consistent with the increased Fe–N(NO) bond distances in the six-coordinate species. Mixing between the stretch and one component of the porphyrin mode ν50 leads to two bands with Fe–NO stretch character. The NRVS spectrum of MbNO shows bands at 452 cm–1 with significant intensity and a much weaker band at 558 cm–1.
A subsequent NRVS study of the two crystalline forms of [Fe(TpFPP)(1-MeIm)(NO)]106 displayed bands at 432 and 433 cm–1 and 560 and 561 cm–1. The 561 cm–1 peak also has a minor contribution at 571 cm–1. The difference between the two species reflects the very small difference (0.004 Å) in the Fe–N(NO) bond distance, Figure 12.
Figure 12.

Figure illustrating the predicted character of the high-frequency FeNO bending mode of monoclinic [Fe(TpFPP)(1-MeIm)(NO)] observed at 561 and 571 cm–1 and predicted at 599 cm–1. Note that there is less observed in-plane motion for this derivative than that of [Fe(TPP)(1-MeIm)(NO)]. Illustration prepared using calculations originally reported in ref (109).
These first studies clearly showed an important difference to be found between the five- vs the six-coordinate species, namely, the reversal of the relative positions of the stretching and bending frequencies depending on coordination number. This is seen to be an important feature for all other Fe(XO) species where analogous five- and six-coordinate species can be prepared and have been analyzed in detail by NRVS studies. This correlation of the pattern of stretching and bending frequencies is applicable to all methods of vibrational analysis including resonance Raman, although the stretching mode is not always observable in rR studies of NO-ligated heme proteins.
These vibrational assignments of the stretching and bending modes have been subsequently confirmed by further NRVS measurements for the isotopomers of [Fe(TPP)(1-MeIm)(14,15N16,18O)] that demonstrate 11 or 12 cm–1 downshifts, but single-crystal data were needed to ensure the correct assignments.107 The single-crystal data for [Fe(TPP)(1-MeIm)(NO)]105 have been further analyzed by a combination of DFT calculations and quantum chemistry-centered normal coordinate analysis (QCC-NCA).108 The most significant results from this study again demonstrate some shortcomings of DFT calculations. The QCC-NCA calculations are expected to provide good estimates of the force constants of the important bonds of the molecule.108 However, the possible effects of not including fits to the entire NRVS spectrum may have an effect on these force constants.
The capping NRVS study for six-coordinate {FeNO}7 derivatives is that of single-crystal measurements in three orthogonal directions for the monoclinic polymorph of [Fe(TpFPP)(1-MeIm)(NO)] along with other single-crystal measurements for the triclinic polymorph of [Fe(TpFPP)(1-MeIm)(NO)] and for [Fe(TpOCH3PP)(1-MeIm)(NO)].109 The Fe–NO stretch is again found at 472 cm–1 in the monoclinic polymorph, and again lower frequency bands mixed with the porphyrin ν50 mode can be assigned. The appearance of two new bands mixed with ν50 is different from that of the TPP derivative where there was only one band mixed with ν50 and emphasizes anew the importance of peripheral substituents.
Another significant difference is in the detailed character of the highest frequency band in the spectrum originally assigned as the Fe–NO stretch. The character of the band that we have assigned as the bend can be partly assessed noting how much, if any, of an in-plane component is observed. In all three of the new complexes studied, the amount of the in-plane component to the total intensity is less than that originally observed for [Fe(TPP)(1-MeIm)(NO)].105,108 The varying character of the highest frequency band results from mixing among the Fe–N–O bending mode, porphyrin modes, and the Fe–NO stretching mode with the differing peripheral substituents leading to a difference in mixing. The majority of motion for the peak remains that of the central nitrogen atom of the FeNO group and is still most consistent as being described as the bending mode.
The x (parallel) and y (perpendicular) in-plane measurements for monoclinic [Fe-(TpFPP)(1-MeIm)(NO)] again demonstrated that the orientation of the FeNO group is paramount in defining the direction of motion. The majority of iron motion for each in-plane mode is either parallel or perpendicular to the FeNO plane similar to the case of the five-coordinate [Fe(OEP)(NO)] and [Fe(DPIX)(NO)] species. However, the effects are not as dominant as in the five-coordinate cases. This may reflect the fact that the Fe–N(NO) bond is slightly weaker in the six-coordinate complexes.
Finally, the lowest frequency region of the spectra (below 200 cm–1) for the monoclinic form of [Fe(TpF-PP)(1-MeIm)(NO)] is especially rich in low-frequency bands with many predicted to have both Fe–Im stretching and imidazole torsional motions. The peak with the major Fe–Im stretch is at 151–153 cm–1, while the doming mode is at 131 cm–1. The measurement of the Fe–Im stretch in six-coordinate heme derivatives is an important observation made possible by NRVS; it is not observed in the resonance Raman spectra. A discussion of the Fe–Im stretches observed in six-coordinate Fe(XO) derivatives will be given in section 3.5.
3.2.3. NRVS of Five-Coordinate {FeNO}6 Species
Out-of-plane and a generalized in-plane oriented single-crystal measurements have been made for the five-coordinate {FeNO}6 complex [Fe(OEP)(NO)]+.110 The intense out-of-plane peak found at 595 cm–1 is assigned as the stretching mode, and an in-plane peak at 402 cm–1 is assigned as the bending mode. This pattern of the stretching frequency found at a higher value than the bend follows that observed for all of the other five-coordinate FeCO and {FeNO}7 species described above. DFT calculations have also been performed to facilitate in the assignments. The in-plane modes are predicted to have iron motion in one degenerate set along the Fe–Np bonds and a second degenerate set roughly halfway between the Fe–Np bonds. A doming mode is observed at 151 cm–1. The values of both the doming mode and the stretching mode in this five-coordinate species and in the related six-coordinate species [Fe(TPP)(1-MeIm)(NO)]+ are very close and suggest the axial Fe–N(NO) bond, which is equal in both derivatives, is the prime determinant of the frequency for the modes in both.
3.2.4. NRVS of Six-Coordinate {FeNO}6 Species
The biological importance of the six-coordinate {FeNO}6 species is less than that of the {FeNO}7 species. Nevertheless, the {FeNO}6 species are significant in NO processing (i.e., fungal NO reductase111) and provided for the biological storage of otherwise labile NO (i.e., the nitrophorins112). The electronic structure of the FeNO unit has been the subject of debate with suggestions varying from FeII(NO+) to FeIV(NO–).
The NRVS spectra of four different {FeNO}6 species have been recorded. The possible frequencies of the Fe–N–O stretch and bend in six-coordinate [Fe(TPP)(1-MeIm)(NO)]+ are observed to be very close at near 600 cm–1.113 This is in distinct contrast to the other diatomic ligated species which have the highest pair of frequencies well separated. Thus, assigning (distinguishing) the stretch and bend is challenging. Even with a comparison with powder spectra obtained with natural abundance and the 15N18O isotopomer, the final assignments of 578 cm–1 for the stretch and 586 cm–1 for the bend are somewhat ambiguous.
The NRVS spectrum of the NO derivative of the isoform nitrophorin 2, an NO-carrying protein, has been recorded and shows a strong band at 594 cm–1 along with a shoulder at 581 cm–1. These were tentatively assigned as the stretch and bend, respectively.114 The porphyrin in the nitrophorin is heme b that has two ionizable carboxylic acid groups. Since the complex is formed at relatively low pH the two carboxylic groups are likely protonated. An interesting feature of this study was a theoretical prediction of the effects of possible protonation states of the carboxylic acid groups and the effects of the protein environment on the spectrum. Substantially different predictions were found, and the authors concluded that heme b was deprotonated in the derivative. A second nitrophorin NO derivative (isoform 7) has also been studied.115 A single band at 589 cm–1 was observed and must correspond to the frequencies of both the stretching and the bending modes.
It is interesting to note that the theoretical frequency predictions of all of the above studies predict larger frequency separations between the stretching and the bending modes than is actually observed. The ambiguity in the assignment of the stretch and bend is clearly a call for the use of our single-crystal NRVS measurements for resolution. Although Ellison and Scheidt116 described the synthesis of several [Fe(OEP)(L)(NO)]+ derivatives where L was a neutral nitrogen donor, none were appropriate for a complete single-crystal NRVS study. Fortunately, crystallization of {FeNO}6 species from a series of various solvents provided single-crystal samples of [Fe(OEP)(2-MeHIm)(NO)]+ appropriate for NRVS measurement of three orthogonal orientations of the crystalline sample.110
The oriented-crystal measurements show a strong out-of-plane mode at 600 cm–1 and two equally intense in-plane modes at 580 and 574 cm–1 in the other two measured directions that are parallel to the porphyrin plane and, respectively, parallel and perpendicular to the imidazole plane. Thus, the stretching and bending modes are clearly identified. Unlike the previously described six-coordinate Fe(XO) species, the frequency of the stretch does not decrease to a value substantially below the bending modes. This probably results because the axial Fe–NO distance is unchanged (not longer) compared to the five-coordinate species, unlike all other Fe(XO) systems. The two bending modes observed at 580 and 574 cm–1 are predicted and observed to be parallel and perpendicular to the axial imidazole and have frequencies much higher than the analogous five-coordinate NO complex. Interestingly, imidazole motions are correlated in the two FeNO bends: the bend perpendicular to imidazole also has imidazole motion perpendicular to the plane, whereas in the bend parallel to the imidazole the ring displays rotational motion in the imidazole plane.
The in-plane iron modes in the ∼300–370 cm–1 range are expected for a low-spin state. They are predicted to have iron motion approximately parallel and perpendicular to the imidazole plane rather than along the Fe–Np directions. This appears to be an effect of the totality of axial ligand bonding of imidazole and NO; the corresponding in-plane motions in [Fe(OEP)(NO)]+ are located both halfway between Fe–Np and along the Fe–Np bonds. There is clearly more in-plane anisotropy in the six-coordinate species.
An important out-of-plane observation in all of the six-coordinate imidazole-ligated Fe(XO) species is the assignment of the Fe–Im stretch. There are typically more than one low-frequency mode that might be assigned. The character of all of the low-frequency bands with both predicted simultaneous iron and imidazole motion must be considered. We examine the predicted character of each calculated frequency and choose the one that best resembles the classical Fe–Im stretch. This “classical” Fe–Im vibration for [Fe(OEP)(2-MeHIm)(NO)]+ is illustrated in Figure 13. Similar motions have been predicted and observed for other six-coordinate Fe(XO) derivatives and will be further discussed as a group in section 3.5.
Figure 13.
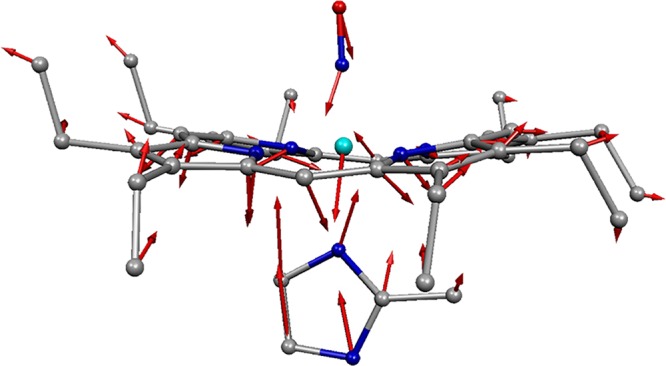
Plot illustrating the predicted character of the mode most characteristic of the Fe–Im stretch in [Fe(OEP)(2-MeHIm)(NO)]+. Predicted frequency is 225 cm–1, and observed frequency is 238 cm–1. Adapted with permission from ref (110). Copyright 2014 American Chemical Society.
3.3. NRVS of Dioxygen (O2) Species
Aerobic life obviously depends on dioxygen and heme proteins dominating its utilization. Important systems include oxygen carriers such as hemoglobin and myoglobin and the systems for the reduction of O2 to H2O that power aerobic metabolism (i.e., cytochrome oxidase). Other important systems are heme-based sensors and many heme–O2 complexes that are important as intermediates in enzymatic processes. Clearly characterization of the vibrational characteristics of heme–oxygen systems is important in developing further understanding.
The study of dioxygen complexes of small molecule iron porphyrin derivatives is complicated by their instability to oxidation by dioxygen. To our knowledge, the only known molecules that have avoided this difficulty, with the isolation of solid-state species, are the picket fence porphyrin derivatives originally synthesized by Collman and co-workers.117
Structure determinations for the dioxygen derivatives of iron(II) picket fence porphyrin117−121 have shown that dioxygen coordinates with an end-on conformation and a Fe–O–O angle of ∼125°. The similarity of the structural features of the dioxygen complexes to that of the {FeNO}7 complexes immediately suggests that there will also be similarities in the vibrational spectra.
NRVS spectra have been measured for three distinct oxyheme complexes with differing trans imidazole ligands, the 1-methyl-, 1-ethyl-, and 2-methylimidazole derivatives.122 There are three high-frequency bands seen that have both significant iron and oxygen contributions. The 1-ethyl- and 2-methylimidazole derivatives have been studied by oriented, single-crystal NRVS measurements. The out-of-plane modes have frequencies of 571, 417, and 393 cm–1 for the 1-ethylimidazole derivative and 563, 419, and 389 cm–1 for the 2-methylimidazole species. These frequencies are similar to those observed in resonance Raman. However, it is to be noted that the assignments from Raman based on 18O2 isotopomers must be regarded as ambiguous. These modes have both in-plane and out-of-plane character from the NRVS single-crystal spectra and are strongly mixed with both bending and stretching contributions to both of the highest frequency bands. The two highest frequencies for the modes showing out-of-plane character are illustrated in Figure 14. As also seen in the {FeNO}7 nitrosyls, the kinetic energy of the highest frequency band is dominated by the central (oxygen) atom, whereas the second mode has kinetic energy predicted to be more uniformly distributed across the FeOO fragment. While the illustrations are based on predictions, the NRVS observed character is in good agreement.
Figure 14.

Predicted vibrational character of the Fe–O2 unit in [Fe(TpivPP)(1-EtIm)(O2)] for the modes observed at 571 and 417 cm–1. Illustration prepared using DFT calculations originally reported in ref (122).
DFT predictions for both of these bands concur with the conclusion of strong mixing between bending and stretching. Moreover, model calculations based on an oxy derivative with only the porphine nucleus also point to strong mixing in the two bands. The third mode with the smallest intensity and at the lowest frequency of the group has only iron out-of-plane motion. This mode had not been observed in the resonance Raman of the oxy derivatives and points to the unique capabilities of the NRVS technique.
Predictions of frequencies for the diamagnetic FeO2 center were made by DFT calculations using both open-shell and closed-shell singlets. Some differences are observed, i.e., frequencies for the nominal Fe–O2 stretch are lower than observed (open shell) or higher than observed (closed shell). Otherwise, neither set of predictions appears to be superior.
The structural similarity of the oxyhemes to the {FeNO}7 hemes suggests that there could be substantial in-plane anisotropy for the oxy species. These bands are observed in the 220–360 cm–1 region. This is indeed strongly suggested by the DFT calculations that show the bands are strongly in-plane polarized and that the majority of the iron motion is either parallel or perpendicular to the FeO2 plane. Oriented crystal measurements parallel and perpendicular to the imidazole plane reveal that significant differences can be observed. Unfortunately, the 4-fold disorder of the dioxygen in the ligand binding pocket does not permit unambiguous conclusions, but it seems likely that the bound dioxygen ligand has a strong effect, similar to NO on the motion of iron.
In the spectral range below ∼220 cm–1 there are peaks with in-plane and out-of-plane character. A major concern for these low-frequency modes is the possible identification of the Fe–N(Im) stretch and doming modes. Predicted modes in this region have simultaneous, significant motion of both iron and imidazole, with many that show sensitivity to imidazole identity. Unlike the situation with resonance Raman, the observation of the Fe–N(Im) stretch in six-coordinate species is possible, a distinct advantage of the NRVS method over Raman spectroscopy. The Fe–N(Im) stretch has been observed and shifts to lower frequency in the order 1-MeIm > 1-EtIm > 2-MeHIm with values of 205, 196, and 187 cm–1. This order is consistent with the differences in mass of the imidazole. With these assignments in hand, the trends in Fe–N(Im) stretches in the entire series of [Fe(porph)(R-Im)(XO)] complexes can now be evaluated and will be discussed more fully in section 3.5
Two oxygenated heme proteins have been studied by NRVS: oxymyoglobin (MbO2) and an engineered myoglobin derivative designed to probe the iron/copper site of cytochrome oxidase.123 The two highest frequencies in the “classical” oxygen storage protein oxymyoglobin are found at 579 and 423 cm–1. The highest frequency band is oxygen centered with the kinetic energy analysis for this band showing that the central oxygen in MbO2 has 77% of the total kinetic energy of the mode consistent with a description as the FeOO bending mode. The 423 cm–1 band is best considered as the Fe–O2 stretching mode. This pattern in similar to that of the {FeNO}7 derivatives. These two frequencies, with higher values relative to the picket fence derivatives, probably result from the known hydrogen bond (from His64) to the coordinated O2.
The directions of the in-plane iron vibrations appear to be either perpendicular or parallel to the FeO2 plane with a 337 cm–1 band that is especially prominent in the NRVS spectrum and that is predicted to be perpendicular to the FeO2 plane.
The engineered protein is based on myoglobin and has two amino acid substitutions to provide three histidines to bind copper just above the oxygen binding site. This derivative is denoted CuBMb. This modified derivative leads to His64 changing from a residue that hydrogen bonds to the coordinated dioxygen to a ligand for the copper. This Cu/Fe derivative with Cu(I) bound mimics the structure of the heme–copper oxygenases and carries out reduction of dioxygen to peroxide.124 Replacement of copper(I) with silver(I) yields an air-stable derivative with bound dioxygen.125 This derivative is denoted Ag(I)–CuBMbO2. The NRVS spectrum of this oxygenated derivative has the two highest frequencies at 588 and 428 cm–1.123 These frequencies are higher than those seen for MbO2 and are thought to result from the influence of the second metal on the oxygenated heme center. It is also noted that the O2 affinity of this derivative is higher than that of deoxyMb even though the hydrogen-bonding histidine is not available. It should also be recognized that the NRVS spectra, especially in the in-plane region, are simpler than those of the picket fence derivatives.
3.4. Cyanide (CN–) Species—A Spin Crossover Complex
The synthesis and characterization of the five-coordinate iron(II) cyanide derivative [Fe-(TPP)(CN)]− revealed an unexpected property. Cyanide, long considered a strong-field ligand, did not provide a sufficiently strong ligand field to give a low-spin species under all conditions. Rather the compound is low spin only at lower temperatures and high spin at higher temperature, i.e., it forms a spin crossover complex (an S = 0 ⇋ S = 2 equilibrium).126 This provided an unusual opportunity, namely, the possibility to track in detail how all the iron-dependent modes of this complex change between the two spin states rather than the typical single vibration. Although the spin-crossover process is fast on the Mössbauer time scale, it is slow on the NRVS (vibrational) time scale and the vibrational components of the two spin states can be resolved. NRVS measurements were made on powder (17 K) and a general oriented single crystal between 116 (totally low spin) and 290 K (spin mixture).127
The difference in the molecular structure for the two spin states is large with equatorial Fe–Np bond distances of 1.986 (LS) vs 2.089 Å (HS), axial Fe–N(CN) distances of 1.878 (LS) vs 2.108 Å (HS), and an out-of-plane displacement from the plane of the four nitrogen atoms of 0.17 (LS) vs 0.45 Å (HS). The high-spin distances may actually be slightly larger as the spin state transition was still not quite complete at the highest achievable temperature for the structure determination. Clearly the difference in frequencies for equivalent vibrations in the two spin states can also be expected to be large. However, the correlations are not quite as simple as might be expected, owing to mixing of iron modes with different porphyrin ring modes at different temperatures and spin state.
The correlation of the out-of-plane modes is relatively straightforward as there is only very modest mixing with porphyrin modes. The observed doming mode shifts from 130 (low spin) to 108 cm–1 (high spin), correlated with the changes in the out-of-plane displacement of iron. This provides an indication of the magnitude of the change that can be expected from the differences in the displacement of iron from the four nitrogen atom plane of 0.17 Å in the low-spin form to ≥0.45 Å in the high-spin form. The observed Fe–CN stretching mode shifts from 461 cm–1 in the low-spin form to 322 cm–1 in the high-spin form. However, the C–N stretches in the two states (obtained by IR, with 2105 cm–1 low spin and 2070 cm–1 high spin) shift in the opposite direction. The inverse correlation of the two stretches is consistent with π-backbonding analogous to that observed in iron carbonyl species. The Fe to ligand π-backbonding is surely more significant in the low-spin state.
The in-plane iron modes are found to be much more mixed with porphyrin ligand modes in the low-spin state than in the high-spin state. Consequently, there is not a one to one correspondence of modes in the two states and the high-spin spectra are less complex.127 The Fe–C–N bending modes display effects from porphyrin ligand mixing. In addition to the degenerate (x and y) bending modes observed at 413.2 and 413.4 cm–1 for the LS species, there is mixing with the porphyrin ligand ν50 modes to yield a degenerate pair with FeCN bending motion predicted at 476.4 and 476.7 cm–1 and observed at 464 cm–1. The two sets of LS FeCN bending modes simplify to a pair of observed modes at 272 cm–1. The differences in the FeCN bending motion between the two spin states is depicted in Figures 15 and 16.
Figure 15.
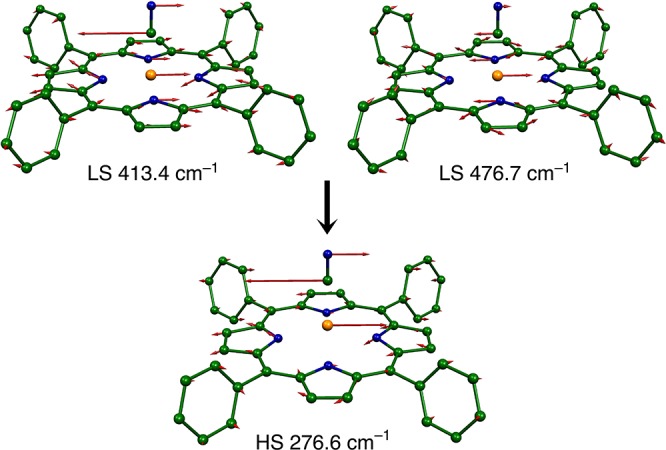
Change in the Fe–C–N bending (y direction) between the low- and the high-spin states of [K(222)][Fe(TPP)(CN)]. Frequencies given are predicted values. Note the substantial FeCN bending contribution to the porphyrin mode ν50 at 476.7 cm–1 that is not found in the high-spin state. Adapted with permission from ref (127). Copyright 2012 American Chemical Society.
Figure 16.

Change in the in-plane (y-direction) modes between the low- and the high-spin states of [K(222)][Fe(TPP)(CN)]. Again, note the differences in porphyrin ring and iron mixing. Adapted with permission from ref (127). Copyright 2012 American Chemical Society.
As is usual for low-spin complexes, the middle frequency range 220–360 cm–1 is dominated by in-plane vibrations. There are a number of bands centered around 244 (four modes) and 304 cm–1 (3 modes with ν53 character). The two regions of in-plane vibrations in the low-spin form simplify to a single region with much less mode mixing with porphyrin ring vibrations in the high-spin state. Thus, the simplification on going to the high-spin state is both a smaller number of modes involving iron motion and a lowering of the frequencies. The frequency differences in both the in-plane and the out-of-plane vibrations is consistent with the increases in both axial and equatorial bond length in the high-spin state. The changes in vibrations and their correlation with the two spin states are shown in Figure 17 with dashed lines connecting the high-spin and low-spin modes with similar iron motion.127
Figure 17.

Diagram illustrating the correlations of iron motion in the high- and low-spin states of [K(222)][Fe(TPP)(CN)]. Color coding is shown in the inset of the high-spin panel. Height of the bars gives the calculated kinetic energy contribution of iron to the mode. Horizontal axes are on the same linear scale; positions of the predicted peaks are somewhat arbitrary in order to avoid overlaps. Reprinted with permission from ref (127). Copyright 2012 American Chemical Society.
The reduced vibrational frequencies lead to a significantly increased vibrational entropy for the high-spin state. Calculations suggest that the vibrational contribution to the overall increase in entropy drives the thermal low-spin → high-spin transition with increasing temperature.
3.5. Significant NRVS-Derived Correlations in Fe(XO) Species
Table 1 provides a summary of selected observed frequencies for vibrations of five- and six-coordinate Fe(XO) species obtained from NRVS measurements. Values are given for the Fe–XO stretch, the Fe–X–O bend, the trans Fe–L stretch, and the doming mode. Values for both coordination numbers are available for {FeCO}6, {FeNO}6, and {FeNO}7 complexes. It should be recognized that the calculated kinetic energy contributions from the three atoms of the various FeXO groups, verified by experimental data when possible, provide the best possible description. As an example of the sometime confusion, the highest frequency vibration for six-coordinate MbNO has been described as a stretch, a bend, or a mixture of both,128−132 but the dominant contribution of the N atom to the kinetic energy distribution indicates that FeNO bending is the verbal description most consistent with the mode character.
Table 1. Summary of Selected Vibrations for Measured Fe(XO) Species.
| compound | measurement typea | Fe–XO stretchb | Fe–X–O bendb | Fe–L stretchb | doming modeb | ref |
|---|---|---|---|---|---|---|
| A. Five-Coordinate Species | ||||||
| [Fe(OEP)(CO)] | Gen-SC | 573 | 505 | c | 157 | (79) |
| [Fe(TMP)(CO)]d | Soln | 580 | 505 | c | c | (69) |
| [Fe(OEP)(NO)] | Spec-SC | 517 | 394 | c | 158 | (99) |
| [Fe(DPIX DME)(NO)] | Spec-SC | 528 | 399 | c | 183 | (100) |
| [Fe(OEP)(NO)]+ | Gen-SC | 595 | 402 | c | 151 | (110) |
| [Fe(TPP(CN)]−(LS) | Gen-SC | 461 | 413 | c | 130 | (127) |
| [Fe(TPP(CN)]−(HS) | Gen-SC | 322 | 277 | c | 108 | (127) |
| B. Six-Coordinate Species | ||||||
| [Fe(OEP)(1-MeIm)(CO)] | Pwd | 499/513 | 582/575 | c | c | (67) |
| [Fe(TPP)(1-MeIm)(CO)] | Gen-SC | 507 | 561/586 | 225 | 64/127 | (67) |
| [Fe(TPP)(1-PhIm)(CO)] | Pwd | 502 | 561/589 | 223 | c | (67) |
| [Fe(TPP)(1,2-DiMeIm)(CO)] | Pwd+ | 506 | 561/583 | 221 | c | (67) |
| MbCO | Soln | 502 | 572 | (68) | ||
| [Fe(ImT3MP)(CO)] | Soln | 470 | 570 | c | 110 | (69) |
| [Fe(TMP)(H2O)(CO)]d | Soln | 520 | 550 | c | c | (69) |
| [Fe(TPP)(1-MeIm)(NO)] | Gen-SC | 472/437 | 566 | 149 | c | (105) |
| mono-[Fe(TpFPP)(1-MeIm)(NO)] | Spec-SC | 473/439/426 | 561 | 153 | 131 | (109) |
| tri-[Fe(TpFPP)(1-MeIm)(NO)] | Gen-SC | 466/432 | 560 | 140 | c | (109) |
| MbNO | Soln | 443 | 547 | 130(?) | (21) | |
| [Fe(OEP)(2-MeHIm)(NO)]+ | Spec-SC | 600 | 574/580 | 238 | 152 | (110) |
| [Fe(TPP)(1-MeIm)(NO)]+ | Pwd | 578(?) | 588(?)e | c | c | (113) |
| NP2-NO | Soln | 594 | 581 | c | c | (114) |
| [Fe(TpivPP)(1-EtIm)(O2)] | Gen-SC | (571,417,393)f | 196 | 118 | (122) | |
| [Fe(TpivPP)(2-MeHIm)(O2)] | Gen-SC | (563,419,389)f | 187 | 119 | (122) | |
| [Fe(TpivPP)(1-MeIm)(O2)] | Pwd | (571,417,393)f | 205 | 118 | (122) | |
| MbO2 | Soln | 423 | 579 | c | c | (123) |
| Ag(I)–CuBMbO2 | Soln | 428 | 588 | c | c | (123) |
Spec-SC, 3 orthogonal single-crystal measurements; Gen-SC, 2 orthogonal single-crystal measurements; Pwd, powder; Pwd+, powder +1 in-plane single-crystal measurement; Soln, frozen solution.
Value in cm–1.
Not available or not assigned.
Suggested assignments, see text.
Stretch and bend assignments possibly reversed.
Stretch and bend strongly mixed.
Figure 18 displays the calculated kinetic energy contributions of the Fe, C, and O atoms of five- and six-coordinate carbonyl derivatives. From the figure it is clear that the Fe-centered mode and central atom-centered modes have reversed frequency ordering in the two coordination number derivatives, consistent with the earlier statement that the ordering of the bend and stretch are reversed between the two coordination numbers and providing a clear marker of coordination number in (all) FeXO species. The pattern for the five- and six-coordinate {FeNO}7 shows similar kinetic energy distribution shifts. In the {FeNO}6 species, the pattern of frequency changes is quite clear for the central atom-centered vibration, but the frequency for the iron-centered vibration is essentially unchanged for the two coordination numbers. Nonetheless, the pattern for distinguishing the two coordination numbers in {FeNO}6 species is quite clear.
Figure 18.

Comparison of the calculated kinetic energy contributions of the Fe, C, and O atoms in [Fe(OEP)(CO)] and [Fe(OEP)(1-MeIm)(CO)]. Vertical axis gives the value of the predicted kinetic energy contribution from each of the three atoms in FeCO at each frequency.
Since the vibrations and the structure of a molecule are expected to be closely coupled, we explore possible relationships between vibrational frequencies and observed structural parameters. A possible complication in attempting such correlations is the mixing of the selected mode with other iron or porphyrin modes that will have different components for each complex. However, there is strong correlation between the observed Fe–XO stretch and the Fe–XO bond length in the characterized six-coordinate species. This is illustrated in Figure 19. A similar correlation for the five-coordinate species is found, and a plot is given in Figure 20.
Figure 19.

Plot showing the correlation of the Fe–XO stretching frequencies determined by NRVS (highest frequency chosen when two are observed) with the observed (X-ray) Fe–X bond lengths for the six-coordinate Fe(Im)(XO) derivatives. Correlation coefficient for the fit for the nonlinear Fe–X–O derivatives is 0.96.
Figure 20.
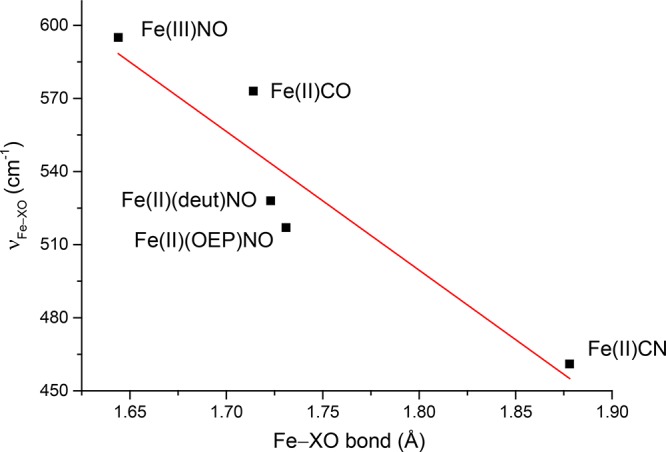
Plot showing the correlation of the Fe–XO stretching frequencies determined by NRVS (highest frequency chosen when two are observed) with the observed (X-ray) Fe–X bond lengths for the five-coordinate Fe(XO) derivatives.. Correlation coefficient for the fit is 0.93.
As we noted earlier for the six-coordinate {FeNO}6 complex, assigning the Fe–Im stretch is challenging. It is likely that the components of the Fe–Im mode are found in more than one NRVS frequency owing to mixing of these low-frequency modes. We believe we have found the most appropriate assignment for each of the six-coordinate Fe(XO) species for which we have an out-of-plane spectrum. In Figure 21 we plotted this assigned Fe–Im frequency against the observed Fe–N(Im) bond distance. As can be seen from the plot of the seven distinct species for which data is available, the correlation found is satisfying. Note that the correlations in Figures 19, 20, and 21 extends over large ranges of distances and frequencies.
Figure 21.

Correlation between Fe–N(Im) bond distances and Fe–Im stretch frequencies (νFe–Im) of six-coordinate [Fe(Porph)(RIm)(XO)] (X = N, C, O) complexes: filled triangle, [Fe(OEP)(2-MeHIm)(NO)]+; filled square, [Fe(TPP)(1-MeIm)(CO)]; open circles, [Fe(TpivPP)(2-MeHIm)(O2)] and [Fe(TpivPP)(1-MeIm)(O2)]; filled circle, [Fe(TPP)(1-MeIm)(NO)], tri-[Fe(TpFPP)(1-MeIm)(NO)], and mono-[Fe(TpFPP)(1-MeIm)(NO)]. Correlation coefficient for the fit is 0.94.
4. Other Ligand Systems and Studies
4.1. NRVS of Imidazole and Imidazolate Complexes
Heme proteins often have a histidine as an axial ligand and a covalent link to the protein. A very common motif in biology is a five-coordinate iron(II) species with the sixth position poised to bind ligands, especially the diatomics NO, CO, and O2. Vibrational methods, most commonly resonance Raman, have been used to study variations in the strength of the axial Fe–Im bond as a way of understanding the differing functions and ligand discrimination of heme proteins. However, other important vibrations such as the in-plane Fe–Np bonds are not resonance Raman active and have only rarely been reported in IR measurements at lower frequencies than commonly employed.133 The use of NRVS promised to provide new information on this important class of molecules that was not previously available.
The preparation of five-coordinate high-spin imidazole-ligated iron(II) porphyrinates requires the use of a sterically hindered imidazole, and 2-methyl- or 1,2-dimethylimidazoles with several porphyrins were used in these studies. A total of six distinct derivatives were studied, with both powder and oriented single-crystal measurements obtained.134 Practical issues allowed oriented-crystal measurements on only two of the species, but the powder spectra do provide important additional information. DFT calculations were carried out in support of the assignments for four of the derivatives.
Initial studies concentrated on deconvolving the strong signals in the 200–300 cm–1 region into the overlapped in-plane and out-of-plane modes.134 Subsequent studies, described later, provide additional details concerning the iron dynamics. The six derivatives all display strong contributions in the 200–300 cm–1 region, with obvious differences between porphyrin species with β-alkyl substituents (OEP and PPIX) or meso-tetraaryl substituents (TPP and TTP). Weaker bands are observed both above and below this region. Differences also occur between derivatives with the axial ligand 2-methylimidazole vs 1,2-dimethylimidazole. The main spectral feature for the β-alkyl substituent species is a very broad band with little feature. The tetraaryl derivatives have three sharp bands in the region when the axial ligand is 2-methylimidazole but increases to six relatively sharp bands when the axial ligand is 1,2-dimethylimidazole. All of these features attest to the importance of using complete (not truncated) molecules in DFT calculations for vibrational predictions. Vibrations from iron motion in the in-plane directions are not resonance Raman active but are accessible in NRVS. Thus, the NRVS studies provide new information on this class.
Two derivatives, [Fe(TPP)(2-MeHIm)] and [Fe(OEP)(2-MeHIm)], for which both powder and oriented single-crystal measurements are available, were selected for detailed analysis with the remaining derivatives yielding buttressing details. Four peaks are seen in the powder NRVS spectrum of [Fe(TPP)(2-MeHIm)] at 224, 248, 294, and 408 cm–1. The single-crystal spectrum clearly shows that the peaks at 408 and 294 cm–1 are in-plane vibrations corresponding to motions associated with ν50 (408 cm–1) and ν53 (294 cm–1), whereas the 248 cm–1 peak is an out-of-plane mode. The largest and broadest peak centered at 220–224 cm–1 is the result of overlapped in-plane and out-of-plane modes. The oriented single-crystal data shows that the in-plane modes are at 206 and 221 cm–1 and out-of-plane modes at 216 and 226 cm–1. Although the presence of the axial imidazole should remove the degeneracy of the x and y in-plane mode directions, none of the in-plane peaks are resolved in the experimental data.
Although the powder spectra of [Fe(OEP)(2-MeHIm)] in the 200–300 cm–1 region are only partially resolved, the single-crystal spectra reveal in-plane modes at 228, 256, 291, 320, 347, and 365 cm–1 and out-of-plane modes at 218 and 230 cm–1. The 291 cm–1 in-plane band corresponds to the similar band at 294 cm–1 in [Fe(TPP)(2-MeHIm)]. The in-plane 228 cm–1 band corresponds to the Fe–Np stretch and the degenerate ν53 pair. The 256 cm–1 band corresponds to in-plane motion of iron perpendicular to the imidazole plane coupled with imidazole translational movement. The overall appearance of the NRVS spectra of [Fe(OEP)(2-MeHIm)] and [Fe(PPIX)(2-MeHIm)] is very similar, again showing the substantial differences between tetraaryl and β-substituted porphyrin derivative. Note however that the effects on the axial modes are quite small.
The conformation of the peripheral ethyl groups in [Fe(OEP)(2-MeHIm)] appears to have substantial effects on the midrange vibrations. DFT-based predictions based on the observed conformation (all ethyl groups down, opposite the imidazole) and a second with four ethyl groups up and four down shows better agreement between the known conformation prediction and the observed spectra. Different spectral features were also predicted for two conformers of [Fe(OEP)(1,2-DiMeIm)] but where the differences were somewhat less conclusive. Similar conclusions were reached for the effect of conformers in [Fe(OEP)(NO)].96
A major point of interest in the characterization of the vibrational spectroscopy of these high-spin five-coordinate imidazole-ligated porphyrinates was to better understand the dynamics of the iron–imidazole group. The axial Fe–Im vibration is Raman active, and a peak at ∼220 cm–1 is observed in deoxymyoglobin and -hemoglobin. A number of isotope-labeling studies and site-directed Mb mutants demonstrate both iron and imidazole character to this band.135,136
The out-of-plane peaks at 216, 226, and 248 cm–1 observed for [Fe(TPP)(2-MeHIm)] and at 218 and 230 cm–1 for [Fe(OEP)(2-MeHIm)] are all predicted to have both iron and imidazole character. The lower frequency peaks in both of these complexes corresponds to the 220 cm–1 peak seen in the deoxy proteins. Is there a second peak in the protein spectra similar to the higher frequency peaks found in the small molecule species? Experimental resonance Raman studies by both Argade et al.135 and Wells et al.136 failed to find evidence for this second vibration, and only the NRVS study reveals this second frequency. The NRVS measurements demonstrate the complexity of the iron–histidine mode and explain the failure of predicting isotope shifts with a two-body model.136 The predicted nature of the out-of-plane modes for [Fe(TPP)(2-MeHIm)] is shown in Figure 22.
Figure 22.
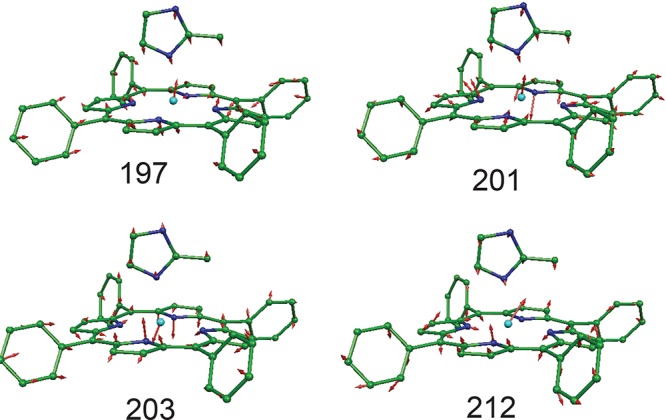
Four DFT-predicted out-of-plane Fe modes contributing to the pair of experimental features at 216 and 226 cm–1 in [Fe(TPP)(2-MeHIm)]. Reprinted with permission from ref (134). Copyright 2012 American Chemical Society.
The complexes with 1,2-dimethylimidazole as the axial ligand generally show shifts of the Fe–Im modes to lower frequency as expected from both the slightly longer Fe–N(Im) bonds and the increased mass of the ligand. The spectral changes and analysis of the [Fe(OEP)(1,2-DiMeIm)] derivative appears more complex, probably as the result of a porphyrin mode mixing with larger shifts observed compared to the tetraaryl series.
The NRVS spectrum of deoxyMb has also been measured.68 An unexpected feature was that the spectrum showed much sharper peaks than either [Fe(OEP)(2-MeHIm)] or [Fe(PPIX)(2-MeHIm)]. Nonetheless, the spectrum is still compressed into the 200–300 cm–1 region with in-plane and out-of-plane modes clearly resolved.
The most significant vibration in the very low frequency region of heme spectra is the doming mode, i.e., a large iron displacement perpendicular to the porphyrin plane with opposing motion of the outer portion of the porphyrin core. Such a motion has particular biological significance in various models of protein control of reactions at the active site of heme proteins, including Perutz’s model for hemoglobin cooperativity.70 This mode is often mixed with with other porphyrin modes. In these complexes, the doming mode is expected to be at the lower end of the ranges of observed doming modes because of the large out-of-plane displacement and the high-spin state of iron. The low-frequency out-of-plane measurement for [Fe(TPP)(2-MeHIm)] reveals features at 83 and 116 cm–1 that correspond to predicted features at 83 and 87 cm–1. The powder data for [Fe(TPP)(1,2-DiMeIm)] displays features at 94 and 128 cm–1. Similar features in [Fe(OEP)(2-MeHIm)] are found at 72 and 139 cm–1. A depiction of some low-frequency modes is given in Figure 23.
Figure 23.

Predicted oop modes at 27, 28, and 83 cm–1 for [Fe(TPP)(2-MeHIm)] (top); predicted mode at 87 cm–1 is similar to the 83 cm–1 mode. Predicted out-of-plane modes at 36 and 116 cm–1 for [Fe(OEP)(2-MeHIm)] (bottom). Reprinted with permission from ref (134). Copyright 2012 American Chemical Society.
A summary of the observed doming mode frequencies is given in Table 2. A few generalizations can be made. The comparison of tetraaryl species with octaethyl derivatives shows that the OEP species are found at higher frequencies. The doming modes in low-spin species typically are found at higher frequencies compared to high-spin species and with mass differences of the axial ligand playing a secondary role. The doming mode in six-coordinate species may be found at slightly higher frequencies than for a related five-coordinate species.
Table 2. Summary of Observed Doming Mode Frequencies.
| complex | doming modea | spin state | ref |
|---|---|---|---|
| five coordinate | |||
| [Fe(TPP)(2-MeHIm)] | 116 | high spin | (134) |
| [Fe(OEP)(2-MeHIm)] | 139 | high spin | (134) |
| [Fe(TPP)(2-MeIm–)]− | 127 | high spin | (138) |
| [Fe(OEP)(2-MeIm–)]− | 125 | high spin | (138) |
| [Fe(TPP)(NO)] | 128 | low spin | (94) |
| [Fe(OEP)(NO)] | 158 | low spin | (94) |
| [Fe(OEP)(CO)] | 157 | low spin | (79) |
| [Fe(OEP)(NO)]+ | 151 | low spin | (110) |
| [Fe(TPP)(CN)]− | 130 | low spin | (127) |
| [Fe(TPP)(CN)]− | 108 | high spin | (127) |
| [Fe(OEP)(Cl)] | 142 | high spin | (38) |
| [Fe(P)(Cl)] | 90 | high spin | (38) |
| [Fe(PPIX)(Cl)] | 135 | high spin | (38) |
| [Fe(P)(Br)] | 69 | high spin | (38) |
| six coordinate | |||
| [Fe(TPP)(1-MeIm)(CO)] | 127 | low spin | (67) |
| [Fe(TpFPP)(1-MeIm)(NO)] | 131 | low spin | (109) |
| [Fe(OEP)(2-MeHIm)(NO)]+ | 152 | low spin | (110) |
| [Fe(TpivPP)(1-EtIm)(O2)] | 118 | low spin | (122) |
| [Fe(TpivPP)(2-MeHIm)(O2)] | 104 | low spin | (122) |
| [Fe(TpivPP)(1-MeIm)(O2)] | 119 | low spin | (122) |
Value in cm–1.
The large variety of histidyl (imidazole) ligated proteins with vastly differing functions immediately raise the question about the nature and possible fine tuning of the heme site. One of the most discussed of the possible effects is the effect of hydrogen bonding to the N–H group of the histidyl ligand. There are a range of hydrogen-bonding properties to the coordinated histidine. These range from weak hydrogen bonding in the O2-binding globins to the peroxidases where an aspartate reside can form a strong hydrogen bond. These differences lead to wide differences in observed ν(Fe–NIm) frequencies from ∼218–221 cm–1 in the globins to ≥240 cm–1 in the peroxidases. Since the limit of a strong hydrogen bond is to form an imidazolate ligand, we investigated this ligand in detail. A series of investigations shows differences in the electronic structure of imidazoles vs imidazolate-ligated iron(II) complexes. These results have been summarized in ref (137). The differences in the molecular structure between imidazoles and imidazolates are shown in Figure 24.
Figure 24.

Diagram illustrating the differences in the coordination group structure and the five-membered ring geometry between the five-coordinate imidazole and imidazolate species. Number to the left in each diagram is the average F–Np distance, middle number is the iron displacement from the four nitrogen plane, and top number is the value of the axial bond distance. Distances and angles for the ligand ring are also displayed. Reproduced from ref (137) with permission. Copyright 2015 Royal Society of Chemistry.
Two pairs of complexes have been compared using NRVS: [Fe(TPP)(2-MeHIm)] and [Fe-(TPP)(2-MeIm–)]− and [Fe(OEP)(2-MeHIm)] and [Fe(OEP)(2-MeIm–)]−.138 In both pair sets, the difference in the vibrational frequencies are found to vary in the direction consistent with the structural differences. However, the magnitude of the changes are different between the two pairs and suggest the importance of differing mixing of the iron modes with the porphyrin modes that are the result of the differing peripheral substituents.
In the TPP pairs in-plane modes are found to decrease by ∼10–15 cm–1 compared to imidazoles, whereas the main out-of-plane mode (the ν(Fe–Im) mode with γ6 (inverse doming)) increases by ∼6 cm–1. The differences are qualitatively consistent with the increase of 0.036 Å in the average Fe–Np bond distance and the decrease of 0.089 Å in the axial Fe–N(Im) bond distance in the imidazolates. In the OEP series, the in-plane mode frequency difference is larger for ν53 by ∼22 cm–1, whereas the out-of-plane difference is an increase of ∼30 cm–1. The differences in the axial Fe–N(Im) stretch are comparable to the differences between the globins and the peroxidases, consistent with at least a strong hydrogen bond to the imidazole. The experimental and predicted frequencies are summarized in Table 3.
Table 3. Comparison of Calculated and Observed Normal Modes of the Imidazole and Imidazolate Derivativesa.
| type | [Fe(TPP)(2-MeHIm)] obsd (calcd) | [K(222)][Fe(TPP)(2-MeIm–)] obsd (calcd) | [Fe(OEP)(2-MeHIm)] obsd (calcd) | [K(222)][Fe(OEP)(2-MeIm–)] obsd (calcd) | description |
|---|---|---|---|---|---|
| ip | 212, 221 (220, 212, 228, 222) | 208 (207, 210, 212, 218) | |||
| ip | 294(292, 295) | 277 (272, 274) | 228 (220, 224, 230b) | 224 (216, 226) | ν53 |
| ip | 408 (407, 411) | 394 (390, 392) | 256, 291 (263, 288, 289) | 244, 266(sh) (250, 256, 260, 271) | ν50 |
| oop | 216, 226 (197, 201, 203, 212) | ||||
| oop | 248 (266) | 254 (260) | 218, 230 (220, 230b) | ∼260 (248, 256, 264) | γ6 (inverse doming) |
| oop | 83, 116 (83, 87) | 58, 103, 127 (80, 85) | 72, 139 (116) | 60, 115, 125 (103) | γ9 (doming) |
| ref | (134) | (138) | (134) | (138) |
All frequencies are expressed in cm–1.
230 cm–1 mode has both ip and oop character.
Although the importance of the orientation of planar axial ligands on the electronic structure of iron porphyrinates has been recognized for some time,139−151 the possible effects of the orientation of planar ligands such as imidazoles on the in-plane iron vibrations had not been established. New NRVS measurements in three orthogonal directions have been made for [Fe(OEP)(2-MeHIm)].152 This five-coordinate complex is found in a crystallographic system that allows such complete orientation studies. The measured spectra in the porphyrin plane in the directions parallel and perpendicular to the imidazole plane are shown in Figure 25 with prominent, but differing, peaks and intensities in the 220–270 cm–1 region. As is clearly evident from these spectra, the axial imidazole has removed the in-plane degeneracy in the x and y directions. This is reminiscent of the loss of in-plane degeneracy in the case of [Fe(OEP)(NO)] described earlier (Figure 10). Indeed the mirror plots show strong similarities. However, unlike the nitrosyl case, where the axial NO ligand orientation had a strong effect on the direction of the in-plane iron motion, the imidazole has only a very modest effect on the directions of the in-plane iron motion. We have compared predictions of the in-plane motions for four-coordinate [Fe(OEP)] with the analogous in-plane motions predicted for [Fe(OEP)(2-MeHIm)]. The in-plane iron motions of [Fe(OEP)] are directly along the Fe–Np bonds, whereas in [Fe(OEP)(2-MeHIm)] there are small deviations from those directions that tend toward being parallel or perpendicular to the imidazole plane, suggesting modest effects from imidazole. The biggest effect of the imidazole is to lift the x, y degeneracy.
Figure 25.

Comparison of the measured Fe VDOS in [Fe(OEP)(2-MeHIm)] in the directions parallel (top) and perpendicular (bottom) to the imidazole plane. Adapted with permission from ref (152). Copyright 2013 American Chemical Society.
Predictions of in-plane iron directionality for the imidazolate complex on the other hand are quite distinct with motion primarily parallel or perpendicular to the imidazolate plane. Unfortunately, we were unable to confirm this experimentally owing to unfavorable crystalline symmetries. The series of the three complexes ([Fe(OEP)(L)], L = NO, imidazolate, and imidazole) for which we have explored iron motion directionality shows that the axial ligand effects on the direction of the in-plane iron motion follows the order NO > imidazolate > imidazole. This series follows the order of the importance of iron to ligand π-backbonding, with the smallest π-bonding in the imidazole complex.153 The axial Fe–N bond distances in the three systems are 1.722,97 2.058,154 and 2.147 Å,155 respectively.
4.2. NRVS with Other Axial Ligands
NRVS spectra have been recorded for two myoglobin derivatives: one derivative has an oxo ligand, formally Fe(IV)=O termed ferryl, and one with hydroxo, formally Fe(III)–OH.156 This study was an effort to better understand the nature of the reactive monooxygenated heme species derived from cleavage of the dioxygen ligand bound to heme iron and whether the oxygen atom of the resulting ferryl species was protonated. The NRVS study provided the frequency of the axial Fe–O in the two species with a value of 805 cm–1 for the stretching vibration in the ferryl complex and 556 cm–1 for the hydroxide complex. DFT calculations suggest that the bond distance difference would be approximately 0.17 Å. These differences are inconsistent with a protonated ferryl species, and the ferryl should be simply Fe—O. The problems with the issues are the result of reduction of the ferryl in X-ray beam during data collection and the resulting structural change.
The second feature in the NRVS spectra consistent with the difference in protonation state comes from the observation of the tilt in the Fe–O bond relative to the porphyrin plane that is coupled to stretching of the in-plane Fe–Np bonds. Protonation of the oxygen would lift the degeneracy of the vibration. The protonated Fe–OH group would lift the nominal degeneracy with the tilting in the Fe–OH plane predicted to have a higher frequency for the vibration relative to the Fe–O tilting perpendicular to the Fe–OH plane. A single vibration at 362 cm–1 is observed in the ferryl, whereas a 30 cm–1 splitting (343 and 376 cm–1) is observed for the hydroxide. This observation is again consistent with an unprotonated ferryl derivative.
These results and conclusions have since been corroborated by a dose-dependent X-ray diffraction study.157 High X-ray doses in the structure determination of the peroxidase ferryl intermediate lead to apparent reduction and a Fe–O bond distance of 1.90 Å, consistent with a hydroxide ligand, whereas the low-dose X-ray study leads to a structure with an Fe–O bond distance of 1.73 Å, consistent with an Fe—O group. The apparent change in oxidation state during the X-ray studies was followed by single-crystal electronic spectra.
4.3. c-Type Cytochromes
The hemes in c-type cytochromes are covalently attached to the cysteine sulfurs of a characteristic CxxCH peptide motif whose cysteine residues form thioether links to the vinyl groups of the heme, whereas the terminal histidine of the CxxCH sequence provides one of two axial ligands to the heme Fe.158,159 The unusual covalent attachment to the protein distorts the heme from planarity, resulting in an unusually asymmetric environment for the heme Fe.
The experimental VDOS presented in Figure 26 resulted from NRVS measurements on reduced horse cytochrome c(160,161) (cyt c), in which the second axial ligand is the sulfur atom of a methionine residue, and illustrates the complex vibrational dynamics of the heme Fe in this environment. Complementary Raman measurements on 57Fe- and 54Fe-enriched cyt c identified at least nine Raman-active bands that contribute to the highly congested 300–450 cm–1 region of the VDOS. Specifically, the frequency shift upon isotope labeling yields a quantitative estimate
| 1 |
for the area that mode α contributes to the VDOS. Together, Raman bands account for 70% of the VDOS in the 300–450 cm–1 region. Several additional unidentified Fe vibrations must be present to account for the remaining area and are not observable by Raman.
Figure 26.
Vibrational densities of states determined from NRVS measurements on oxidized and reduced cytochrome c. Upper panel compares the contributions expected from vibrations observed in Raman spectra, based on frequency shifts between 54Fe- and 57Fe-enriched samples according to eq 1. Solid black curve in the upper panel is the sum of contributions from individual Raman-active modes, indicated as dashed curves.161
NRVS results reported for a cyt c from the bacterium H. thermophilus(162,163) as well as for cyt f,164 in which the amino terminus of the polypeptide provides the second axial ligand to the heme Fe, exhibit similarly complex spectra. Although isotope shift data were unavailable, comparison with Raman data led to the identification of 15 unresolved frequencies ranging from 284 to 445 cm–1 for cyt f.164 A similar number of bands resulted from Gaussian deconvolution of the VDOS of H. thermophilus cyt c, unconstrained by Raman results.162
The 5 cm–1 57Fe/54Fe isotope shift of the 372 cm–1 mode in horse cyt c indicates a large Fe contribution to the atomic motion (eFe2 = 0.47). Leu et al. raised the possibility that this vibration might be associated with stretching of the Fe–S bond to methionine,161 but calculations performed to model NRVS results on H. thermophilus cyt c suggest that the stretching of the axial Fe–S and Fe–N bonds is distributed among many modes in the 145–325 cm–1 range.162
Alterations in the experimental signal confirm the sensitivity of the VDOS to structural changes as a function of oxidation state,160,164 point mutations,163 or species differences. Simulations can reproduce qualitative changes observed for cyt c mutants by varying the heme distortion and the force constants of the Fe–His link and the CxxCH peptide.163 However, the spectral complexity renders it difficult to identify unambiguous markers for specific structural changes.
4.4. Model-Independent Extraction of Experimental Force Constants
The data on c-type cytochromes illustrate a fundamental limitation of traditional approaches to vibrational spectroscopy for characterizing highly asymmetric metal environments. The large number of vibrations contributing to the vibrational dynamics of the Fe atom indicates that it is not always possible to establish a one-to-one correspondence between Fe–ligand bonds and vibrational markers that directly measure the strength of those bonds. In fact, NRVS lacks the frequency resolution to experimentally resolve all of the contributing vibrations. Even when the majority of vibrational modes with Fe–ligand character are resolved using Raman measurements, it is not straightforward to use this set of frequencies to characterize Fe coordination.
The unusually quantitative information content of NRVS data provide a direct experimental means to characterize the strength of the coordination sphere even when it is not possible to associate individual modes with Fe–ligand vibrations. Lipkin24 identified an effective force constant
| 2 |
determined by averaging the quantity mjω2 over frequency, weighted by the VDOS of atom j. We dubbed this quantity the stiffness(161) and showed that, for a harmonic system, ks quantifies the force F = ksxj required to displace atom j with other atoms fixed at their equilibrium positions.166 As an alternative to eq 2, the stiffness can also be determined directly from the raw data, without extracting the VDOS.24
Stiffnesses reported for iron porphyrinates and heme proteins illustrate its ability to measure Fe coordination (Table 4). The largest changes reflect the strong correlation of bond strength with spin state, with stiffnesses in the 170–200 and 280–370 pN/pm ranges reported for high-spin and low-spin hemes, respectively. This trend is expected because of the stronger coordination and shorter Fe–ligand bonds observed for low-spin complexes. The stiffness may be able to gauge more subtle coordination changes as well. For example, reduction of cyt c increases the stiffness by approximately 40 pN/pm, which is consistent with a 4 pm shortening of the axial Fe–S bond.161
Table 4. Summary of Stiffness of Heme Fe in Various Environments.
| stiffness (pN/pm) | spin | oxidation | axial ligands | ref | |
|---|---|---|---|---|---|
| cyt c | 322 ± 17 | 0 | +2 | His, Met | (161) |
| cyt c | 284 ± 17 | 1/2 | +3 | His, Met | (161) |
| cyt f | 342 ± 18 | 0 | +2 | His, N-term | (164) |
| cyt f | 313 ± 34 | 1/2 | +3 | His, N-term | (164) |
| MbNO | 368 ± 10 | 1/2 | +2 | His, NO | (105) |
| Zn(II)–FeBMbNO | 321 ± 21 | 1/2 | +2 | NO | (165) |
| MbO2 | 371 ± 9 | 0 | +2 | His, O2 | (123) |
| Ag(I)–CuBMbO2 | 373 ± 13 | 0 | +2 | His, O2 | (123) |
| deoxyMb | 190 ± 20 | 2 | +2 | His | (160), (161) |
| Fe(II)–FeBMb | 182 ± 9 | 2 | +2 | His | (165) |
| metMb | 235 ± 4 | 5/2 | +2 | His, H2O | (123) |
| [Fe(P)(Cl)] | 192 ± 2 | 5/2 | +3 | Cl– | (38) |
| [Fe(OEP)(Cl)] | 196 ± 6 | 5/2 | +3 | Cl– | (38) |
| [Fe(PPIX)(Cl)] | 197 ± 4 | 5/2 | +3 | Cl– | (38) |
| [Fe(TPP)(2-MeHIm)] | 188 ± 1 | 2 | +2 | 2-MeHIm | (138) |
| [Fe(TPP)(2-MeIm–)]− | 171 ± 5 | 2 | +2 | 2-MeIm– | (138) |
It is equally important to note the relative insensitivity of the stiffness to structural changes more distant from the Fe. Although the VDOS of a series of ferric porphyrin chloride complexes with differing peripheral substituents as well as a series of five-coordinate {FeNO}7 complexes94 exhibit very noticeable qualitative variations, the stiffnesses show no significant quantitative difference.38 Similarly, the stiffnesses reported for imidazole-ligated ferrous iron porphyrins and histidine-ligated ferrous heme proteins fall within a 10 pN/pm range that is narrower than the experimental uncertainties. On the other hand, a stiffness decrease of nearly 20 pN/pm upon imidazole deprotonation is consistent with significant but anticorrelated changes reported for both axial and equatorial Fe–N bond lengths.138
The resilience
| 3 |
has the same dimensions as the stiffness but an entirely distinct interpretation. For a harmonic system, the resilience kr quantifies the force F = krxj required to displace an atom by a distance xjwith the surrounding atoms free to respond but the center of mass fixed.166 The integrands of the integrals in eqs 2 and 3, displayed in Figure 27, highlight the distinct frequency regions that determine the stiffness and resilience. For heme proteins, the stiffness depends primarily on frequencies above 200 cm–1, where we expect to find localized Fe–ligand vibrations. In contrast, frequencies well below 100 cm–1 dominate the integrand of eq 2.
Figure 27.

Plots of integrands in eqs 2 and 3 illustrate the relative importance of high- and low-frequency regions to determining the stiffness and resilience, respectively.
Vibrations in this low-frequency region have a character distinct from the localized Fe–ligand vibrations that determine the stiffness. For proteins, MD simulations indicate a significant contribution from rigid motion of the heme in response to oscillations of the surrounding protein.160 Although there is no realistic opportunity to resolve the large number of contributing vibrations experimentally, the value of identifying individual modes in this region is questionable in any case. However, the resilience offers an averaged quantitative measure that may be a useful way to quantify the matrix in which the heme is embedded. In the language of inorganic chemistry, the resilience may be thought of as an “outer-sphere” force constant, in contrast with the “inner-sphere” force constant provided by the stiffness.
The resilience has been reported for only a few heme proteins. Values of approximately 20 pN/pm were determined both for native deoxyMb and for FeBMb, a mutant with a second nonheme metal site engineered near the heme to mimic the active site of NO reductase.160,165 It is noteworthy that the resiliences determined for the neighboring heme and nonheme Fe sites are experimentally indistinguishable,165 consistent with the notion that kr quantifies a property of the common matrix in which the two sites are embedded. In contrast, the resilience determined for both oxidized and reduced cyt c is approximately 50% larger than for Mb.160 A more rigid protein structure and the high degree of covalent linkage to the polypeptide are both factors that may contribute to the enhanced cyt c resilience.
Accurate experimental determinations of both stiffness and resilience require some care in data analysis. In particular, accurate subtraction of any frequency-independent background signal is essential to reduce systematic errors in determination of the stiffness. The experimental uncertainties reported in Table 4 reflect only random errors due to experimental counting statistics. On the other hand, the resilience is sensitive to anomalies at very low frequency that result from careless subtraction of the recoilless resonance that dominates the experimental signal. Such errors can be minimized when a reliable measurement of the experimental resolution function is available.
In principle, both stiffness and resilience are tensor quantities, with values that vary with molecular orientation. To our knowledge, the only investigation where this has been taken advantage of involved measurements of oriented crystals of [Fe(TPP)(2-MeHim)].94 In this case, the stiffness value 209 ± 6 determined from measurements parallel to the porphyrin plane was significantly larger than the 153 ± 9 value determined perpendicular to the plane.138 Notice that these values appropriately bracket the value determined for a polycrystalline powder (Table 4).
4.5. Nonvibrational Contributions to Fe Dynamics
The low-frequency vibrations that determine the resilience also dominate the spontaneous thermal fluctuations of the Fe on sufficiently fast time scales or at sufficiently low temperatures. The solid line in Figure 28 indicates the vibrational contribution to the mean-squared-displacement (MSD) ⟨xFe2⟩ of the Fe in oxidized cyt c as a function of temperature, as calculated from the VDOS determined from NRVS measurements at 68 K. The original definition of the resilience in terms of the temperature dependence of the MSD160,175 emphasizes this connection.
Figure 28.

Comparison of effective MSDs obtained from Mössbauer measurements with the pure vibrational MSDs calculated from NRVS measurements on oxidized cyt c.
Other experimental methods for probing atomic fluctuations include elastic scattering and conventional Mössbauer spectroscopy. Quantitative comparison with the vibrational MSD determined using NRVS holds interest because of the differing time scales of the various measurements. Data points in Figure 28 are “effective” MSD values
| 4 |
that were determined from integrated spectral areas A observed in Mössbauer measurements on a bacterial cyt c.174 Here, k = 2π/λ (λ = 0.86 Å for 57Fe). Equation 4 yields the actual MSD ⟨xFe2⟩ under the assumption of an isotropic, Gaussian distribution of atomic positions, as expected for harmonic vibrations. The nearly linear variation of the effective MSDs with temperature below 200 K has been interpreted as evidence for purely vibrational contributions, and the arbitrary constant in eq 4 is adjusted so that the effective MSDs extrapolate to zero at T = 0. The fact that the effective MSDs track the vibrational predictions from NRVS measurements below 200 K confirms this interpretation. Moreover, this comparison establishes a quantitative baseline for identifying contributions from additional dynamical processes.
Substantial deviations from the harmonic extrapolation to higher temperatures, as illustrated in Figure 28, have been attributed to additional dynamics resulting from conformational fluctuations of the protein.174 The measurements include contributions from all atomic motions faster than the 150 ns lifetime of the 57Fe excited state. It is important to note that these additional motions do not lead to a distribution of atomic positions that is either Gaussian or isotropic in general, and thus, the experimental quantity δ2 defined by eq 4 is no longer a quantitative measure of the actual MSD of these motions.
The “dynamical transition” near 200 K was originally identified in Mössbauer measurements on myoglobin.176,177 A subsequent report of a similar transition using (elastic) neutron scattering178 opened an investigation to a wider range of biomolecules, including nucleic acids181 as well as both soluble and membrane proteins. The nature of the additional dynamics at higher temperature remains the subject of ongoing investigation and discussion, although there is general agreement that they are activated by fluctuations in the surrounding solvent.179,180,182
In light of the great attention that these dynamics measurements have received in complex biomolecules, it is somewhat surprising how little they have been applied to understand simpler dynamical processes in smaller molecules. Some time ago we observed that the Mössbauer spectrum of the five-coordinate nitrosyl derivative [Fe(TPP)(NO)] unexpectedly showed a much larger temperature dependence on the signal intensity than was usually observed, with very low intensity at room temperature and below. This was confirmed by comparison with the temperature dependence of another five-coordinate nitrosyl [Fe(OEP)(NO)]. We hypothesized that this intensity effect in [Fe(TPP)(NO)] was the result of a profound crystalline disorder that is likely to be a dynamical process. Further study of [Fe(TPP)(NO)] led to additional interesting observations. We found through temperature-dependent X-ray diffraction studies that the complex underwent an important phase change that substantially decreases or eliminates the FeNO group disorder. In the high-temperature phase, the FeNO group has eight distinct orientations (4/m symmetry),167 whereas after the phase change there only two (Ci symmetry)168 and no movement of the FeNO group is likely in the low-temperature phase. The phase change was found to be gradual and occur between ∼200 and 245 K. Similar motions and an order/disorder phase change have also been observed in a five-coordinate cobalt nitrosyl species.169−171 Observations on the temperature-dependent ordering of the FeNO group in several six-coordinate species makes clear that the order/disorder of the nitrosyl group must involve rotation around the Fe–N–O bond.172,173
How can the FeNO disorder (rotation) affect the apparent mean-squared-displacement of iron? Since a standard feature of the Fe–N–O group in hemes is an off-axis tilt as well as a consequent asymmetry in the equatorial Fe–N bonds,97,98 the 4-fold disorder of the FeNO group on each side of the porphyrin plane leads to a shift from the average position of the iron with every Fe–N–O rotational jump. This yields a “smearing” of the iron atom position and a larger value of the effective MSD.
However, NRVS measurements provide a way to predict the mean-squared displacement of iron in [Fe(TPP)(NO)] in the absence of this high-temperature disorder and thus provide at least a qualitative explanation into the temperature-dependent Mössbauer signal.
A plot comparing the temperature-dependent effective iron MSDs from the NRVS and Mössbauer experiments for [Fe(TPP)(NO)] is shown in Figure 29. The effective iron MSDs between the two measurements for [Fe(TPP)(NO)] only begin to deviate above ∼200 K with the differences becoming larger with increasing temperatures. We believe that the additional contribution to δ2 in the Mössbauer experiment is related to the reversible phase change for [Fe(TPP)(NO)] and the concomitant order/disorder of the FeNO group.
Figure 29.

Comparison of the predicted MSDs obtained from the powder NRVS spectrum and the effective MSDs obtained obtained from temperature-dependent Mössbauer measurements for [Fe(TPP)(NO)].
The effective iron MSD values for [Fe(OEP)(NO)] from the Mössbauer and NRVS experiments are seen to be comparable over the entire temperature range and show that the two measurements have a close correspondence over the complete temperature range, in distinct contrast to that seen for [Fe(TPP)(NO)]. Two separate sets of effective MSDs from independent Mössbauer measurements are shown in Figure 30. We can conclude that the values of δ2 determined from both sets of measurements represent essentially only vibrational contributions from iron, and the basis for such comparisons is secure.
Figure 30.

Comparison of the predicted MSDs obtained from the powder NRVS spectrum and the effective MSDs obtained from a temperature-dependent Mössbauer measurement for Fe(OEP)(NO).
Although the differences in the effective MSD values of [Fe(TPP)(NO)] require further investigation to yield a quantitative explanation, the importance of the effects of the FeNO disorder on the Mössbauer intensities is quite clear.
5. Summary and Outlook
The NRVS technique provides unique site selectivity and vibrational spectral completeness for Mössbauer-active nuclei. For iron-containing samples, every vibrational mode contributes to the experimental signal in direct proportion to the mean-squared displacement of the 57Fe atom in which iron motion is present. We detail how and why this is important in the succeeding paragraphs.
Perhaps the most consequential feature of NRVS is that there are no arbitrary optical selection rules. This means that important vibrations forbidden or otherwise not seen by other methods will be observed by NRVS. One outstanding example is that of the Fe–Im stretch in six-coordinate species with various diatomic XO ligands trans to imidazole that allowed further quantitative understanding of the effect of the XO ligand on the Fe–N(Im) bond. Although the strength of this interaction is a significant feature that in principle provides unique information for understanding the mechanism and bonding, the Fe–N(Im) frequency is unavailable from Raman or IR experiments on six-coordinate hemes.
NRVS provides for convenient exploration of the low-frequency modes including the doming mode, i.e., the perpendicular motion of iron out-of-plane with porphyrin ring atom motions in the opposite direction. Such motions are significant for understanding the nature of axial ligand association/dissociation and possible ligand sensing.
In favorable cases, NRVS measurements on oriented single-crystal samples give unequivocal information about the character of each observed signal. The combination of oriented single-crystal NRVS measurements coupled with DFT-predicted frequencies provides the most definitive assignment of modes possible. The combination also provides for a powerful examination of mode localization. An important example was given in the characterization of the highest frequency mode observed in the six-coordinate {FeNO}7 nitrosyls that differs significantly from the conventional description given in Raman studies and is quite different from that arising from the 14NO/15NO isotope-labeled study. The NRVS-defined atom motions were found to be quite different from that of a classical Fe–N(NO) stretch as inferred by some from Raman spectra.
It is important to note the power of oriented single-crystal measurements relative to that of isotopic substitution of, for example, an axial ligand. This results because the single-crystal measurement provides information on the nature of the mode and possible assignment over the entire range of iron-based frequencies, whereas isotopic substitution provides useful (and occasionally misleading) information over a limited set of vibrations.
The selectivity of NRVS that stems from a Mössbauer-active nucleus allows the specific labeling of one site in a system with multiple sites to allow exploration of the importance of individual sites in a complex system.
Finally, and fundamentally, the strength of the NRVS technique results from its uniquely quantitative nature. The experimental NRVS signal for each resolved frequency yields an absolute measure of the mean-squared displacement of the probe atom (here, 57Fe) along the direction of the X-ray beam for the corresponding vibrational mode.
The utility of NRVS has already led to substantial oversubscription for time available for experimental measurements. However, there are currently planned upgrades for the APS, ESRF, and PETRA III lines. The exact improvements to be made are not completely certain but are expected to include better focusing, enhanced beam intensities, and detector enhancements. These will certainly allow measurements for samples with lower concentrations of iron, including importantly more protein samples, less measurement time per sample, and increased throughput. More speculatively, advanced microfocusing could permit closer study of select regions of samples, perhaps even cells. More sophisticated single-crystal experiments, including perhaps proteins, would be possible. These expected improvements might even provide the means for examining enzymatic systems during reaction cycles or other reactive species. Exciting times are ahead!
Acknowledgments
We thank our collaborators in this research whose names are given in the citations. Research reported in this publication was supported by the National Institutes of Health under Grant GM-38401 to W.R.S. and the National Science Foundation under CHE-1026369 to J.T.S. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We especially thank the members of the technical staff of Sector 3 for their assistance during our work at the beamline.
Glossary
Abbreviations
- 222
4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane
- Cp
cyclopentadienyl
- DFT
density functional theory
- EPR
electron paramagnetic resonance
- FTIR
Fourier transform infrared spectroscopy
- 1,2-DiMeIm
1,2-dimethylimidazole
- DPIX DME
dianion of deuteroporphyrin IX dimethyl ester
- 1-EtIm
1-ethylimidazole
- 1-MeIm
1-methylimidazole
- HOMO
highest occupied molecular orbital
- ImT3MP
dianion of imidazole-tailed-trimesitylporphyrin
- L
general ligand
- 2-MeHIm
2-methylimidazole
- 4-MeHIm
4-methylimidazole
- Nax
nitrogen of axial ligands
- NIm
imidazole nitrogen
- Np
porphyrinato nitrogen
- NMR
nuclear magnetic resonance
- NRVS
nuclear resonance vibrational spectroscopy
- OEP
dianion of 2,3,7,8,12,13,17,18-octaethylporphyrin
- OEPOH
dianion of octaethyl-meso-hydroxyporphyrin
- QCC-NCA
quantum chemistry at the center of the conventional coordinate analysis.
- 1-PhIm
1-phenylimidazole
- Porph
generalized porphyrin dianion
- PPIX
dianion of protoporphyrin IX
- PPIX DME
dianion of protoporphyrin IX dimethyl ester
- Py
pyridine
- R-Im and/or Im
generalized imidazole
- rR
resonance Raman
- THF
tetrahydrofuran
- TpFPP
dianion of tetra-p-fluorophenylporphyrin
- TMP
meso-tetramesitylporphyrin dianion
- TpivPP
dianion of α,α,α,α-tetrakis(o-pivalamidophenyl)porphyrin (picket fence)
- TpOCH3PP
dianion of tetra(p-methoxyphenyl)porphyrin
- TPP
dianion of meso-tetraphenylporphyrin
- TTP
dianion of meso-tetratolylporphyrin
- VDOS
vibrational density of states
Biographies
W. Robert Scheidt received his B.S. degree from the University of Missouri—Columbia and Ph.D. degree in Inorganic Chemistry from the University of Michigan with Paul Rasmussen. He had a 2 year postdoctoral stay with Prof. J. L. Hoard at Cornell University, where he studied higher coordination number complexes and began studies into metalloporphyrin synthesis and physical characterization (over the years, X-ray, magnetic susceptibility, Mössbauer, EPR, and other spectroscopies, including NRVS). He joined the faculty in Chemistry and Biochemistry at the University of Notre Dame, where he rose through the ranks and became the Wm. K. Warren Professor of Chemistry and Biochemistry in 1999. He now continues as Emeritus and Research Professor.
Jianfeng Li was born in Shanxi, China (1974). He received his B.S. degree in 1996 and Ph.D. degree in 2003 from Shanxi University under the supervision of Prof. Diansheng Liu. Then he joined Prof. W. Robert Scheidt’s group as a postdoctoral associate in the Department of Chemistry and Biochemistry at the University of Notre Dame. He returned to Beijing, China in 2011 and became a Professor at the University of Chinese Academy of Sciences (UCAS). His research activity is focused on the geometric and electronic structures of model compounds which undertake the functions of the metalloproteins.
J. Timothy Sage received his B.S. degree in Physics from Carnegie-Mellon University in 1979 and his Ph.D. degree in Physics from the University of Illinois at Urbana—Champaign in 1986 under the supervision of Prof. Peter G. Debrunner. Following several years of postdoctoral experience, he joined the Physics faculty at Northeastern University in 1994. He develops and applies spectroscopic approaches to investigate the structure and dynamics of proteins and other biological molecules.
The authors declare no competing financial interest.
References
- Seto M.; Yoda Y.; Kikuta S.; Zhang X. W.; Ando M. Observation of Nuclear Resonant Scattering Accompanied by Phonon Excitation Using Synchrotron Radiation. Phys. Rev. Lett. 1995, 74, 3828–3831. 10.1103/PhysRevLett.74.3828. [DOI] [PubMed] [Google Scholar]
- Sturhahn W.; Toellner T. S.; Alp E. E.; Zhang X.; Ando M.; Yoda Y.; Kikuta S.; Seto M.; Kimball C. W.; Dabrowski B. Phonon Density of States Measured by Inelastic Nuclear Resonant Scattering. Phys. Rev. Lett. 1995, 74, 3832–3835. 10.1103/PhysRevLett.74.3832. [DOI] [PubMed] [Google Scholar]
- Keppler C.; Achterhold K.; Ostermann A.; van Bürck U.; Potzel W.; Chumakov A. I.; Baron A. Q.; Rüffer R.; Parak F. Determination of the Phonon Spectrum of Iron in Myoglobin Using Inelastic X-Ray Scattering of Synchrotron Radiation. Eur. Biophys. J. 1997, 25, 221–224. 10.1007/s002490050034. [DOI] [PubMed] [Google Scholar]
- Paulsen H.; Winkler H.; Trautwein A. X.; Grünsteudel H.; Rusanov V.; Toftlund H. Measurement and Simulation of Nuclear Inelastic-Scattering Spectra of Molecular Crystals. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 975–984. 10.1103/PhysRevB.59.975. [DOI] [Google Scholar]
- Leupold O.; Chumakov A. I.; Alp E. E.; Sturhahn W.; Baron A. Q. R. Noniron Isotopes. Hyperfine Interact. 1999, 123/124, 611–631. 10.1023/A:1017000713255. [DOI] [Google Scholar]
- Paulsen H.; Benda R.; Herta C.; Schünemann V.; Chumakov A. I.; Duelund L.; Winkler H.; Toftlund H.; Trautwein A. X. Anisotropic Nuclear Inelastic Scattering of an Iron (II) Molecular Crystal. Phys. Rev. Lett. 2001, 86, 1351–1354. 10.1103/PhysRevLett.86.1351. [DOI] [PubMed] [Google Scholar]
- Mao H. K.; Xu J.; Struzhkin V. V.; Shu J.; Hemley R. J.; Sturhahn W.; Hu M. Y.; Alp E. E.; Vocadlo L.; Alfe D.; et al. Phonon Density of States of Iron up to 153 Gigapascals. Science 2001, 292, 914–916. 10.1126/science.1057670. [DOI] [PubMed] [Google Scholar]
- Paulsen H.; Rusanov V.; Benda R.; Herta C.; Schünemann V.; Janiak C.; Dorn T.; Chumakov A. I.; Winkler H.; Trautwein A. X. Metastable Isonitrosyl Structure of the Nitroprusside Anion Confirmed by Nuclear Inelastic Scattering. J. Am. Chem. Soc. 2002, 124, 3007–3011. 10.1021/ja016239c. [DOI] [PubMed] [Google Scholar]
- Parak F.; Achterhold K. Protein Dynamics Studied on Myoglobin. Hyperfine Interact. 1999, 123/124, 825–840. 10.1023/A:1017004814164. [DOI] [Google Scholar]
- Bergmann U.; Sturhahn W.; Linn D. E. Jr.; Jenney F. E. Jr.; Adams M. W.; Rupnik K.; Hales B. J.; Alp E. E.; Mayse A.; Cramer S. P. Observation of Fe-H/D Modes by Nuclear Resonant Vibrational Spectroscopy. J. Am. Chem. Soc. 2003, 125, 4016–4017. 10.1021/ja028767+. [DOI] [PubMed] [Google Scholar]
- Achterhold K.; Parak F. G. Protein Dynamics: Determination of Anisotropic Vibrations at the Haem Iron of Myoglobin. J. Phys.: Condens. Matter 2003, 15, S1683–S1692. 10.1088/0953-8984/15/18/302. [DOI] [Google Scholar]
- Sturhahn W.; Jackson J.. Geophysical applications of nuclear resonant spectroscopy. In Advances in High-Pressure Mineralogy; Ohtan E., Ed.; Geological Society of America: McLean, VA, 2007; Vol. 421; pp 157–174. [Google Scholar]
- Achterhold K.; Keppler C.; Ostermann A.; van Bürck U.; Sturhahn W.; Alp E. E.; Parak F. G. Vibrational Dynamics of Myoglobin Determined by the Phonon-Assisted Mössbauer Effect. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2002, 65, 051916. 10.1103/PhysRevE.65.051916. [DOI] [PubMed] [Google Scholar]
- Alp E. E.; Sturhahn W.; Toellner T. S.; Zhao J.; Leu B. M.. Nuclear Resonance Scattering of Synchrotron Radiation as a Unique Electronic, Structural, and Thermodynamic Probe. In The Rudolf Mössbauer Story; Kalvius M., Kienle P.; Eds.; Springer-Verlag: Berlin, Heidelberg, 2012; pp 339–356. [Google Scholar]
- Cvetkovic A.; Menon A. L.; Thorgersen M. P.; Scott J. W.; Poole F. L. II; Jenney F. E. Jr.; Lancaster W. A.; Praissman J. L.; Shanmukh S.; Vaccaro B. J.; et al. Microbial Mtalloproteomes are Largely Uncharacterized. Nature 2010, 466, 779–782. 10.1038/nature09265. [DOI] [PubMed] [Google Scholar]
- Visscher W. M. Study of Lattice Vibrations by Resonance Absorption of Nuclear Gamma Rays. Ann. Phys. 1960, 9, 194–210. 10.1016/0003-4916(60)90028-2. [DOI] [Google Scholar]
- Singwi K. S.; Sjölander A. Resonance Absorption of Nuclear Gamma Rays and the Dynamics of Atomic Motions. Phys. Rev. 1960, 120, 1093–1102. 10.1103/PhysRev.120.1093. [DOI] [Google Scholar]
- Mössbauer R. L. Nuclear Resonance Fluorescence of Gamma Radiation in 191Ir. Eur. Phys. J. A 1958, 151, 124–143. 10.1007/BF01344210. [DOI] [Google Scholar]
- Ogata H.; Krämer T.; Wang H.; Schilter D.; Pelmenschikov V.; van Gastel M.; Neese F.; Rauchfuss T. B.; Gee L. B.; Scott A. D.; et al. Hydride Bridge in [NiFe]-Hydrogenase Observed by Nuclear Resonance Vibrational Spectroscopy. Nat. Commun. 2015, 6, 7890. 10.1038/ncomms8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt W. R.; Durbin S. M.; Sage J. T. Nuclear Resonance Vibrational Spectroscopy-NRVS. J. Inorg. Biochem. 2005, 99, 60–71. 10.1016/j.jinorgbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Zeng W.; Silvernail N. J.; Scheidt W. R.; Sage J. T.. Nuclear Resonance Vibrational Spectroscopy (NRVS). In Applications of Physical Methods to Inorganic and Bioinorganic Chemistry; Scott R. A., Lukehart C. M., Eds.; John Wiley and Sons, Ltd.; Chichester, UK, 2007; pp 401–421. [Google Scholar]
- A possible exception might be a sample where a characteristic vibration is at an energy unlikely to be present in any impurity. An example might be the vibration from the terminal Fe=O bond.
- Sage J. T.; Paxson C.; Wyllie G. R. A.; Sturhahn W.; Durbin S. M.; Champion P. M.; Alp E. E.; Scheidt W. R. Nuclear Resonance Vibrational Spectroscopy of a Protein Active-Site Mimic. J. Phys.: Condens. Matter 2001, 13, 7707–7722. 10.1088/0953-8984/13/34/315. [DOI] [Google Scholar]
- Lipkin H. J. Mössbauer Sum Rules for Use with Synchrotron Sources. Phys. Rev. B: Condens. Matter Mater. Phys. 1995, 52, 10073. 10.1103/PhysRevB.52.10073. [DOI] [PubMed] [Google Scholar]
- Sturhahn W. CONUSS and PHOENIX: Evaluation of Nuclear Resonant Scattering Data. Hyperfine Interact. 2000, 125, 149–172. 10.1023/A:1012681503686. [DOI] [Google Scholar]
- Leu B. M.; Zgierski M. Z.; Wyllie G. R. A.; Ellison M. K.; Scheidt W. R.; Sturhahn W.; Alp E. E.; Durbin S. M.; Sage J. T. Vibrational Dynamics of Biological Molecules: Multi-quantum Contributions. J. Phys. Chem. Solids 2005, 66, 2250–2256. 10.1016/j.jpcs.2005.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai B. K.; Durbin S. M.; Prohofsky E. W.; Sage J. T.; Wyllie G. R. A.; Scheidt W. R.; Sturhahn W.; Alp E. E. Iron Normal Mode Dynamics in (Nitrosyl)iron(II)-tetra-phenyl-porphyrin from X-ray Nuclear Resonance Data. Biophys. J. 2002, 82, 2951–2963. 10.1016/S0006-3495(02)75636-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson E. B.; Decius J. C.; Cross P. C.. Molecular Vibrations; McGraw-Hill, New York, 1955. [Google Scholar]
- Li X.-Y.; Czernuszewicz R. S.; Kincaid J. R.; Spiro T. G. Consistent Porphyrin Force Field. 3. Out-of-Plane Modes in the Resonance Raman Spectra of Planar and Ruffled Nickel Octaethylporphyrin. J. Am. Chem. Soc. 1989, 111, 7012–7023. 10.1021/ja00200a018. [DOI] [Google Scholar]
- Rush T. S. III.; Kozlowski P. M.; Piffat C. A.; Kumble R.; Zgierski M. Z.; Spiro T. G. Computational Modeling of Metalloporphyrin Structure and Vibrational Spectra: Porphyrin Ruffling in NiTPP. J. Phys. Chem. B 2000, 104, 5020–5034. 10.1021/jp000266s. [DOI] [Google Scholar]
- Rai B. K.; Durbin S. M.; Prohofsky E. W.; Sage J. T.; Ellison M. K.; Scheidt W. R.; Sturhahn W.; Alp E. E. Iron Normal Mode Dynamics in a Porphyrin-Imidazole Model for Deoxyheme Proteins. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2002, 66, 051904. 10.1103/PhysRevE.66.051904. [DOI] [PubMed] [Google Scholar]
- Rai B. K.; Durbin S. M.; Prohofsky E. W.; Sage J. T.; Ellison M. K.; Roth A.; Scheidt W. R.; Sturhahn W.; Alp E. E. Direct Determination of the Complete Set of Iron Normal Modes in a Porphyrin-Imidazole Model for Carbonmonoxy-Heme Proteins: [Fe(TPP)(CO)(1-MeIm)]. J. Am. Chem. Soc. 2003, 125, 6927–6936. 10.1021/ja028219w. [DOI] [PubMed] [Google Scholar]
- Starovoitova V.; Budarz T. E.; Wyllie G. R. A.; Scheidt W. R.; Sturhahn W.; Alp E. E.; Prohofsky E. W.; Durbin S. M. Vibrational Spectroscopy and Normal Mode Analysis of Fe(II) Octaethylporphyrin. J. Phys. Chem. B 2006, 110, 13277–13282. 10.1021/jp060345p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starovoitova V.; Wyllie G. R. A.; Scheidt W. R.; Sturhahn W.; Alp E. E.; Durbin S. M. Intermolecular Dynamics in Crystalline Iron Octaethylporphyrin (FeOEP). J. Phys. Chem. B 2008, 112, 12656–12661. 10.1021/jp806215r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q.; Pavlik J. W.; Scheidt W. R.; Wiest O. Predicting Nuclear Resonance Vibrational Spectra of [Fe(OEP)(NO)] - A Methods Study. J. Chem. Theory Comput. 2012, 8, 214–223. 10.1021/ct2006456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe M.; Kitagawa T.; Kyogoku Y. Resonance Raman Spectra of Octaethylporphyrinato-Ni(II) and Meso-Deuterated and 15N-Substituted Derivatives. II. A Normal Coordinate Analysis. J. Chem. Phys. 1978, 69, 4526–4534. 10.1063/1.436450. [DOI] [Google Scholar]
- Kozlowski P. M.; Spiro T. G.; Bérces A.; Zgierski M. Z. Low-Lying Spin States of Iron(II) Porphine. J. Phys. Chem. B 1998, 102, 2603–2608. 10.1021/jp973346d. [DOI] [Google Scholar]
- Barabanschikov A.; Demidov A.; Kubo M.; Champion P. M.; Sage J. T.; Zhao J.; Sturhahn W.; Alp E. E. Spectroscopic Identification of Reactive Porphyrin Motions. J. Chem. Phys. 2011, 135, 015101. 10.1063/1.3598473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portmann S.; Lúthi H. P. MOLEKEL: an Interactive Molecular Graphics Tool. Chimia (Aarau) 2000, 54, 766–770. [Google Scholar]
- Spiro T. G.; Soldatova A. V.; Balakrishnan G. CO, NO and O2 as Vibrational Probes of Heme Protein Interactions. Coord. Chem. Rev. 2013, 257, 511–527. 10.1016/j.ccr.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro T. G.; Wasbotten I. H. CO as a Vibrational Probe of Heme Protein Active Sites. J. Inorg. Biochem. 2005, 99, 34–44. 10.1016/j.jinorgbio.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Linder D. P.; Rodgers K. R.; Banister J.; Wyllie G. R. A.; Ellison M. K.; Scheidt W. R. Five-Coordinate FeIIINO and FeIICO Porphyrinates: Where are the Electrons and Why does it Matter?. J. Am. Chem. Soc. 2004, 126, 14136–14148. 10.1021/ja046942b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel K. M.; Kozlowski P. M.; Zgierski M. Z.; Spiro T. G. Determinants of the FeXO (X = C, N, O) Vibrational Frequencies in Heme Adducts from Experiment and Density Functional Theory. J. Am. Chem. Soc. 1999, 121, 9915–9921. 10.1021/ja990042r. [DOI] [Google Scholar]
- Streit B. R.; Blanc B.; Lukat-Rodgers G. S.; Rodgers K. R.; DuBois J. L. How Active Site Protonation State Influences the Reactivity and Ligation of the Heme in Chlorite Dismutase. J. Am. Chem. Soc. 2010, 132, 5711–5724. 10.1021/ja9082182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukat-Rodgers G. S.; Rodgers K. R.; Caillet-Saguy C.; Izadi-Pruneyre N.; Lecroisey A. Novel Heme Ligand Displacement by CO in the Soluble Hemophore HasA and Its Proximal Ligand Mutants: Implications for Heme Uptake and Release. Biochemistry 2008, 47, 2087–2098. 10.1021/bi7019518. [DOI] [PubMed] [Google Scholar]
- Although the expected changes in the Fe–C vs C–O bond distances have been observed,47 the detection of the vibrational changes is both an easier experiment and more sensitive.
- Silvernail N. J.; Roth A.; Schulz C. E.; Noll B. C.; Scheidt W. R. Heme Carbonyls: Environmental Effects on νC–O and Fe–C/C–O Bond Length Correlations. J. Am. Chem. Soc. 2005, 127, 14422–14433. 10.1021/ja053148x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X.; Demidov A.; Champion P. M. Measurements of the Photodissociation Quantum Yields of MbNO and MbO2 and the Vibrational Relaxation of the Six-coordinate Heme Species. J. Am. Chem. Soc. 2002, 124, 5914–5924. 10.1021/ja017359n. [DOI] [PubMed] [Google Scholar]
- Lim M.; Jackson T. A.; Anfinrud P. A. Nonexponential Protein Relaxation: Dynamics of Conformational Change in Myoglobin. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5801–6804. 10.1073/pnas.90.12.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J. T.; Schomacker K. T.; Champion P. M. Solvent-Dependent Structure and Dynamics in Myoglobin. J. Phys. Chem. 1995, 99, 3394–3405. 10.1021/j100010a059. [DOI] [Google Scholar]
- Mizutani Y.; Kitagawa T. Ultrafast Structural Relaxation of Myoglobin Following Photodissociation of Carbon Monoxide Probed by Time-Resolved Resonance Raman Spectroscopy. J. Phys. Chem. B 2001, 105, 10992–10999. 10.1021/jp010923w. [DOI] [Google Scholar]
- Srajer V.; Reinisch L.; Champion P. M. Investigation of Laser-Induced Long-Lived States of Photolyzed MbCO. Biochemistry 1991, 30, 4886–4895. 10.1021/bi00234a008. [DOI] [PubMed] [Google Scholar]
- Bourgeois D.; Vallone B.; Schotte F.; Arcovito A.; Miele A. E.; Sciara G.; Wulff M.; Anfinrud P.; Brunori M. Complex Landscape of Protein Structural Dynamics Unveiled by Nanosecond Laue Crystallography. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 8704–8709. 10.1073/pnas.1430900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.; Nienhaus K.; Pahl R.; Krasselt A.; Anderson S.; Parak F.; Nienhaus G. U.; Srajer V. Ligand Migration Pathway and Protein Dynamics in Myoglobin: A Time-Resolved Crystallographic Study on L29W MbCO. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11704–11709. 10.1073/pnas.0504932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W. D.; Sage J. T.; Champion P. M.; Chien E.; Sligar S. G. Probing Heme Protein Conformational Equilibration Rates with Kinetic Selection. Biochemistry 1996, 35, 3487–3502. 10.1021/bi952474u. [DOI] [PubMed] [Google Scholar]
- Matsui T.; Unno M.; Ikeda-Saito M. Heme Oxygenase Reveals Its Strategy for Catalyzing Three Successive Oxygenation Reactions. Acc. Chem. Res. 2010, 43, 240–247. 10.1021/ar9001685. [DOI] [PubMed] [Google Scholar]
- Watkins C. C.; Boehning D.; Kaplin A. I.; Rao M.; Ferris C. D.; Snyder S. H. Carbon Monoxide Mediates Vasoactive Intestinal Polypeptide-Associated Nonadrenergic/Noncholinergic Neurotransmission. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 2631–2635. 10.1073/pnas.0308695100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. E.; Wootton P.; Mason H. S.; Bould J.; Iles D. E.; Riccardi D.; Peers C.; Kemp P. J. Hemoxygenase-2 Is an Oxygen Sensor for a Calcium-Sensitive Potassium Channel. Science 2004, 306, 2093–2097. 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- Springer B. A.; Sligar S. G.; Olson J. S.; Phillips G. N. Jr. Mechanisms of Ligand Recognition in Myoglobin. Chem. Rev. 1994, 94, 699–714. 10.1021/cr00027a007. [DOI] [Google Scholar]
- Stone J. R.; Marletta M. A. Soluble Guanylate Cyclase from Bovine Lung: Activation with Nitric Oxide and Carbon Monoxide and Spectral Characterization of the Ferrous and Ferric States. Biochemistry 1994, 33, 5636–5640. 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- Sharma V. S.; Magde D. Activation of Soluble Guanylate Cyclase by Carbon Monoxide and Nitric Oxide: A Mechanistic Model. Methods 1999, 19, 494–505. 10.1006/meth.1999.0892. [DOI] [PubMed] [Google Scholar]
- Dioum E. M.; Rutter J.; Tuckerman J. R.; Gonzalez G.; Gilles-Gonzalez M.-A.; McKnight S. L. NPAS2: A Gas-Responsive Transcription Factor. Science 2002, 298, 2385–2387. 10.1126/science.1078456. [DOI] [PubMed] [Google Scholar]
- Lukat-Rodgers G. S.; Correia C.; Botuyan M. V.; Mer G.; Rodgers K. R. Heme-Based Sensing by the Mammalian Circadian Protein, CLOCK. Inorg. Chem. 2010, 49, 6349–6365. 10.1021/ic902388q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran V.; Benabbas A.; Youn H.; Champion P. M. Vibrational Coherence Spectroscopy of the Heme Domain in the CO-Sensing Transcriptional Activator CooA. J. Am. Chem. Soc. 2011, 133, 18816–18827. 10.1021/ja206152m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin K. A.; Kerby R. L.; Youn H.; Roberts G. P.; Burstyn J. N. The Transcription Regulator RcoM-2 from Burkholderia Xenovorans is a Cysteine-Ligated Hemoprotein that Undergoes a Redox-Mediated Ligand Switch. Biochemistry 2008, 47, 9016–9028. 10.1021/bi800486x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelver D.; Thorsteinsson M. V.; Kerby R. L.; Chung S. Y.; Roberts G. P.; Reynolds M. F.; Parks R. B.; Burstyn J. N. Identification of Two Important Heme Site Residues (Cysteine 75 and Histidine 77) in CooA, the CO-Sensing Transcription Factor of Rhodospirillum Rubrum. Biochemistry 1999, 38, 2669–2678. 10.1021/bi982658j. [DOI] [PubMed] [Google Scholar]
- Leu B. M.; Silvernail N. J.; Zgierski M. Z.; Wyllie G. R. A.; Ellison M. K.; Scheidt W. R.; Zhao J.; Sturhahn W.; Alp E. E.; Durbin S. M.; et al. Quantitative Vibrational Dynamics of Iron in Carbonyl Porphyrins. Biophys. J. 2007, 92, 3764–3783. 10.1529/biophysj.106.093773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J. T.; Durbin S. M.; Sturhahn W.; Wharton D. C.; Champion P. M.; Hession P.; Sutter J.; Alp E. E. Long-Range Reactive Dynamics in Myoglobin. Phys. Rev. Lett. 2001, 86, 4966–4969. 10.1103/PhysRevLett.86.4966. [DOI] [PubMed] [Google Scholar]
- Ohta T.; Liu J.-G.; Saito M.; Kobayashi Y.; Yoda Y.; Seto M.; Naruta Y. Axial Ligand Effects on Vibrational Dynamics of Iron in Heme Carbonyl Studied by Nuclear Resonance Vibrational Spectroscopy. J. Phys. Chem. B 2012, 116, 13831–13838. 10.1021/jp304398g. [DOI] [PubMed] [Google Scholar]
- Perutz M. F. Stereochemistry of Cooperative Effects in Haemoglobin: Haem-Haem Interaction and the Problem of Allostery. Nature 1970, 228, 726–734. 10.1038/228726a0. [DOI] [PubMed] [Google Scholar]
- Srajer V.; Reinisch L.; Champion P. M. Protein Fluctuations, Distributed Coupling, and the Binding of Ligands to Heme Proteins. J. Am. Chem. Soc. 1988, 110, 6656–6670. 10.1021/ja00228a009. [DOI] [Google Scholar]
- Zhu L.; Sage J. T.; Champion P. M. Observation of Coherent Reaction Dynamics in Heme. Science 1994, 266, 629–632. 10.1126/science.7939716. [DOI] [PubMed] [Google Scholar]
- Klug D. D.; Zgierski M. Z.; Tse J. S.; Liu Z.; Kincaid J. R.; Czarnecki K.; Hemley R. J. Doming Modes and Dynamics of Model Heme Compounds. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12526–12530. 10.1073/pnas.152464699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.; Bocian D. F. Carbonyl Tilting and Bending Potential Energy Surface of Carbon Monoxyhemes. J. Phys. Chem. 1996, 100, 6363–6367. 10.1021/jp953543h. [DOI] [Google Scholar]
- Kozlowski P. M.; Vogel K. M.; Zgierskia M. Z.; Spiro T. G. Steric Contributions to CO Binding in Heme Proteins: A Density Functional Analysis of FeCO Vibrations and Deformability. J. Porphyrins Phthalocyanines 2001, 5, 312–322. 10.1002/jpp.318. [DOI] [Google Scholar]
- Rougee M.; Brault D. Influence of Trans Weak or Strong Field Ligands Upon the Affinity of Deuteroheme for Carbon Monoxide. Monoimidazoleheme as a Reference for Unconstrained Five-Coordinate Hemoproteins. Biochemistry 1975, 14, 4100–4106. 10.1021/bi00689a028. [DOI] [Google Scholar]
- Wayland B. B.; Mehne L. F.; Swartz J. Mono- and Biscarbonyl Complexes of Iron (II) Tetraphenylporphyrin. J. Am. Chem. Soc. 1978, 100, 2379–2383. 10.1021/ja00476a020. [DOI] [Google Scholar]
- Strauss S. H.; Holm R. H. Carbon Monoxide Affinities of Iron (II) Octaethylporphyrin, Octaethylchlorin, and Octaethylisobacteriochlorin. Inorg. Chem. 1982, 21, 863–868. 10.1021/ic00133a005. [DOI] [Google Scholar]
- Linder D. P.; Silvernail N. J.; Barabanschikov A.; Zhao J.; Alp E. E.; Sturhahn W.; Sage J. T.; Scheidt W. R.; Rodgers K. R. The Diagnostic Vibrational Signature of Pentacoordination in Heme Carbonyls. J. Am. Chem. Soc. 2014, 136, 9818–9821. 10.1021/ja503191z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvernail N. J.; Noll B. C.; Schulz C. E.; Scheidt W. R. Coordination of Diatomic Ligands to Heme: Simply CO. Inorg. Chem. 2006, 45, 7050–7052. 10.1021/ic0613356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt W. R.Stereochemical Systematics for Porphyrins and Metalloporphyrins. In Handbook of Porphyrin Science; Kadish K. M., Smith K., Guilard R., Eds.; World Scientific: Singapore and Hackensack, NJ, 2012; Vol. 24 (Coordination Chemistry and Materials), pp 1–180. [Google Scholar]
- Vogel K. M.; Kozlowski P. M.; Zgierski M. Z.; Spiro T. G. Role of the Axial Ligand in Heme-CO Backbonding; DFT Analysis of Vibrational Data. Inorg. Chim. Acta 2000, 297, 11–17. 10.1016/S0020-1693(99)00253-4. [DOI] [Google Scholar]
- Das T. K.; Tomson F. L.; Gennis R. B.; Gordon M.; Rousseau D. L. pH-Dependent Structural Changes at the Heme-Copper Binuclear Center of Cytochrome c Oxidase. Biophys. J. 2001, 80, 2039–2045. 10.1016/S0006-3495(01)76177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S.; Kincaid J. R. Resonance Raman Studies of the Carbonmonoxy Form of Catalase Evidence for and Effects of Phenolate Ligation. FEBS Lett. 1992, 314, 293–296. 10.1016/0014-5793(92)81492-5. [DOI] [PubMed] [Google Scholar]
- Eakanunkul S.; Lukat-Rodgers G. S.; Sumithran S.; Ghosh A.; Rodgers K. R.; Dawson J. H.; Wilks A. Characterization of the Periplasmic Heme-Binding Protein Shut from the Heme Uptake System of Shigella Dysenteriae. Biochemistry 2005, 44, 13179–13191. 10.1021/bi050422r. [DOI] [PubMed] [Google Scholar]
- Wang J.; Takahashi S.; Hosler J. P.; Mitchell D. M.; Ferguson-Miller S.; Gennis R. B.; Rousseau D. L. Two Conformations of the Catalytic Site in the aa3-Type Cytochrome c Oxidase from Rhodobacter Sphaeroides. Biochemistry 1995, 34, 9819–9825. 10.1021/bi00031a001. [DOI] [PubMed] [Google Scholar]
- It should also be noted that the DFT calculation reported in ref (82) predicted values of νFe–C for model porphine complexes that were 50–60 cm–1 higher than those reported in the paper, again consistent with the idea that these species have a weak-field trans ligand.
- The {FeNO}n notation is that of Enemark and Feltham,89 where n is the number of d electrons plus the number of electrons in the π* orbitals of NO. The notation is designed to strongly emphasize the highly covalent nature of the triatomic FeNO unit.
- Enemark J. H.; Feltham R. D. Principles of Structure, Bonding, and Reactivity for Metal Nitrosyl Complexes. Coord. Chem. Rev. 1974, 13, 339–406. 10.1016/S0010-8545(00)80259-3. [DOI] [Google Scholar]
- Karow D. S.; Pan D.; Tran R.; Pellicena P.; Presley A.; Mathies R. A.; Marletta M. A. Spectroscopic Characterization of the Soluble Guanylate Cyclase-Like Heme Domains from Vibrio Cholerae and Thermoanaerobacter Tengcongensis. Biochemistry 2004, 43, 10203–10211. 10.1021/bi049374l. [DOI] [PubMed] [Google Scholar]
- Walker F. A. Nitric Oxide Interaction with Insect Nitrophorins and Thoughts on the Electron Configuration of the {FeNO}6 complex. J. Inorg. Biochem. 2005, 99, 216–236. 10.1016/j.jinorgbio.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Fülöp V.; Moir J. W. B.; Ferguson S. J.; Hajdu J. The Anatomy of a Bifunctional Enzyme: Structural Basis for Reduction of Oxygen to Water and Synthesis of Nitric Oxide by Cytochrome cd1. Cell 1995, 81, 369–377. 10.1016/0092-8674(95)90390-9. [DOI] [PubMed] [Google Scholar]
- Wasser I. M.; de Vries S.; Moenne-Loccoz P.; Schroder I.; Karlin K. D. Nitric Oxide in Biological Denitrification: Fe/Cu Metalloenzyme and Metal Complex NOx Redox Chemistry. Chem. Rev. 2002, 102, 1201–1234. 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- Leu B.; Zgierski M.; Wyllie G. R. A.; Scheidt W. R.; Sturhahn W.; Alp E. E.; Durbin S. M.; Sage J. T. Quantitative Vibrational Dynamics of Iron in Nitrosyl Porphyrins. J. Am. Chem. Soc. 2004, 126, 4211–4227. 10.1021/ja038526h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt W. R.; Barabanschikov A.; Pavlik J. W.; Silvernail N. J.; Sage J. T. Electronic Structure and Dynamics of Nitrosyl Porphyrins. Inorg. Chem. 2010, 49, 6240–6252. 10.1021/ic100261b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert N.; Galinato M. G. I.; Paulat F.; Richter-Addo G. B.; Sturhahn W.; Xu N.; Zhao J. Y. Nuclear Resonance Vibrational Spectroscopy Applied to [Fe(OEP)(NO)]: The Vibrational Assignments of Five-Coordinate Ferrous Heme Nitrosyls and Implications for Electronic Structure. Inorg. Chem. 2010, 49, 4133–4148. 10.1021/ic902181e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison M. K.; Scheidt W. R. Structural Distortion in Five-Coordinate Nitrosyl Iron Porphyrins. Axial Ligand Tilting and Its Effect on Equatorial Geometry. J. Am. Chem. Soc. 1997, 119, 7404–7405. 10.1021/ja971140k. [DOI] [Google Scholar]
- Scheidt W. R.; Duval H. F.; Neal T. J.; Ellison M. K. Intrinsic Structural Distortions in Five-Coordinate (nitrosyl)iron(II) Porphyrinate Derivatives. J. Am. Chem. Soc. 2000, 122, 4651–4659. 10.1021/ja993995y. [DOI] [Google Scholar]
- Pavlik J. W.; Barabanschikov A.; Oliver A. G.; Alp E. E.; Sturhahn W.; Zhao J.; Sage J. T.; Scheidt W. R. Probing Directionally Specific Iron Motion with Nuclear Resonance Vibrational Spectroscopy. Angew. Chem., Int. Ed. 2010, 49, 4400–4404. 10.1002/anie.201000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlik J. W.; Peng Q.; Silvernail N. J.; Alp E. E.; Hu M. Y.; Zhao J.; Sage J. T.; Scheidt W. R. Anisotropic Iron Motion in Nitrosyl Iron Porphyrinates: Natural and Synthetic Hemes. Inorg. Chem. 2014, 53, 2582–2590. 10.1021/ic4028964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piciulo P. L.; Rupprecht G.; Scheidt W. R. Stereochemistry of Nitrosylmetalloporphyrins: Nitrosyl-α,β,γ,-δ-tetraphenylporphinato(1-methylimidazole)iron and Nitrosyl-α, β,γ,δ-tetraphenylporphinato(4-methylpiperidine) manganese. J. Am. Chem. Soc. 1974, 96, 5293–5295. 10.1021/ja00823a067. [DOI] [Google Scholar]
- Scheidt W. R.; Piciulo P. L. Nitrosylmetalloporphyrins. III. Synthesis and Molecular Stereo-Chemistry of Nitrosyl-α,β,γ,δ-tetraphenylporphinato(1-methylimidazole)iron(II). J. Am. Chem. Soc. 1976, 98, 1913–1919. 10.1021/ja00423a044. [DOI] [PubMed] [Google Scholar]
- Scheidt W. R.; Brinegar A. C.; Ferro E. B.; Kirner J. F. Nitrosylmetalloporphyrins. 4. Molecular Stereochemistry of Two Crystalline Forms of Nitrosyl-α,-β,-γ,-δ-tetraphenylpor-phinato(4-methylpiperidine)iron(II). A Structural Correlation with ν(NO). J. Am. Chem. Soc. 1977, 99, 7315–7322. 10.1021/ja00464a035. [DOI] [Google Scholar]
- Traylor T. G.; Sharma V. S. Why Nitric Oxide?. Biochemistry 1992, 31, 2847–2849. 10.1021/bi00126a001. [DOI] [PubMed] [Google Scholar]
- Zeng W.; Silvernail N. J.; Wharton D. C.; Georgiev G. Y.; Leu B. M.; Scheidt W. R.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. Direct Probe of Functional Fe Vibrations Elucidates NO Activation of Heme Proteins. J. Am. Chem. Soc. 2005, 127, 11200–11201. 10.1021/ja051052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvernail N. J.; Barabanschikov A.; Pavlik J. W.; Noll B. C.; Zhao J.; Alp E. E.; Sturhahn W.; Sage J. T.; Scheidt W. R. Interplay of Structure and Vibrational Dynamics in Six-Coordinate Heme Nitrosyls. J. Am. Chem. Soc. 2007, 129, 2200–2201. 10.1021/ja066869k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulat F.; Berto T. C.; DeBeer George S.; Goodrich L.; Praneeth V. K. K.; Sulok C. D.; Lehnert N. Vibrational Assignments of Six-Coordinate Ferrous Heme Nitrosyls: New Insight from Nuclear Resonance Vibrational Spectroscopy. Inorg. Chem. 2008, 47, 11449–11451. 10.1021/ic801626w. [DOI] [PubMed] [Google Scholar]
- Lehnert N.; Sage J. T.; Silvernail N. J.; Scheidt W. R.; Alp E. E.; Sturhahn W.; Zhao J. Oriented Single-Crystal Nuclear Resonance Vibrational Spectroscopy of [Fe(TPP)(MI)(NO)]: Quantitative Assessment of the trans Effect of NO. Inorg. Chem. 2010, 49, 7197–7215. 10.1021/ic1010677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q.; Pavlik J. W.; Silvernail N. J.; Alp E. E.; Hu M. Y.; Zhao J.; Sage J. T.; Scheidt W. R. 3-D Motions of Iron in Six-Coordinate {FeNO}7 Hemes by Nuclear Resonance Vibrational Spectroscopy. Chem. - Eur. J. 2016, 22, 6323–6332. 10.1002/chem.201505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Peng Q.; Oliver A. G.; Alp E. E.; Hu M. Y.; Zhao J.; Sage J. T.; Scheidt W. R. Comprehensive Fe-Ligand Vibration Identification in {FeNO}6 Hemes. J. Am. Chem. Soc. 2014, 136, 18100–18110. 10.1021/ja5105766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.-Y.; Shimizu H.; Adachi S.; Nakagawa A.; Tanaka I.; Nakahara K.; Shoun H.; Obayashi E.; Nakamura H.; Iizuka T.; et al. Crystal Structure of Nitric Oxide Reductase from Denitrifying Fungus Fusarium Oxysporum. Nat. Struct. Biol. 1997, 4, 827–832. 10.1038/nsb1097-827. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. M. C.; Hazzard J. M. H.; Nussenzveig R. H.; Champagne D. E.; Walker F. A. Reversible Binding of Nitric Oxide by a Salivary Heme Protein from a Bloodsucking Insect. Science 1993, 260, 539–542. 10.1126/science.8386393. [DOI] [PubMed] [Google Scholar]
- Praneeth V. K. K.; Paulat F.; Berto T. C.; George S. D.; Naether C.; Sulok C. D.; Lehnert N. Electronic Structure of Six-Coordinate Iron (III) - Porphyrin NO Adducts: The Elusive Iron (III) - NO (radical) State and Its Influence on the Properties of These Complexes. J. Am. Chem. Soc. 2008, 130, 15288–15303. 10.1021/ja801860u. [DOI] [PubMed] [Google Scholar]
- Moeser B.; Janoschka A.; Wolny J. A.; Paulsen H.; Filippov I.; Berry R. E.; Zhang H.; Chumakov A. I.; Walker F. A.; Schünemann V. Nuclear Inelastic Scattering and Mössbauer Spectroscopy as Local Probes for Ligand Binding Modes and Electronic Properties in Proteins: Vibrational Behavior of a Ferriheme Center Inside a β-Barrel Protein. J. Am. Chem. Soc. 2012, 134, 4216–4228. 10.1021/ja210067t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach H.; Faus I.; Rackwitz S.; Wolny J. A.; Walker F. A.; Chumakov A. I.; Ogata H.; Knipp M.; Schünemann V. Nitric Oxide Heme Interactions in Nitrophorin 7 Investigated by Nuclear Inelastic Scattering. Hyperfine Interact. 2014, 226, 439–443. 10.1007/s10751-013-0982-7. [DOI] [Google Scholar]
- Ellison M. K.; Scheidt W. R. Synthesis, Molecular Structures, and Properties of Six-Coordinate [Fe(OEP)(L)(NO)]+ Derivatives. Elusive Nitrosyl Ferric Porphyrins. J. Am. Chem. Soc. 1999, 121, 5210–5219. 10.1021/ja984308q. [DOI] [Google Scholar]
- Collman J. P.; Gagne R. R.; Reed C.; Halbert T. R.; Lang G.; Robinson W. T. Picket Fence Porphyrins. Synthetic Models for Oxygen Binding Hemoproteins. J. Am. Chem. Soc. 1975, 97, 1427–1439. 10.1021/ja00839a026. [DOI] [PubMed] [Google Scholar]
- Collman J. P.; Gagne R. R.; Reed C. A.; Robinson W. T.; Rodley G. A. Structure of an Iron (II) Dioxygen Complex; A Model for Oxygen Carrying Hemeproteins. Proc. Natl. Acad. Sci. U. S. A. 1974, 71, 1326–1329. 10.1073/pnas.71.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson G. B.; Rodley G. A.; Robinson W. T.; Gagne R. R.; Reed C. A.; Collman J. P. Structure of a Dioxygen Adduct of (1-Methylimidazole)-Meso-Tetrakis (α, α, α, α-o-pivalamidophenyl)porphinatoiron (II). An Iron Dioxygen Model for the Heme Component of Oxymyoglobin. Inorg. Chem. 1978, 17, 850–857. 10.1021/ic50182a012. [DOI] [Google Scholar]
- Collman J. P.; Gagne R. R.; Halbert T. R.; Marchon J. C.; Reed C. A. Reversible Oxygen Adduct Formation in Ferrous Complexes Derived from a Picket Fence Porphyrin. Model for Oxymyoglobin. J. Am. Chem. Soc. 1973, 95, 7868–7870. 10.1021/ja00804a054. [DOI] [PubMed] [Google Scholar]
- Li J.; Noll B. C.; Oliver A. G.; Schulz C. E.; Scheidt W. R. Correlated Ligand Dynamics in Oxyiron Picket Fence Porphyrins: Structural and Mössbauer Investigations. J. Am. Chem. Soc. 2013, 135, 15627–15641. 10.1021/ja408431z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.-F.; Peng Q.; Barabanschikov A.; Pavlik J. W.; Alp E. E.; Sturhahn W.; Zhao J.; Schulz C. E.; Sage J. T.; Scheidt W. R. New Perspectives on Iron-Ligand Vibrations of Oxyheme Complexes. Chem. - Eur. J. 2011, 17, 11178–11185. 10.1002/chem.201101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W.; Barabanschikov A.; Wang N.; Lu Y.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. Vibrational Dynamics of Oxygenated Heme Proteins. Chem. Commun. 2012, 48, 6340–6342. 10.1039/c2cc31239e. [DOI] [PubMed] [Google Scholar]
- Sigman J. A.; Kwok B. C.; Lu Y. From Myoglobin to Heme-Copper Oxidase: Design and Engineering of a CuB Center into Sperm Whale Myoglobin. J. Am. Chem. Soc. 2000, 122, 8192–8196. 10.1021/ja0015343. [DOI] [Google Scholar]
- Sigman J. A.; Kim H. K.; Zhao X.; Carey J. R.; Lu Y. The Role of Copper and Protons in Heme-Copper Oxidases: Kinetic Study of an Engineered Heme-Copper Center in Myoglobin. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3629–3634. 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Lord R. L.; Noll B. C.; Baik M.-H.; Schulz C. E.; Scheidt W. R. Cyanide: A Strong Field Ligand for Ferrohemes and Hemoproteins?. Angew. Chem., Int. Ed. 2008, 47, 10144–10146. 10.1002/anie.200804116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.-F.; Peng Q.; Barabanschikov A.; Pavlik J. W.; Alp E. E.; Sturhahn W.; Zhao J.; Sage J. T.; Scheidt W. R. Vibrational Probes and Determinants of the S = 0 ⇌ S = 2 Spin Crossover in Five-Coordinate [Fe(TPP)(CN)]−. Inorg. Chem. 2012, 51, 11769–11778. 10.1021/ic301719v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chottard G.; Mansuy D. Resonance Raman Studies of Hemoglobin Complexes with Nitric Oxide, Nitrosobenzene and Nitrosomethane: Observation of the Metal-Ligand Vibrations. Biochem. Biophys. Res. Commun. 1977, 77, 1333–1338. 10.1016/S0006-291X(77)80125-3. [DOI] [PubMed] [Google Scholar]
- Benko B.; Yu N. T. Resonance Raman Studies of Nitric Oxide Binding to Ferric and Ferrous Hemoproteins: Detection of Fe(III)-NO Stretching, Fe(III)-N-O Bending, and Fe(II)-N-O Bending Vibrations. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 7042–7046. 10.1073/pnas.80.22.7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb L. A.; Lee B. S.; Yu N. T. Resonance Raman Investigation of Nitric Oxide Bonding in Iron Porphyrins: Detection of the Fe–NO Stretching Vibration. Inorg. Chem. 1993, 32, 281–286. 10.1021/ic00055a009. [DOI] [Google Scholar]
- Coyle C. M.; Vogel K. M.; Rush T. S. III; Kozlowski P. M.; Williams R.; Spiro G.; Dou T. G.; Ikeda-Saito Y.; Olson M.; Zgierski J. S.; FeNO M. Z. FeNO Structure in Distal Pocket Mutants of Myoglobin Based on Resonance Raman Spectroscopy. Biochemistry 2003, 42, 4896–4903. 10.1021/bi026395b. [DOI] [PubMed] [Google Scholar]
- Hu S.; Kincaid J. R. Resonance Raman Spectra of the Nitric Oxide Adducts of Ferrous Cytochrome P450cam in the Presence of Various Substrates. J. Am. Chem. Soc. 1991, 113, 9760–9766. 10.1021/ja00026a008. [DOI] [Google Scholar]
- Ogoshi H.; Watanbe E.; Yoshida Z.; Kincaid J.; Nakamoto K. Synthesis and Far-Infrared Spectra of Ferric Octaethylporphine Complexes. J. Am. Chem. Soc. 1973, 95, 2845–2849. 10.1021/ja00790a017. [DOI] [Google Scholar]
- Hu C.; Barabanschikov A.; Ellison M. K.; Zhao J.; Alp E. E.; Sturhahn W.; Zgierski M. Z.; Sage J. T.; Scheidt W. R. Nuclear Resonance Vibrational Spectra of Five-Coordinate Imidazole-Ligated Iron(II) Porphyrinates. Inorg. Chem. 2012, 51, 1359–1370. 10.1021/ic201580v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argade P. V.; Sassardi M.; Rousseau D. L.; Inubushi T.; Ikeda-Saito M.; Lapidot A. Confirmation of the Assignment of the Iron-Histidine Stretching Mode in Myoglobin. J. Am. Chem. Soc. 1984, 106, 6593–6596. 10.1021/ja00334a024. [DOI] [Google Scholar]
- Wells A.; Sage J. T.; Morikis D.; Champion P. M.; Chiu M. L.; Sligar S. G. The Iron-Histidine Mode of Myoglobin Revisited: Resonance Raman Studies of Isotopically Labeled Escherichia Coli Expressed Myoglobin. J. Am. Chem. Soc. 1991, 113, 9655–9660. 10.1021/ja00025a034. [DOI] [Google Scholar]
- Hu C.; Schulz C. E.; Scheidt W. R. All High-Spin (S = 2) Iron(II) Hemes are NOT Alike. Dalton Trans. 2015, 44, 18301–18310. 10.1039/C5DT02795K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.; Peng Q.; Silvernail N. J.; Barabanschikov A.; Zhao J.; Alp E. E.; Sturhahn W. J.; Sage J. T.; Scheidt W. R. Effects of Imidazole Deprotonation on Vibrational Spectra of High-Spin Iron(II) Porphyrinates. Inorg. Chem. 2013, 52, 3170–3177. 10.1021/ic3026396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt W. R.; Geiger D. K. The Molecular Stereochemistry of a ‘Spin-Equilibrium’ (S = 1/2, S = 5/2) Hemichrome Salt: Bis(3-chloropyridine)octaethylporphinatoiron(III) Perchlorate. J. Chem. Soc., Chem. Commun. 1979, 1154–1155. 10.1039/C39790001154. [DOI] [Google Scholar]
- Scheidt W. R.; Geiger D. K.; Haller K. J. Structural Characterization of a Variable-Spin Porphinatoiron(III) Complex. Molecular Stereochemistry of Bis(3-chloropyridine)-octa-ethylporphinato-iron(III) Perchlorate at 98 K (S = 1/2) and 293 K (S = 1/2, S = 5/2). J. Am. Chem. Soc. 1982, 104, 495–499. 10.1021/ja00366a019. [DOI] [Google Scholar]
- Scheidt W. R.; Geiger D. K.; Hayes R. G.; Lang G. Control of Spin State in Porphinatoiron(III) Complexes. An Axial Ligand Orientation Effect Leading to an Intermediate-Spin Complex. Molecular Structure and Physical Characterization of the Monoclinic Form of Bis(3-chloropyridine)(octaethylporphinato)iron(III) Perchlorate. J. Am. Chem. Soc. 1983, 105, 2625–2632. 10.1021/ja00347a018. [DOI] [Google Scholar]
- Migita C. T.; Iwaizumi M. Low-Temperature EPR Studies of Highly Anisotropic Low-Spin (Protoporphyrinato) Iron (III) complexes. J. Am. Chem. Soc. 1981, 103, 4378–4381. 10.1021/ja00405a015. [DOI] [Google Scholar]
- Carter K. R.; Ah-lim T.; Graham P. The Coordination Environment of Mitochondrial Cytochromes b. FEBS Lett. 1981, 132, 243–246. 10.1016/0014-5793(81)81170-2. [DOI] [PubMed] [Google Scholar]
- Walker F. A.; Reis D.; Balke V. L. Models of the Cytochromes b. 5. EPR Studies of Low-Spin Iron(III) Tetraphenylporphyrins. J. Am. Chem. Soc. 1984, 106, 6888–6898. 10.1021/ja00335a003. [DOI] [Google Scholar]
- Walker F. A.; Huynh B. H.; Scheidt W. R.; Osvath S. R. Models of the Cytochromes b. Effect of Axial Ligand Plane Orientation on the EPR and Mössbauer Spectra of Low-Spin Ferrihemes. J. Am. Chem. Soc. 1986, 108, 5288–5297. 10.1021/ja00277a038. [DOI] [Google Scholar]
- Scheidt W. R.; Kirner J. F.; Hoard J. L.; Reed C. A. Unusual Orientation of Axial Ligands in Metalloporphyrins. The Molecular Structure of Low-Spin Bis(2-Methylimidazole)(meso-tetraphenylporphinato)iron(III) Perchlorate. J. Am. Chem. Soc. 1987, 109, 1963–1968. 10.1021/ja00241a011. [DOI] [Google Scholar]
- Scheidt W. R.; Osvath S. R.; Lee Y. J. Crystal and Molecular Structure of Bis(Imidazole)(Meso-Tetraphenylporphinato)iron(III) Chloride. A Classic Molecule Revisited. J. Am. Chem. Soc. 1987, 109, 1958–1963. 10.1021/ja00241a010. [DOI] [Google Scholar]
- Soltis S. M.; Strouse C. E. Electronic Structure of Low-Spin Ferric Porphyrins: Single-Crystal EPR Evidence for Pseudo-Jahn-Teller Distortion in (Tetraphenylporphinato)Iron(III) Bis(Imidazole) Cations. J. Am. Chem. Soc. 1988, 110, 2824–2829. 10.1021/ja00217a022. [DOI] [Google Scholar]
- Safo M. K.; Gupta G. P.; Walker F. A.; Scheidt W. R. Models of the Cytochromes b. Control of Axial Ligand Orientation with a “Hindered” Porphyrin System. J. Am. Chem. Soc. 1991, 113, 5497–5510. 10.1021/ja00015a001. [DOI] [Google Scholar]
- Safo M. K.; Gupta G. P.; Watson C. T.; Simonis U.; Walker F. A.; Scheidt W. R. Models of the Cytochromes b. Low-Spin Bis-Ligated (Porphinato) Iron (III) Complexes with Unusual Molecular Structures and NMR, EPR, and Mössbauer Spectra. J. Am. Chem. Soc. 1992, 114, 7066–7075. 10.1021/ja00044a017. [DOI] [Google Scholar]
- Safo M. K.; Walker F. A.; Raitsimring A. M.; Walters W. P.; Dolata D. P.; Debrunner P. G.; Scheidt W. R. Axial Ligand Orientation in Iron(III) Porphyrinates: Effect of Axial π-Acceptors. Characterization of the Low-Spin Complex [Fe(TPP)(4-CNPy)2]ClO4. J. Am. Chem. Soc. 1994, 116, 7760–7770. 10.1021/ja00096a037. [DOI] [Google Scholar]
- Peng Q.; Li M.; Hu C.; Pavlik J. W.; Oliver A.; Alp E. E.; Hu M. Y.; Zhao J.; Sage J. T.; Scheidt W. R. Probing Heme Vibrational Anisotropy: An Imidazole Orientation Effect?. Inorg. Chem. 2013, 52, 11361–11369. 10.1021/ic401644g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.; Sulok C. D.; Paulat F.; Lehnert N.; Twigg A. I.; Hendrich M. P.; Schulz C. E.; Scheidt W. R. Just a Proton: Distinguishing the Two Electronic States of Five-Coordinate High-Spin Iron(II) Porphyrinates with Imidazole/ate Coordination. J. Am. Chem. Soc. 2010, 132, 3737–3750. 10.1021/ja907584x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.; Noll B. C.; Schulz C. E.; Scheidt W. R. Proton-Mediated Electron Configuration Change in High-Spin Iron(II) Porphyrinates. J. Am. Chem. Soc. 2005, 127, 15018–15019. 10.1021/ja055129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.; An J.; Noll B. C.; Schulz C. E.; Scheidt W. R. Electronic Configuration of High-Spin Imidazole-Ligated Iron(II) Octaethylporphyrinates. Inorg. Chem. 2006, 45, 4177–4185. 10.1021/ic052194v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W.; Barabanschikov A.; Zhang Y.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. Synchrotron-Derived Vibrational Data Confirm Unprotonated Oxo Ligand in Myoglobin Compound II. J. Am. Chem. Soc. 2008, 130, 1816–1817. 10.1021/ja077823+. [DOI] [PubMed] [Google Scholar]
- Meharenna Y. T.; Doukov T.; Li H.; Soltis S. M.; Poulos T. L. Crystallographic and Single Crystal Spectral Analysis of the Peroxidase Ferryl Intermediate. Biochemistry 2010, 49, 2984–2986. 10.1021/bi100238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg R.; Barrett J.. Cytochromes; Academic Press: London, New York, 1973; pp 127–217. [Google Scholar]
- Scott R. A.; Mauk A. G.. Cytochrome c: A Multidisciplinary Approach; University Science Books: Sausalito, CA, 1996. [Google Scholar]
- Leu B. M.; Zhang Y.; Bu L.; Straub J. E.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. Resilience of the Iron Environment in Heme Proteins. Biophys. J. 2008, 95, 5874–5889. 10.1529/biophysj.108.138198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu B. M.; Ching T. H.; Zhao J.; Sturhahn W.; Alp E. E.; Sage J. T. Vibrational Dynamics of Iron in Cytochrome c. J. Phys. Chem. B 2009, 113, 2193–2200. 10.1021/jp806574t. [DOI] [PubMed] [Google Scholar]
- Galinato M. G. I.; Kleingardner J. G.; Bowman S. E. J.; Alp E. E.; Zhao J.; Bren K. L.; Lehnert N. Heme-Protein Vibrational Couplings in Cytochrome c Provide a Dynamic Link that Connects the Heme-Iron and the Protein Surface. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (23), 8896–8900. 10.1073/pnas.1200345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinato M. G. I.; Bowman S. E. J.; Kleingardner J. G.; Martin S.; Zhao J.; Sturhahn W.; Alp E. E.; Bren K. L.; Lehnert N. Effects of Protein Structure on Iron-Polypeptide Vibrational Dynamic Coupling in Cytochrome c. Biochemistry 2015, 54, 1064–1076. 10.1021/bi501430z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams K. L.; Tsoi S.; Yan J.; Durbin S. M.; Ramdas A. K.; Cramer W. A.; Sturhahn W.; Alp E. E.; Schulz C. E. Fe Vibrational Spectroscopy of Myoglobin and Cytochrome f. J. Phys. Chem. B 2006, 110, 530–536. 10.1021/jp053440r. [DOI] [PubMed] [Google Scholar]
- Chakraborty S.; Reed J.; Sage J. T.; Branagan N. C.; Petrik I. D.; Miner K. D.; Hu M. Y.; Zhao J.; Alp E. E.; Lu Y. Recent Advances in Biosynthetic Modeling of Nitric Oxide Reductases and Insights Gained from Nuclear Resonance Vibrational and Other Spectroscopic Studies. Inorg. Chem. 2015, 54, 9317–9329. 10.1021/acs.inorgchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu B. M.; Sage J. T. Stiffness, resilience, compressibility. Atomic Scale Force Spectroscopy of Biomolecules. Hyperfine Interact. 2016, 237, 87. 10.1007/s10751-016-1275-8. [DOI] [Google Scholar]
- Scheidt W. R.; Frisse M. E. Nitrosylmetalloporphyrins. II. Synthesis and Molecular Stereochemistry of Nitrosyl-α,β,γ,δ-tetraphenylporphinatoiron(II). J. Am. Chem. Soc. 1975, 97, 17–21. 10.1021/ja00834a005. [DOI] [PubMed] [Google Scholar]
- Silvernail N. J.; Olmstead M. M.; Noll B. C.; Scheidt W. R. Tetragonal to Triclinic – A Phase Change for [Fe(TPP)(NO)]. Inorg. Chem. 2009, 48, 971–977. 10.1021/ic801617q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt W. R.; Hoard J. L. Stereochemistry of Low-Spin Cobalt Porphyrins. I. Structure and Bonding in a Nitrosylcobalt Porphyrin and Their Bearing on One Rational Model for the Oxygenated Protoheme. J. Am. Chem. Soc. 1973, 95, 8281–8288. 10.1021/ja00806a013. [DOI] [PubMed] [Google Scholar]
- Grande L. M.; Oliver A. G.; Noll B. C.; Scheidt W. R. Dynamics of NO Motion in Solid-State [Co(TPP)(NO)]. Inorg. Chem. 2010, 49, 6552–6557. 10.1021/ic1003462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groombridge C. J.; Larkworthy L. F.; Mason J. M. Swinging of the Bent Nitrosyl Ligand in [Co(15NO)(TPP)]: A Solid-State Motion Detected by 15N CPMAS NMR Spectroscopy. Inorg. Chem. 1993, 32, 379–380. 10.1021/ic00056a006. [DOI] [Google Scholar]
- Silvernail N. J.; Pavlik J. W.; Noll B. C.; Schulz C. E.; Scheidt W. R. Reversible NO Motion in Crystalline [Fe(Porph)(1-MeIm)(NO)] Derivatives. Inorg. Chem. 2008, 47, 912–920. 10.1021/ic701700p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvernail N. J.; Barabanschikov A.; Sage J. T.; Noll B. C.; Scheidt W. R. Mapping NO Movements in Crystalline [Fe(Porph)(NO)(1-MeIm)]. J. Am. Chem. Soc. 2009, 131, 2131–2140. 10.1021/ja8055613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov E. N.; Gvosdev R.; Goldanskii V. I.; Parak F. G. Differences in the Dynamics of Oxidized and Reduced Cytochrome c Measured by Mössbauer Spectroscopy. JBIC, J. Biol. Inorg. Chem. 1997, 2, 710–713. 10.1007/s007750050187. [DOI] [Google Scholar]
- Zaccai G. How Soft is a Protein? A Protein Dynamics Force Constant Measured by Neutron Scattering. Science 2000, 288, 1604–1607. 10.1126/science.288.5471.1604. [DOI] [PubMed] [Google Scholar]
- Keller H.; Debrunner P. G. Differences in the Dynamics of Oxidized and Reduced Cytochrome c Measured by Mössbauer Spectroscopy. Phys. Rev. Lett. 1980, 45, 68–71. 10.1103/PhysRevLett.45.68. [DOI] [Google Scholar]
- Parak F. G.; Frolov E. N.; Mössbauer R. L.; Goldanskii V. I. Dynamics of Metmyoglobin Crystals Investigated by Nuclear Gamma Resonance Absorption. J. Mol. Biol. 1981, 145, 825–833. 10.1016/0022-2836(81)90317-X. [DOI] [PubMed] [Google Scholar]
- Doster W.; Cusack S.; Petry W. Dynamical Transition of Myoglobin Revealed by Inelastic Neutron Scattering. Nature 1989, 337, 754–756. 10.1038/337754a0. [DOI] [PubMed] [Google Scholar]
- Doster W. The Dynamical Transition of Proteins, Concepts and Misconceptions. Eur. Biophys. J. 2008, 37, 591–602. 10.1007/s00249-008-0274-3. [DOI] [PubMed] [Google Scholar]
- Young R. D.; Frauenfelder H.; Fenimore P. W. Mössbauer Effect in Proteins. Phys. Rev. Lett. 2011, 107, 158102. 10.1103/PhysRevLett.107.158102. [DOI] [PubMed] [Google Scholar]
- Caliskan G.; Briber R. M.; Thirumalai D.; Garcia-Sakai V.; Woodson S. A.; Sokolov A. P. Dynamic Transition in tRNA is Solvent Induced. J. Am. Chem. Soc. 2006, 128, 32–33. 10.1021/ja056444i. [DOI] [PubMed] [Google Scholar]
- Bellissent-Funel M.-C.; Hassanali A.; Havenith M.; Henchman R.; Pohl P.; Sterpone F.; van der Spoel D.; Xu Y.; Garcia A. E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. 10.1021/acs.chemrev.5b00664. [DOI] [PMC free article] [PubMed] [Google Scholar]