

Abstract
Rationale
Vascular maturation plays an important role in wound repair post-myocardial infarction (MI). The Notch3 is critical for pericyte recruitment and vascular maturation during embryonic development.
Objective
This study is to test whether Notch3 deficiency impairs vascular maturation and blunts cardiac functional recovery post-MI.
Approach and results
Wild type (WT) and Notch3 knockout (Notch3KO) mice were subjected to MI by the ligation of left anterior descending coronary artery (LAD). Cardiac function and coronary blood flow reserve (CFR) were measured by echocardiography. The expression of angiogenic growth factor, pericyte/capillary coverage and arteriolar formation were analyzed. Loss of Notch3 in mice resulted in a significant reduction of pericytes and small arterioles. Notch3 KO mice had impaired pericyte/capillary coverage and CFR compared to WT mice. Notch3 KO mice were more prone to ischemic injury with larger infarcted size and higher rates of mortality. The expression of CXCR-4 and VEGF/Ang-1 was significantly decreased in Notch3 KO mice. Notch3 KO mice also had few NG2+/Sca1+ and NG2+/c-kit+ progenitor cells in the ischemic area and exhibited worse cardiac function recovery at 2 weeks after MI. These were accompanied by a significant reduction of pericyte/capillary coverage and arteriolar maturation. Furthermore, Notch3 KO mice subjected to MI had increased intracellular adhesion molecule-2 (ICAM-2) expression and CD11b+ macrophage infiltration into ischemic areas compared to that of WT mice.
Conclusion
Notch3 mutation impairs recovery of cardiac function post-MI by the mechanisms involving the preexisting coronary microvascular dysfunction conditions, and impairment of pericyte/progenitor cell recruitment and microvascular maturation.
Keywords: Notch3 deficiency, Pericytes, Vascular maturation, Myocardial infarction, Vascular progenitor cells
1. Introduction
Myocardial infarction (MI) affects millions of individuals each year and represents a considerable economic burden to healthcare systems in United States. Despite advances in therapy, treatment remains difficult and our understanding of the molecular basis of disease remains in flux. One of the emerging approaches for treatment of heart failure post-MI is the promotion of coronary angiogenesis [1,2]. Angiogenesis is the key to resolving myocardial ischemia. The survival of the ischemic heart is increased by promoting new blood vessel formation in ischemic areas, limiting regions of damage and ultimately preserving cardiac function [3–8]. Although promotion of cardiac angiogenesis is a promising therapeutic strategy in post-MI, the clinical trials using proangiogenic factors have shown limited benefits. Therefore, the development of a successful angiogenic therapy for post-MI requires an in-depth understanding of the molecular mechanisms of coronary angiogenesis. Accumulating evidence demonstrates that the controlling neovessel maturation and the maintenance of vasculature integrity and stabilization is essential for angiogenic therapy. During angiogenesis, the neovessels of the ischemic area acquire a muscular coat to form a mature vasculature and stabilize capillaries, whereas uncoated neovessels undergo capillaries regression. Previous studies have revealed that endothelial cells can be implanted in vivo, forming neovessels; however, these vessels rapidly regress without the support of SMC or pericytes (PC) [9,10]. Therefore, the recruitment of SMC/PC is crucial for the growth of mature vessels and the maintenance of already formed capillaries in post-MI. So far, little is known about the molecular signaling and vascular maturation in post-MI. Unraveling the molecular mechanisms of neovessel maturation and angiogenesis in post-MI will provide novel approaches to prevent and/or reverse ischemic heart failure.
The Notch3 receptor is located on the surface of the mural cells (vascular smooth muscle cells or pericytes) that surround blood vessels. The Notch3 receptor is specifically expressed in arteries. NOTCH3 mutations cause cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) in human [11]. Mutations of Notch3 also result in the reduction of artery caliber and capillary density in the brain [12]. Impairment of Notch3 has been shown to disrupt VSMC maturation/differentiation and is correlated with increased infarction size in ischemic stroke [12–16]. In addition, Notch3 mutation carriers have been shown to have increased risk of early myocardial infarction [17]. Taken together, these studies suggest that loss of Notch3 receptor may increase the risk for ischemic injury. However, the mechanism by which Notch3 gene mutation causes the exacerbation of myocardial ischemic injury remains unclear. In the present study, we hypothesize that Notch3 deficiency reduces myocardial angiogenesis and coronary flow reserve through the impairment of pericyte/progenitor cell recruitment and vascular maturation in post-MI.
2. Methods
2.1. Experimental animals
Male Notch3tm1Grid mice and their respective wild-type (WT) control mice were obtained from Jackson Laboratories (Bar Harbor, ME). All animals were fed with laboratory standard food and water, and housed in individually ventilated cages in the Laboratory Animal Facilities at the University of Mississippi Medical Center. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Mississippi Medical Center (Protocol ID: 1280A) and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85-23, Revised 1996).
2.2. Mouse myocardial ischemia model
Notch3 tm1Grid mice and WT mice at age of 7–9 month old were subjected to myocardial ischemia by the ligation of left anterior descending coronary artery (LAD) for 24 h as acute myocardial ischemia model and 2 weeks for post-myocardial ischemia model. Briefly, mice were anesthetized with the mixture of ketamine (100 mg/kg) and xylazine (15 mg/kg). The mice were intubated and connected to artificial ventilation in ambient air. Left thoracotomy was performed to expose the heart. LAD was ligated using an 8-0 nylon suture [18,19]. Twenty four hours or 2 weeks after surgery, mice were sacrificed and heart tissue was collected.
2.3. Echocardiography and cardiac hypertrophy
Transthoracic echocardiograms were performed on mice using a Vevo770 High-Resolution In Vivo Micro-Imaging System equipped with a RMV 710B scan head (VisualSonics Inc., Canada). The experimental mouse was anesthetized by inhalation of 1.5–2% isoflurane mixed with 100% medical oxygen in an isolated chamber. Anesthesia was kept with 1–1.5% isoflurane, maintaining the heart rate between 400 and 450 beats per minute. M-mode cine loops 1were recorded and analyzed by High-Frequency Ultrasound Imaging software (VisualSonics Inc., Canada) to assess myocardial parameters and cardiac functions of left ventricle, including left ventricle end-systolic diameter (LVESD), left ventricle end-diastolic diameter (LVEDD), left ventricle end-systolic volume (LVESV), left ventricle end-diastolic volume (LVEDV), thickness of the left ventricle anterior wall (LVAW) and posterior wall (LVPW) at end systole and end diastole, and stroke volume (SV), cardiac output (CO), as well as ejection fraction (EF%) and fractional shortening (FS%) [18,19].
A cine loop of left proximal coronary artery (LCA) was recorded in pulsed-wave Doppler-mode at baseline (inhalation of 1% isoflurane), and under hyperemic conditions (inhalation of 2.5% isoflurane). The coronary flow reserve (CFR) was calculated as the ratio of peak blood flow velocity during hyperemia to peak blood flow velocity at baseline [20,21]. To determine the cardiac hypertrophy, heart weight (HW), heart/body weight (HW/WB) ratio and heart weight/tibia length ratio were measured at the end of experiments.
2.4. Histology and immunofluorescence
Left ventricular sections were dissected from border zone of ischemic area (3 mm–4 mm below the ligation site, to the apex side) and stained with fluorescein-labeled Griffonia Bandeiraea Simplicifolia Isolectin B4 (1:50; IB4, Invitrogen, OR, USA) to visualize capillaries. Pericytes were labeled with mouse monoclonal antibody specific to neural/glial antigen-2 (NG2 proteoglycan) (1:200; Abcam, MA, USA). The coverage of pericyte/capillary was expressed as the ratio of the number of NG2-positive cells to the number of IB4-positive cells. Smooth muscles were labeled with mouse monoclonal antibody specific α-smooth muscle actin (SMA) (1:250; Sigma, MO, USA). The coverage of SMA/capillary was expressed as the ratio of the number of SMA-positive cells to the number of IB4-positive cells. Small arterioles were counted and calculated as arteriole number per fields (40×). Monocyte/Macrophage infiltration was stained with fluorescein-labeled rat anti-mouse CD11b (1:50; BD, CA, USA). The intercellular adhesion molecule-2 (ICAM-2) was stained with rabbit polyclonal antibody (1:50; Santa Cruz, CA, USA).
2.5. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and cardiomyocyte apoptosis
In situ DeadEnd™ Colorimetric Apoptosis Detection System (Promega, Madison, WI, USA) was performed to detect apoptotic cells in frozen heart sections of ischemic area according to the manufacturer’s instructions. 4″, 6-diamidino-2-phenylindole (DAPI) was used to visualize nuclei. To determine the cardiomyocyte apoptosis, heart sections were double immunostained with cardiac troponin T (1:100, ThermoFisher) and TUNEL.
2.6. Myocardial progenitor cell Sca1, c-kit, NG2+/c-kit+ and NG2+/Sca1+ immunostaining
Heart tissue sections (8 μm) were incubated with progenitor cell marker Sca1 and c-kit antibodies overnight. Sca1 and c-kit were visualized using FITC labeled goat anti-mouse IgG antibodies; NG2+/c-kit+ and NG2+/Sca1+ were visualized with Fluorolink™ Cy™3 labeled goat anti-mouse IgG antibodies (1:200). Sections were counterstained with DAPI. Myocardial C-kit+, Sca-1+, NG2+/c-kit+ and NG2+/Sca1+ were assessed by measure the area of positive cells per field (10×) [22].
2.7. Western blot analysis
Left ventricle samples from border zone of ischemic area were homogenized in lysis buffer with protease inhibitor cocktail. The Polyvinylidene difluoride membranes were probed with antibodies specific to cleaved Notch3, angiopoietin-1 (ANG1), CXCR-4, SDF-1α and vascular endothelial growth factor (VEGF) (Santa Cruz, CA, USA). The membranes were then washed and incubated with an anti-rabbit or anti-mouse secondary antibody conjugated with horseradish peroxidase. Densitometries were analyzed using TINA 2.0 image analysis software.
2.8. Assessment of infarct size and risk area
The ischemic hearts were cut transversely into 1-mm-thick slices and stained with 1% triphenyltetrazolium chloride (TTC) in PBS (pH 7.4) for 30 min at 37 °C in a water bath. After fixation for 1 h in 10% neutral buffered formaldehyde, each slice was photographed. Viable myocardium remained red and infarct area stained white. Infarct area and total left ventricle (LV) area were measured using Image J software, and expressed as a percentage of the total LV area [23]. In addition, ischemic heart was stained with Evens blue and TTC to examine the infarcted area (Evens blue and TTC unstained), risk area (Evens blue unstained, TTC stained) and health area (Evens blue stained).
2.9. Retinal angiogenesis
C57Bl/6J mice and Notch3 KO mice were sacrificed. The retinas of mice were harvested and dry mounts were prepared. The retinal pericyte (NG+), endothelial cells (IB4+) and VSMCs (SMA+) in retinal arterioles were measured by immunofluorescence analysis as described above.
2.10. Statistical analysis
The data were expressed as Mean ± SEM. Statistical analysis was performed using Student’s unpaired two-tailed t-test for comparisons between two groups, on-way followed by post hoc test for multiple comparisons. Fisher’s exact test was used for the mortality comparisons between two groups. For the statistical analysis of the echocardiography parameters, we performed a one-way ANOVA followed by post hoc test to examine the differences between multi-groups. P value <0.05 was considered as statistically significant.
3. Results
3.1. Knockout of Notch3 reduces pericyte coverage and VSMC in vivo in a mouse model of retinal angiogenesis and in the mouse heart
Since Notch3 has a critical role in the recruitment of pericytes as well as VSMC proliferation [24,25], we first examined the role of Notch3 deficiency on pericyte coverage and VSMC in vivo using the mouse model of retinal angiogenesis. As shown in Fig. 1A, pericytes were labeled with pericyte marker NG2 (red), VSMC were labeled with α-smooth muscle actin (SMA) and the endothelial cells were labeled with IB4 (green). Knockout of Notch3 resulted in a dramatic reduction of pericyte coverage compared to WT control mice. The SMA coverage was also reduced in Notch3 KO mice compared to WT control mice.
Fig. 1.
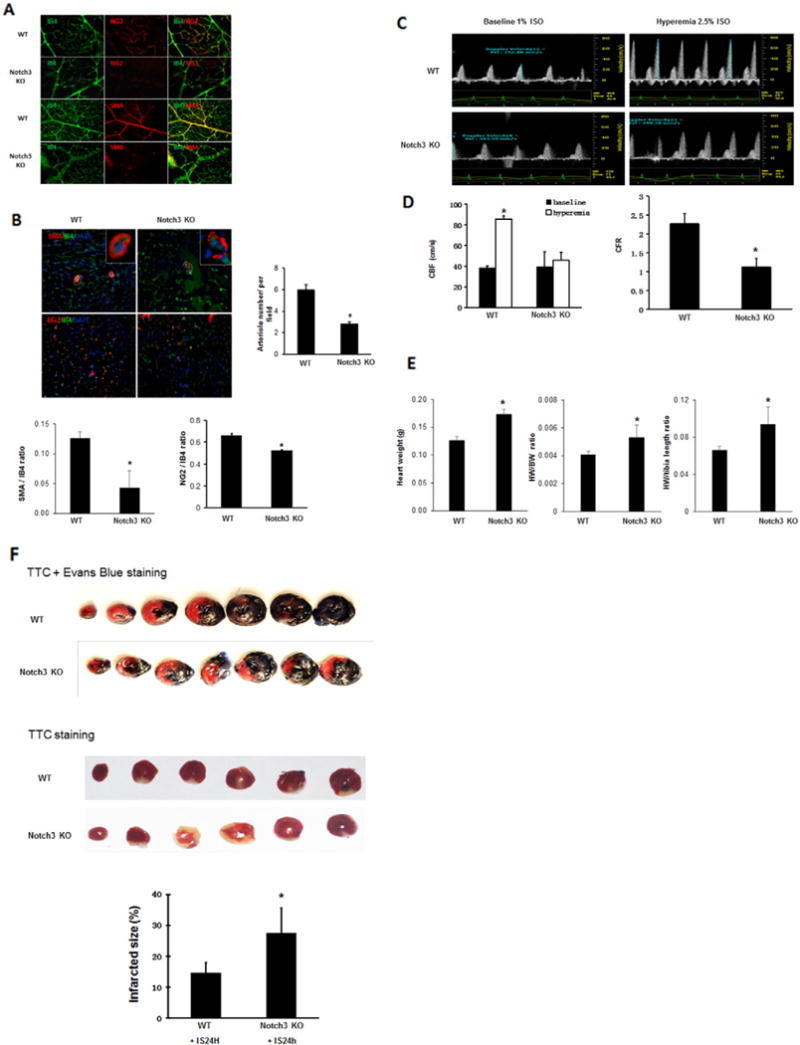
Knockout of Notch3 reduces pericyte/VSMC coverage and coronary flow reserve and increases infarcted size. A. In the retina of both WT and Notch3 KO mouse, pericytes were labeled with pericyte marker NG2 (red), VSMC were labeled with α-smooth muscle actin (SMA, red) and the endothelial cells were labeled with IB4 (green). Knockout of Notch3 resulted in a dramatic reduction of pericyte and SMA coverage compared to WT control mice. B. Knockout of Notch3 resulted in a significant reduction of the number of SMA and cardiac pericytes and small arterioles with abnormal vessel wall in the heart compared to WT mice at basal levels. *P < 0.05. 40×. C and D. Pulsed-wave Doppler images showed that the peak hyperemic diastolic blood flow velocity and CFR were significantly decreased in Noch3 KO mice (n = 4) than that of WT mice (n = 8). Quantification of CBF and CFR. *P < 0.05, compared with baseline respectively. #P < 0.05, compared to WT mice. CBF, coronary blood flow. CFR, coronary flow reserve. E. Measurements of heart weight (HW), heart weight/body weight ratio (HW/BW) and heart weight (HW)/tibia length ratio. The HW, HW/BW ratio and HW/tibia length ratio were significantly increased in the Notch3KO mice compared to WT control mice. *P < 0.05, n = 5–6 mice. F. Mouse heart was stained with Evens blue and 1% 2,3,5-triphenyltetrazolium chloride (TTC). The risk area (Evens blue unstained, TTC stained) was increased in Notch3KO mice subjected to MI compared to WT mice. Mouse heart sections with 1% 2,3,5-triphenyltetrazolium chloride (TTC) staining showed that myocardial infarcted area was significantly increased in the Notch3KO mice compared to WT mice. n = 6 mice, *P < 0.05.
To further determine the effects of Notch3 deficiency on coronary angiogenesis, the number of cardiac pericyte and small arterioles were measured in the hearts of Notch3KO mice. Knockout of Notch3 resulted in a significant reduction of cardiac SMA/pericytes and small arterioles in the heart compared to WT mice at basal levels (Fig. 1B).
3.2. Knockout of Notch3 in mice reduces CFR at baseline and is prone to MI injury
Since Notch3 KO mice exhibited decreased VSMC and small arterioles, we hypothesized that these abnormalities may lead to an impairment of coronary blood flow/reserve and cardiac hypertrophy. Echocardiography examination revealed that knockout of Notch3 did not result in significant difference in cardiac function compared to WT mice at baseline (Table 1). However, pulsed-wave Doppler showed that the hyperemic peak diastolic blood flow velocity of LCA was significantly decreased in Notch3 KO mice, leading to a significant reduction in CFR (Fig. 1C and D). Furthermore, knockout of Notch3 in mice led to cardiac hypertrophy as evidenced by the increased heart weight (HW), heart/body weight (HW/WB) ratio and heart weight/tibia length ratio (Fig. 1E). These data suggest a preexisting coronary microvascular dysfunction and cardiac hypertrophy in the Notch3 KO mice.
Table 1.
Morphological and cardiac function parameters.
| Parameters | WT (n = 10) | WT + IS2w (n = 6) | Notch3 KO (n = 5) | Notch3 KO + IS2w (n = 5) |
|---|---|---|---|---|
| LVESD (mm) | 1.97 ± 0.11 | 2.57 + 0.14* | 2.41 ± 0.33 | 3.28 ± 0.10*,# |
| LVEDD (mm) | 3.38 ± 0.12 | 3.83 + 0.14 | 3.84 ± 0.29 | 4.47 ± 0.10*,# |
| LVESV (μl) | 13.00 ± 1.71 | 24.42 + 3.17* | 23.55 ± 8.07 | 43.76 ± 3.18*,# |
| LVEDV (μl) | 47.74 ± 4.00 | 63.66 + 5.47* | 66.13 ± 12.22‡ | 91.03 ± 4.67*,# |
| SV (μl) | 34.84 ± 2.60 | 39.26 + 2.58 | 42.82 ± 5.25 | 47.56 ± 2.33 |
| CO (μl/min) | 13.57 ± 0.57 | 18.34 + 1.47* | 18.48 ± 2.07‡ | 17.72 ± 2.99 |
| LVAW, s (mm) | 1.23 ± 0.06 | 0.98 + 0.02* | 1.21 ± 0.02 | 1.08 ± 0.06 |
| LVAW, d (mm) | 0.73 ± 0.04 | 0.64 + 0.01 | 0.74 ± 0.05 | 0.75 ± 0.06 |
| LVPW, s (mm) | 1.02 ± 0.05 | 0.87 + 0.02 | 1.06 ± 0.07 | 0.87 ± 0.11 |
| LVPW, d (mm) | 0.59 ± 0.04 | 0.52 + 0.01 | 0.72 ± 0.06 | 0.69 ± 0.08# |
Data are means ± SEM. LVESD, left ventricle end-systolic diameter. LVEDD, left ventricle end-diastolic diameter. LVESV, left ventricle end-systolic volume. LVEDV, left ventricle end-diastolic volume. SV, stroke volume. CO, cardiac output. LVAW, s, left ventricle systolic anterior wall thickness. LVAW, d, left ventricle diastolic anterior wall thickness. LVPW, s, left ventricle systolic posterior wall thickness. LVPW, d, left ventricle diastolic posterior wall thickness.
P < 0.05, compared with respective baseline.
P < 0.05, compared with respective WT + IS2w.
P < 0.05, compared with respective WT baseline.
Next, we investigated the consequences of Notch3 deficiency and impaired CFR in response to myocardial ischemic injury. WT mice subjected to acute myocardial ischemia resulted in 14.3% (n = 14 mice) mortality within 24 h. Notch3 KO mice had a significant higher mortality (43.8%, n = 16 mice, P < 0.05) within that same time span. As shown in Fig. 1F, the risk area was increased in Notch3KO mice subjected to MI compared to WT mice. Moreover, Notch3KO mice displayed a larger infarcted size compared to WT mice (Fig. 1F), suggesting loss of Notch3 may exacerbate ischemic injury.
3.3. Knockout of Notch3 blunts the expression of angiogenic growth factor in response to ischemia
WT mice subjected to MI for 24 h resulted in a significant increase in the expression of Notch3 compared to WT mice without ischemia (Fig. 2A). WT mice subjected to myocardial ischemia for 24 h resulted in a significant elevation of ANG-1 expression. Knockout of Notch3 in the mice failed to increase expression of ANG-1 in the border zone of ischemia (Fig. 2B). Similar, myocardial ischemia caused a significant increase in the expression of VEGF in the WT mice, but this was not seen in the Notch3KO mice (Fig. 2C).
Fig. 2.
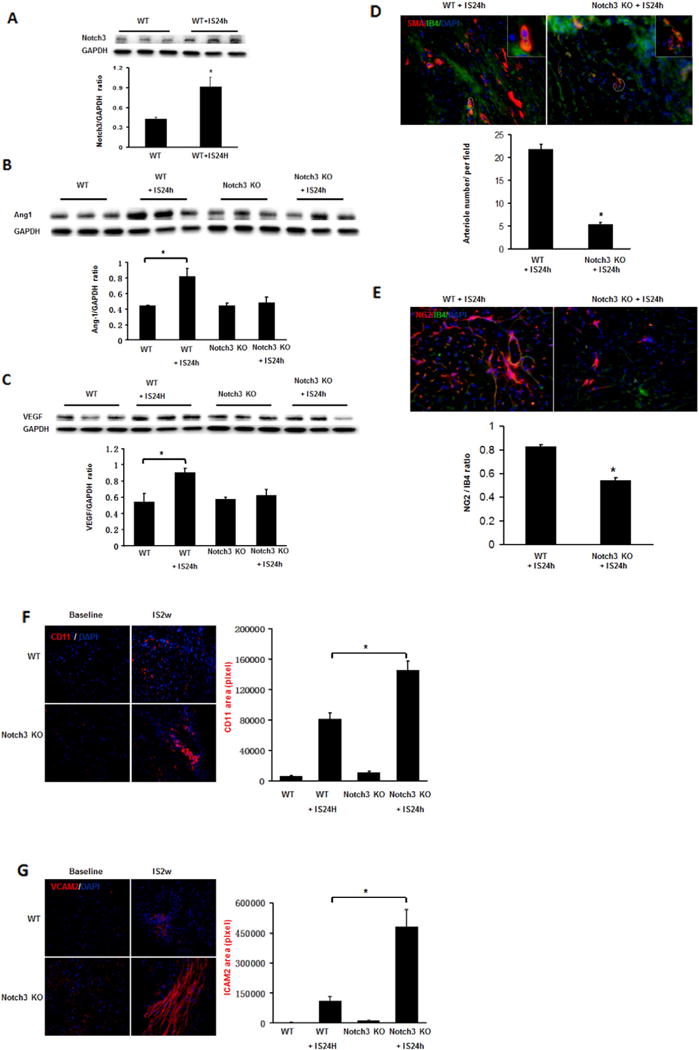
Loss of Notch3 fails to upregulate angiogenic growth factors and stimulate smooth muscle cell and pericyte formation in the post-MI. A. Western blot analysis and quantification revealed that WT mice subjected to MI for 24 h resulted in a significant increase of Notch3. B. Western blot analysis and quantification of Ang-1 normalized to GAPDH showed that the expression of Ang-1 was significantly reduced in the Notch3 KO post-MI mice. C. Western blot analysis and quantification of VEGF normalized to GAPDH the expression of VEGF was significantly reduced in the Notch3 KO post-MI mice. n = 3 mice, *P < 0.05. D. Immunofluorescence image and quantification of mouse heart section 24 h after infarction, smooth muscle cells were labeled with anti-SMA in vasculature and endothelial cells were labeled with IB4 (green). *P < 0.05. E. Immunofluorescence image and quantification of mouse heart section 24 h after infarction, pericytes were labeled with pericyte marker NG2 (red), endothelial cells were labeled with IB4 (green). *P < 0.05. 40×. F. Immunofluorescence image and quantification of mouse heart section, CD11 (red) area was calculated. *P < 0.05. 40×. G. Immunofluorescence image and quantification of mouse heart section, ICAM2 (red) area was calculated. *P < 0.05. 40×.
3.4. Notch3 KO mice fail to stimulate arteriole formation post-MI
We then further examined whether reduction of angiogenic growth factor would result in impairment of arteriole formation in response to ischemia in the Notch3 KO mice. At 2 weeks, WT mice subjected to MI had a robust arteriole formation. Notch3 KO post-MI mice had less smooth muscle cell (labeled with anti-SMA) in vasculature. The smooth muscle cells also exhibited irregular coverages, leading to incomplete arteriole walls and immature neovessel formation. Moreover, the number of small arterioles in the border zone of ischemic area was significantly reduced in the Notch3 KO post-MI mice compared to WT post-MI mice (Fig. 2D).
3.5. Knockout of Notch3 reduces the pericytes/capillary coverage and increases inflammation post-MI
Notch3 KO-post-MI mice also had significantly less pericyte coverage per capillary (labeled with NG2 and IB4) in the coronary microvasculature compared to WT (Fig. 2E). We further tested if loss of pericyte coverage in capillaries led to vascular leakage and inflammation. The results of immunostaining with CD11b, an antigen expressed on monocyte/macrophage, revealed that Notch3 KO-post-MI mice had a significant higher number of CD11+ cell in the ischemic area (Fig. 2F). Similarly, ICAM-2, a protein localized at endothelial junctions and involved in vascular permeability, was significantly increased in Notch3 KO post-MI mice (Fig. 2G). Taken together, these results suggest loss of Notch3 disrupts EC/pericyte interactions which may contribute to microvascular leakage and inflammation in post-MI.
3.6. Knockout of Notch3 reduces CXCR-4 expression and recruitment of progenitor cells under ischemic conditions
The expression of CXCR-4 was significantly reduced whereas SDF-1α expression was not altered in the ischemic heart of Notch3KO mice (Fig. 3A–B). Next, we investigated whether knockout of Notch3 affected the progenitor cells in the infarcted mouse hearts. Sca1+ and c-kit+ cells were examined in the border zone of infarction at 14 days after myocardial ischemia. The numbers of Sca1+ and c-kit+ cells were decreased in infarcted hearts at day 14 post-MI in the Notch3KO mice. Moreover, knockout of Notch3 resulted in a significant reduction of NG2+/Sca1+ and NG2+/c-kit+ cells compared to controls at day 14 of post-MI (Fig. 3C).
Fig. 3.
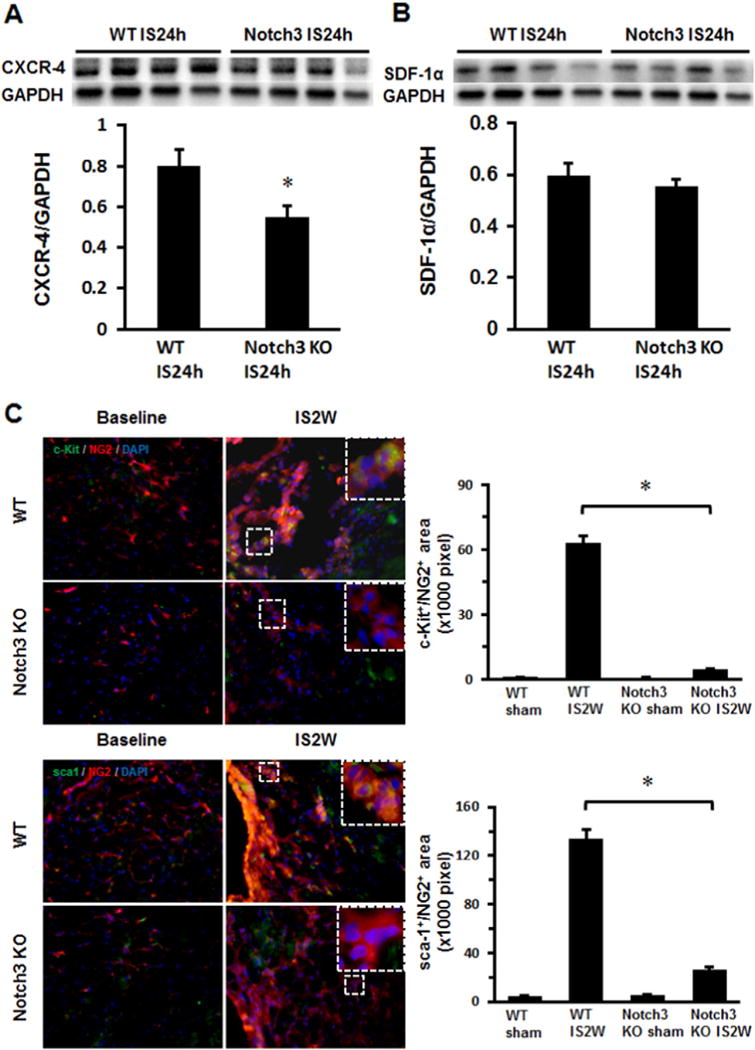
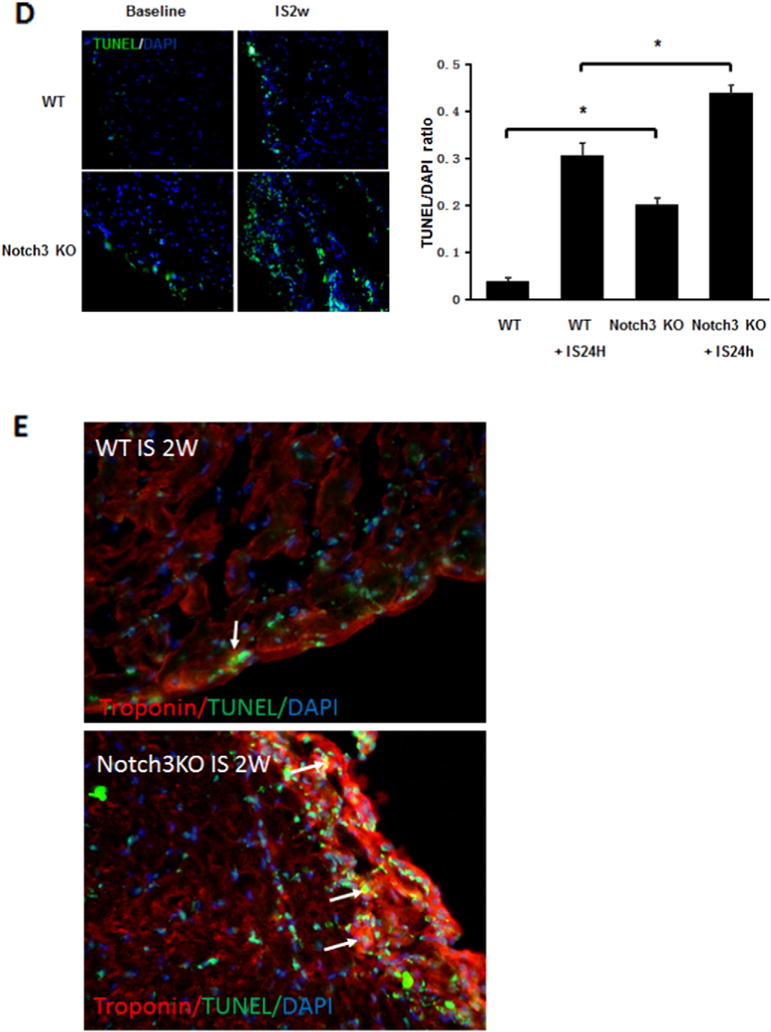
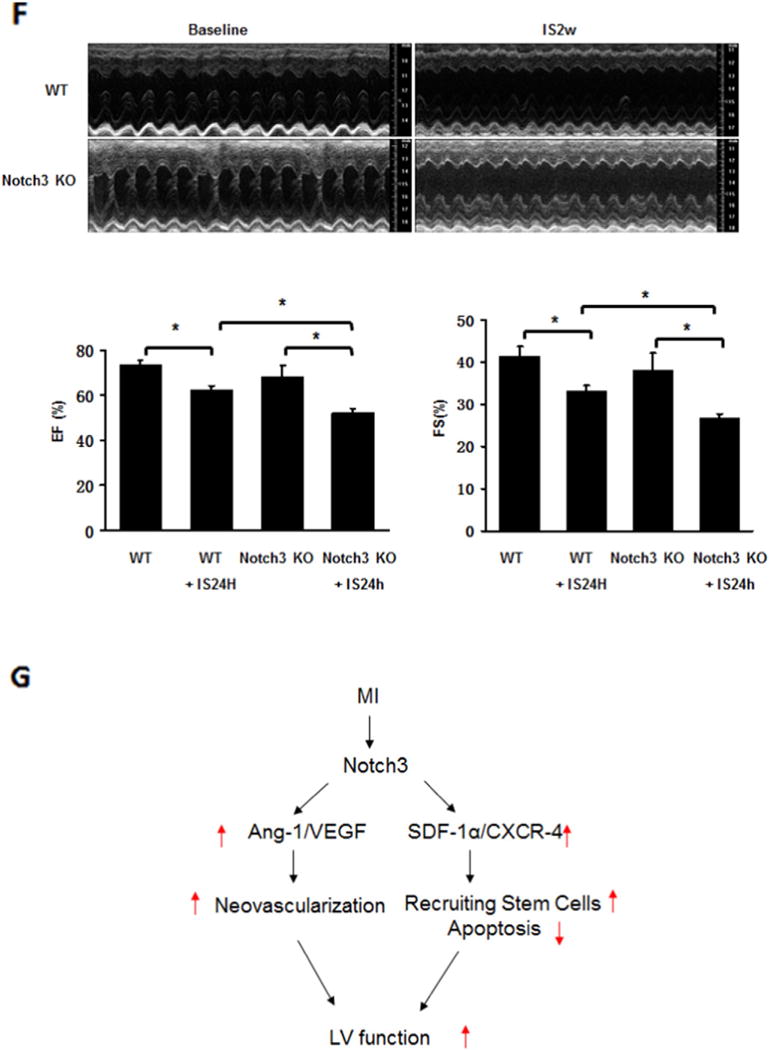
Knockout of Notch3 reduces progenitor cells and worse cardiac function in post-MI. A and B. Western blot analysis of the expression of CXCR-4 and SDF-1α expression revealed that the expression of CXCR4 was significantly reduced in the ischemic heart of Notch3 KO mice (n = 4 mice, *P < 0.05). C. Immunostaining analysis showed that the area of c-kit+/NG2+ and Sca-1+/NG2+ was significantly reduced in the Notch3 KO post-MI mice (n = 6–7 mice, *P < 0.05). D. TUNEL and quantification measured cardiac apoptosis of mouse, TUNEL (green), 40×. The number of TUNEL positive cells was significantly increased in Notch3 KO post-MI compared to WT post-MI (n = 6–7 mice, *P < 0.05). E. Heart sections were immunostained with cardiomyocyte marker Troponin T (red) and apoptosis marker TUNEL (green). Knockout of Notch3 resulted in a dramatic increase in number of Troponin+/TUNEL+ cells (white arrows). F. Echocardiography and quantification showed FS and EF change of post-MI. Notch3 deficiency exacerbated cardiac dysfunction in post-MI (n = 10–5 mice *P < 0.05). G. The proposed working model. MI activates Notch3 and leads to upregulation of angiogenic growth factor and progenitor recruiting factor expression, these result in increase in neovascularization and stem cell recruitment into ischemic area, reduces cardiomyocyte apoptosis and improves LV functional recovery in post-MI.
3.7. Knockout of Notch3 increases cardiomyocyte apoptosis and impedes cardiac functional recovery post-MI
Two weeks post-MI, Notch3KO mice had significantly higher rates of apoptosis in the ischemic region compared to WT mice (Fig. 3D). Moreover, knockout of Notch3 resulted in a dramatic increase in number of Troponin T+/TUNEL+ cells (white arrows), suggesting apoptosis in the cardiomyocyte (Fig. 3E).
Echocardiography revealed worsen EF and FS value in Notch3 mice compared to WT (Fig. 3F). Other echocardiographic parameters such as left ventricle end-systolic diameter (LVESD); left ventricle end-diastolic diameter (LVEDD); left ventricle end-systolic volume (LVESV); and left ventricle end-diastolic volume (LVEDV) were also significantly increased in the Notch3KO post-MI mice, indicating development of dilated cardiomyopathy (Table 1).
4. Discussion
Our previous study suggests that pre-existing coronary microvascular dysfunction and established microvascular rarefaction may exacerbate post-MI cardiac dysfunction and impair post-MI repair and recovery [22]. In this study, we further demonstrated that loss of Notch3 reduced cardiac pericyte coverage, impaired coronary blood flow reserve, and ultimately increased the area of ischemic injury. Our study also showed that loss of Notch3 reduced the numbers of NG2+/Sca1+/c-kit+ progenitor cells and diminished microvascular maturation, thus promoting ischemia-induced microvascular leakage and inflammation. Our data strongly suggests that Notch3 is necessary to maintain both the pericyte/VSMC differentiation and the integrity of coronary microvasculature in response to myocardial ischemia.
Accumulating evidence indicates that capillary rarefaction has a causal role in the onset and progression of heart failure [26,27]. Capillaries are the primary determinant of myocardial perfusion assessed by coronary flow reserve (CFR) [28]. Microvascular rarefaction and impairment of CFR are associated with heart failure. Human studies have demonstrated that CFR is impaired in hypertrophic cardiomyopathy, dilated cardiomyopathy and heart failure with preserved ejection fraction (HFpEF) [29–32]. One recent human study also provided evidence of a strong relationship between microvascular rarefaction and HFpEF [26]. In the present study, we found that the number of small arterioles and CFR were significantly decreased in the Notch3 KO mice compared to their control mice at baseline, suggesting a reduction in coronary microvasculature and possible impairment of myocardial perfusion under stressed conditions. We further demonstrated that Notch3 KO mice were more prone to ischemic injury with a larger infarct size and higher mortality within 24 h. Notch3 plays an important role in the regulation of smooth muscle maturation, modulation of vascular function, and response to ischemia [33–35]. Knockout of Notch3 in mice has been shown to cause structural defects with enlargement of the vessels and impaired smooth muscle formation in the distal arteries [34]. Moreover, Notch3 mutation is associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) [36,37]. A recent study demonstrates that loss of Notch3 in mice alters coronary microvasculature and exacerbates angiotensin II-induced heart failure [38]. These data strongly suggest that preexisting coronary microvascular dysfunction and microvascular rarefaction may contribute to exacerbation of ischemic injury in the Notch3 KO mice.
Our previous study revealed that Notch3 expression was significantly increased in ischemic heart of post-MI mice [39]. Consistent with our previous findings, the present study also demonstrated that mice subjected MI for 24 h resulted in upregulation of Notch3 expression. Interestingly, knockout of Notch3 in mice exhibited more severe post-MI cardiac dysfunction and impaired post-MI repair and recovery. Our data indicate that activation of Notch3 may be necessary to preserve cardiac function in response to myocardial ischemia. We further explored the mechanisms by which Notch3 deficiency interfered with cardiac functional recovery post-MI. Our previous study demonstrates that diabetic insufficient angiogenesis is caused by impaired VSMC-EC communication and formation of immature vasculature, which results in neovessel regression [40]. Here, we showed that coverage of cardiac pericytes (labeled with NG2)/capillary were reduced in response to ischemia in the Notch3 KO mice. The size of arterioles around the ischemic area was dramatically decreased in Notch3 KO post-MI mice. Furthermore, Notch3 KO post-MI mice had more immature vessel formation. Using mouse retinal angiogenesis model, we further confirmed that loss of Notch3 impaired pericyte/EC coverage and vascular maturation. Knockout of Notch3 in mice led to increased inflammatory cell (CD11b and ICAM-2) infiltration into the ischemic area. These abnormalities might be caused by the malformation of artery mural cells like pericytes or smooth muscle cells. Overall, we speculate that reduction of Notch3 impairs pericyte/smooth muscle cells recruitment and proliferation, impedes microvascular maturation, and increases leakage of inflammatory cells. These changes impede recovery of cardiac function after an ischemic event, and may ultimately cause progression to dilated cardiomyopathy.
Our exciting data strongly suggest that reduction of pericyte/VSMC coverage and formation of immature neovessels might be novel mechanisms responsible for the exacerbation of ischemic injury in the Notch3KO mice. We further explored the molecular basis by which loss of Notch3 impairs VSMC/pericytes differentiation by examining the interactions between the Notch3 and pericyte recruitment related signaling pathways. In general, pericyte recruitment is modified by the crosstalk between ligands/receptors, such as ANG1/Tie2, VEGF/VEGFR2, platelet derived growth factor-BB/platelet derived growth factor-β and Jagged1/Notch. Our data showed that the expression of VEGF and Ang-1 was significantly reduced in the Notch3 KO mice, suggesting an involvement of Notch3/VEGF/Ang-1 signaling pathway. In addition, accumulating evidence demonstrates that pericytes may act as multi-potent stem cells, which can differentiate into fibroblasts, smooth muscle cells, macrophage/dendritic cells, endothelial cells, adipocytes or chondrocytes [41–43]. Pericytes are specialized mural cells that maintain support structure for capillary endothelial cells. They also have contractile property to regulate blood flow in capillaries [44,45]. In response to vascular injury or at late phase vascular remodeling, pericytes also have the potential of differentiation into endothelial cells [46,47]. In the present study, we found that knockout of Notch3 significantly reduced the CXCR-4 expression and decreased the number of NG2+/Sca1+ and NG2+/c-kit+ progenitor cells in ischemic hearts. These results indicate that Notch3 deficiency may impede wound repair by impairment of stem cells during ischemic injury (Fig. 3G).
In conclusion, the current study reveals the mechanism by which Notch3 mutation deteriorates cardiac function and impairs cardiac recovery post-MI. These effects occur through coronary microvascular dysfunction, loss of pericytes and impaired neovessel maturation in response to ischemia. The results from our studies would lead to the development of new therapeutic approaches to ischemic heart failure centered at increasing pericyte coverage, thereby preventing capillary regression and preserving cardiac function.
Acknowledgments
This study was supported by grants from NIH grant 2R01HL102042-05 and University of Mississippi Medical Center Intramural Research Support Program to J.X. Chen
Footnotes
Conflict of interest
The authors report no relationships that could be construed as a conflict of interest.
References
- 1.Oka T, Akazawa H, Naito AT, et al. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114:565–571. doi: 10.1161/CIRCRESAHA.114.300507. [DOI] [PubMed] [Google Scholar]
- 2.Hou J, Kang YJ. Regression of pathological cardiac hypertrophy: signaling pathways and therapeutic targets. Pharmacol Ther. 2012;135:337–354. doi: 10.1016/j.pharmthera.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shyu KG, Chang CC, Wang BW, et al. Increased expression of angiopoietin-2 and Tie2 receptor in a rat model of myocardial ischaemia/reperfusion. Clin Sci (Lond) 2003;105:287–294. doi: 10.1042/CS20030025. [DOI] [PubMed] [Google Scholar]
- 4.Banai S, Shweiki D, Pinson A, et al. Upregulation of vascular endothelial growth factor expression induced by myocardial ischaemia: implications for coronary angiogenesis. Cardiovasc Res. 1994;28:1176–1179. doi: 10.1093/cvr/28.8.1176. [DOI] [PubMed] [Google Scholar]
- 5.Ray PS, Estrada-Hernandez T, Sasaki H, et al. Early effects of hypoxia/reoxygenation on VEGF, ang-1, ang-2 and their receptors in the rat myocardium: implications for myocardial angiogenesis. Mol Cell Biochem. 2000;213:145–153. doi: 10.1023/a:1007180518474. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki H, Ray PS, Zhu L, et al. Oxidative stress due to hypoxia/reoxygenation induces angiogenic factor VEGF in adult rat myocardium: possible role of NFkappaB. Toxicology. 2000;155:27–35. doi: 10.1016/s0300-483x(00)00274-2. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki H, Ray PS, Zhu L, et al. Hypoxia/reoxygenation promotes myocardial angiogenesis via an NF kappa B-dependent mechanism in a rat model of chronic myocardial infarction. J Mol Cell Cardiol. 2001;33:283–294. doi: 10.1006/jmcc.2000.1299. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki H, Fukuda S, Otani H, et al. Hypoxic preconditioning triggers myocardial angiogenesis: a novel approach to enhance contractile functional reserve in rat with myocardial infarction. J Mol Cell Cardiol. 2002;34:335–348. doi: 10.1006/jmcc.2001.1516. [DOI] [PubMed] [Google Scholar]
- 9.Fruttiger M. Development of the retinal vasculature. Angiogenesis. 2007;10:77–88. doi: 10.1007/s10456-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 10.Wang ZZ, Au P, Chen T, et al. Endothelial cells derived from human embryonic stem cells form durable blood vessels in vivo. Nat Biotechnol. 2007;25:317–318. doi: 10.1038/nbt1287. [DOI] [PubMed] [Google Scholar]
- 11.Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 12.Joutel A, Monet-Lepretre M, Gosele C, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest. 2010;120:433–445. doi: 10.1172/JCI39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dichgans M. Genetics of ischaemic stroke. Lancet Neurol. 2007;6:149–161. doi: 10.1016/S1474-4422(07)70028-5. [DOI] [PubMed] [Google Scholar]
- 14.Lacombe P, Oligo C, Domenga V, et al. Impaired cerebral vasoreactivity in a transgenic mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy arteriopathy. Stroke. 2005;36:1053–1058. doi: 10.1161/01.STR.0000163080.82766.eb. [DOI] [PubMed] [Google Scholar]
- 15.Rboleda-Velasquez JF, Zhou Z, Shin HK, et al. Linking Notch signaling to ischemic stroke. Proc Natl Acad Sci U S A. 2008;105:4856–4861. doi: 10.1073/pnas.0709867105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verreault S, Joutel A, Riant F, et al. A novel hereditary small vessel disease of the brain. Ann Neurol. 2006;59:353–357. doi: 10.1002/ana.20775. [DOI] [PubMed] [Google Scholar]
- 17.Lesnik Oberstein SA, Jukema JW, Van Duinen SG, et al. Myocardial infarction in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) Medicine (Baltimore) 2003;82:251–256. doi: 10.1097/01.md.0000085054.63483.40. [DOI] [PubMed] [Google Scholar]
- 18.Hou X, Zeng H, He X, et al. Sirt3 is essential for apelin-induced angiogenesis in post-myocardial infarction of diabetes. J Cell Mol Med. 2015;19:53–61. doi: 10.1111/jcmm.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He X, Zeng H, Chen JX. Ablation of SIRT3 causes coronary microvascular dysfunction and impairs cardiac recovery post myocardial ischemia. Int J Cardiol. 2016;215:349–357. doi: 10.1016/j.ijcard.2016.04.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.You J, Wu J, Ge J, et al. Comparison between adenosine and isoflurane for assessing the coronary flow reserve in mouse models of left ventricular pressure and volume overload. Am J Physiol Heart Circ Physiol. 2012;303:H1199–H1207. doi: 10.1152/ajpheart.00612.2012. [DOI] [PubMed] [Google Scholar]
- 21.Chang WT, Fisch S, Chen M, et al. Ultrasound based assessment of coronary artery flow and coronary flow reserve using the pressure overload model in mice. J Vis Exp. 2015:e52598. doi: 10.3791/52598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng H, Li L, Chen JX. Overexpression of angiopoietin-1 increases CD133+/c-kit+ cells and reduces myocardial apoptosis in db/db mouse infarcted hearts. PLoS One. 2012;7:e35905. doi: 10.1371/journal.pone.0035905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuo QH, Zeng H, Stinnett A, et al. Critical role of angiopoietins/Tie-2 in hyperglycemic exacerbation of myocardial infarction and impaired angiogenesis. Am J Physiol Heart Circ Physiol. 2008;294:H2547–H2557. doi: 10.1152/ajpheart.01250.2007. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Pan L, Moens CB, et al. Notch3 establishes brain vascular integrity by regulating pericyte number. Development. 2014;141:307–317. doi: 10.1242/dev.096107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu X, Liu XY, Fagan A, et al. Ultrastructural changes in cerebral capillary pericytes in aged Notch3 mutant transgenic mice. Ultrastruct Pathol. 2012;36:48–55. doi: 10.3109/01913123.2011.620220. [DOI] [PubMed] [Google Scholar]
- 26.Mohammed SF, Hussain S, Mirzoyev SA, et al. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 28.Kaul S, Jayaweera AR. Myocardial capillaries and coronary flow reserve. J Am Coll Cardiol. 2008;52:1399–1401. doi: 10.1016/j.jacc.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 29.Tsagalou EP, Anastasiou-Nana M, Agapitos E, et al. Depressed coronary flow reserve is associated with decreased myocardial capillary density in patients with heart failure due to idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2008;52:1391–1398. doi: 10.1016/j.jacc.2008.05.064. [DOI] [PubMed] [Google Scholar]
- 30.Kawada N, Sakuma H, Yamakado T, et al. Hypertrophic cardiomyopathy: MR measurement of coronary blood flow and vasodilator flow reserve in patients and healthy subjects. Radiology. 1999;211:129–135. doi: 10.1148/radiology.211.1.r99ap36129. [DOI] [PubMed] [Google Scholar]
- 31.Kato S, Saito N, Kirigaya H, et al. Impairment of coronary flow reserve evaluated by phase contrast cine-magnetic resonance imaging in patients with heart failure with preserved ejection fraction. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.115.002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watzinger N, Lund GK, Saeed M, et al. Myocardial blood flow in patients with dilated cardiomyopathy: quantitative assessment with velocity-encoded cine magnetic resonance imaging of the coronary sinus. J Magn Reson Imaging. 2005;21:347–353. doi: 10.1002/jmri.20274. [DOI] [PubMed] [Google Scholar]
- 33.Belin de Chantemele EJ, Retailleau K, Pinaud F, et al. Notch3 is a major regulator of vascular tone in cerebral and tail resistance arteries. Arterioscler Thromb Vasc Biol. 2008;28:2216–2224. doi: 10.1161/ATVBAHA.108.171751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Domenga V, Fardoux P, Lacombe P, et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–2735. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arboleda-Velasquez JF, Zhou Z, Shin HK, et al. Linking Notch signaling to ischemic stroke. Proc Natl Acad Sci U S A. 2008;105:4856–4861. doi: 10.1073/pnas.0709867105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ayata C. CADASIL: experimental insights from animal models. Stroke. 2010;41:S129–S134. doi: 10.1161/STROKEAHA.110.595207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto Y, Craggs L, Baumann M, et al. Review: molecular genetics and pathology of hereditary small vessel diseases of the brain. Neuropathol Appl Neurobiol. 2011;37:94–113. doi: 10.1111/j.1365-2990.2010.01147.x. [DOI] [PubMed] [Google Scholar]
- 38.Ragot H, Monfort A, Baudet M, et al. Loss of Notch3 signaling in vascular smooth muscle cells promotes severe heart failure upon hypertension. Hypertension. 2016;68:392–400. doi: 10.1161/HYPERTENSIONAHA.116.07694. [DOI] [PubMed] [Google Scholar]
- 39.Li L, Zeng H, Chen JX. Apelin-13 increases myocardial progenitor cells and improves repair postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2012;303:H605–H618. doi: 10.1152/ajpheart.00366.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen JX, Stinnett A. Disruption of Ang-1/Tie-2 signaling contributes to the impaired myocardial vascular maturation and angiogenesis in type II diabetic mice. Arterioscler Thromb Vasc Biol. 2008;28:1606–1613. doi: 10.1161/ATVBAHA.108.169235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dore-Duffy P. Pericytes: pluripotent cells of the blood brain barrier. Curr Pharm Des. 2008;14:1581–1593. doi: 10.2174/138161208784705469. [DOI] [PubMed] [Google Scholar]
- 42.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 43.Sa-Pereira I, Brites D, Brito MA. Neurovascular unit: a focus on pericytes. Mol Neurobiol. 2012;45:327–347. doi: 10.1007/s12035-012-8244-2. [DOI] [PubMed] [Google Scholar]
- 44.Dore-Duffy P, Cleary K. Morphology and properties of pericytes. Methods Mol Biol. 2011;686:49–68. doi: 10.1007/978-1-60761-938-3_2. [DOI] [PubMed] [Google Scholar]
- 45.Hirschi KK, D’Amore PA. Pericytes in the microvasculature. Cardiovasc Res. 1996;32:687–698. [PubMed] [Google Scholar]
- 46.Nag S. Morphology and molecular properties of cellular components of normal cerebral vessels. Methods Mol Med. 2003;89:3–36. doi: 10.1385/1-59259-419-0:3. [DOI] [PubMed] [Google Scholar]
- 47.Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23. doi: 10.1007/s00441-003-0745-x. [DOI] [PubMed] [Google Scholar]