

Abstract
Atomic-scale thickness, molecular impermeability, low atomic number, and mechanical strength make graphene an ideal electron-transparent membrane for material characterization in liquids and gases with scanning electron microscopy and spectroscopy. Here, we present a novel sample platform made of an array of thousands of identical isolated graphene-capped micro-channels with high aspect ratio. A combination of a global wide field of view with high resolution local imaging of the array allows for high throughput in situ studies as well as for combinatorial screening of solutions, liquid interfaces and immersed samples. We demonstrate the capabilities of this platform by studying a pure water sample in comparison with alkali halide solutions, a model electrochemical plating process and beam induced crystal growth in liquid electrolyte. Spectroscopic characterization of liquid interfaces and immersed objects with Auger and X-ray fluorescence analysis through the graphene membrane are also demonstrated
Keywords: graphene, ambient pressure electron microscopy, liquid cell, electrochemistry, Auger
TOC figure
Introduction
The development of in situ high throughput parallel screening of micro objects and processes in liquids with nanoscale spatial, high temporal and spectroscopic resolution is a current necessity in research related to materials genome, combinatorial chemistry, drug discovery and etc. Microfabricated fluidic or enclosed liquid cells equipped with electron transparent membrane windows a few tens of nanometers thick enable modern high resolution transmission electron microscopy (TEM) in liquid media.1–4 Despite the continuing research and development efforts,5–9 this highly successful approach has been difficult to apply to more accessible scanning electron microscopy (SEM), mainly due to the significantly larger electron scattering by the membrane at typical SEM beam energies of 1 keV to 30 keV. Attempts to overcome the attenuated signal and reduced spatial resolution by increasing beam current and/or energy usually result in undesirable beam-induced effects such as radiolysis and sample alteration.
In contrast to conventional membranes, the electron inelastic mean free paths in a free standing two-dimensional (2D) material such as graphene (Gr) exceed its thickness.10 Therefore, a membrane made of a 2D material is nearly transparent to electrons in a wide energy range,11 eliminating the aforementioned limitations. The successful application of graphene liquid cells (Gr-cells) has recently been demonstrated for both SEM12 and high resolution TEM (HRTEM)13–14. An alternative implementation of graphene windows via isolation of the entire SEM column from the ambient with electron transparent membrane has also recently been reported.15
Reliable and high-yield integration of chemical vapor deposited (CVD) graphene into the Gr-cell microfabrication process remains a challenging task. The major difficulty is a limited (yet very high) mechanical strength of the grain boundaries of CVD graphene. Having a breaking strength on the order of 90 GPa, suspended single layer CVD graphene is capable of sustaining a pressure differential in excess of 105 Pa, provided its lateral dimensions do not exceed a few micrometers.16 Therefore, the early Gr-cell designs were single aperture devices with a field of view (FOV) of only a few micrometers. The recent demonstration of common-chamber multi-orifice Gr-cells was a significant step forward in atmospheric pressure electron spectroscopy;17–18 however, the probability of a catastrophic liquid release into a high vacuum (HV) chamber due to an accidental (or beam induced) graphene rupture increases proportionally with the number of orifices. Therefore, this approach is currently used in combination with sophisticated interlocks and differential pumping stages.
In this report we describe a new liquid cell platform made from an ordered densely packed array of thousands of identical isolated microchannels capped with Gr. These high-aspect-ratio microchambers are filled with a few picoliters of liquid, thus an accidental rupture of even a large number of cells would not affect the high-vacuum environment of the SEM. The measured lifetime of a water sample in the array exceeds several hours, enabling sufficient time to perform routine SEM studies. The simultaneous presence of multiple channels filled with either liquid or vapor, or empty channels, in the same FOV also makes it possible to study the SEM contrast mechanisms. We demonstrate the possibility of performing X-ray energy dispersive spectroscopy (EDS) and Auger electron spectroscopy (AES) of liquid and immersed samples, along with the corresponding chemical mapping. Using a model electrochemical reaction, we observe the early stages of Cu electroplating on the Gr surface in real time. We envision that a microchannel array (MCA) such as described here will be employed as a platform for high yield combinatorial in operando SEM studies of liquid-gas-solid interfaces relevant to electrochemical or biomedical applications.
Results and discussion
The fabrication details of Gr capped MCA (Figure 1) are described in the Methods section. To perform electrochemical, electrophoretic or electrical measurements, the top (Au) and the bottom (Pt) electrodes were deposited onto a micro-channeled silica glass surface using sputtering and atomic layer deposition (ALD), respectively (Figure 1a). A Gr bilayer was transferred onto the Au electrode using poly(methyl methacrylate) (PMMA) as a sacrificial layer.19
Figure 1.

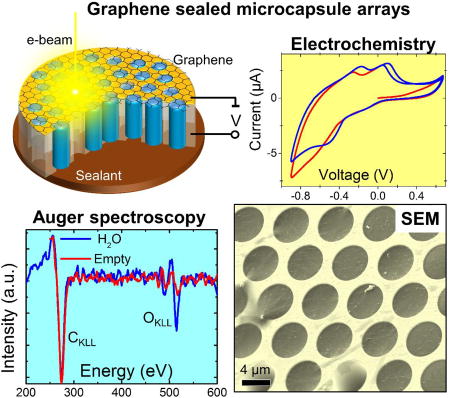
The fabrication and filling of the MCA matrix with an analyte. (a) A 200 nm/10 nm Au/Cr film was sputtered sequentially onto the front side of the MCA silica matrix for metallization and reliable adhesion of graphene. For electrochemical measurements, the interior of the MCA channels was coated with 40 nm of Pt as a counter-electrode using atomic layer deposition (ALD). Bilayer graphene was transferred onto the front side of the MCA using PMMA as a sacrificial layer. The inset shows a cross-sectional SEM image of the MCA half-coated with Pt. (b) PMMA was dissolved in an acetone bath followed by a gradual substitution with isopropyl alcohol (IPA) and water. (c) Depending on application, the sample was sealed either with ultraviolet (UV) curable adhesive or liquid metal (galinstan). The inset demonstrates the SEM image (3 keV) of water filled MCA channels. Experimental voltammograms recorded in a 1 mol/L CuSO4 solution are shown here to demonstrate the electrochemical capabilities of the MCA platform.
The high aspect ratio (1:80) microchannels were filled with a liquid sample via sequential substitution of acetone first with isopropyl alcohol and finally with a solution of interest. (Figure 1b). Depending on the application, the backside of the Gr-cell was sealed with either a water immiscible conductive GaInSn eutectic alloy or an ultraviolet (UV)-curable epoxy. The resultant cell contains thousands of identical vacuum-tight microchannels filled with a liquid of interest (Figure 1d).
Characterization of liquid MCA samples using SEM
To explore the behavior of our new system, we first study SEM image contrast in a water filled MCA sample. The MCA platform enables studying the channel content comparatively and quantitatively due to the simultaneous SEM imaging and analysis of cells with different filling status while being probed under the same imaging conditions.
When an MCA is filled with water, the observed SEM gray scale values (GSV) can be partitioned into 4 typical groups (Figure 2a): (i) open channels with completely or partially broken graphene membranes (bright); (ii) empty channels covered with a suspended graphene (bright); (iii) channels containing water vapor and yielding the lowest signal (dark); and (iv) channels filled with water that generate the intermediate gray values in SEM images. More quantitatively this partitioning can be represented by GSV histograms depicted in the Figure 2a and Figures S1a, b. Interestingly, the Monte-Carlo20 (MC) electron trajectory simulations of MCA of all aforementioned groups predict the smallest number of the secondary (SE) and backscattered electrons (BSEs) collected from empty channels (Figure 2b). This discrepancy between the observed and the MC-simulated images originates from the negative charging of MCA silica walls of empty or partly graphene covered channels under the primary e-beam irradiation, which leads to higher secondary electrons yield.21 The SEM signal from channels with liquid excludes a contribution from the channel walls and is formed by SE’s and BSE’s from both the graphene membrane and water (Figure 2b). Hence, the SEM image of liquid channel is uniformly gray. The electron scattering inside vapor filled channel is reduced and, therefore, the interaction volume of electrons with vapor significantly exceeds the one in a liquid channel. Different from the empty channel scenario, electrons and ions inside the vapor filled channels neutralize the charging of the glass walls, and SE signal originates only from the graphene membrane and a low-density vapor inside the channel. As a result, the vapor filled channels yield the lowest SE intensity among all other covered cells. The intermediate contrasts can also be observed due to fast transient processes such as bubble formation, beam induced radiolysis, re-condensation or impurities segregation.
Figure 2.

(a) SEM image (Ebeam=10 keV) of empty, water-filled and vapor-filled MCA channels, all sealed with bilayer graphene. Bright (framed with black square), uniformly gray (framed with blue square), and uniformly dark (framed with red square) channels correspond to empty, water and vapor filled channels, respectively; The color-coded histogram on the right shows the distinctly different distributions of the gray scale values (GSV) inside the corresponding square frames. (b) Monte-Carlo trajectories simulations of 8 keV electron beam interacting with an empty (vacuum-filled) channel, a water-vapor-filled channel (assuming 3.17 kPa water saturated vapor pressure at 25°C), a liquid-water-filled channel, and the gold MCA surface. The number of BSEs is proportional to the density and Z-number of the sample, and varies inversely with the probing depth. (c) The H2O to Au signal ratio of scattered electrons vs. electron beam energy. Error bars show the standard deviation of gray scale values obtained from more than 5 different SEM images recorded under the same beam energy (d) SEM images of the MCA (contrast and coloration were adjusted to highlight water filled versus empty or vapor-filled channels), showing a decrease in the number of channels filled with liquid over time (vacuum ≈ 10−4 Pa). Bright circles correspond to microchannels filled with water, whereas dark circles are empty ones. The scale bar corresponds to 50 µm (e) Experimentally recorded time evolution of the filling factor, i.e. the ratio of filled channels to all channels (blue circles). The data were obtained from consecutive SEM images of the same FOV (2.5 × 104 µm2 corresponding to ≈ 900 micro-channels). The beam was blanked between the measurements to avoid radiolytic bubble formation inside the channels. Error bars are due to uncertainty in the filling status of the channels at the very perimeter of the FOV. The red curve depicts data fitting based on a normal distribution of defects across channels. (f) The sequence of SEM (Ebeam=2 keV) images showing the typical steps of the channel drying process: water filled (top panel), bubble (middle panel) and vapor containing (bottom panel) channel. The arrows indicate the transport of water under the graphene towards few newly formed water blisters (dark)
It is important to note that different from TEM and prior SEM studies of liquid media enclosed in liquid cells22 the graphene’s high electron transparency offers the capability of imaging the liquid-solid/liquid-gas interfaces via collecting true secondary low energy electrons (see Fig S3a and corresponding description in supporting material). This opens new possibilities for monitoring the spatio-temporal evolutions of the electrified interfaces within only one-two nanometers from the graphene surface. On the contrary, monitoring high energy back scattered electrons allows probing the objects immersed in liquid few microns deep below the capping graphene membrane. Therefore, the choice of the primary electron beam energy and detector type are important for optimal SEM imaging conditions of liquid samples. Since the MCA platform allows for simultaneous recording of the SEM signals from graphene capped channels that are water-filled (Swater) and vapor-filled (Svapor), as well as from the Au surface (SAu), it is possible to deduce a measurable parameter that is independent of the pre-selected SEM brightness and contrast settings (see supporting information (SI) for details). We introduced an effective contrast value between a liquid sample and Au surface:
| (1) |
This measurable parameter reflects the ratio of total electron yields from liquid and Au substrate which depends on a particular liquid-substrate combination and graphene thickness and is not influenced by specific contrast/brightness settings of the electron detector. The experimental values of β as a function of the electron beam energy are depicted in Figure 2c. At high primary beam energy, the BSE contribution dominates in the SEM signal and is not attenuated by the capping membrane. Therefore, at high primary beam energies, β approximately follows the ratio of the corresponding BSE coefficients of water and the Au surface. On the other hand, at very low energies, the SE yield from the graphene membrane itself becomes comparable to or even exceeds the attenuated SE emission from a liquid and a substrate, and the effective contrast value approaches one (see SI for further details).
Lifetime of liquid samples in vacuum conditions
The lifetime of a liquid sample in an MCA under vacuum conditions is one of the crucial experimental parameters defining the overall time allowed for imaging and analysis. It depends on the water leakage rate through intrinsic defects in a graphene membrane and/or through diffusion runaway along the Gr-MCA interface. The as-grown graphene quality,16, 23–24 the interface preparation, and the graphene transfer process can significantly affect both the intrinsic porosity (the areal ratio of holes to a Gr window) of the resultant membrane and the interfacial leakage.16 We used bilayer graphene to reduce the inherent CVD graphene permeability.24–25 We assume that the backside of the MCA sample is vacuum tight sealed with an adhesive and the MCA matrix is also water impermeable. As it will be shown below and in the supporting file, the lifetime of the liquid sample inside the MCA is predominantly controlled by the density of native defects, tears and wrinkles in the covering graphene.
Since the MCA is comprised of identical channels, the lifetime of the liquid sample can be evaluated via measuring the filling factor (the ratio of filled channels to their total number in a FOV) as a function of time. The distinct difference in SE and BSE signals from liquid and dried channels allows us to use a simple threshold image processing algorithm to discriminate between these two moieties and determine the filling factor within the FOV. The sequence of SEM images in the Figure 2d demonstrates typical water loss in an MCA under vacuum conditions over several hours: from ≈ 86% filled channels in the beginning of observation to ≈ 23% after 5.1 hours. The measured filling factor as a function of time is depicted in the Figure 2e and indicates that the half-life time – the time when the filling factor halves – for the sample t1/2 ≈ 2.7 h.
To analyze the impact of leakage on the filling factor, we developed a model based on a water permeable defects having normal distribution of effective areas across all channels. In this model, we assume that the volume of liquid phase in the channel decreases with time due to the leaks through: (i) nanoscopic holes in the graphene and (ii) the graphene-substrate interface. The latter leakage channel is clearly evidenced in the low voltage SEM sequence in the Figure 2f as appearance and growth of “under carpet” water blisters surrounding the evaporating channel. The liquid remains adhered to graphene membrane all the time until its thickness reaches the onset of capillary instability due to hydrophobic nature of the graphene (middle panel in the Figure 2f). The free space released in the channel as a result of gradual liquid phase loss is filled with the water vapor. Assuming molecular flow and a normal distribution of the nano-holes with an effective area s, a mean value s0, and a standard deviation σ for a 5 µm diameter microchannel in the MCA sample, the filling factor in Fig. 2e can be fit using a Gaussian distribution function with s0 = 4.3 × 103 nm2 and σ = 2.1 × 104 nm2 (see SI for details). The reported porosity of the pristine CVD graphene varies between 0.012% and 0.61%.25 Accepting the average porosity numbers for our case, the total open area of defects in the bilayer graphene per orifice will be less than 7.3 × 102 nm2. Comparing the obtained number with an experimentally determined total s0, it is reasonable to conclude that these are not graphene defects but the interfacial leakage that determines the lifetime of the MCA liquid sample under HV conditions. This result is not surprising since the standard graphene transfer procedures unavoidably results in a network of percolating wrinkles and microscopic tears at the sample interface (see topography S4a and low voltage SEM images S4b in supporting material).
Spectroscopy
In addition to imaging, transparency of the graphene membranes to electrons and photons enables chemical analysis and elemental mapping of liquids and immersed micro-objects under the ambient pressure conditions. Below we discuss the application of two of the most commonly used electron spectroscopies integrated with SEM – energy-dispersive X-ray spectroscopy (EDS) and Auger electron spectroscopy (AES) – in liquids. Generally, the inelastic mean free paths for outgoing electrons is orders of magnitude smaller than the penetration depth for similar energy X-ray photons, making the AES probing depth TW on the order of 1 nm to 2 nm while an EDS signal can be recorded from hundreds of nanometers deep into the water. Therefore, these “electron in-electron out” and “electron in-photon out” spectroscopies (Figure 3a) are not equivalent but rather complementary to each other.
Figure 3.

Electron spectroscopies in a liquid-filled MCA. (a) The experimental setup for spectroscopic (EDS and AES) data collection. The yellow bulb defines the electron interaction volume. (b) EDS spectra (log scale) recorded in water filled (blue) and empty (red) channel acquired through a double layer graphene membrane. The difference in the O Kα peak intensity is due to the presence of liquid water inside the channel. The Si Kα and Au Kα peaks originate from the Au coated MCA matrix, which is made of silica; (c) An SEM image (top) and EDS maps of an MCA with water, depicting Au and O element distribution. The white circles depict the empty channels, while dashed ones denote the channels where water evaporated during map acquisition; (d) AES OKLL spectra exhibiting characteristic K-VV transitions typical for liquid water. For comparison, off-resonance AES of water jet excited with synchrotron soft X-rays from ref.26 is shown in a gray color. (e) Differential AES spectra recorded from two adjacent water filled (blue curve) and empty (red curve) MCA channels. (f) The calculated dependence of OKLL to CKLL peak intensities ratio as a function of the thickness of water under the graphene and a number of the graphene layers. The right panel shows experimental I0/IC data recorded from ten water filled channels capped with a bilayer graphene. The measured average ratio (dotted line) corresponds to an effective membrane thickness to be approximately 3 graphene layers
EDS analysis
Figure 3b depicts two EDS spectra acquired through the graphene membrane from the water filled (blue line) MCA channel and the same channel after liquid was dried (red line). The spectra contain the major O Kα peak at 0.53 keV showing the signature of liquid water below the graphene membrane and three minor peaks: C Kα, Au Mα andSi Kα originating from the graphene and surface/walls of the MCA matrix. One can notice that despite the attenuation of outgoing X-rays by water, the scattering of primary electrons in water enhances C Kα, Au Mα and the Si Kα emission in the wafer filled channels compared with the empty ones. This is due to geometry of the sample and EDS setup, where 15 keV beam penetrates microns deep into the empty channel before it hits the walls. The generated X-rays have no direct line of sight to the detector and become strongly attenuated by MCA matrix. Figure 3c shows the corresponding SEM images and elemental maps obtained through the graphene membrane. The channels filled with water exhibit a more prominent O Kα intensity compared with the empty (or vapor filled) channels, which marked with white circles in Figure 3c. EDS is not sufficiently sensitive to distinguish between empty and vapor filled channels. Note, that the contribution of the graphene membrane to the total EDS signal is negligible, making the developed MCA platform an excellent candidate for analysis of samples immersed in gaseous and liquid environments.
AES analysis
Unlike EDS spectro-microscopy, which can probe water immersed objects hundreds nanometers deep27, AES is a surface sensitive technique due to strong attenuation of the 100 eV–1000 eV Auger electrons in the condensed matter. Therefore, scanning AES was used exclusively to study solid surfaces under high or ultra-high vacuum conditions28. In this work we demonstrate that ultrathin membranes can be used to extend the standard laboratory based scanning AES metrology to the realm of liquid interfaces and immersed objects. Figure 3d shows a normal Auger electron spectrum recorded in an individual water-filled MCA channel through a bilayer graphene cap. For comparison, off-resonance AES of a water jet excited with synchrotron soft x-rays is shown in gray color.26 The overall shape of the recorded AES spectra, arising from the superposition of multiple K-VV type Auger decays, correlates well with the synchrotron results. In this case, a water oxygen K core hole is filled with electrons from a few available valence levels (V) with the corresponding Auger electron emission from the same moiety of the valence levels (V). The broadening and the energy shifts of the liquid water AES bands originate from the combination of the polarization screening and delocalization effects occurring due to numerous intermolecular decay channels available in liquid water.29–30 The elevated noise level in our scanning AES stems from the short acquisition time, which was chosen to mitigate strong radiolysis of water. Figure 3e shows two differential AES spectra collected from water-filled and empty channels. Both spectra have identical CKLL peaks associated with the graphene membranes but only the spectrum collected from the liquid-containing channel exhibits a prominent OKLL component, confirming the presence of water. We use the experimental AES intensity ratio, , and the attenuation formalism (see ref.31 and corresponding SI material) to estimate both the effective graphene thickness, TG, and the water probing depth, TW. The ratio between O and C peaks under particular detection angle θ can be written:
| (2) |
Here, σCKLL and σOKLL are the cross sections for K shell ionization of C and O atoms; γCKLL and γOKLL represent corresponding Auger probabilities; F(ECKLL) and F(EOKLL) are the transmission efficiencies of the electron spectrometer at the two energies; λCG, λOG, and λOW, represent the attenuation lengths of the CKLL and OKLL Auger electrons in graphene (G) and water (W), respectively. NG and NW correspond to atom densities of the graphene membrane and water, respectively (see supporting material for the parameters used). Figure 3f shows the calculated OKLL to CKLL intensity ratio as a function of water depth for several thicknesses of the graphene membrane. The curves demonstrate the saturation of the ratio, occurring once the water layer exceeds the AES probing depth. In particular, the signal for a water layer thickness of 3 nm nearly reaches the constant bulk-like level, independent of the graphene thickness. Interestingly, the experimental data collected from water-filled MCA cells yield , which corresponds to the signal for bulk water covered in average with approximately 3 graphene layers (Figure 3f, right panel). Here the experimental uncertainty is calculated on the base of twelve tested channels. This deviation from a nominal bilayer graphene coverage is likely due to the presence of residual hydrocarbon and oxygen containing contaminants on the transferred CVD graphene membranes, which increases their effective thickness and attenuation of the AES signal. These preexisting -OH, -O containing contaminants contribute to OKLL spectrum of the dry graphene (red curve in the Fig. 3 e) and preclude reliable discrimination between the vapor filled and empty channels during Auger characterization.
Application examples
In addition to unique electron and X-ray imaging and spectroscopy capabilities, the graphene-capped MCA liquid sample platform has yet another advantages allowing both: (i) high magnification stereomicroscopy in individual liquid channels and (ii) simultaneous monitoring of thousands of independent microchambers in real time when set for a large FOV. The platform, therefore, combines the advantages of high resolution SEM studies of local phenomena in liquids with the advantages of powerful image processing, pattern recognition, and data mining algorithms when applied to large FOV.32 Below, we describe few examples of what can be routinely performed with this setup.
Electrochemical measurements
As an example of probing electrochemistry at (meso-) and microscale, we examine here classical copper electroplating from an aqueous 1 mol/L CuSO4 solution. In our two-electrode system, the graphene-covered gold coating on the front (imaging) side of the MCA plate acts as a working electrode, and the back side platinum coating serves as a counter and pseudo-reference electrode, as shown in Figure 1. Real time SEM visualization of the electroplating process in individual microchannels is correlated in Figure 4 with the global voltammogram simultaneously recorded for the whole MCA sample. The system starts out with a clear graphene windows at positive potential (Figure 5a, inset 1). At around −0.3 V the voltammogram shows a broad peak that corresponds to the onset of copper nucleation at the graphene-gold-electrolyte interface on the channels’ periphery (Figure 4a, inset 2). This peak is very similar to the α-peak observed during copper electroplating on gold in the presence of ppm-level traces of disulfide and chlorine additives. It may arise in our system due to the presence of small amounts of contaminants in the electrolyte and also have some contribution from dissolved oxygen reduction and hydrogen adsorption on the working electrode. Clearly, this peak corresponds to underpotential deposition of Cu nanoparticles since it is followed by a main copper deposition peak (Figure 4a, inset 3) related to growth of larger copper crystals. Interestingly, at this stage the previously-formed nanocrystals start dissolving, presumably in an Ostwald ripening-type process (Figure 4a, inset 3, channel’s periphery). It has been previously shown33 that the α-peak can be assigned to underpotential deposition of Cu nanocrystals. Upon reversal of the potential sweep, copper is stripped from the working electrode in a series of anodic peaks, leaving graphene windows empty again (Figure 4a, inset 4). In order to ascertain the chemical composition of the observed deposits, we performed EDS mapping of microchannels, following application of cathodic and anodic potentials. As Figure 4b confirms, a large microparticle that was grown during deposition is made of copper. It dissolves during stripping leaving behind a copper sulfate solution with a much weaker EDS-Cu signal. This model system demonstrates the usefulness of the MCA approach in studying electrochemical processes in situ.
Figure 4.

Copper electroplating and stripping: individual cells FOV. (a) Cyclic voltammogram of Cu deposition and stripping at the graphene electrode in ca 1 mol/L aqueous CuSO4 electrolyte. The voltammogram was obtained at 1 mV/s scanning rate; potential was swept from positive to negative polarity and back. Platinum was used as a pseudo-reference electrode. (b) SEM images and corresponding EDS Cu maps showing deposition and stripping of a copper particle
Figure 5.

Cesium iodide crystal growth: individual cells FOV. (a)–(c) consecutive screen shots from SEM video of the beam-induced nucleation and growth process of two CsI crystals from saturated aqueous solution (see video SV1 in SI). Although crystals originally nucleate at the wall, at later stages (panel c) they become disconnected from it due to partial dissolution. Beam energy was 15 kV. The scale bar is 1 micron. (d) The SEM image (gray) and EDS maps (colored) of gold, iodine, and cesium recorded from a region with grown CsI crystals. Channels are of the same size as in a). (e) Crystal area vs. time curves extracted from videos SV1–SV3 of SI demonstrates nearly linear dependency. The yellow curve interrupts twice for periods when the beam was blanked. Interestingly, the crystal size neither increases, nor decreases during these periods.
Beam Induced Crystal growth
Another illustration of MCA usage is the investigation of the complex process of electron beam induced precipitation during SEM studies in liquid solutions. For this experiment, the MCA was filled with a saturated aqueous solution of CsI, and its front-side graphene electrode was grounded. While solution below the graphene membrane was stable in a vacuum, high-beam-current SEM imaging of the sample led to precipitation of cubic crystals inside the channels, as illustrated in Figure 5a–c. Most of the crystals were observed to nucleate at the microchannel walls (Figure 5a), and only few grew on the graphene membrane itself. This observation can be explained by two factors: higher density of nucleation sites on the walls, and enhanced yield of secondary electrons from the thick solid walls as compared to the liquid electrolyte and thin graphene membrane. After thermalization, the solvated electrons must be co-reactants in the crystal growth, as without irradiation no crystallization occurs. This fact rules out the possibility of precipitation due to simple water evaporation in a vacuum (i.e. leaky channels). Deposition and dissolution of metals (Au, Ag, Pt) and colloidal particles under electron beam irradiation has been reported previously by multiple groups.35–39 However, only one paper, to our knowledge, has reported a beam-induced synthesis of inorganic salt crystals: Na2S2O8 was decomposed under the beam into Na2SO4, and the latter precipitated due to its lower solubility in water. In our experiments, EDS mapping of the grown crystals clearly shows that they contain cesium and iodine, and not gold (Figure 5 d) despite the fact that the gold coating slowly corrodes at the perimeter of the channels and Au3+ ions must be present in solution (3I2+2Au→2AuI3, see below). Several inorganic salts (CsIOn, n = 2,3,4) can form as a reaction with water radiolysis products and CsI according to:
| (3a) |
| (3b) |
| (3c) |
Or, overall:
| (3d) |
The CsIOn iodates (which are also cubic, as are CsI and gold) are less soluble in water than CsI, and should readily precipitate, if formed. However, as follows from equation 3, their synthesis must be accompanied by release of large amount of molecular hydrogen. Simple estimates based on the crystal size (from Figure 5 a–c), hydrogen solubility in saturated CsI solution (Sechenov coefficients) and amount of released hydrogen show that large hydrogen bubbles must be formed during CsIOn growth – a phenomenon that is not observed. Thus, we conclude that the growing crystals must be made of pristine cesium iodide. The mechanism of their precipitation from solution must necessarily be different from the growth of noble metal crystals and Na2SO4 (electrochemical reduction of precursor) and colloidal particle aggregation (alteration of zeta potential that determines their stability in solution). CsI is a strong electrolyte that doesn’t form colloidal solutions. It is also clear that its composition is not changed during crystallization.
Thus, the CsI precipitation from solution must be determined by the change in the activity coefficients of Cs+ and I− ions due to alteration of the ionic strength of the solution by the water radiolysis products. The main steady-state water radiation products are hydrogen gas, hydrogen peroxide and protons. Hydrogen is a strong reduction agent (especially in statu nascendi molecular (H2) or atomic (H) states). It cannot significantly influence the solubility of CsI, as it can neither reduce this salt, nor change the solution ionic strength (not being an ion itself). Hydrogen peroxide is an oxidizer and, being a good solvent similar to water, it is unlikely it may decrease the solubility of CsI. On the contrary, solvated protons are ions that can significantly alter the ionic strength of the solution, and, through that, change the CsI solubility. Note, that low pH typically increases solubility of inorganic compounds (e.g. insoluble carbonates, sulfides, phosphates, hydroxides, etc.). Hence, the most plausible mechanism that can account for the crystal precipitation, growth and dissolution is the complex dynamics of local ionic strength of the solution mostly caused by solvated protons. Depending on the radiation dose, initial CsI concentration, and channel geometry (which limits diffusion), the radiation products shift the local chemical equilibrium favoring nucleation, growth or dissolution of the CsI crystals at different locations and times. Elucidation of the exact mechanism of this process would require solving a system of radiolysis reaction-diffusion equations under scanning beam excitation and specific geometry as well as extension of Debye-Huckel theory43 to saturated CsI electrolyte. This, however, is outside the scope of the present paper. As a concluding remark, we note that the rate of CsI crystal growth is almost linear and depends on the beam energy and current (Figure 5e). The crystals have a clear non-dendritic shape and stop growing when the electron beam is blanked (yellow curve, Figure 5e). It is also noteworthy that the growing fronts of two adjacent crystals merge as they grow (Figure 5b–c) and are not repelled due to precursor depletion as was observed for metallic particles before. Taken together, all these observations suggest a kinetic-limited, rather than diffusion-limited growth mechanism.
High throughput SEM imaging and data analysis
The new feature and main advantage of the MCA platform for SEM studies in liquids is the possibility of monitoring dozens and hundreds of cells and accumulating statistics on their behavior in one experiment. Figure 6a shows a large multichannel SEM FOV of the electrochemical copper deposition process, where 57 individual cells (channels) were randomly selected. This image is a frame from a video (see SM Vid), capturing the process of copper deposition and stripping on graphene membrane, as a constant bias is applied to it. The growth of copper crystals (seen in cells # 20, 30, 25, etc.) is stochastic, not homogeneous across the cell perimeter and leads to an increase of the overall cell brightness (metallic copper has higher secondary electron yield than the electrolyte). To analyze the behavior of the entire MCA FOV, we used channels perimeters recognition software. SEM signal intensity was then averaged within the boundaries of each of the 57 identified cells and plotted as a function of time in Figure 6b. Initially the graphene electrode is unbiased (open circuit) and then, from 15 s to 280 s a negative 2.2 V voltage (copper plating conditions) is applied to it. After 280 s its potential is changed to +1 V (copper stripping conditions). Several dark blue and dark red lines in the diagram of Figure 6b belong to, the cells with ruptured graphene membrane (e.g. #51) or contaminated with bright deposit (e.g. #57), which show a constant low and high signal, respectively. The rest of the channels exhibit an increase in signal during copper plating and a decrease during stripping (e.g. #20) cycles. Statistical distribution of this data is shown in Figure 6c, where for each moment of time a histogram (number of cells with a given mean intensity vs. mean intensity) is plotted in the form of a 2D colored diagram. All histograms were sampled at 100 bins. Initial distribution (at t = 1 s) shows a maximum around 4 intensity units, and some scattered pixels in the range 1–7 units (range limits correspond, of course, to cells with ruptured membranes and deposits, respectively, as described above). Application of −2.2 V potential leads to a shift of the distribution maximum to ca. 3 units and its slow drift to larger values (bright yellow line in Fig. 6c). During the electroplating stage, the histogram pixels corresponding to some cells move very rapidly to the right side of the diagram, manifesting strong copper crystal growth. The onset of anodic dissolution can be observed just past 280 s, when at +1 V, pixels of many cells drift back to lower intensity values (Fig. 6c). The complex non-uniform behavior map observed even in such a simple electrochemical system demonstrates the necessity of simultaneous screening of a large number of samples, when studying nanoscale processes. Only accumulation of a large amount of statistical data reveals the behavioral complexity and may help determine the underlying mechanisms. Current approaches in liquid cell electron microscopy only allow measuring one sample at a time. The presented MCA platform clearly resolves the high throughput sampling problem in data acquisition and analysis with high veracity.
Figure 6.

Copper electroplating and stripping: multichannel FOV. (a) an SEM image of a 48 µm × 34 µm area of an MCA filled with 1 mol/L CuSO4 electrolyte with 57 randomly selected cells. The image is a captured frame from a video file that recorded copper deposition and stripping process. It presents a moment at time t= 206 s, when a negative bias of 2.2 V was applied to the top (graphene) electrode and copper deposition took place. Cell # 20 shows large grown copper crystals. The image was used for high throughput analysis. (b) Color diagram of the mean SEM signal intensity within each of the 57 cells shown in (a) as a function time and cell number. Color bar is in arbitrary units of mean SEM intensity. Bias applied to graphene was changed in steps over time and is shown on the right of the diagram by dashed lines. Note how intensity of cell 20 changes as copper is plated at negative and stripped at positive potential. (c) Color-coded histogram representing distribution of mean SEM signal intensity within 57 cells plotted vs. time. Colorbar shows number of cells with a given mean intensity. The maximum of the histogram changes only slightly, as Cu deposition in most cells was limited, however several cells show strong increase of the signal during deposition (lines of the histogram that migrate to the right). The white patterned strip between 350 s and 400 s in panels (b) and (c) is a gap in data due to video recording freeze during the experiment.
Combinatorial SEM studies
Similar to the microarray bioassays approach, the platform enables combinatorial/comparative SEM analysis of different liquid analytes simultaneously present in the MCA matrix (Figure 7). Here, three sections of an MCA sample were filled with water and solution of NaBr and LiBr. SEM imaging of all 3 sample sectors under the same conditions and detector settings allows their direct and quantitative comparison. After identifying individual cells in each of the images (Fig. 7b–d) with an image recognition algorithm, we calculated mean signal intensity within the boundaries of each cell and plotted histograms of these mean cell intensities. Regions filled with water and NaBr electrolyte have very few empty or broken cells, and their histograms have one maximum at ca 100–104 units (Fig. 7b, 7d), whereas the LiBr histogram (Fig. 7c) breaks into two distinct distributions: empty cells with a maximum around 72 units and filled cells with a maximum around 97 units. Note that a direct quantitative comparison of the gray scale values maxima: 97 units for LiBr, 100 units for water and 104 units for NaBr allows for prompt discrimination between different analytes using SEM images.
Figure 7.

An example of combinatorial SEM imaging (Ebeam = 5 keV) using MCA platform. Three section of the bilayer graphene MCA sample were filled with water and ca. 1 mol/L solutions of LiBr and NaBr and imaged with E–T detector under the same contrast brightness settings. (a) a schematic of the MCA sample with colored regions filled with different electrolytes. (b)–(d) SEM images and cell histograms of each region. Histograms show distribution of number of cells with a given mean cell signal intensity. Note, that the gold-coated matrix GSV of the images were excluded from analysis and are not reflected in the histograms.
Conclusions and outlook
In summary, we have developed and tested a novel sample platform for SEM experiments in liquids. The platform is based on a graphene-capped ordered array of microcapsules filled with liquid analyte(s). The array is capable of retaining liquid samples for hours under high vacuum, which is sufficient for routine electron imaging and spectroscopy experiments. In particular, we show that high electron transparency and mechanical strength of the bilayer graphene allow high resolution SEM imaging (including low voltage SEM), Auger and EDS spectroscopies to be done on samples of practical importance such as water and other electrolytes. Typical experiments such as electrochemical plating and crystal growth were demonstrated as examples. Moreover, since the MCA platform is composed of a lattice of identical microcapsules, it can be used in conjunction with powerful statistical analysis, data mining, and pattern recognition methods. The latter allows for the study of the complex spectro-temporal and spatiotemporal behaviors at liquid-solid interfaces. Finally, this platform is not limited to SEM metrology but can be used in laboratory stand-alone or synchrotron based X-ray photoelectron spectroscopy (XPS)45, photoelectron emission microscopy (PEEM)32, and low energy electron microscopy (LEEM) setups. We also successfully applied a variety of scanning probe microscopy techniques to the MCA liquid sample platform, which will be reported in forthcoming publications.
Methods
Sample preparation
Graphene was grown on a Cu foil using a high pressure modification of the commonly used CVD method. As-grown graphene was transferred onto one side of commercially available glass multichannel arrays using PMMA as a sacrificial layer (Figure 1 a).19 Briefly, a 200 nm PMMA film was spin-coated onto a graphene/Cu stack followed by etching copper in ammonium persulfate solution (APS) at 40 °C for 2 h. Then, the graphene monolayer was rinsed three times in deionized (DI) water and transferred onto another graphene/Cu foil. After annealing the sample on a hot plate for 2 h at 180 °C, etching in APS and rinsing in DI water were repeated again. A PMMA/bilayer graphene stack was transferred onto a MCA consisting of thousands of hollow straight channels with a diameter of 5 µm and 1:80 aspect ratio. Prior to graphene transfer, the MCA front surface was pre-coated with 200 nm/10 nm Au/Cr film serving as an adhesive layer for graphene and to minimize substrate charging during the SEM imaging. Then, the sample was annealed on the hot plate for 2 h at 180 °C again. For electrochemical measurements, the inner part of the channels at the backside of MCA was covered with 40 nm Pt layer using ALD (Figure 1a and SI). After the transfer, the PMMA was dissolved in acetone bath at 70 °C. Then, the acetone was gradually substituted with the IPA solution at 80 °C and then with DI water (Figure 1b). For studies involving electrolytes, a droplet of the electrolyte was drop-casted onto the backside of the MCA. After few minutes required for establishing concentration equilibrium, the excess of the droplet was removed with a filter paper and a UV curable adhesive or liquid metal such as galinstan was applied to seal the liquid containing MCA channels (Figure 1c). The backside of the sample dedicated to combinatorial SEM imaging was pre-patterned with strips of a hydrophobic layer before the liquid filling. This prevented cross-contamination between the analytes during application.
SEM imaging
The sample was observed using a field emission scanning electron microscope. An Everhart-Thornley (E–T) detector sensitive to both SE and backscattered electrons (BSEs) was employed for SEM imaging. The SEM base pressure was ≈ 10−4 Pa. The gray scale value of the SEM images was linearly proportional to the intensity of scattered and secondary electrons collected by the detector. For lifetime measurements, the SEM images were obtained at 5 keV and few tens pA primary electron beam. For EDS spectroscopy and mapping, the sample was probed by a 15 keV primary beam. In terms of Fig. 3a: θ=45°, α=90° for EDS.
AES analysis
The AES analysis was performed at room temperature in an ultra-high vacuum (UHV) chamber at a base pressure of ≈ 10−7 Pa. The AES spectra were collected at 3 keV or 5 keV primary electron beam energies and 400 pA current followed by spectra averaging over 19.6 µm2 area of MCA channels. In terms of Fig. 3a θ=85°, α=65°. Radiolysis effects, such as hydrogen bubble formation, may strongly affect AES spectra collection. Only those filled channels which did not change their composition during AES acquisition were used for analysis. The peak-to-peak intensity was deduced from the differential spectra. To calculate the thicknesses of graphene and water layers, the attenuation formula31 and corresponding set of parameters (see supporting information) were used.
Supplementary Material
Acknowledgments
AY and HG contributed to the manuscript equally. AY, HG, and ES acknowledge support under the Cooperative Research Agreement between the University of Maryland and the National Institute of Standards and Technology Center for Nanoscale Science and Technology, Award 70NANB14H209, through the University of Maryland. Authors are thankful to Dr. N. Zhitenev, Dr. J. McClelland, Dr. M. Stiles, S. Stranick (all at NIST) for constructive feedback on the manuscript and to Dr. C. Wang and Dr. R. Sharma (all at NIST) for graphene characterization with (HR)TEM.
Footnotes
Estimation of liquid water retention time in an MCA under vacuum conditions, liquid water contrast in SEM, AES analysis in water, graphene wrinkles at water diffusion channels, TEM graphene characterization, examples of the combinatorial SEM using graphene capped MCA platform, ALD parameters for Pt counter electrode deposition
References
- 1.de Jonge N, Ross FM. Electron Microscopy of Specimens in Liquid. Nature nanotechnology. 2011;6(11):695–704. doi: 10.1038/nnano.2011.161. [DOI] [PubMed] [Google Scholar]
- 2.Grogan JM, Bau HH. The Nanoaquarium: A Platform for in Situ Transmission Electron Microscopy in Liquid Media. Journal of Microelectromechanical Systems. 2010;19(4):885–894. [Google Scholar]
- 3.Jensen E, Burrows A, Mølhave K. Monolithic Chip System with a Microfluidic Channel for in Situ Electron Microscopy of Liquids. Microsc. Microanal. 2014;20(02):445–451. doi: 10.1017/S1431927614000300. [DOI] [PubMed] [Google Scholar]
- 4.Tanase M, Winterstein J, Sharma R, Aksyuk V, Holland G, Liddle JA. High-Resolution Imaging and Spectroscopy at High Pressure: A Novel Liquid Cell for the Transmission Electron Microscope. Microscopy and Microanalysis. 2015;21(06):1629–1638. doi: 10.1017/S1431927615015482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiberge S, Zik O, Moses E. An Apparatus for Imaging Liquids, Cells, and Other Wet Samples in the Scanning Electron Microscopy. Review of Scientific Instruments. 2004;75(7):2280–2289. [Google Scholar]
- 6.Suga M, Nishiyama H, Konyuba Y, Iwamatsu S, Watanabe Y, Yoshiura C, Ueda T, Sato C. The Atmospheric Scanning Electron Microscope with Open Sample Space Observes Dynamic Phenomena in Liquid or Gas. Ultramicroscopy. 2011;111(12):1650–1658. doi: 10.1016/j.ultramic.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Jensen E, Købler C, Jensen PS, Mølhave K. In-Situ Sem Microchip Setup for Electrochemical Experiments with Water Based Solutions. Ultramicroscopy. 2013;129:63–69. doi: 10.1016/j.ultramic.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Al-Asadi AS, Zhang J, Li J, Potyrailo RA, Kolmakov A. Design and Application of Variable Temperature Setup for Scanning Electron Microscopy in Gases and Liquids at Ambient Conditions. Microscopy and Microanalysis. 2015;21(03):765–770. doi: 10.1017/S1431927615000732. [DOI] [PubMed] [Google Scholar]
- 9.Liv N, van Oosten Slingeland DS, Baudoin J-P, Kruit P, Piston DW, Hoogenboom JP. Electron Microscopy of Living Cells During in Situ Fluorescence Microscopy. ACS nano. 2015;10(1):265–273. doi: 10.1021/acsnano.5b03970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanuma S, Powell C, Penn D. Calculations of Electron Inelastic Mean Free Paths. Ix. Data for 41 Elemental Solids over the 50 Ev to 30 Kev Range. Surface and Interface Analysis. 2011;43(3):689–713. [Google Scholar]
- 11.Kraus J, Reichelt R, Günther S, Gregoratti L, Amati M, Kiskinova M, Yulaev A, Vlassiouk I, Kolmakov A. Photoelectron Spectroscopy of Wet and Gaseous Samples through Graphene Membranes. Nanoscale. 2014;6(23):14394–14403. doi: 10.1039/c4nr03561e. [DOI] [PubMed] [Google Scholar]
- 12.Stoll JD, Kolmakov A. Electron Transparent Graphene Windows for Environmental Scanning Electron Microscopy in Liquids and Dense Gases. Nanotechnology. 2012;23(50):505704. doi: 10.1088/0957-4484/23/50/505704. [DOI] [PubMed] [Google Scholar]
- 13.Yuk JM, Park J, Ercius P, Kim K, Hellebusch DJ, Crommie MF, Lee JY, Zettl A, Alivisatos AP. High-Resolution Em of Colloidal Nanocrystal Growth Using Graphene Liquid Cells. Science. 2012;336(6077):61–64. doi: 10.1126/science.1217654. [DOI] [PubMed] [Google Scholar]
- 14.Wang C, Qiao Q, Shokuhfar T, Klie RF. High-Resolution Electron Microscopy and Spectroscopy of Ferritin in Biocompatible Graphene Liquid Cells and Graphene Sandwiches. Adv. Mater. 2014;26(21):3410–3414. doi: 10.1002/adma.201306069. [DOI] [PubMed] [Google Scholar]
- 15.Han Y, Nguyen K, Ogawa Y, Shi H, Park J, Muller DA. Electron Microscopy in Air: Transparent Atomic Membranes and Imaging Modes. Microscopy and Microanalysis. 2015;21(S3):1111–1112. [Google Scholar]
- 16.Bunch JS, Verbridge SS, Alden JS, van der Zande AM, Parpia JM, Craighead HG, McEuen PL. Impermeable Atomic Membranes from Graphene Sheets. Nano Letters. 2008;8(8):2458–2462. doi: 10.1021/nl801457b. [DOI] [PubMed] [Google Scholar]
- 17.Weatherup RS, Eren B, Hao Y, Bluhm H, Salmeron MB. Graphene Membranes for Atmospheric Pressure Photoelectron Spectroscopy. The journal of physical chemistry letters. 2016;7(9):1622–1627. doi: 10.1021/acs.jpclett.6b00640. [DOI] [PubMed] [Google Scholar]
- 18.Velasco-Velez JJ, Pfeifer V, Hävecker M, Weatherup RS, Arrigo R, Chuang CH, Stotz E, Weinberg G, Salmeron M, Schlögl R. Photoelectron Spectroscopy at the Graphene–Liquid Interface Reveals the Electronic Structure of an Electrodeposited Cobalt/Graphene Electrocatalyst. Angew. Chem. Int. Ed. 2015;54(48):14554–14558. doi: 10.1002/anie.201506044. [DOI] [PubMed] [Google Scholar]
- 19.Suk JW, Kitt A, Magnuson CW, Hao Y, Ahmed S, An J, Swan AK, Goldberg BB, Ruoff RS. Transfer of Cvd-Grown Monolayer Graphene onto Arbitrary Substrates. ACS Nano. 2011;5(9):6916–6924. doi: 10.1021/nn201207c. [DOI] [PubMed] [Google Scholar]
- 20.Demers H, Poirier-Demers N, Couture AR, Joly D, Guilmain M, de Jonge N, Drouin D. Three-Dimensional Electron Microscopy Simulation with the Casino Monte Carlo Software. Scanning. 2011;33(3):135–146. doi: 10.1002/sca.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstein J, Newbury D, Joy D, Lyman C, Echlin P, Lifshin E, Sawyer L, Michael J. Scanning Electron Microscopy and X-Ray Microanalysis. 2003 [Google Scholar]
- 22.Ross FM. Liquid Cell Electron Microscopy. Cambridge University Press; 2016. [Google Scholar]
- 23.O’Hern SC, Jang D, Bose S, Idrobo J-C, Song Y, Laoui T, Kong J, Karnik R. Nanofiltration across Defect-Sealed Nanoporous Monolayer Graphene. Nano letters. 2015;15(5):3254–3260. doi: 10.1021/acs.nanolett.5b00456. [DOI] [PubMed] [Google Scholar]
- 24.Boutilier MS, Sun C, O’Hern SC, Au H, Hadjiconstantinou NG, Karnik R. Implications of Permeation through Intrinsic Defects in Graphene on the Design of Defect-Tolerant Membranes for Gas Separation. ACS nano. 2014;8(1):841–849. doi: 10.1021/nn405537u. [DOI] [PubMed] [Google Scholar]
- 25.O’Hern SC, Stewart CA, Boutilier MS, Idrobo J-C, Bhaviripudi S, Das SK, Kong J, Laoui T, Atieh M, Karnik R. Selective Molecular Transport through Intrinsic Defects in a Single Layer of Cvd Graphene. ACS nano. 2012;6(11):10130–10138. doi: 10.1021/nn303869m. [DOI] [PubMed] [Google Scholar]
- 26.Winter B, Hergenhahn U, Faubel M, Bjorneholm O, Hertel IV. Hydrogen Bonding in Liquid Water Probed by Resonant Auger-Electron Spectroscopy. Journal of Chemical Physics. 2007;127(9):94501–94501. doi: 10.1063/1.2770457. [DOI] [PubMed] [Google Scholar]
- 27.Kirz J, Jacobsen C, Howells M. Soft X-Ray Microscopes and Their Biological Applications. Quarterly reviews of biophysics. 1995;28(01):33–130. doi: 10.1017/s0033583500003139. [DOI] [PubMed] [Google Scholar]
- 28.Seah MP, Briggs D. Practical Surface Analysis: Auger and X-Ray Photoelectron Spectroscopy. John Wiley & Sons; 1990. [Google Scholar]
- 29.Slavíček P, Kryzhevoi NV, Aziz EF, Winter B. Relaxation Processes in Aqueous Systems Upon X-Ray Ionization: Entanglement of Electronic and Nuclear Dynamics. The Journal of Physical Chemistry Letters. 2016;7(2):234–243. doi: 10.1021/acs.jpclett.5b02665. [DOI] [PubMed] [Google Scholar]
- 30.Jahnke T, Sann H, Havermeier T, Kreidi K, Stuck C, Meckel M, Schöffler M, Neumann N, Wallauer R, Voss S. Ultrafast Energy Transfer between Water Molecules. Nature Physics. 2010;6(2):139–142. [Google Scholar]
- 31.Briggs D, Seah MP. Practical Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy. John Wiley & Sons Ltd; 1983. [Google Scholar]
- 32.Guo H, Strelcov E, Yulaev A, Wang J, Appathurai N, Urquhart S, Vinson J, Sahu S, Zwolak M, Kolmakov A. Enabling Photoemission Electron Microscopy in Liquids Via Graphene-Capped Microchannel Arrays. Nano letters. 2017;17(2):1034–1041. doi: 10.1021/acs.nanolett.6b04460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiu Y-D, Dow W-P, Huang S-M, Yau S-L, Lee Y-L. Sensitivity Enhancement for Quantitative Electrochemical Determination of a Trace Amount of Accelerator in Copper Plating Solutions. Journal of The Electrochemical Society. 2011;158(5):D290–D297. [Google Scholar]
- 34.Chiu Y-D, Dow W-P. Accelerator Screening by Cyclic Voltammetry for Microvia Filling by Copper Electroplating. Journal of The Electrochemical Society. 2013;160(12):D3021–D3027. [Google Scholar]
- 35.Kolmakova N, Kolmakov A. Scanning Electron Microscopy for in Situ Monitoring of Semiconductor– Liquid Interfacial Processes: Electron Assisted Reduction of Ag Ions from Aqueous Solution on the Surface of Tio2 Rutile Nanowire†. The Journal of Physical Chemistry C. 2010;114(40):17233–17237. [Google Scholar]
- 36.Donev EU, Hastings JT. Electron-Beam-Induced Deposition of Platinum from a Liquid Precursor. Nano letters. 2009;9(7):2715–2718. doi: 10.1021/nl9012216. [DOI] [PubMed] [Google Scholar]
- 37.Park JH, Schneider NM, Grogan JM, Reuter MC, Bau HH, Kodambaka S, Ross FM. Control of Electron Beam-Induced Au Nanocrystal Growth Kinetics through Solution Chemistry. Nano letters. 2015;15(8):5314–5320. doi: 10.1021/acs.nanolett.5b01677. [DOI] [PubMed] [Google Scholar]
- 38.Unocic RR, Lupini AR, Borisevich AY, Cullen DA, Kalinin SV, Jesse S. Direct-Write Liquid Phase Transformations with a Scanning Transmission Electron Microscope. Nanoscale. 2016;8(34):15581–15588. doi: 10.1039/c6nr04994j. [DOI] [PubMed] [Google Scholar]
- 39.Noh KW, Liu Y, Sun L, Dillon SJ. Challenges Associated with in-Situ Tem in Environmental Systems: The Case of Silver in Aqueous Solutions. Ultramicroscopy. 2012;116:34–38. [Google Scholar]
- 40.Yuk JM, Zhou Q, Chang J, Ercius P, Alivisatos AP, Zettl A. Real-Time Observation of Water-Soluble Mineral Precipitation in Aqueous Solution by in Situ High-Resolution Electron Microscopy. ACS nano. 2015;10(1):88–92. doi: 10.1021/acsnano.5b04064. [DOI] [PubMed] [Google Scholar]
- 41.Pawlikowski EM, Prausnitz JM. Estimation of Setchenow Constants for Nonpolar Gases in Aqueous Solutions of Common Salts at Moderate Temperatures. Ind. Eng. Chem. Fundam. 1983;22(1):86–90. [Google Scholar]
- 42.Schneider NM, Norton MM, Mendel BJ, Grogan JM, Ross FM, Bau HH. Electron–Water Interactions and Implications for Liquid Cell Electron Microscopy. The Journal of Physical Chemistry C. 2014;118(38):22373–22382. [Google Scholar]
- 43.Wright MR. An Introduction to Aqueous Electrolyte Solutions. John Wiley & Sons, Ltd; Chichester, UK: 2007. [Google Scholar]
- 44.Ievlev AV, Jesse S, Cochell TJ, Unocic RR, Protopopescu VA, Kalinin SV. Quantitative Description of Crystal Nucleation and Growth from in Situ Liquid Scanning Transmission Electron Microscopy. ACS nano. 2015;9(12):11784–11791. doi: 10.1021/acsnano.5b03720. [DOI] [PubMed] [Google Scholar]
- 45.Nappini S, Matruglio A, Naumenko D, Dal Zilio S, Bondino F, Lazzarino M, Magnano E. Graphene Nanobubbles on Tio 2 for in-Operando Electron Spectroscopy of Liquid-Phase Chemistry. Nanoscale. 2017;9:4456–4466. doi: 10.1039/c6nr09061c. [DOI] [PubMed] [Google Scholar]
- 46.Vlassiouk I, Regmi M, Fulvio P, Dai S, Datskos P, Eres G, Smirnov S. Role of Hydrogen in Chemical Vapor Deposition Growth of Large Single-Crystal Graphene. ACS Nano. 2011;5(7):6069–6076. doi: 10.1021/nn201978y. [DOI] [PubMed] [Google Scholar]
- 47.Li X, Cai W, An J, Kim S, Nah J, Yang D, Piner R, Velamakanni A, Jung I, Tutuc E, Banerjee SK, Colombo L, Ruoff RS. Large-Area Synthesis of High-Quality and Uniform Graphene Films on Copper Foils. Science. 2009;324(5932):1312–1314. doi: 10.1126/science.1171245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.