

ABSTRACT
α1-Antichymotrypsin (α1-ACT) belongs to a kind of acute-phase inflammatory protein. Recently, such protein has been proved exist in the amyloid deposits which is the hallmark of Alzheimer's disease, but limitedly reported in prion disease. To estimate the change of α1-ACT during prion infection, the levels of α1-ACT in the brain tissues of scrapie agents 263K-, 139A- and ME7-infected rodents were analyzed, respectively. Results shown that α1-ACT levels were significantly increased in the brain tissues of the three kinds of scrapie-infected rodents, displaying a time-dependent manner during prion infection. Immunohistochemistry assays revealed the increased α1-ACT mainly accumulated in some cerebral regions of rodents infected with prion, such as cortex, thalamus and cerebellum. Immunofluorescent assays illustrated ubiquitously localization of α1-ACT with GFAP positive astrocytes, Iba1-positive microglia and NeuN-positive neurons. Moreover, double-stained immunofluorescent assays and immunohistochemistry assays using series of brain slices demonstrated close morphological colocalization of α1-ACT signals with that of PrP and PrPSc in the brain slices of 263K-infected hamster. However, co-immunoprecipitation does not identify any detectable molecular interaction between the endogenous α1-ACT and PrP either in the brain homogenates of 263K-infected hamsters or in the lysates of prion-infected cultured cells. Our data here imply that brain α1-ACT is increased abnormally in various scrapie-infected rodent models. Direct molecular interaction between α1-ACT and PrP seems not to be essential for the morphological colocalization of those two proteins in the brain tissues of prion infection.
KEYWORDS: astrocytes, microglia, prion disease, α1-Antichymotrypsin, PrP
INTRODUCTION
Prion diseases (PrD) are a group of chronic and fatal neurodegenerative disorders usually infect the central nervous system (CNS) of human beings and other mammals, for instance, scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle, as well as chronic wasting disease (CWD) in mule, deer and elk. Human prion diseases consist of Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), Gerstmann-Sträussler-Scheinker syndrome (GSS) and Kuru.1 The causative of these diseases known as prion, an unconventional and unique pathogen, which conformational conversion from a normal prion protein (PrPC) to a pathogenic PrPSc form which is characterized by fiber-like polymers, aggregation, partially resistance to protease digestion, and resistance to detergent solubilization. Besides of accumulation of PrPSc in the affected tissues during its extremely long incubation period, the primary neuropathological characteristics are neuron loss, spongiform degeneration and astrogytosis, therefore, these kinds of diseases also called transmissible spongiform encephalopathies (TSEs).2
α1-antichymotrypsin (α1-ACT) is one of the serine proteinase inhibitors (serpin), which consists of 3 β-sheets, 8 α-helices and one active site situated within a hypervariable reactive center loop (RCL).3,4 Usually, α1-ACT exerts their function by binding and forms equimolar complexes with its target proteinases to inhibit specific serine proteases.5 α1-ACT is widely expressed in multiple tissue types, such as liver, heart, lung, skeletal muscle, brain, spinal cord and prostate.6 Physiologically, α1-ACT presents a low level in brain tissues.7 In some pathological conditions, the expression of α1-ACT may alter abnormally. In the patients with Alzheimer's disease (AD), α1-ACT has been found surrounding astrocytes and is able to form a complex with toxic beta-amyloid (Aβ)1-42 peptide which is main component of the senile plaques.8,9 However, the situation of α1-ACT in the CNS tissues of prion diseases is unclear. Recently, our proteomic studies have proposed that the level of α1-ACT remarkably increases in the brain tissues of the prion experimental rodents and sCJD patients, but decreases in the cerebrospinal fluid (CSF) of sCJD patients,10,11 highlighting that the brain α1-ACT may undergo abnormal process during the progression of prion diseases.
In the present study, we found remarkable up-regulations of α1-ACT in the brain tissues of scrapie-infected rodent models, especially in the regions of cortex, thalamus and cerebellum where severe astrogliosis was observable. Increased brain α1-ACT displayed a time-dependent manner during prion infection. The signals of α1-ACT were ubiquitously colocalized with the glial fibrillary acidic protein (GFAP)-positive astrocytes, Iba1-p-ositive microglia and NeuN-positive neurons. Moreover, α1-ACT showed close colocalization with the signals of PrP and PrPSc in the brain tissues of scrapie agent 263K-infected hamster. However, no detectable molecular interaction was identified between α1-ACT and PrP complexes either in the brain homogenates of scrapie agent 263K-infected hamsters or in the lysates of prion-infected cultured cells.
RESULTS
Significant Increase of α1-ACT in the Brain Tissues of the Scrapie-Infected Rodent Models at End Stage
α1-ACT mainly expresses in the astrocytes of central nerve system (CNS) and displays multiple biological functions.8 To see the possible changes of brain α1-ACT level during prion infection, the brain homogenates of the scrapie agent 263K-infected hamsters at terminal stage were subjected into Western blot with α1-ACT mAb. Compared with that of healthy controls, the α1-ACT-specific signals were markedly stronger in the preparations of three 263K-infected hamsters, showing significantly increased in the analysis of the mean gray values of α1-ACT (Fig. 1A). Remarkable increases of brain α1-ACT were also detected in the Western blots of other two scrapie-infected mouse models, 139A- and ME7-infected mice, at end stage (Fig. 1A). Further, the expressions of α1-ACT in the brains of those scrapie-infected rodents at end stage were evaluated by α1-ACT-specific IHC assays. As expected, guanidine-treated PrP-specific IHC revealed widespread various-size granular PrPSc-positive signals in brain sections of the cortex regions of 263K-infected hamsters, 139A- and ME7-infected mice, but not in that of uninfected control. Numerous large brown α1-ACT-positive signals were observed in the brain tissues of those three kinds of scrapie-infected rodents, predominantly in the gray matters of cortex, thalamus and cerebellum, whereas only obviously less, smaller and weaker the α1-ACT stainings were seen in the brains of healthy rodents (Fig. 1B and 1C). It implies that the brain α1-ACT levels were markedly increased in the scrapie-infected rodent models at end stage.
FIGURE 1.
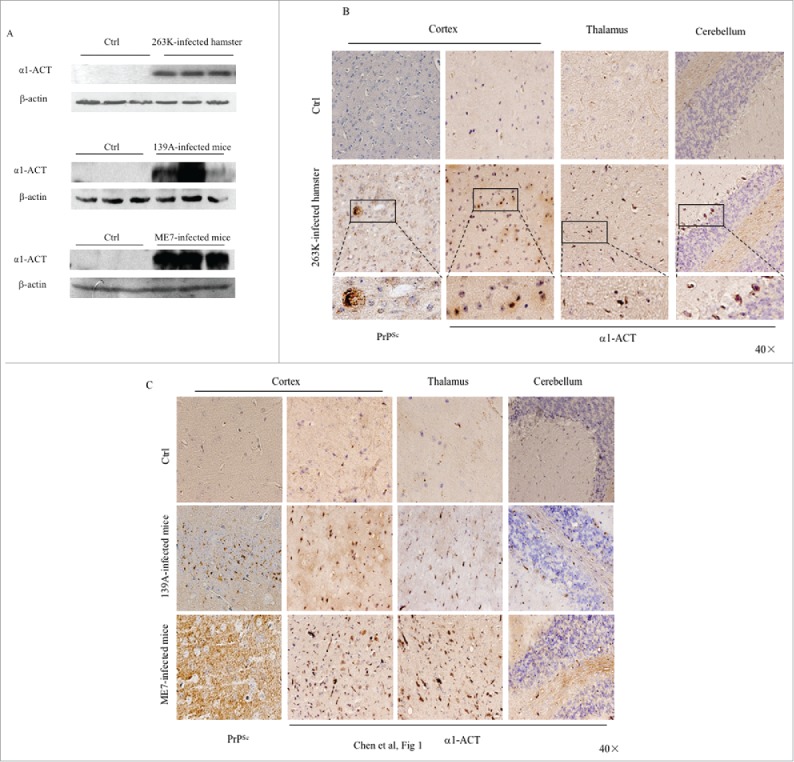
Comparative analyses of the levels of α1-ACT in the brains of the scrapie-infected rodent models. a Western blots of the brain homogenates of various scrapie-infected rodent models at the terminal stage and the individual age-matched normal ones. Each group contains three individual animals. The scrapie strains are indicated on the top of each graph and the quantitative assays of the various blots normalized with the data of β-actin are indicated on the left. b Representative IHC assays of α1-ACT in the sections of various brain regions of 263K-infected hamsters at terminal stage, including the cortex, thalamus and cerebellum (presenting on the top side). The relevant brain sections of normal hamsters are used as control (40 ×). The magnified images are shown under each picture. c Representative IHC analyses of α1-ACT in the sections of various brain regions of 139A-infected (upper panel) and ME7-infected (lower panel) mice at terminal stage (40 ×).
Time-Dependent Increases of α1-ACT During the Incubation Period of Scrapie Infection
To evaluate the dynamic alterations of α1-ACT during scrapie infection, the brain samples of 263K-infected hamster collected on the 10th, 30th, 50th and terminal stage (70th) dpi, and the brain samples of 139A and ME7-infected mouse collected on the 86th, 117th, 147th and terminal stage (179th) dpi were comparatively analyzed by α1-ACT specific Western blot. Meanwhile, the presences of PrPSc in those samples were tested by PK-treated PrP specific Western blot. It revealed that the α1-ACT levels in the brain samples of 263K-infected hamster were clearly up-regulated in the samples of 10 dpi, maintained at high level in those collected at the subsequent time-points (30 and 50 dpi) till the end of disease (70 dpi). Meanwhile, PrPSc signals were observable in brains of 263K-infected hamsters of 50 dpi and much stronger in that of 70 dpi (Fig. 2A). Similarly, the levels of the brain α1-ACT in ME7-infected mice were continually increased in the samples of 117 and 147 dpi and reached to the top in that of 179 dpi, while the PrPSc signals were detectable in the samples since 117 dpi and increased steadily in the following ones (Fig. 2B). It displays a time-dependent increase of brain α1-ACT during prion infection.
FIGURE 2.
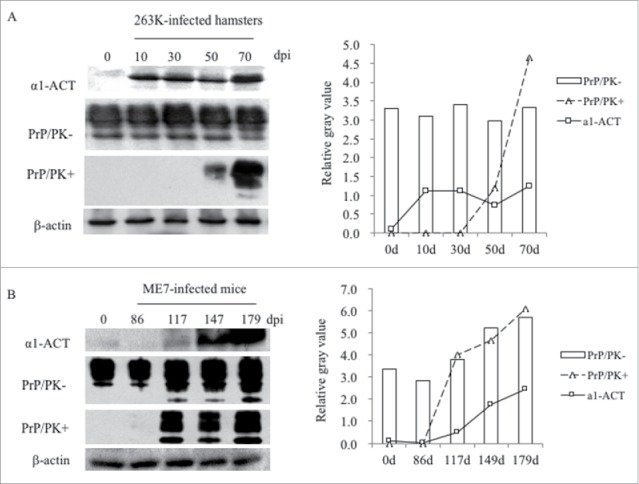
Dynamic assays of the alterations of α1-ACT, total PrP and PrPSc in the brains of prion-infected rodent models during incubation periods. a Western blots for α1-ACT, total PrP and PrPSc in the brain of 263K-infected hamsters. The brain samples were collected 0, 20, 40, 60, 80 dpi. b Western blots for α1-ACT, total PrP and PrPSc in the brain of ME7-infected mice. The brain samples were collected 0, 86, 117, 147, 179 dpi. Various blots are indicated on the left. The quantitative analysis of gray values of total PrP and PrPSc is presented on the right.
Comprehensive Distributions of α1-ACT in the Proliferative Astrocytes and Microglia, as well as the Damaged Neurons in the Brain of Scrapie-Infected Hamsters
To assess the distributions of brain α1-ACT after prion infection, the brain slices of the 263K-infected hamsters at the end stage were immunofluorescently double-stained by α1-ACT antibody together with the antibodies against the biomarker for astrocytes (GFAP), microglia (Iba1) or neuron (NeuN) individually. Robustly stronger α1-ACT signals (green) were observed in the brain sections of 263K-infected hamsters than those of normal ones under confocal microscopy (Fig. 3). Large amounts of α1-ACT signals (green) co-localized with the GFAP- and Iba1-positive cells (red) in the slices of 263K-infected hamsters, whereas much less colocalization between α1-ACT and GFAP or Iba1 signals was visible in those of the normal hamsters (Fig. 3A and B). In the brain slices of 263K-infected hamsters, the increased α1-ACT signals were also overlapped with the NeuN-positive cells showing different sizes and shapes while in those of normal hamsters much less and weaker α1-ACT signals seemed not to overlap with the large and round NeuN-stained cells (Fig. 3C). It illustrates a comprehensive distribution of α1-ACT in the reactively proliferative astrocytes and microglia, as well as damaged neurons in the brains of scrapie infected hamsters.
FIGURE 3.
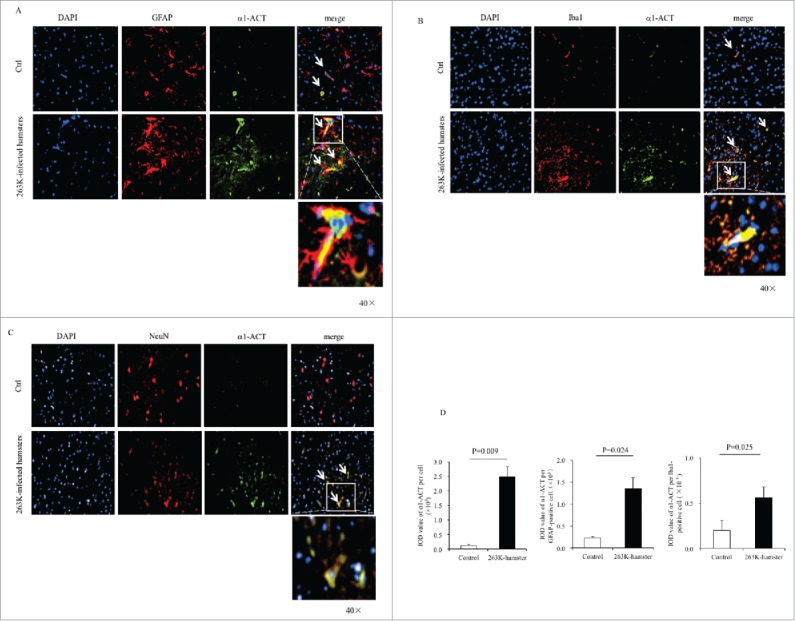
Immunofluorescent analyses of the relationships of α1-ACT with various sorts of cells in the brains of 263K-infected hamsters at the end of disease and the age-matched normal hamsters as control. a-c Representative images of the hamsters' brains double-immunostained with the antibodies of α1-ACT (green) and GFAP, (red) (a), Iba1 (red) (b), or NeuN (red) (c) (100 ×). The zoomed-in pictures are shown on the bottom. d Analyses of the IOD values with the software in Operatta. The average IOD values of α1-ACT per cell (× 105) (left panel), per GFAP-positive cell (× 105) (middle panel) as well as per Iba1-positive cell (× 105) (right panel). Statistical differences between infected and normal animals are indicated above.
To evaluate the difference in fluorescent value of α1-ACT per cell between uninfected and 263K-infected hamsters, the integrated optical density (IOD) of α1-ACT per cell from three slices each group were automatically captured. Compared with that of uninfected control after balanced the cell numbers, the average IOD value α1-ACT per cell in the brain slices of 263K-infected hamsters was remarkably higher (Fig. 3D, left panel). Subsequently, the IOD values of α1-ACT per GFAP- and Iba1-postive cells were separately counted. Significant higher IOD values of α1-ACT both per GFAP-positive cell (Fig. 3D, middle panel) and per Iba1-positive cell (Fig. 3D, right panel) were also obtained in the brain slices of 263K-infected hamsters. It suggests that the increase of brain α1-ACT in prion infected hamsters at the end course may result mainly from not only the actively proliferated astrocytes and microglia.
The Association of the Increased α1-ACT with the Deposits of PrPSc in Brain Tissues of 263K-Infected Hamsters
α1-ACT has been described to be a major component of amyloid brain deposits in the brains of the patients with AD.8,9 To explore the possible association between α1-ACT and PrPSc deposits in prion-infected animals, the brain slices of 263K-infected and normal hamsters were double-stained with α1-ACT and PrP antibodies in IFA. As shown in Fig. 4A, large amounts of colocalization between α1-ACT and PrP signals were observed in the brain slices of 263K-infected hamsters, showing asteroid or irregular polygon shapes morphologically. Colocalization signals between α1-ACT and PrP were observed in the brain slices of normal hamsters, with significantly less amount and small size.
FIGURE 4.
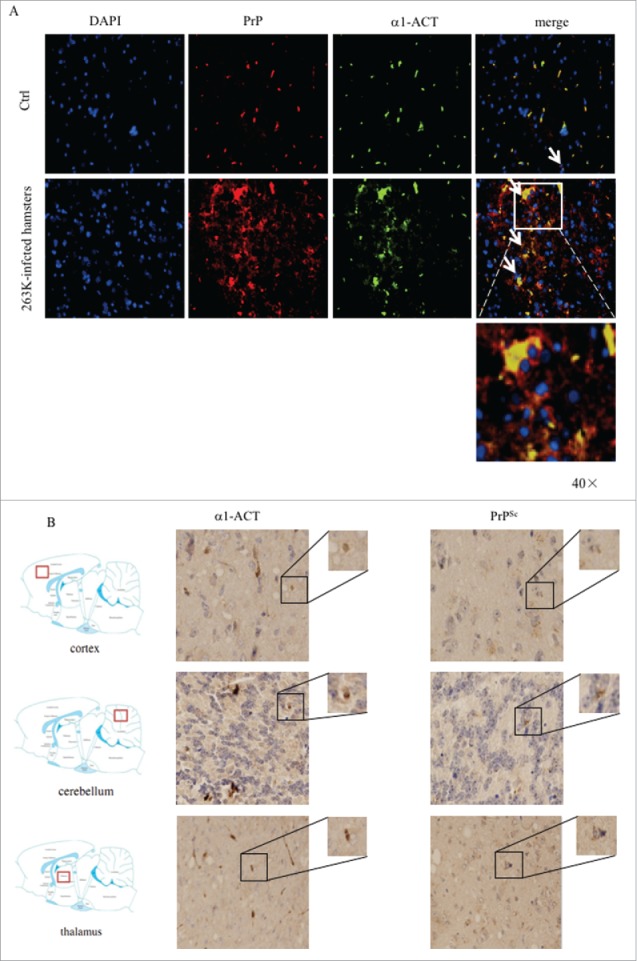
Analyses of the association of α1-ACT with PrP in brain tissues. a Representative images of double-stained IFA assays with the antibodies of α1-ACT (green) and PrP (red) on the brain sections of normal and 263K-infected hamsters (100 ×). The zoomed-in pictures are shown on the bottom. b Representative images of IHC assays for α1-ACT and PrPSc in the serial sections of different brain regions of 263K-infected hamsters, including cortex, thalamus and cerebellum (indicated above). The magnified images are shown on the top right of each picture and the positions of the observing fields within the whole brain are indicated on the left.
Furthermore, the association of α1-ACT and PrPSc deposits in the brains of 263K-infected hamster was evaluated by IHC assays using serial brain sections. Prior to staining of PrP specific antibody, the brain sections were exposed to GdnSCN for the purpose of removing normal PrPC. As expected, large amounts of α1-ACT and PrPSc specific signals were identified in various brain regions of 263K-infected hamsters, including cortex, thalamus and cerebellum. Careful analyses of the serial brain sections of 263K-infected hamsters illustrated that many brown signals of α1-ACT and PrPSc were at the same positions (Fig. 4B). It suggests that the increased α1-ACT may accumulate together with PrPSc deposits during prion infection.
No Detectable Molecular Interaction Between α1-ACT and PrP in the Brain Homogenates of Scrapie-Infected Hamsters or the Lysates of Prion-Infected Cell Line
To assess the possible molecular interaction between endogenous PrP and α1-ACT in the brains, 10% brain homogenates of 263K-infected and uninfected hamsters were performed by immunoprecipitation tests using pAb α1-ACT as the capturing ones and anti-PrP (mAb 3F4) as the detecting one. Fig. 5A revealed that the endogenous α1-ACT was precipitated with pAb α1-ACT, but no PrP-specific signal was observed in the precipitations with 3F4 mAb, showing similar blot patterns as that of mouse IgG control.
FIGURE 5.
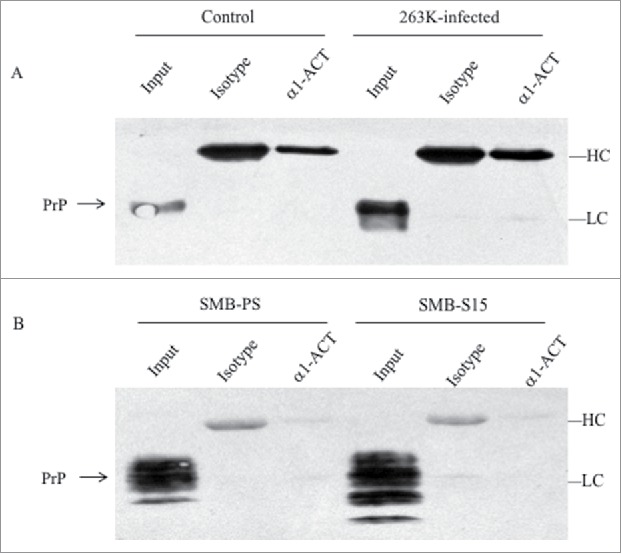
Evaluation of the molecular interaction between endogenous α1-ACT and PrP with immunoprecipitation assays. a Brain homogenates of normal and 263K-infected hamsters. The proteins in homogenates were precipitated with pAbs against α1-ACT or rabbit IgG (marked as isotype) and subsequently evaluated by PrP-specific Western blots. b Lysates of SMB-S15 and SMB-PS cells. The proteins in cell lysates were precipitated with pAb α1-ACT or rabbit IgG (marked as isotype) and subsequently evaluated by PrP-specific Western blots. Aliquots of brain homogenates or cell lysates (marked as input) were directly loaded into SDS-PAGE as internal controls. HC and LC represent the heavy chain and light chain of IgG.
To get more information of molecular interaction between endogenous PrP and α1-ACT in the scrapie-infected cell model, scrapie agent Chandler-infected cell line SMB-S15 and its control cell line SMB-PS were subjected into immunoprecipitation. PrP-specific (mAb 6D11) Western blot illustrated the similar results that no PrP signal was identified in the products precipitated by pAb α1-ACT (Fig. 5B). Those data indicate that endogenous α1-ACT and PrP, either in the brain tissues or in the cultured cells, seems not to be able to form detectable complex.
DISCUSSION
α1-ACT is the member of serine protease inhibitor superfamily of proteins, of which no less than 700 have been identified in eukaryotes and prokaryotes.5 In this study, we have found the abnormally upregulated α1-ACT in the brains of scrapie infected experimental rodents. Actually, the increased brain α1-ACT has not only been found in the senile plaques and surrounding astrocytes, but also been identified to form the complex with toxic Aβ1-42 peptide in patients with AD.12,13 Moreover, polymorphism of the α1-ACT gene is a susceptibility factor for some neurological illnesses, such as α1-ACT gene might be related to the early onset of PD and MSA.14,15 Those data indicate that the upregulation of α1-ACT may be a common neuropathological characteristic for different neurodegenerative diseases.
Global microarray screens have identified the transcriptional levels of α1-ACT are remarkably elevated not only in the brains of different scrapie-infected mouse models, but also in the occipital cortices of sCJD patients.16 Previous study has reported that the transcription of α1-ACT was highly upregulated in the brains of scrapie-infected mice and increased progressively during prion infections.16 It suggests that the elevation of brain α1-ACT transcription and expression may occur in prion diseases of different species. Our IHC data here exhibit that the increased α1-ACT occurs mainly in some cerebral regions of 263K-infected hamsters, 139A and ME7-infected mice, such as cortex, thalamus and cerebellum, where exhibit severe astrogliosis. IFA data illustrate that the α1-ACT signals predominately colocalize with the GFAP-positive astrocytes and Iba1-positive microglia, especially in the brains of scrapie infected mice at terminal stage. Quantitative analysis reveals that the increased brain α1-ACT intensity shows close association not only with the increased cell numbers of astrocytes and microglia, but also with the increased α1-ACT expressing quantity per cell during prion infection. It strongly indicates that the major sources for overexpressing α1-ACT are the reactive proliferating astrocytes and microglia during prion pathogenesis.
Astrocytes is one of the most abundant cell types in brain tissues and more easily to present reactive astrogliosis during prion infection. Microglia activation is also repeatedly reported in many neurodegenerative diseases.17-19 Once activation, microglia has the ability to transform into phagocytes and release various substrates such as cytokines/chemokines, nitric oxide, free radicals, etc.20 As a kind of acute-phase inflammatory protein, the expression of α1-ACT can be stimulated by the presences of cytokines, such as IL-1, IL-6 and TNF-α.21-23 More studies illustrate the levels of some cytokines are elevated in the brains of rodents infected with prion.24,25 Therefore, according to the data here and many other observations, we speculate that upon the propagation and accumulation of PrPSc in the brains during prion pathogenesis, the astrocytes and microglia in the brains are reactively activated from the steady state to active proliferation, followed by releasing some cytokines and stimulating the microglia and astrocytes to synthesis α1-ACT.
The association with Aβ amyloid deposits with the increased α1-ACT has been addressed pathologically and molecularly.12,13,26 Study of AD with transgenic mouse models also demonstrates that the tg mice expressing both ACT and APP contain more age-related plaque deposition in the brains than that expressing APP alone.9 Using serial sections of brain tissues, the specific stainings of α1-ACT and PrPSc in IHC seems to locate at the same positions. IFA data also illustrate colocalization of α1-ACT signals with PrP proteins. Moreover, unlike the merged signals of α1-ACT and PrP in the normal brain slices with cell-like morphology, the merged signals in the infected brain slices were much larger, rough and irregular, which may indicate a colocalization of α1-ACT with PrPSc deposits. PrP protein illustrates active molecular interaction with other proteins and more than 20 various PrP-partner proteins have been identified in the past two decades.27 Actually, α1-ACT has the ability to interact with some proteins, such as cathepsin G, human glandular kallikrein 2, mast cell chymases, pancreatic cationic elastase, kallikrein 3 (prostate specific antigen) and Aβ. However, under our experimental condition, we fail to figure out the α1-ACT-PrP complex either in the brain homogenates of normal and 263K-infected hamsters, or in the lysates of prion infected cell lines SMB-S15 and its normal control SMB-PS that contain detectable endogenous α1-ACT. Albeit the experimental bias cannot be absolutely excluded, no detectable molecular interaction between the native α1-ACT and PrP in brain tissues and cultured cells suggest that direct molecular binding of those two proteins may not be essential for the histological colocalization of those two signals observed in IHC and IFA assays. Whether such morphological colocalization between α1-ACT and PrP in the brains of prion infected animals is via the potential third partner element deserves further exploration.
Our IFA results here have also illustrated the overlapping images of α1-ACT signals with the neurons, even the morphologically shrunken neurons, in the brains infected with agent 263K, but very rare with that in the normal control. One of the possibilities for the colocalization of α1-ACT with neurons in the scrapie-infected brains might be the increased expression of α1-ACT in the neurons due to prion replication or accumulation. However, the evaluations of α1-ACT by Western blot, real time PCR and IFA in the prion infected cell lines SMB-S15 and its normal control SMB-PS do not identify distinguishable difference (Fig. S1), highlighting that the PrPSc replication and accumulation in neuron-derived cells, at least in vitro, does not significantly change the transcription and expression of the cellular α1-ACT. Another possibility is that the α1-ACT overlapped on neurons in the scrapie- infected brains comes from active secretion of α1-ACT of the proliferating astrocytes during prion infection. Besides of the overexpression of α1-ACT surround of Alzheimer amyloid plaques, clearly elevated α1-ACT have been reported in cerebrospinal fluid and plasma of AD patients.28-31 Additionally, the levels of α1-ACT have been reported to be substantially upregulated in urine of patients with sCJD and the animals with natural prion disease, such as cattle with BSE and deer with CWD.16 Those data indicate that the overexpressed α1-ACT can secret and distribute extensively in CNS tissues and other body fluids, which eventually deposits onto neurons via unknown pathway.
Hyperphosphorylation and accumulation of tau leading to formation of neurofibrillary tangles (NFT) in neurons and tau aggregation in glial cells are the main pathological hallmarks of AD as well as other tauopathies. The purified α1-ACT has ability to induce tau phosphorylation and apoptosis in primary mouse and human neurons.32 Various kinds of abnormal tau pathology have been also described in different types of prion diseases, such as the aberrant phosphorylation of tau, alterations of tau isoforms.33-36 Increase of tau in CSF samples is used as diagnostic criteria for sCJD in many countries. However, large amount of NFT in brain is not frequently observed in prion diseases. Obviously, different neurodegeneration diseases may have different tau pathology. The exact impact of increased α1-ACT on tau pathology and pathogenesis in prion disease deserved to further study.
MATERIALS AND METHODS
Ethics Statement
Usage of animal specimens in this study was approved by the Ethical Committee of National Institute for Viral Disease Prevention and Control, China CDC. Animal housing and experimental protocols were in accordance with the Chinese Regulations for the Administration of Affairs Concerning Experimental Animals.
Cell Culture
Scrapie agent Chandler-infected cell line SMB-S15 and its cured cell line SMB-PS were obtained from Roslin Institute, UK. SMB-S15 was originally from the brain of a scrapie strain Chandler-infected mouse and can support scrapie prions to replicate continuously in vitro, which possesses almost same biochemical characteristics as its original PrPSc in brain tissues.37 SMB-PS is the cured permanently SMB cells by pentosan sulfate (PS), which removes the replication and infectivity of PrPSc thoroughly.38 Cells were cultured in DMEM with 10% fetal calf serum, under a 5% CO2 and 33°C humidified atmospheres. Cells were collected, counted and stored at −20°C for further study.
Reagents and Antibodies
The following primary antibodies were enrolled in the present study, including anti-Alpha 1-Antichymotrypsin (α1-ACT) rabbit-derived polyclonal antibody (pAb) (PAB015Mu01, Cloud-Clone Corp, USA), anti-β-actin mouse-derived monoclonal antibody (mAb) (Sc-47778, Santa Cruz Biotechnology, USA), anti-NeuN mouse-derived mAb (MAB377, Millipore Corporation, USA), anti-Iba1 mouse-derived mAb (ab15690, Abcom, USA), anti-Glial fibrillary acidic protein (GFAP) mouse-derived mAb (#3670, Cell Signaling Technology, USA), PrP specific mAb 3F4 (MAB1562, Millipore) and 6D11 (Sc-58581, Santa Cruz Biotechnology, USA).
Horseradish peroxidase (HRP)-conjugated goat-derived anti-mouse (#31430) or anti-rabbit IgG (#31460) were purchased from Thermo. Alexa Fluor probe labeled secondary antibodies (A-11034 and A-11004) were purchased from Invitrogen. DAPI (D9542) was from Sigma. Protease inhibitor cocktail set III (539134) were from Merck. HRP-conjugated secondary antibody for immunohistochemical staining was from BOSTER (SV0002, China). Cell protein extraction reagent (CW0889) was purchased from CWBIO. Enhanced ChemoLuminescence (ECL) system (NEL103E001EA) was the product of PerkinElmer.
Preparation of Brain Homogenates
Brain samples from scrapie agent 263K-infected hamsters, and from scrapie agents 139A- and ME7-infected mice were enrolled in this study. The clinical and neuropathological features of these rodent models have been described elsewhere.39 Briefly, two weeks of postnatal rodents were intracerebrally inoculated with various scrapie agents under halothane anaesthesia, respectively. The clinical symptoms were recorded for statistically. The incubation time was calculated from the inoculation to the onset of clinical manifestations. The incubation periods of the 263K-infected hamsters varied from 45 to 82 days (66.7±1.1 days). The incubation periods of the 139A and ME7-infected mice were from 154 to 226 days (183±23.1 days) and from 165 to 193 days (184.2±11.8 days), respectively. The brains of those scrapie-infected rodents were harvested surgically either under euthanasia with intraperitoneal injection of chloral hydrate at the moribund time or postmortem after death. Meanwhile, age-matched uninfected healthy rodents were used as the controls. For dynamic assays, brain samples of 263K-infected hamsters were separately harvested at the different time points of prion infection: 0, 10, 30, 50, and 70-day post inoculation (dpi), and brain samples of the 139A and ME7-infected mice harvested on the 0th, 86th, 117th, 149th and 179th dpi were included as well. The samples of 70 dpi of 263K-infected hamsters and that of 179 dpi of 139A and ME7 represented the samples of terminal stage.
Fresh collected brain tissues were washed in Tris-buffered saline (TBS, 10 mM Tris-HCl, 133 mM NaCl, pH7.4) for three times, and then homogenized in lysis buffer (100 mM NaCl, 10 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate, 10 mM Tris, pH 7.4) containing protease inhibitor cocktail set III. The homogenates were centrifuged at 2000 × g for 10 min, and the supernatant fractions were aliquoted for further experiments.
Western Blots
Aliquots of brain homogenates were separated by 12% sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred onto nitrocellulose membranes. Membranes were blocked with 5% (w/v) skimmed milk in 1 × Tris-buffered saline containing 0.1% Tween 20 (TBST) at room temperature (RT) for 30 min and incubated with individual primary antibodies at 4°C overnight. After washing with TBST, membranes were subsequently incubated with individual HRP-conjugated secondary antibodies and reactive signals were developed using a commercial ECL kit. Images were captured by ChemiDocTM XRS+ Imager (Bio-Rad, USA).
To test the protein K (PK)-resistant of PrP in brain tissues of scrapie-infected rodents, the brain homogenates of various scrapie-infected rodents were treated with a final concentration of 25 μg/ml PK at 37°C for 60 min prior to Western blot analyses. The PK digestion was stopped by incubating the samples at 100°C for 10 min.
Immunohistochemical Staining (IHC)
Brain tissues were fixed in 10% buffered formalin solution and paraffin sections (5 μm in thickness) were prepared routinely. For PrPSc immunohistochemical test, the prepared brain sections were deparaffinized and immersed in guanidine isothiocyanate for 1h at 4°C. PrP-specific mAb (1:1000 diluted in 1% normal goat serum) was incubated with the sections at 4°C overnight. For α1-ACT immunohistochemical test, the sections were incubated with 1:100-diluted anti-α1-ACT pAb at 4°C overnight. Subsequently, the sections were incubated with 1:250-diluted HRP-conjugated goat-derived anti-rabbit or anti-mouse secondary antibody at 37°C for 1h, and visualized by incubation with 3, 3′-diaminobenzidine tetrahydrochloride (DAB). The slices were counterstained with hematoxylin for 1 min, dehydrated and routinely mounted. The images were captured with an attached digital color camera (DP70, Olympus Optical).
Immunofluorescence Assay (IFA)
Brain sections were subjected to permeate with 0.3% Triton X-100 in phosphate-buffered saline (PBS) for 30 min and blocked with normal goat serum for 1 h. After blocked, sections were incubated with 1:50-diluted anti-α1-ACT and 1:50-diluted anti-GFAP, 1:50-diluted anti-Iba1 or 1:50-diluted anti-NeuN, in dilution solution (PBS with 2% BSA and 0.3% Triton X-100) at 4°C overnight. The sections were subsequently incubated with 1:200-diluted Alexa Fluor 488-labeled goat-derived anti-rabbit and Alexa Fluor 568-labeled goat-derived anti-mouse secondary antibodies at 37°C for 1 h. After removing secondary antibodies, DAPI were used to stain the nucleus at final concentration of 1 μg/ml at RT for 30 min. The slices were then mounted with permount and viewed using a PerkinElmer microscopy.
Immunoprecipitation (IP)
2 μg of α1-ACT captured pAb was mixed with 50 μl Dynabeads®-coated Protein G (#100.04D, Invitrogen, USA) at 4°C overnight and subsequently incubated with 400 μg of total protein of the prepared 10% brain homogenate of normal and 263K-infected hamster or whole SMB cell lysate at 4°C overnight. The immunocomplexes were collected by separating on the magnet and washed for five times in washing buffer before being separated by 12% SDS-PAGE. The immunocomplexes were detected by PrP-specific (3F4 or 6D11 mAb) Western blots.
Quantitative Real-Time PCR (qRT-PCR)
Real-time PCR in this study was performed in a Bio Rad CFX96 Real-Time System (Bio Rad, USA). Total cell RNAs were extracted from SMB-PS and SMB-S15 cells with Trizol reagent and were subjected to first-strand cDNA synthesis with reverse transcription system (Invitrogen, USA) according to the manufacturer's protocols. The specific primers were designed based on the sequences of mouse α1-ACT gene (SerpinA3N) in GenBank (NM_009252.2), including 5′- CCCTGAGGAGTGGAAGAAT-3′ and 5′-CCTGATGCCCAGCTTTGAAA-3′, respectively. PCR amplification was performed in triplicate with a total of 40 cycles (3 s at 95°C, 30 s at 52°C and 30 s at 72°C). The comparative Ct (the fractional cycle number at which the amount of amplified target reached a fixed threshold) method was used for the relative quantitative detection of the expressions of the target gene. The relative Ct for the target gene was subtracted from the Ct for the β-actin gene.
Statistical Analysis
In the present study, all experiments were performed at least three times. Statistical analysis was conducted by the SPSS 17.0 statistical package. Quantitative analysis of Western blot and quantification of colocalization were processed with Image J software. The final results were presented as mean ± stand error of mean (SEM). The student's t test was evaluated for statistical analysis and P <0.05 was considered significant.
Supplementary Material
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
FUNDING
This work was supported by Chinese National Natural Science Foundation Grants (81401670, 81630062), National Key Research and Development Plan (2016YFC1202700), SKLID Development Grant (2012SKLID102, 2016SKLID603) and the Young Scholar Scientific Research Foundation of China CDC (2016A101).
REFERENCES
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;93:337–48. doi: 10.1016/S0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 3.Huber R, Carrell RW. Implications of the three-dimensional structure of alpha 1-antitrypsin for structure and function of serpins. Biochemistry. 1989;28:8951–66. doi: 10.1021/bi00449a001. [DOI] [PubMed] [Google Scholar]
- 4.Janciauskiene S. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim Biophys Acta. 2001;1535:221–35. doi: 10.1016/S0925-4439(01)00025-4. [DOI] [PubMed] [Google Scholar]
- 5.Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, Gettins PG, Irving JA, Lomas DA, Luke CJ, Moyer RW, et al.. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. 2001;276:33293–6. doi: 10.1074/jbc.R100016200. PMID:11435447 [DOI] [PubMed] [Google Scholar]
- 6.Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al.. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–9. doi: 10.1093/bioinformatics/bti042. PMID:15388519 [DOI] [PubMed] [Google Scholar]
- 7.Gopalan S, Kasza A, Xu W, Kiss DL, Wilczynska KM, Rydel RE, Kordula T. Astrocyte- and hepatocyte-specific expression of genes from the distal serpin subcluster at 14q32.1 associates with tissue-specific chromatin structures. J Neurochem. 2005;94:763–73. doi: 10.1111/j.1471-4159.2005.03204.x. PMID:15969742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mucke L, Yu GQ, McConlogue L, Rockenstein EM, Abraham CR, Masliah E. Astroglial expression of human alpha(1)-antichymotrypsin enhances alzheimer-like pathology in amyloid protein precursor transgenic mice. Am J Pathol. 2000;157:2003–10. doi: 10.1016/S0002-9440(10)64839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilsson LN, Bales KR, DiCarlo G, Gordon MN, Morgan D, Paul SM, Potter H. Alpha-1-antichymotrypsin promotes beta-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:1444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C, Xiao D, Zhou W, Shi Q, Zhang HF, Zhang J, Tian C, Zhang JZ, Dong XP. Global Protein Differential Expression Profiling of Cerebrospinal Fluid Samples Pooled from Chinese Sporadic CJD and non-CJD Patients. Mol Neurobiol. 2014;49:290–302. doi: 10.1007/s12035-013-8519-2. [DOI] [PubMed] [Google Scholar]
- 11.Shi Q, Chen LN, Zhang BY, Xiao K, Zhou W, Chen C, Zhang XM, Tian C, Gao C, Wang J, et al.. Proteomics analyses for the global proteins in the brain tissues of different human prion diseases. Mol Cell Proteomics. 2015;14:854–69. doi: 10.1074/mcp.M114.038018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor alpha 1-antichymotrypsin in the brain amyloid deposits of Alzheimer's disease. Cell. 1988;52:487–501. doi: 10.1016/0092-8674(88)90462-X. [DOI] [PubMed] [Google Scholar]
- 13.Abraham CR, Shirahama T, Potter H. Alpha 1-antichymotrypsin is associated solely with amyloid deposits containing the beta-protein. Amyloid and cell localization of alpha 1-antichymotrypsin. Neurobiol Aging. 1990;11:123–9. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto M, Kondo I, Ogawa N, Asanuma M, Yamashita Y, Mizuno Y. Genetic association between susceptibility to Parkinson's disease and alpha1-antichymotrypsin polymorphism. Brain Res. 1997;759:153–5. doi: 10.1016/S0006-8993(97)00330-2. [DOI] [PubMed] [Google Scholar]
- 15.Furiya Y, Hirano M, Kurumatani N, Nakamuro T, Matsumura R, Futamura N, Ueno S. Alpha-1-antichymotrypsin gene polymorphism and susceptibility to multiple system atrophy (MSA). Brain Res Mol Brain Res. 2005;138:178–81. doi: 10.1016/j.molbrainres.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 16.Miele G, Seeger H, Marino D, Eberhard R, Heikenwalder M, Stoeck K, Basagni M, Knight R, Green A, Chianini F, et al.. Urinary alpha1-antichymotrypsin: a biomarker of prion infection. PLoS One. 2008;3:e3870. doi: 10.1371/journal.pone.0003870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guiroy DC, Wakayama I, Liberski PP, Gajdusek DC. Relationship of microglia and scrapie amyloid-immunoreactive plaques in kuru, Creutzfeldt-Jakob disease and Gerstmann-Straussler syndrome. Acta Neuropathol. 1994;87:526–30. doi: 10.1007/BF00294180. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki A, Hirato J, Nakazato Y. Immunohistochemical study of microglia in the Creutzfeldt-Jakob diseased brain. Acta Neuropathol. 1993;86:337–44. doi: 10.1007/BF00369445. [DOI] [PubMed] [Google Scholar]
- 19.Du L, Zhang Y, Chen Y, Zhu J, Yang Y, Zhang HL. Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol Neurobiol. 2016;1–18. [DOI] [PubMed] [Google Scholar]
- 20.Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annual Review of Physiology. 2017;79:619–43. [DOI] [PubMed] [Google Scholar]
- 21.Castell JV, Gomez-Lechon MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989;242:237–9. doi: 10.1016/0014-5793(89)80476-4. [DOI] [PubMed] [Google Scholar]
- 22.Baumann U, Huber R, Bode W, Grosse D, Lesjak M, Laurell CB. Crystal structure of cleaved human alpha 1-antichymotrypsin at 2.7 A resolution and its comparison with other serpins. J Mol Biol. 1991;218:595–606. doi: 10.1016/0022-2836(91)90704-A. [DOI] [PubMed] [Google Scholar]
- 23.Kordula T, Bugno M, Rydel RE, Travis J. Mechanism of interleukin-1- and tumor necrosis factor alpha-dependent regulation of the alpha 1-antichymotrypsin gene in human astrocytes. J Neurosci. 2000;20:7510–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Q, Xie WL, Zhang B, Chen LN, Xu Y, Wang K, Ren K, Zhang XM, Chen C, Zhang J, et al.. Brain microglia were activated in sporadic CJD but almost unchanged in fatal familial insomnia and G114V genetic CJD. Virol J. 2013;10:216. doi: 10.1186/1743-422X-10-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie WL, Shi Q, Zhang J, Zhang BY, Gong HS, Guo Y, Wang SB, Xu Y, Wang K, Chen C, et al.. Abnormal activation of microglia accompanied with disrupted CX3CR1/CX3CL1 pathway in the brains of the hamsters infected with scrapie agent 263K. J Mol Neurosci. 2013;51(3):919–32. doi: 10.1007/s12031-013-0002-z. [DOI] [PubMed] [Google Scholar]
- 26.Sun YX, Wright HT, Janciauskiene S. Glioma cell activation by Alzheimer's peptide Abeta1-42, alpha1-antichymotrypsin, and their mixture. Cell Mol Life Sci. 2002;59:1734–43. doi: 10.1007/PL00012501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strom A, Diecke S, Hunsmann G, Stuke AW. Identification of prion protein binding proteins by combined use of far-Western immunoblotting, two dimensional gel electrophoresis and mass spectrometry. Proteomics. 2006;6:26–34. doi: 10.1002/pmic.200500066. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, DeKosky ST, Ikonomovic MD, Kamboh MI. Distribution of plasma alpha 1-antichymotrypsin levels in Alzheimer disease patients and controls and their genetic controls. Neurobiol Aging. 2002;23:377–82. doi: 10.1016/S0197-4580(01)00322-0. [DOI] [PubMed] [Google Scholar]
- 29.DeKosky ST, Ikonomovic MD, Wang X, Farlow M, Wisniewski S, Lopez OL, Becker JT, Saxton J, Klunk WE, Sweet R, et al.. Plasma and cerebrospinal fluid alpha1-antichymotrypsin levels in Alzheimer's disease: correlation with cognitive impairment. Ann Neurol. 2003;53:81–90. doi: 10.1002/ana.10414. [DOI] [PubMed] [Google Scholar]
- 30.Sun YX, Minthon L, Wallmark A, Warkentin S, Blennow K, Janciauskiene S. Inflammatory markers in matched plasma and cerebrospinal fluid from patients with Alzheimer's disease. Dement Geriatr Cogn Disord. 2003;16:136–44. doi: 10.1159/000071001. [DOI] [PubMed] [Google Scholar]
- 31.Licastro F, Parnetti L, Morini MC, Davis LJ, Cucinotta D, Gaiti A, Senin U. Acute phase reactant alpha 1-antichymotrypsin is increased in cerebrospinal fluid and serum of patients with probable Alzheimer disease. Alzheimer Dis Assoc Disord. 1995;9:112–8. doi: 10.1097/00002093-199509020-00009. [DOI] [PubMed] [Google Scholar]
- 32.Padmanabhan J, Levy M, Dickson DW, Potter H. Alpha1-antichymotrypsin, an inflammatory protein overexpressed in Alzheimer's disease brain, induces tau phosphorylation in neurons. Brain. 2006;129:3020–34. doi: 10.1093/brain/awl255. [DOI] [PubMed] [Google Scholar]
- 33.Chen C, Shi Q, Zhang BY, Wang GR, Zhou W, Gao C, Tian C, Mei GY, Han YL, Han J, et al.. The prepared tau exon-specific antibodies revealed distinct profiles of tau in CSF of the patients with Creutzfeldt-Jakob disease. PLoS One. 2010;5:e11886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang GR, Gao C, Shi Q, Zhou W, Chen JM, Dong CF, Shi S, Wang X, Wei Y, Jiang HY, et al.. Elevated levels of tau protein in cerebrospinal fluid of patients with probable Creutzfeldt-Jakob disease. Am J Med Sci. 2010;340:291–5. doi: 10.1097/MAJ.0b013e3181e92a1f. [DOI] [PubMed] [Google Scholar]
- 35.Wang GR, Shi S, Gao C, Zhang BY, Tian C, Dong CF, Zhou RM, Li XL, Chen C, Han J, et al.. Changes of tau profiles in brains of the hamsters infected with scrapie strains 263 K or 139 A possibly associated with the alteration of phosphate kinases. BMC Infect Dis. 2010;10:86. doi: 10.1186/1471-2334-10-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen C, Zhou W, Lv Y, Shi Q, Wang J, Xiao K, Chen LN, Zhang BY, Dong XP. The Levels of Tau Isoforms Containing Exon-2 and Exon-10 Segments Increased in the Cerebrospinal Fluids of the Patients with Sporadic Creutzfeldt-Jakob Disease. Mol Neurobiol. 2016;53:3999–4009. doi: 10.1007/s12035-015-9348-2. [DOI] [PubMed] [Google Scholar]
- 37.Clarke MC, Haig DA. Multiplication of scrapie agent in cell culture. Res Vet Sci. 1970;11:500–1. [PubMed] [Google Scholar]
- 38.Birkett CR, Hennion RM, Bembridge DA, Clarke MC, Chree A, Bruce ME, Bostock CJ. Scrapie strains maintain biological phenotypes on propagation in a cell line in culture. EMBO J. 2001;20:3351–8. doi: 10.1093/emboj/20.13.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Q, Zhang BY, Gao C, Zhang J, Jiang HY, Chen C, et al.. Mouse-adapted scrapie strains 139A and ME7 overcome species barrier to induce experimental scrapie in hamsters and changed their pathogenic features. Virol J. 2012;9:63. doi: 10.1186/1743-422X-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.