

ABSTRACT
The variability of culturable bacterial diversity and distribution was studied by phylogenetic analysis of 16S rRNA sequences. Seventeen water samples were examined and were collected, from different depths in the range of 5 m to 2700 m at 3 sampling sites (CTD06, CTD10 and CTD11) in the South Atlantic Ocean. Phylogenetic analysis of 16S rRNA gene sequences revealed a significant diversity of culturable bacteria. A total of 247 strains clustered into 8 classes: γ-Proteobacteria, α-Proteobacteria, Actinobacteria, Actinomycetales, Bacilli, Flavobacteria, Opitutae and Sphingobacteria. The 17 water samples were dominated by populations of strains belonging to the genus Erythrobacter (16.60%). Of the 247 strains, 10 were potential new species and might form a minor population in the deep sea. To our knowledge, this is the first report to analyze the diversity of culturable bacteria in the South Atlantic Ocean from different depths across the water column.
KEYWORDS: 16S rRNA, bacterial diversity, deep sea, phylogenetic analysis, South Atlantic Ocean
Introduction
The oceans of the world are teeming with microscopic life forms. Nominal cell counts of >105 cells per ml in surface sea water predict that the oceans harbor 3.6 × 1029 microbial cells with a total cellular carbon content of approximately 3 × 1017 g.1 Communities of bacteria, archaea, protists and unicellular fungi account for most of the oceanic biomass. Given the enormous number of microbes and their vast metabolic diversity, the accumulation of mutations during the past 3.5 billion years should have led to very high levels of genetic and phenotypic variation.2
Marine bacteria can be divided into 2 groups depending on their response to nutrient supply. One group can respond quickly to changes in the quantity and type of nutrient input.3 These bacteria may be indigenous to the deep sea, or they may be surface-derived and settle to depths with sedimenting particulates.4 Bacteria in the second group are more specific in their nutrient requirements and slower to adapt to changes in nutrient input.5 This group may include microorganisms that can grow preferentially at high pressures and in an oligotrophic environment.
Due to the difficulties of exploring the deep sea, defining its diversity and the distribution of natural culturable microbial communities has been a long-standing challenge in the field of microbial ecology and evolution. Gene sequences encoding rRNAs provide a basis for estimating microbial phylogenetic diversity and generating taxonomic inventories of marine microbial populations.6,7 Evolutionary distances between orthologous sequences or similarities to database entries identified through BLAST, FASTA or Bayesian classifiers identify operational taxonomic units (OTUs) that correspond to species or kinds of organisms.8 A variety of parametric and nonparametric methods extrapolate information from observed frequencies of OTUs or species abundance curves to predict the number of different microbial taxa in a local sample.2,9 Richness estimates of marine microbial communities through comparisons of rRNAs range from a few hundred phylotypes per ml in the water column to as many as 3,000 from marine sediments.10 One of the largest water column surveys (1,000 PCR amplicons) described the presence of only 516 unique sequences and estimated the occurrence of approximately 1,600 coexisting ribotypes in a coastal bacterioplankton community.11
In the present study we investigated the microbial diversity of the sea, and especially deep-sea environments, by isolating and characterizing microorganisms from sea-water samples. We examined 17 sea-water samples obtained from the South Atlantic Ocean at different depths (5–2,700 m). Phylogenetic analysis of 16S rRNA sequences of 247 strains from water samples collected at different depths revealed a significant degree of microbial diversity. The results demonstrated that some bacteria were common to all sample sites, whereas other bacteria were uniquely detected at certain sites. To our knowledge, this is the first report to analyze the bacterial diversity of culturable bacteria in the South Atlantic Ocean from different depths of the water column.
Materials and methods
Study sites and sample collection
Sea-water samples from different depths (5–2,700 m) of the South Atlantic Ocean were collected from 3 sites: CTD06 (W13.3°/S15.2°), CTD10 (W13.3°/ S15.1°) and CTD11 (W13.4°/ S15.2°) during the No. 26 ocean Cruise by R/V Dayang 1 in 2012. The details of sampling sites and depths are presented in Table 1. Samples were collected from the water column of approximately 3000 m using SBE-911 plus CTD. Once retrieved, samples were immediately stored at −4°C and microorganisms were isolated in the on-board laboratory.
Table 1.
Locations of sampling stations, sample distribution, and strain number of different depths and isolation media.
| Station | Sampling location | Dive date | Depth (m) | Strain No. | 2216E medium | R2A medium |
|---|---|---|---|---|---|---|
| CTD06 | South Atlantic Ocean (W13.3°/S15.2°) | 01082012 | 5 | 12 | 7 | 5 |
| 50 | 17 | 10 | 7 | |||
| 100 | 12 | 6 | 6 | |||
| 125 | 17 | 9 | 8 | |||
| 150 | 18 | 9 | 9 | |||
| 200 | 9 | 5 | 4 | |||
| 500 | 17 | 7 | 10 | |||
| 1000 | 12 | 6 | 6 | |||
| 1500 | 11 | 6 | 5 | |||
| 2000 | 14 | 8 | 6 | |||
| 2500 | 13 | 7 | 6 | |||
| 2700 | 18 | 10 | 8 | |||
| CTD10 | South Atlantic Ocean (W13.3°/ S15.1°) | 04082012 | 2400 | 9 | 5 | 4 |
| 2500 | 9 | 5 | 4 | |||
| 2600 | 20 | 10 | 10 | |||
| 2700 | 19 | 13 | 6 | |||
| CTD11 | South Atlantic Ocean (W13.4°/ S15.2°) | 06082012 | 2700 m | 20 | 12 | 8 |
The microorganisms from sea-water samples were isolated using 2216E and R2A media. All plates were incubated at 20°C-22°C and bacterial strains were obtained across 3–7 d. Pure culture isolates were preserved for further research.
16SrRNA sequencing and phylogenetic analysis
Genomic DNA from the microorganisms was extracted using a bacterial genomic DNA FastPrep Extraction Kit (SaiBaiSheng Gene, Shanghai), according to the manufacturer's protocol.
Polymerase chain reaction (PCR) amplification of the nearly full-length 16S rRNA gene was performed using the universal primers 16SF (5′-AGAGTTTGATCCTGGCTCAG) and 16SR (5′-ACGGCTACCTTGTTACGACT). PCR was performed using the extracted highly purified genomic DNA as template under the following conditions: 94°C for 5 min, followed by 94°C for 40 s, 55°C for 40 s, and 72°C for 1 min for 32 cycles with a final 10 min extension at 72°C. The primer for PCR sequencing was P300 (5′-CCAGACTCCTACGGGAGGCAGC).
Similarity searches of all the sequences were performed in the GenBank database using BLASTn and the EzBiocloud database using Eztaxon server 2.1. The sequence alignments were performed using CLUSTAL_X, and the phylogenetic trees were constructed from evolutionary distances using the neighbor-joining method of the MEGA 5 program package.
Nucleotide sequence accession number
The partial and full 16S rRNA gene sequences of the strains were submitted to the NCBI GenBank database under the accession numbers KJ399591-KJ399837.
Results
Results of the isolated strains
Bacterial populations were successfully isolated from sea-water samples using 2216E and R2A media. The 3 study sites were CTD06, CTD10 and CTD11 from the South Atlantic Ocean. Numerous colonies of bacteria were observed on 2 kinds of media plates after incubation for 3 to 7 d. A total of 247 strains from the 17 water samples were obtained from different depths, as shown in Table 1. At station CTD06, 12 samples were collected from the water column of 5–2700 m, and 170 strains were obtained. At station CTD10, 4 samples were collected from the water column of 2400–2700 m, and 57 strains were isolated. The only water sample from 2700 m was collected at station CTD11, and the number of strains was 20. From all 17 water samples, 135 strains were obtained using 2216E medium, while 112 strains were obtained using R2A medium. For most of these samples, the number of strains isolated using 2216E medium was slightly greater than that obtained using R2A medium, but the difference was not significant.
Phylogenetic analysis of culturable strains by 16S rRNA gene sequence
A total of 247 strains from the 17 water samples obtained from different depths were analyzed based on their 16S rRNA gene sequence. The strains were classified into 8 classes: γ-Proteobacteria, α-Proteobacteria, Actinobacteria, Actinomycetales, Bacilli, Flavobacteria, Opitutae and Sphingobacteria.
The 17 water samples from different depths were dominated by populations of strains belonging to the genus Erythrobacter (16.60%). Members of the genus Erythrobacter were observed in all 3 study sites, and notably in all of the water samples examined from the water column of 5–2700 m at the CTD06 sample station. The 16S rRNA gene of these strains was 99.43% to 100% identical to that of Erythrobacter sp., E.citreus, E. flavus or E. pelagi.
The second most common group of strains identified in the present study was from the genus Alteromonas. In total, 29 strains were obtained from 9 water samples. These strains were 98.99% to 99.88% identical to Alteromonas sp, A. australica, A. marina, A. genovensis, A. macleodii or A. litorea. The next most common group of strains was identical to the genus Marinobacter. A total of 26 strains were obtained from 10 water samples. The strains were 98.86% to 99.86% identical to the Marinobacter sp, M. algicola, M. hydrocarbonoclasticus, M. lacisalsi or M. vinifirmus.
Diversity of culturable strains from different isolation medium
From all 17 water samples at different depths, 135 strains were obtained using 2216E medium. Analyzed by their 16S rRNA gene sequence, these strains were classified into 42 genera, as shown in Table 2. Among them, the strains belonging to 18 genera were only observed on 2216E medium, and the number of strains of each genus were very similar. Meanwhile, 112 strains were obtained by using R2A medium and classified into 29 genera. Among them, the strains belonging to 11 genera were observed only on R2A medium. For most of these 11 genera, there was only one strain belonging to the genus. In total, there were 24 genera obtained on both 2216E and R2A media. The most common group of strains belonging to the genus Erythrobacter, Alteromonas and Marinobacter were all observed on both kinds of isolation media.
Table 2.
Genera of the bacterial strains isolated by different media.
| Genus | Isolation medium | Genus | Isolation medium | Genus | Isolation medium |
|---|---|---|---|---|---|
| Echinicola | 2216E | Alcanivorax | Ma | Aestuariibacter | R2A |
| Gordonia | 2216E | Alteromonas | Ma | Aquimarina | R2A |
| Hyphomonas | 2216E | Brevibacterium | Ma | Aurantimonas | R2A |
| Idiomarina | 2216E | Citromicrobium | Ma | Bacillus | R2A |
| Litorisediminicola | 2216E | Coraliomargarita | Ma | Leeuwenhoekiella | R2A |
| Maricaulis | 2216E | Erythrobacter | Ma | Marinicauda | R2A |
| Martelella | 2216E | Gramella | Ma | Mycobacterium | R2A |
| Mesonia | 2216E | Halomonas | Ma | Ponticaulis | R2A |
| Micrococcus | 2216E | Henriciella | Ma | Roseivivax | R2A |
| Oceanicaulis | 2216E | Kangiella | Ma | Salinisphaera | R2A |
| Pelagibaca | 2216E | Marinobacter | Ma | Sinobacterium | R2A |
| Polycyclovorans | 2216E | Marivirga | Ma | Salegentibacter | Ma |
| Psychrobacter | 2216E | Microbacterium | Ma | Salinicola | Ma |
| Roseivirga | 2216E | Muricauda | Ma | Spongiibacter | Ma |
| Ruegeria | 2216E | Oceanicola | Ma | Stakelama | Ma |
| Stappia | 2216E | Pseudoalteromonas | Ma | Sulfitobacter | Ma |
| Thalassospira | 2216E | Roseovarius | Ma | Zunongwangia | Ma |
| Winogradskyella | 2216E | Sagittula | Ma |
Ma, Strains isolated from both 2216E and R2A media
Diversity of culturable strains from the whole water column at station CTD06
Bacteria strains were isolated from the whole water column of 5–2700 m at station CTD06. A total of 170 strains were obtained from all 12 water samples at different depths. For the purpose of analysis, the whole water column was divided into 3 layers: the upper layer (5 m∼100 m), lower layer (125 m∼1000 m) and deeper layer (1500 m∼2700 m).
The phylogenetic tree in Fig. 1 shows the diversity of strains isolated from water samples of the upper layer at station CTD06. These 41 strains clustered into 4 major lineages of bacteria: γ-Proteobacteria, α-Proteobacteria, Flavobacteria and Sphingobacteria. In total, these strains belonged to 15 genera, and the most common groups found in the upper layer were from the genera Erythrobacter (21.95%) and Alteromonas (21.95%). The strain M14 clustered within the Sphingobacteria group. Eztaxon searches revealed that M14 showed 95.97% homology with Marivirga sericea (AB078081), and is a potential new species. M6 and M7 clustered within the Flavobacteria group, and showed 99.87% homology with Salegentibacter mishustinae (AY576653) and 99.42% homology with Mesonia mobilis (DQ367409), respectively.
Figure 1.

Neighbor-joining tree based on the 16S rRNA sequences of strains from the water samples of the upper layer at station CTD06. The numbers on the tree indicate the bootstrap percentage based on 1,000 replications and are shown for branches with more than 50% support. The scale bar represents 0.05 nucleotide substitutions per sequence position.
The phylogenetic tree in Fig. 2 shows the diversity of strains isolated from the water samples of the lower layer at station CTD06. These 73 strains clustered into 6 major lineages of bacteria: γ-Proteobacteria, α-Proteobacteria, Actinobacteria, Actinomycetales, Flavobacteria and Sphingobacteria. In all, these strains belonged to 34 genera, and the most common group of strains found in the lower layer was the genus Erythrobacter (21.92%). These strains were 99.44% to 100% identical to Erythrobacter citreus, E. flavus or E. pelagi.
Figure 2.

Neighbor-joining tree based on the 16S rRNA sequences of strains from the water samples of the lower layer at station CTD06. The numbers on the tree indicate the bootstrap percentage based on 1,000 replications and are shown for branches with more than 50% support. The scale bar represents 0.05 nucleotide substitutions per sequence position.
The strains M36, M42 and M43 clustered within the Actinobacteria group. Eztaxon searches revealed that strain M36 showed 99.35% homology with Gordonia bronchialis DSM 43247T (CP001802). M42 was identified as having 99.72% homology with Micrococcus yunnanensis YIM 65004T (FJ214355), while M43 had 99.74% homology with Micrococcus cohnii WS4601T (FR832424). R29 clustered within the Actinomycetales group and was identified as having 100% homology with Mycobacterium poriferae ATCC 35087T (AF480589).
The phylogenetic tree in Fig. 3 shows the diversity of strains isolated from the water samples of the deeper layer at station CTD06. These 56 strains clustered into 6 major lineages of bacteria: γ-Proteobacteria, α-Proteobacteria, Opitutae, Actinomycetales, Flavobacteria and Sphingobacteria. In all, these strains belonged to 20 genera and the most common group of strains found in the deeper layer was from the genus Erythrobacter (16.07%). These strains were 99.44% to 100% identical to Erythrobacter citreus, E. flavus or E. pelagi.
Figure 3.

Neighbor-joining tree based on the 16S rRNA sequences of strains from the water samples of the deeper layer at station CTD06. The numbers on the tree indicate the bootstrap percentage based on 1,000 replications and are shown for branches with more than 50% support. The scale bar represents 0.05 nucleotide substitutions per sequence position.
Strains M63 and R64 clustered within the Opitutae group. Eztaxon searches revealed that strain M63 and R64 were 96.23% to 96.28% identical to Coraliomargarita akajimensis DSM 45221T (CP001998), and are potential new species. Strain M63 and R64 were isolated only from the water sample obtained at a depth of 1500 m at station CTD06. R71 clustered within the Sphingobacteria group and was identified as having 99.87% homology with Leeuwenhoekiella blandensis MED217T (AANC01000011).
In all, a total of 170 strains were obtained from the whole water column at station CTD06, and these strains belonged to 49 genera. Among them, the genera Alcanivorax, Alteromonas, Citromicrobium, Erythrobacter, Oceanicola and Salegentibacter were observed in samples from all 3 levels of the water column, as shown in Table 3. The genera Aurantimonas, Mesonia, Oceanicaulis, and Polycyclovorans were isolated only from the upper level of the water column. Meanwhile, the genera Brevibacterium, Coraliomargarita, Hyphomonas, Idiomarina, Maricaulis, Marinicauda, Muricauda and Pseudoalteromonas were observed only in samples from the deeper level.
Table 3.
Genera of the bacterial strains isolated from the water column at station CTD06.
| Genus | Water level | Genus | Water level | Genus | Water level |
|---|---|---|---|---|---|
| Aestuariibacter | Lb | Litorisediminicola | Ua, Lb | Roseivirga | Lb |
| Alcanivorax | Ua, Lb, Dc | Maricaulis | Dc | Roseovarius | Ua, Lb |
| Alteromonas | Ua, Lb, Dc | Marinicauda | Dc | Ruegeria | Lb |
| Aquimarina | Lb | Marinobacter | Lb, Dc | Sagittula | Lb |
| Aurantimonas | Ua | Marivirga | Ua, Lb | Salegentibacter | Ua, Lb, Dc |
| Brevibacterium | Dc | Martelella | Lb | Salinicola | Lb |
| Citromicrobium | Ua, Lb, Dc | Mesonia | Ua | Salinisphaera | Lb |
| Coraliomargarita | Dc | Microbacterium | Lb | Sinobacterium | Lb |
| Echinicola | Lb | Micrococcus | Lb | Spongiibacter | Lb |
| Erythrobacter | Ua, Lb, Dc | Muricauda | Dc | Stakelama | Lb |
| Gordonia | Lb | Mycobacterium | Lb | Stappia | Lb |
| Gramella | Lb, Dc | Oceanicaulis | Ua | Sulfitobacter | Ua, Dc |
| Halomonas | Ua, Dc | Oceanicola | Ua, Lb, Dc | Thalassospira | Lb |
| Henriciella | Lb | Polycyclovorans | Ua | Winogradskyella | Lb |
| Hyphomonas | Dc | Ponticaulis | Lb | Zunongwangia | Lb |
| Idiomarina | Dc | Pseudoalteromonas | Dc | ||
| Leeuwenhoekiella | Lb, Dc | Psychrobacter | Dc |
U, Upper level of the water column at the CTD06 station
L, Lower level of the water column at the CTD06 station
D, Deeper level of the water column at the CTD06 station
Diversity of culturable strains from the deeper water layer at the CTD10 station
The phylogenetic tree in Fig. 4 shows the diversity of strains isolated from the 4 water samples from 2400 m - 2700 m at station CTD10. These 57 strains clustered into 5 major lineages of bacteria: γ-Proteobacteria, α-Proteobacteria, Actinomycetales, Flavobacteria and Sphingobacteria. The most common group of strains found in the 4 water samples of 2400 m - 2700 m at station CTD10 was from the genus Marinobacter (24.56%). The strains were 99.29% to 99.75% identical to Marinobacter algicola.
Figure 4.
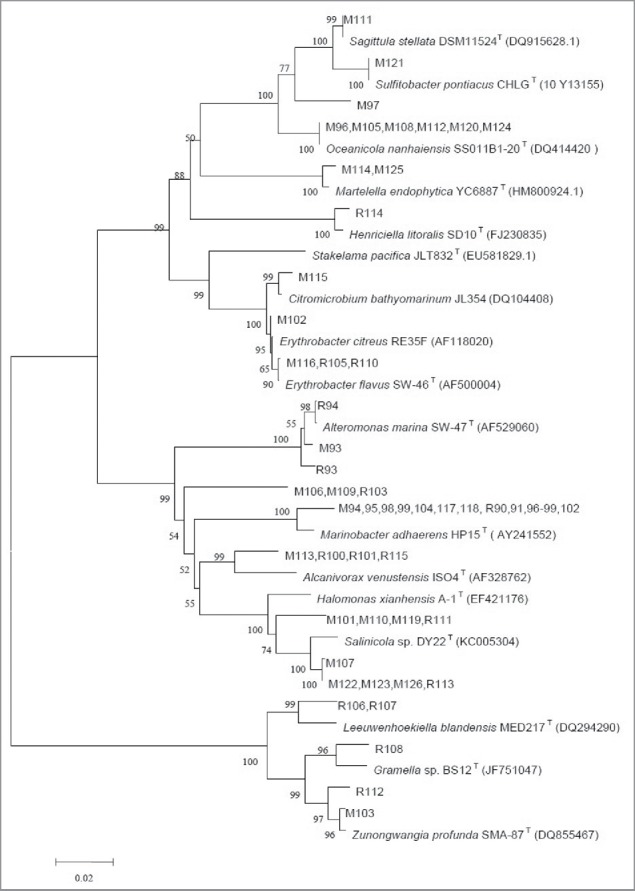
Neighbor-joining tree based on the 16S rRNA sequences of strains from the water samples at station CTD10. The numbers on the tree indicate the bootstrap percentage based on 1,000 replications and are shown for branches with more than 50% support. The scale bar represents 0.02 nucleotide substitutions per sequence position.
Strains R108, R112 and M103 clustered within the Flavobacteria group. M103 and R112 showed 99.35% homology with Zunongwangia profunda SM-A87T (CP001650) and 99.74% homology with Zunongwangia mangrove P2E16T (KC907713), respectively, while R108 had 100% homology with Gramella flava JLT2011T (JX397931). R106 and R107 clustered within the Sphingobacteria group and were identified as having 95.64% homology with Leeuwenhoekiella blandensis MED217T (AANC01000011), and are potential new species.
Diversity of culturable strains from the deeper water layer at the CTD11 station
The phylogenetic tree in Fig. 5 shows the diversity of strains isolated from the water samples from 2700 m at station CTD11. These 20 strains clustered into 3 major lineages of bacteria: γ-Proteobacteria, α-Proteobacteria and Bacilli. The most common group of strains found in the water sample of 2700 m at CTD11 station was from the genus Alteromonas (35.00%). The strains were 99.08% ∼99.88% identical to Alteromonas marina, A. litorea or A. macleodii. The strain R120 clustered within the Bacilli group. Eztaxon searches revealed that strain R120 was 100% identical to Bacillus safensis FO-036bT (AF234854). Out of all the water samples, R120 was isolated only from the water sample obtained at a depth of 2700 m at station CTD11.
Figure 5.

Neighbor-joining tree based on the 16S rRNA sequences of strains from the water samples at station CTD11. The numbers on the tree indicate the bootstrap percentage based on 1,000 replications and are shown for branches with more than 50% support. The scale bar represents 0.02 nucleotide substitutions per sequence position.
Diversity of culturable bacteria isolated from the same depth among different stations
A total of 22 strains were obtained from water samples at a depth of 2500 m at both the CTD06 and CTD10 stations. Among the strains, 13 were isolated at CTD06, while 9 were obtained from water samples at CTD10. These 22 strains clustered into 3 major lineages of bacteria: γ-Proteobacteria, α-Proteobacteria and Flavobacteria.
Eztaxon searches revealed that the 13 strains at CTD06 were identical to 8 genera: Alcanivorax, Alteromonas, Erythrobacter, Halomonas, Hyphomonas, Maricaulis, Muricauda and Salegentibacter. Among them the most common groups were from the genera Halomonas (23.08%) and Muricauda (23.08%). The 9 strains at CTD10 were identical to 4 genera: Erythrobacter, Halomonas, Marinobacter and Zunongwangia. Among these 9 strains, the most common were from the genus Marinobacter (66.67%) and showed 99.43–99.75% homology with Marinobacter algicola DG893T (ABCP01000031).
Only 2 genera of Erythrobacter and Halomonas were observed at depth 2500 m at both the CTD06 and CTD10 stations, and the 16S rRNA gene sequence similarities between these strains are shown in Table 4. M76 and M102 were isolated from the CTD06 and CTD10 stations, respectively. They were identical or very similar to the genus Erythrobacter (99.7%). M75, M80, R80 and M101 were all identical to the genus Halomonas. Among them, only M101 was isolated from station CTD10. These 4 strains were quite similar to each other (99.8% similarity).
Table 4.
Similarity matrix comparing 16S rRNA sequences of strains observed at both CTD06 and CTD10 at a depth of 2500 m.
A total of 55 strains were obtained from water samples at a depth of 2700 m among CTD06, CTD10 and CTD11. Among them, 16 strains were isolated at CTD06, 19 strains were isolated at CTD10, and 20 strains were obtained at CTD11. These 55 strains clustered into 4 major lineages of bacteria: γ-Proteobacteria, α-Proteobacteria, Bacilli and Flavobacteria.
Eztaxon searches revealed that 16 strains at CTD06 were identical to 10 genera: Alcanivorax, Alteromonas, Citromicrobium, Erythrobacter, Gramella, Maricaulis, Marinicauda, Marinobacter, Oceanicola and Psychrobacter. There were similar numbers of strains from each genus. The largest strain number from a single genus was 3, and these strains were identical to the genus Erythrobacter. The 19 strains at CTD10 were identical to 11 genera: Alcanivorax, Citromicrobium, Erythrobacter, Halomonas, Henriciella, Marinobacter, Martelella, Oceanicola, Salinicola, Sulfitobacter and Zunongwangia. Among the 19 strains, the largest number of strains from a single genus was 4; these strains were identical to genus Salinicola and showed 99.59–99.61% homology with Salinicola salaries M27T (AM229316). The number of strains from other genera were also very similar. The 20 strains at CTD11 were identical to 8 genera: Alteromonas, Bacillus, Erythrobacter, Marinobacter, Martelella, Oceanicola, Salinicola and Sulfitobacter. Among the 20 strains, the largest number of strains from a single genus was 7, sharing identity with the genus Alteromonas.
The genus Erythrobacter, Marinobacter and Oceanicola were all observed at stations CTD06, CTD10 and CTD11. Among them, the number of strains from genus Erythrobacter was 8, and these strains showed 99.73–100% homology with Erythrobacter flavus SW-46T (AF500004). The 16S rRNA gene sequence similarities between these strains are shown in Table 5. Among them, M90 and R82 were isolated from CTD06. These 2 strains are quite similar to each other (100% similarity) and may represent the same strain. However, they are more distantly related to the strains from other sample stations (95.5–95.6% similarity). M117 and M118 were both observed at CTD10 and are quite similar to each other (99.9%). M137, R116, R117 and R121 were all isolated from CTD11. These 4 strains are quite similar to each other (99.8–100% similarity). The number of strains from genus Oceanicola was 6, and these strains all showed 100% homology with Oceanicola nanhaiensis SS011B1–20T (DQ414420).
Table 5.
Similarity matrix comparing 16S rRNA sequences of the genus Marinobacter found at CTD06, CTD10 and CTD11 at a depth of 2700 m.
| % Similarity |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strains | M.a.Da | M90 | R82 | M117 | M118 | M137 | R116 | R117 | R121 |
| M90 | 95.8 | 100 | |||||||
| R82 | 95.8 | 100 | 100 | ||||||
| M117 | 99.5 | 95.6 | 95.6 | 100 | |||||
| M118 | 99.6 | 95.5 | 95.5 | 99.9 | 100 | ||||
| M137 | 99.8 | 95.6 | 95.6 | 99.8 | 99.9 | 100 | |||
| R116 | 99.5 | 95.6 | 95.6 | 100 | 99.9 | 99.8 | 100 | ||
| R117 | 99.8 | 95.6 | 95.6 | 99.8 | 99.9 | 100 | 99.8 | 100 | |
| R121 | 99.5 | 95.6 | 95.6 | 100 | 99.9 | 99.8 | 100 | 99.8 | 100 |
M.a.D, Marinobacter algicola DG893 (AY258110)
Discussion
The deep sea is the largest ecosystem and supports some of the highest levels of diversity on Earth.12,13 Because of exploration difficulties, some environments remain under studied and poorly understood. The aphotic zone of the water column (>500 m depth) is in permanent darkness and thus contains an allochtonous ecosystem which is mainly driven by the deposition of organic matter from the euphotic zone. Deep-sea bacteria play a significant role in organic matter recycling and are responsible for approximately half of the global net mineralization of organic matter in marine ecosystems.14,15 Hydrostatic pressure in the marine water column increases by approximately 0.1 MPa per 10 m of depth, such that microorganisms that can function in the deep ocean must do so at pressures considerably higher than those at the surface.16 Thus, there must be a large number of diverse microorganisms adapting to the increasing pressure at different depths. In the present study, we examined 17 sea-water samples obtained from the South Atlantic Ocean at different depths (5–2,700 m). Phylogenetic analysis of 16S rRNA sequences from water samples collected at different depths revealed a significant degree of microbial diversity.
A total of 247 different 16S rRNA gene sequences analyzed clustered into 8 classes: γ-Proteobacteria, α-Proteobacteria, Actinobacteria, Actinomycetales, Bacilli, Flavobacteria, Opitutae and Sphingobacteria. The 17 water samples were dominated by populations of strains belonging to the class γ-Proteobacteria (44.53%). The dominance of γ-Proteobacteria sequences is commonly reported, and the same result has been found at many different locations, such as the Mid-Atlantic Ridge,17 the North Atlantic,18 the South China Sea,19,20 the Pacific nodule province 21 and the Japan Trench.22,23 Furthermore, their abundance was shown to increase with depth in the North Atlantic,18 and our investigation obtained the same results at CTD 06 and CTD 10.
However, the most dominant genus among all of the strains was Erythrobacter (16.60%), which is a member of the α-Proteobacteria. Strains identical to the genus Erythrobacter were observed in all the 3 sampling sites, and notably in all samples across the whole water column of 5–2700 m at CTD06. These strains were most closely related to Erythrobacter citreus from the western Mediterranean Sea,24 E. flavus from the East Sea in Korea 25 and E. pelagi from the Red Sea.26 The second most common group of strains was identical to the genus Alteromonas. In total, 29 of such strains were obtained from 9 water samples. These strains were very close to A. australica from the Tasman Sea,27 A. marina from the East Sea in Korea,28 A. genovensis from a marine electroactive biofilm in the port of Genoa, Italy,29 A. macleodii in the Mediterranean Sea 30 and A. litorea from an intertidal sediment of the Yellow Sea in Korea.31
R120, obtained from CTD11 at a depth of 2700 m, was the only strain belonging to the class of Bacilli, and showed 100% homology with Bacillus safensis FO-036bT (AF234854). B. safensis was first identified in 2006 as a contaminant from spacecraft-assembly facilities (SAF) in the USA, from which it derived its specific epithet safensis.32 B. safensis are salt-tolerating, plant-growth- promoting rhizobacteria.33,34 The production of industrially important enzymes such as endoinulinase,35 β-galactosidase36 and lipases37 by some strains of B. safensis have been reported. However, there are few reports of B. safensis strains isolated from deep-sea water.
The 2 strains M63 and R64 from CTD06 at 1500 m clustered within the Opitutae group and were very closely related to Coraliomargarita akajimensis, which is an obligately aerobic, gram-negative, non-spore-forming, non-motile, spherical bacterium from seawater in Japan.38 R106 and R107 isolated from CTD10 at 2600 m clustered within the Sphingobacteria group and were identified as having 95.64% homology with Leeuwenhoekiella blandensis MED217T (AANC01000011), which is a heterotrophic, rod-shaped, aerobic, yellow-pigmented and gliding bacterium from the north-western Mediterranean Sea.39 These strains might be a minor population in the deep sea and are potential new species.
An analysis of the difference in strains between the sampling sites at depths of 2500 m and 2700 m was also performed. The results demonstrated that the deep-sea bacteria diversity of the 3 sampling sites was not significantly different. Strains belonging to the same genus at all 3 sampling sites were quite similar to each other.
In the present study, the diversity and distribution of culturable sea-water bacteria from the South Atlantic Ocean was assessed by collecting strains from different sampling sites. The results revealed a significant degree of microbial diversity and demonstrated that some bacteria were common to all sample sites, whereas other bacteria were unique to specific samples. For example, the genera Erythrobacter, Marinobacter and Oceanicola were all observed at stations CTD06, CTD10 and CTD11. Meanwhile, the strains belonging to the genera Maricaulis, Marinicauda and Psychrobacter were observed only at the CTD06 station. To our knowledge this is the first report to analyze the diversity of culturable bacteria in the South Atlantic Ocean from different depths across the water column. Further efforts to quantify and track these bacteria as well as to identify their physiologic characteristics will be necessary to fully understand the complex environment of the deep sea in the South Atlantic Ocean.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Supported by COMRA Project (Grand No. DY125–15-R-01 and DY135–15-R-01), Foundation for Outstanding Young Scientist in Shandong Province (Grand No. BS2014NY012), China Postdoctoral Science Foundation (Grand No. 2015M581456) and Guidance Funds for Discipline Construction (Grand No. WH20140202).
References
- [1].Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci 1998; 95(12):6578-83; PMID:9618454; http://dx.doi.org/ 10.1073/pnas.95.12.6578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci 2006; 103(32):12115-20; PMID:16880384; http://dx.doi.org/ 10.1073/pnas.0605127103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Church MJ. The trophic tapestry of the sea. Proc Natl Acad Sci 2009; 106(37):15519-20; PMID:19805206; http://dx.doi.org/ 10.1073/pnas.0908881106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Grossart HP, Gust G. Hydrostatic pressure affects physiology and community structure of marine bacteria during settling to 4000 m: an experimental approach. Mar Ecol Prog Ser 2009; 390:97-104; http://dx.doi.org/ 10.3354/meps08201 [DOI] [Google Scholar]
- [5].Langenheder S, Lindström ES, Tranvik LJ. Weak coupling between community composition and functioning of aquatic bacteria. Limnol Oceanogr 2005; 50(3):957-67. [Google Scholar]
- [6].Knittel K, Lösekann T, Boetius A, Kort R, Amann R. Diversity and distribution of methanotrophic archaea at cold seeps. Appl Environ Microbiol 2005; 71(1):467-79; PMID:15640223; http://dx.doi.org/ 10.1128/AEM.71.1.467-479.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol 2003; 57(1):369-94; PMID:14527284; http://dx.doi.org/ 10.1146/annurev.micro.57.030502.090759 [DOI] [PubMed] [Google Scholar]
- [8].Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 2005; 71(3):1501-6; PMID:15746353; http://dx.doi.org/ 10.1128/AEM.71.3.1501-1506.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Curtis TP, Sloan WT, Scannell JW. Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci 2002; 99(16):10494-9; PMID:12097644; http://dx.doi.org/ 10.1073/pnas.142680199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kemp PF, Aller JY. Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol Ecol 2004; 47(2):161-77; PMID:19712332; http://dx.doi.org/ 10.1016/S0168-6496(03)00257-5 [DOI] [PubMed] [Google Scholar]
- [11].Acinas SG, Klepac-Ceraj V, Hunt DE, Pharino C, Ceraj I, Distel DL, Polz MF. Fine-scale phylogenetic architecture of a complex bacterial community. Nature 2004; 430(6999):551-4; PMID:15282603; http://dx.doi.org/ 10.1038/nature02649 [DOI] [PubMed] [Google Scholar]
- [12].Danovaro R, Company JB, Corinaldesi C, D'Onghia Galil B, Gambi C, Gooday AJ, Lampadariou N, Luna GM, Morigi C, Olu K, Polymenakou P, Ramirez-Llodra E, Sabbatini A, Sard F, Sibuet M, Tselepides A. Deep-sea biodiversity in the Mediterranean Sea: the known, the unknown, and the unknowable. PLoS ONE 2010; 5(8):e11832; PMID:20689848; http://dx.doi.org/ 10.1371/journal.pone.0011832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ramirez-Llodra E, Brandt A, Danovaro R, et al.. Deep, diverse and definitely different: unique attributes of the world's largest ecosystem. Biogeosciences 2010; 7(9):2851-99; http://dx.doi.org/ 10.5194/bg-7-2851-2010 [DOI] [Google Scholar]
- [14].Yokokawa T, Nagata T. Linking bacterial community structure to carbon fluxes in marine environments. J Oceanogr 2010; 66(1):1-12; http://dx.doi.org/ 10.1007/s10872-010-0001-4 [DOI] [Google Scholar]
- [15].Ogawa H, Amagai Y, Koike I, Kaiser K, Benner R. Production of refractory dissolved organic matter by bacteria. Science 2001; 292(5518):917-20; PMID:11340202; http://dx.doi.org/ 10.1126/science.1057627 [DOI] [PubMed] [Google Scholar]
- [16].Egan ST, McCarthy DM, Patching JW, et al.. An investigation of the physiology and potential role of components of the deep ocean bacterial community (of the NE Atlantic) by enrichments carried out under minimal environmental change. Deep Sea Research Part I: Oceanographic Research Papers 2012; 61:11-20; http://dx.doi.org/ 10.1016/j.dsr.2011.11.005 [DOI] [Google Scholar]
- [17].Nercessian O, Fouquet Y, Pierre C, Prieur D, Jeanthon C. Diversity of Bacteria and Archaea associated with a carbonate rich metalliferous sediment sample from the Rainbow vent field on the Mid Atlantic Ridge. Environ Microbiol 2005; 7(5):698-714; PMID:15819852; http://dx.doi.org/ 10.1111/j.1462-2920.2005.00744.x [DOI] [PubMed] [Google Scholar]
- [18].Agogué H, Lamy D, Neal P R, Sogin ML, Herndl GJ. Water mass specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol Ecol 2011; 20(2):258-74; PMID:21143328; http://dx.doi.org/ 10.1111/j.1365-294X.2010.04932.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lai X, Zeng X, Fang S, et al.. Denaturing gradient gel electrophoresis (DGGE) analysis of bacterial community composition in deep-sea sediments of the South China Sea. World J Microb Biot 2006; 22(12):1337-45; http://dx.doi.org/ 10.1007/s11274-006-9181-x [DOI] [Google Scholar]
- [20].Jiang H, Dong H, Ji S, et al.. Microbial diversity in the deep marine sediments from the Qiongdongnan Basin in South China Sea. Geomicrobiol J 2007; 24(6):505-17; http://dx.doi.org/ 10.1080/01490450701572473 [DOI] [Google Scholar]
- [21].Xu M, Wang P, Wang F, et al.. Microbial diversity at a deep-sea station of the Pacific nodule province. Biodivers Conserv 2005; 14(14):3363-80; http://dx.doi.org/ 10.1007/s10531-004-0544-z [DOI] [Google Scholar]
- [22].Li L, Kato C, Horikoshi K. Microbial diversity in sediments collected from the deepest cold-seep area, the Japan Trench. Mar Biotechnol 1999; 1(4):391-400; PMID:10489418; http://dx.doi.org/ 10.1007/PL00011793 [DOI] [PubMed] [Google Scholar]
- [23].Li L, Kato C, Horikoshi K. Bacterial diversity in deep-sea sediments from different depths. Biodivers Conserv 1999; 8(5):659-77; http://dx.doi.org/ 10.1023/A:1008848203739 [DOI] [Google Scholar]
- [24].Denner EBM, Vybiral D, Koblízek M, Kämpfer P, Busse HJ, Velimirov B. Erythrobacter citreus sp. nov., a yellow-pigmented bacterium that lacks bacteriochlorophyll a, isolated from the western Mediterranean Sea. Int J Syst Evol Microbiol 2002; 52(5):1655-61; PMID:12361270; http://dx.doi.org/ 10.1099/00207713-52-5-1655 [DOI] [PubMed] [Google Scholar]
- [25].Yoon JH, Kim H, Kim IG, Kang KH, Park YH. Erythrobacter flavus sp. nov., a slight halophile from the East Sea in Korea. Int J Syst Evol Microbiol 2003; 53(4):1169-74; PMID:12892146; http://dx.doi.org/ 10.1099/ijs.0.02510-0 [DOI] [PubMed] [Google Scholar]
- [26].Wu H, Lai PY, Lee OO, Zhou XJ, Miao L, Wang H, Qian PY. Erythrobacter pelagi sp. nov., a member of the family Erythrobacteraceae isolated from the Red Sea. Int J Syst Evol Microbiol 2012; 62(Pt 6):1348-53; PMID:21828015; http://dx.doi.org/ 10.1099/ijs.0.029561-0 [DOI] [PubMed] [Google Scholar]
- [27].Ivanova EP, Ng HJ, Webb HK, Kurilenko VV, Zhukova NV, Mikhailov VV, Ponamoreva ON, Crawford RJ. Alteromonas australica sp. nov., isolated from the Tasman Sea. Antonie Van Leeuwenhoek 2013; 103(4):877-84; PMID:23291832; http://dx.doi.org/ 10.1007/s10482-012-9869-x [DOI] [PubMed] [Google Scholar]
- [28].Yoon JH, Kim IG, Kang KH, Oh TK, Park YH. Alteromonas marina sp. nov., isolated from sea water of the East Sea in Korea. Int J Syst Evol Microbiol 2003; 53(5):1625-30; PMID:13130060; http://dx.doi.org/ 10.1099/ijs.0.02536-0 [DOI] [PubMed] [Google Scholar]
- [29].Vandecandelaere I, Nercessian O, Segaert E, Achouak W, Mollica A, Faimali M, De Vos P, Vandamme P. Alteromonas genovensis sp. nov., isolated from a marine electroactive biofilm and emended description of Alteromonas macleodii Baumann et al. 1972 (Approved Lists 1980). Int J Syst Evol Microbiol 2008; 58(11):2589-96; PMID:18984698; http://dx.doi.org/ 10.1099/ijs.0.65691-0 [DOI] [PubMed] [Google Scholar]
- [30].López‐López A, Bartual SG, Stal L, Onyshchenko O, Rodríguez-Valera F. Genetic analysis of housekeeping genes reveals a deep-sea ecotype of Alteromonas macleodii in the Mediterranean Sea. Environ Microbiol 2005; 7(5):649-59; PMID:15819847; http://dx.doi.org/ 10.1111/j.1462-2920.2005.00733.x [DOI] [PubMed] [Google Scholar]
- [31].Yoon JH, Yeo SH, Oh TK, Park YH. Alteromonas litorea sp. nov., a slightly halophilic bacterium isolated from an intertidal sediment of the Yellow Sea in Korea. Int J Syst Evol Microbiol 2004; 54(4):1197-201; PMID:15280291; http://dx.doi.org/ 10.1099/ijs.0.63079-0 [DOI] [PubMed] [Google Scholar]
- [32].Satomi M, La Duc MT, Venkateswaran K. Bacillus safensis sp. nov., isolated from spacecraft and assembly-facility surfaces. Int J Syst Evol Microbiol 2006; 56(8):1735-40; PMID:16902000; http://dx.doi.org/ 10.1099/ijs.0.64189-0 [DOI] [PubMed] [Google Scholar]
- [33].Chakraborty U, Chakraborty B N, Chakraborty A P, Dey PL. Water stress amelioration and plant growth promotion in wheat plants by osmotic stress tolerant bacteria. World J Microbiol Biotechnol 2013; 29(5):789-803; PMID:23239372; http://dx.doi.org/ 10.1007/s11274-012-1234-8 [DOI] [PubMed] [Google Scholar]
- [34].Kothari VV, Kothari RK, Kothari CR, Bhatt VD, Nathani NM, Koringa PG, Joshi CG, Vyas BR. Genome sequence of salt-tolerant Bacillus safensis strain VK, isolated from saline desert area of Gujarat, India. Genome Announc 2013; 1(5):e00671-13; PMID:24009116; http://dx.doi.org/ 10.1128/genomeA.00671-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Singh RS, Singh RP, Yadav M. Molecular and biochemical characterization of a new endoinulinase producing bacterial strain of Bacillus safensis AS-08. Biologia 2013; 68(6):1028-33; http://dx.doi.org/ 10.2478/s11756-013-0259-2 [DOI] [Google Scholar]
- [36].Nath A, Chakrabarty S, Sarkar S, et al.. Purification and characterization of β-galactosidase synthesized from Bacillus safensis (JUCHE 1). Ind Eng Che Res 2013; 52(33):11663-72; http://dx.doi.org/ 10.1021/ie4008584 [DOI] [Google Scholar]
- [37].Kumar D, Parshad R, Gupta VK. Application of a statistically enhanced, novel, organic solvent stable lipase from Bacillus safensis DVL-43. Int J Biol Macromol 2014; 66:97-107; PMID:24534493; http://dx.doi.org/ 10.1016/j.ijbiomac.2014.02.015 [DOI] [PubMed] [Google Scholar]
- [38].Yoon J, Yasumoto-Hirose M, Katsuta A, Sekiguchi H, Matsuda S, Kasai H, Yokota A. Coraliomargarita akajimensis gen. nov., sp. nov., a novel member of the phylum ‘Verrucomicrobia’isolated from seawater in Japan. Int J Syst Evol Microbiol 2007; 57(5):959-63; PMID:17473241; http://dx.doi.org/ 10.1099/ijs.0.64755-0 [DOI] [PubMed] [Google Scholar]
- [39].Pinhassi J, Bowman JP, Nedashkovskaya OI, Lekunberri I, Gomez-Consarnau L, Pedrós-Alió C. Leeuwenhoekiella blandensis sp. nov., a genome-sequenced marine member of the family Flavobacteriaceae. Int J Syst Evol Microbiol 2006; 56(7):1489-93; PMID:16825617; http://dx.doi.org/ 10.1099/ijs.0.64232-0 [DOI] [PubMed] [Google Scholar]