

ABSTRACT
The aberrant alterations of calmodulin (CaM) and its downstream substrates have been reported in some neurodegenerative diseases, but rarely described in prion disease. In this study, the potential changes of Ca2+/CaM and its associated agents in the brains of scrapie agent 263K-infected hamsters and the prion infected cell line SMB-S15 were evaluated by various methodologies. We found that the level of CaM in the brains of 263K-infected hamsters started to increase at early stage and maintained at high level till terminal stage. The increased CaM mainly accumulated in the regions of cortex, thalamus and cerebellum of 263K-infected hamsters and well localization of CaM with NeuN positive cells. However, the related kinases such as total and phosphorylated forms of CaMKII and CaMKIV, as well as the downstream proteins such as CREB and BDNF in the brain of 263K-infected hamsters were decreased. Further analysis showed a remarkable increase of S-nitrosylated (SNO) form of CaM in the brains of 263K-infected hamsters. Dynamic analysis of S-nitrosylated CaM showed the SNO form of CaM abnormally increases in a time-dependent manner during prion infection. Compared with that of the normal partner cell line SMB-PS, the CaM level in SMB-S15 cells was increased, meanwhile, the downstream proteins, such as CaMKII, p-CaMKII, CREB, as well as BDNF, were also increased, especially in the nucleic fraction. No SNO-CaM was detected in the cell lines SMB-S15 and SMB-PS. Our data indicate an aberrant increase of CaM during prion infection in vivo and in vitro.
KEYWORDS: calmodulin, prion infection, S-nitrosylation, CaMKII, CaMKIV
INTRODUCTION
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are a unique family of invariably lethal chronic neurodegenerative diseases that affect humans (Creutzfeldt-Jakob disease, CJD; Gerstmann-Sträussler-Scheinker disease, GSS; fatal familial insomnia, FFI; Kuru), and animals such as cattle (bovine spongiform encephalopathy, BSE or ‘mad cow disease’), goat and sheep (scrapie), deer, elk and moose (chronic wasting disease, CWD).1 Human prion diseases can be infectious (acquired), inherited (genetic), or sporadic. The etiological agent is prion, whose propagation is believed to be conformational conversion from the cellular prion protein (PrPC) to the pathological form (PrPSc).2 The neuropathological hallmarks of prion diseases are deposits of PrPSc, spongiform degeneration, neuron loss and reactive astrogliosis in the central nerve system (CNS).3
Calmodulin (CaM) is a ubiquitous calcium-binding protein that can bind up to four calcium ions and functions as a major transducer of calcium signals.4 Binding of Ca2+ induces conformational change in CaM, i.e., exposing its hydrophobic residues, which promotes interactions of the Ca2+/CaM complex with numerous targeting proteins and subsequently regulates their functionalities.5,6 Many of these Ca2+/CaM targets modulate cellular signaling pathways, such as various Ser/Thr protein kinases (Ca2+/CaM binding protein kinase, CaMKs), which are particularly abundant in brain and induce phosphorylation on their downstream substrates after binding of Ca2+/CaM. Activations of CaMKs are described to be able to regulate numerous activities in neuronal development, plasticity and behavior.7,8 Some studies have reported the dysregulation of Ca2+/CaM and its associated kinases involve in neurodegenerative disorders, e.g. Alzherimer's disease (AD) and Parkinson's disease (PD).9-11 Our proteomic results have revealed that CaM level is elevated in the cortex and CSF of sCJD patients.12,13 Nevertheless, the statuses of CaM and the relevant kinases in prion infected brains are limitedly reported.
To figure out the possible alterations of Ca2+/CaM and its associated agents in the brains during prion infection, the levels of CaM and its downstream factors in the brains of hamsters infected with scrapie agent 263K and the prion infected cell line SMB-S15 were evaluated by various methodologies. We found that the level of CaM in the brains of 263K-infected hamsters started to increase at early stage and maintained at high level till the terminal stage. However, the related kinases such as total and phosphorylated forms of CaMKII and CaMKIV, as well as the downstream proteins such as cAMP-response element binding protein (CREB) and brain derived neurotrophic factor (BDNF) in the brains of 263K-infected animals were decreased. Further analysis showed a remarkable elevation of S-nitrosylated CaM in the brain tissues of hamsters infected with scrapie agent 263K, which might relate with the inhibition of CaM activity.
RESULTS
The Level of CaM Was Markedly Increased in the Brains of Scrapie Agent 263K-Infected Hamsters, with a Time-Dependent Phenomenon
To see the potential alteration of CaM in the brain tissues of TSE, 10% brain homogenates of three scrapie strain 263K-infected hamsters and three age-matched control ones were evaluated by Western blots with the CaM-specific antibody. A 17-kDa large CaM-specific band was observed in the brain homogenates of all three tested 263K-infected hamsters, while almost undetectable in the normal ones (Fig. 1A). Analysis of the relative gray values of CaM after normalized to that of the individual β-actin revealed that significantly higher CaM levels in 263K-infected hamsters compared with that of normal control (P = 0.0001).
FIGURE 1.
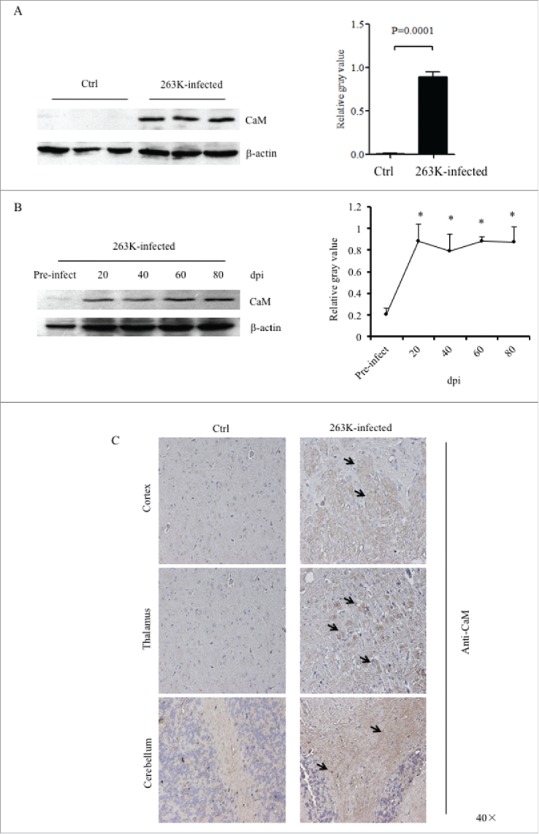
Upregulation of CaM in the brains of scrapie-infected rodents. A. Western blot analysis of CaM in the brain homogenates of scrapie agents 263K-infected hamsters collected at terminal stage. The same amounts of individual brain homogenate were loaded and analyzed by 12% SDS-PAGE. β-actin was used as an internal control. The scrapie agent 263K as well as normal control is shown on the top of the graphs. The densities of signals are determined by densitometry and shown relative to CaM/β-actin. Graphical data denote mean+SD (n = 3). Statistical differences compared with controls are illustrated on the top. B. Dynamic assays of CaM in the brain tissues of normal and 263K-infected hamsters during the incubation period. Experiments were repeated three times. Various specific immunoblots are marked on the right and the time-point of the day post-inoculation is shown as dpi. The gray values of each line are shown relative to CaM/β-actin. C. Representative IHC assays of CaM in the cortex, thalamus and cerebellum of 263K-infected hamsters and normal controls (40 ×).
To access the changes of the brain CaM during prion infection, the brain specimens of hamsters infected with scrapie agent 263K collected at different time points during incubation period were comparatively subjected into Western blots. Prior to the dynamic test, the levels of CaM in the brain tissues of the young and adult healthy hamsters that were at the same ages as the animals of pre-inoculation (roughly 3 weeks after weaning) and at terminal stage were evaluated. Weak, but clear, CaM-specific bands were detected by Western blots, which showed quite similar levels between young and adult healthy animals (Supplementary Fig. 1), indicating a stable expression of CaM in brain tissues. As shown in Fig. 1B, the signals of CaM became markedly stronger in the brain sample of the hamsters 20 days post-inoculation (dpi) and maintained at the comparatively level in the subsequent samples. Quantitative analyses of the average gray values of CaM after normalized with the individual β-actin illustrated rapid increase of brain CaM level in the early stage of prion infection (20 dpi).
To get more detailed insight into the alteration of CaM in various brain regions at terminal stage of prion infection, brain sections of 263K-infected and controls were stained by CaM-specific IHC assays. Large quantities of the CaM-specific brown signals, with irregular and variably sized astral shapes morphologically, were observed in the regions of cortex, thalamus and cerebellum of 263K-infected hamsters, (Fig. 1C). However, in the regions of hippocampus, rhombencephalon and olfactory bulb of 263K-infected hamsters, only few of the CaM stained signals were identified, which were comparable with that of normal controls (Supplementary Fig. 2).
FIGURE 2.
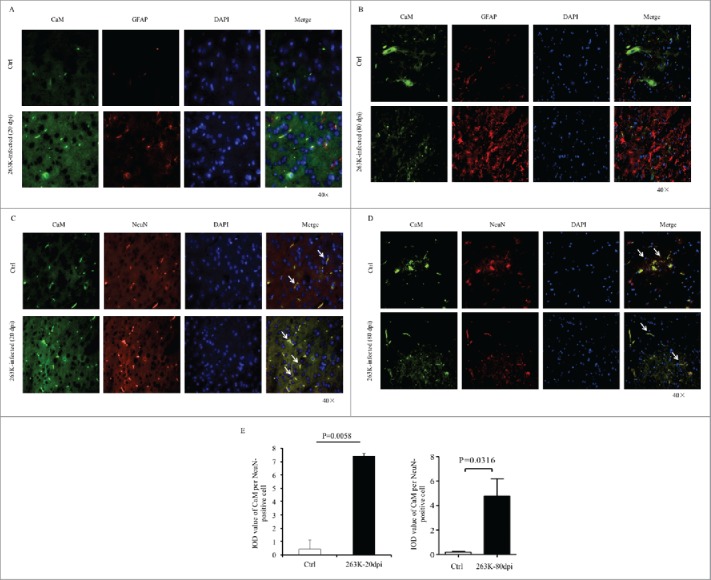
Immunofluorescent analysis of the associations of CaM with different kinds of CNS cells of normal and scrapie-infected hamsters collected at early and terminal stage. Representative images double-immunostained with the antibodies for CaM (green) and GFAP (red) on the slices from the hamsters of 20 dpi (A) and 80 dpi (B), or that for NeuN (red) from the hamsters of 20 dpi (C) and 80 dpi (D) (× 40). The normal controls are the individual age-matched healthy hamsters. The merged signals are shown with the white arrow. E. Analyses of the IOD values with the software in Operatta. The average IOD values of CaM per NeuN-positive cell (× 105). Left panel: the infected hamsters of 20 dpi. Right panel: the infected hamsters of 80 dpi. Statistical differences between the infected animals and the individual normal controls are indicated above.
The Increased CaM Localized Well in Neurons, but not in Proliferative Astrocytes in the Brains of 263K-Infected Hamsters
To further identify the distributing feature of the CaM positive signals during prion infection, the brain sections of 263K-infected hamsters collected at early (20 dpi) and terminal stage (80 dpi) were immunofluorescently double-stained with CaM and GFAP- or NeuN-specific antibodies, respectively. Compared with that of the individual age-matched normal hamsters, much more CaM positive signals (green) were observable in the brain slices of 263K-infected hamsters of both 20 and 80 dpi (Fig. 2). In the preparations stained with CaM and NeuN antibodies, the CaM signals looked to colocalize well with the NeuN positive cells (red) both in the infected and normal hamster brains (Fig. 2A). In contrary, CaM signals seemed colocalize neither with the GFAP positive cells in the normal controls nor with the proliferative GFAP positive cells in 263K-infected hamsters (Fig. 2B). Further, the integrated optical density (IOD) of CaM per NeuN positive cell from three slices from the 263K-infected or normal hamsters was counted. It showed a significantly increased average IOD CaM in each NeuN positive cell of the 263K-infected hamsters collected at early and terminal stage than that of normal ones (Fig. 2C). It may highlight that the increased brain CaM in the scrapie-infected hamsters possibly derives mainly from the remained neurons.
Alterations of CaMKII and CaMKIV in the Brains of 263K-Infected Hamsters at End Stage
To assess the influences of the elevated brain CaM on the downstream elements of Ca2+-signaling during prion infection, the expressions of CaMKII and CaMKIV in the brains of 263K-infected hamsters were separately investigated by Western blots. Compared with the data of normal hamsters, the levels of CaMKIIα and CaMKIIβ in the brains of 263K-infected hamsters were significantly decreased (Fig. 3A). Meanwhile, the levels of the phosphorylated for CaMKIIα at Threonine 286 (p-CaMKIIα, T286) were reduced (Fig. 3A). Further analysis of the ratios of p-CaMKIIα to CaMKIIα revealed significant reduction in the brain tissues of 263K-infected hamsters (P<0.05).
FIGURE 3.
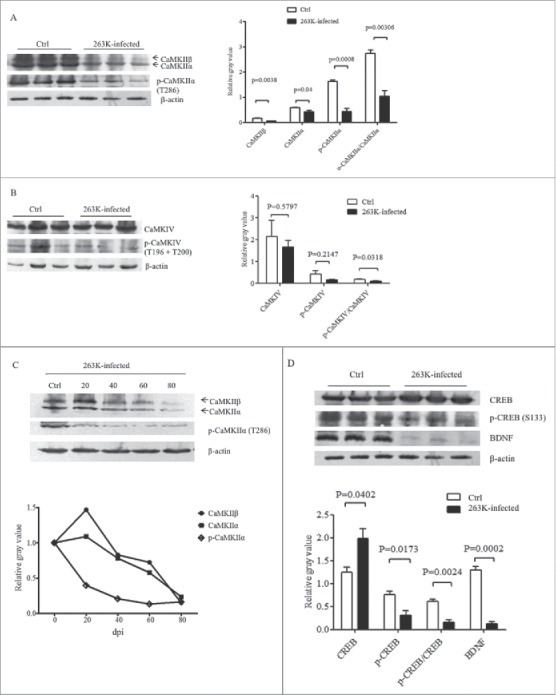
Downregulation of CaMKs and phospho-CaMKs (p-CaMKs) in the brains of scrapie infected hamsters. Western blots analysis of CaMKIIα, CaMKIIβ and p-CaMKIIα (T286) (A), as well as CaMKIV and p-CaMKIV (T196+T200) (B) in the brain homogenates of scrapie agent 263K-infected hamsters collected at moribund stage. C. Dynamic analysis of the CaMKIIα, CaMKIIβ and p-CaMKII (T286) in the brains of 263K-infected hamsters during incubation periods. The brain samples were collected 0, 20, 40, 60, 80 dpi and pooled with three individual hamsters. D. Western blots for CREB, p-CREB (S133) and BDNF in the brain homogenates of scrapie agents 263K-infected hamsters collected at end stage. The same amounts of individual brain homogenate were loaded and analyzed by 12% SDS-PAGE. β-actin was used as an internal control. The quantitative analysis of the signal densities of CaMKs, p-CaMKs, CREB, p-CREB and BDNF were normalized with that of β-actin. Graphical data denote mean+SD.
The levels of brain CaMKIV and its phosphorylated form of CaMKIV at Threonine 196 and 200 (p-CaMKIV, T196 and T200) between the infected and normal hamsters were comparatively tested by Western blots. Although both CaMKIV and p-CaMKIV levels in the infected brains were slightly lower than that of normal ones, no statistical difference was addressed (Fig. 3B). However, the ratio of p-CaMKIV to CaMKIV at the end stage of prion infection was significantly lower than that in the normal control.
To evaluate the dynamic alterations of CaMKII and p-CaMKII during scrapie infection, the brain samples of 263K-infected hamsters collected on the 20th, 40th, 60th, 80th dpi were comparatively evaluated. Both brain levels of CaMKIIα and CaMKIIβ showed time-dependent downtrend during the incubation periods, that CaMKIIα and CaMKIIβ decreased markedly since 40 and 60 dpi, respectively (Fig. 3C). Meanwhile, the level of phospho-CaMKIIα (p-CaMKIIα) dropped down even more notably, showing significant reduction since 20 dpi (Fig. 3C).
Further, the amounts of brain CREB and BDNF, which are the substrates of CaMK, were analyzed by Western blots. As shown in Fig. 3D, the amounts of CREB in the brains of 263K infected animals increased, but that of the phosphorylated form of CREB (at Ser 133), which represents the activated form, were obviously decreased. In line with our previous observation,14 the brain levels of BDNF of the hamsters infected with scrapie agent 263K were remarkably lower than that of the normal control (Fig. 3D). Those data indicate a downregulated situation of CaMKs in the brain tissues during prion infection.
The Alterations of CaM and CaMKs in Prion-Infected Cell Line SMB-S15
The levels of CaM and its downstream kinases in prion-infected cell line SMB-S15 and its partner normal cell line SMB-PS were comparative evaluated. Western blots showed an increase of CaM levels in SMB-S15 cells, with statistical difference compared with SMB-PS cells (Fig. 4A). Similar as our previous study,15 only CaMKIIα could be detected by Western blot in SMB cells. In contrast to the data of scrapie infected brains, both total CaMKIIα and phosphorylated CaMKIIα in SMB-S15 cells were slightly, but significantly, increased (Fig. 4B). Quantitative assays of the gray values also demonstrated marked increase of the ratio of p-CaMKIIα/CaMKIIα in SMB-S15 cells. The levels of total CaMKIV and p-CaMKIV in SMB-S15 cells were decreased, meanwhile, the ratio of p-CaMKIV/CaMKIV of SMB-S15 cells was also declined (Fig. 4C). Moreover, the level of CREB of SMB-S15 cells was higher than that of SMB-PS cells, whereas that of p-CREB of SMB-S15 cells was lower (Fig. 4D). In line with our previous observation,14 slight increase of BDNF was also observed in SMB-S15 cells (Fig. 4D).
FIGURE 4.
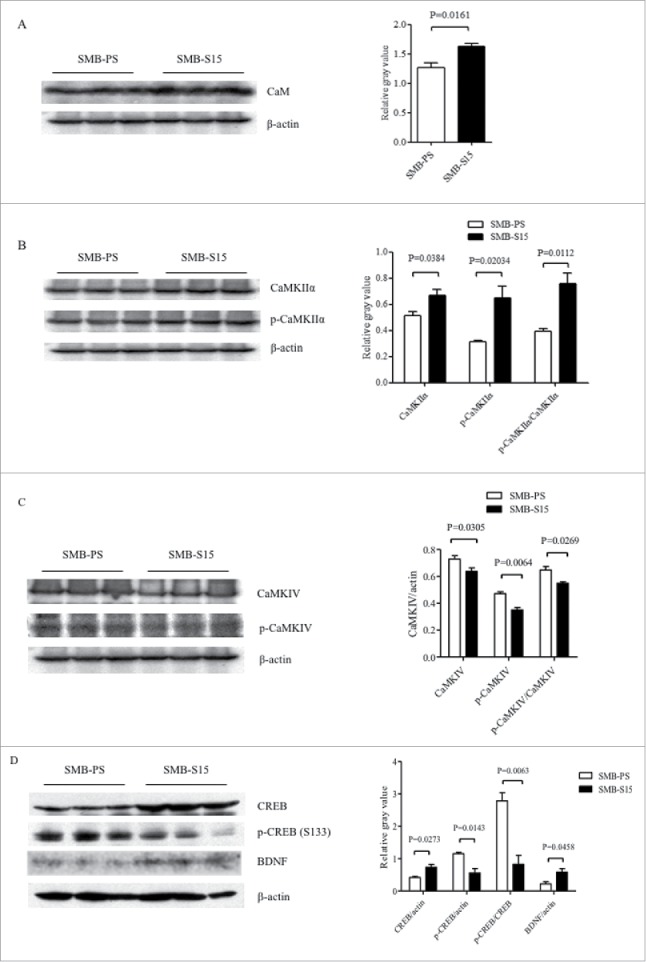
Evaluations of the levels of CaM and associated agents in scrapie cell line SMB-S15 and its normal cell line SMB-PS. Western blots for CaM (A), CaMKIIα and p-CaMKIIα (B), CaMKIV and p-CaMKIV (C), as well as CREB, p-CREB and BDNF (D). β-actin was used as an internal control and illustrated under each blot. The quantitative analyses of gray values of each blot after normalized with the individual β-actin are shown on the right. The average relative gray value is calculated from three independent blots and presented as mean+SD.
To figure out the distributions of above factors in the cultured cells, the cellular fractions of cytoplasm and nucleus of SMB-S15 and -PS cells were separately prepared and subjected into the individual Western blots. Besides of large amounts in cytoplasm, much more CaMKIIα and p-CaMKIIα were identified in the fraction of nucleus of SMB-S15 cells than that of SMB-PS cells (Fig. 5A). The signals of CaMKIV and p-CaMKIV were only detected in the preparations of cytoplasm of both SMB-S15 and -PS cells (Fig. 5A). Although CREB, p-CREB and BDNF distributed in the fractions of cytoplasm and nucleus of both SMB-S15 and -PS cells, more CREB, p-CREB and BDNF were in the nucleus of S15 cells (Fig. 5B). Above results highlight that the increased CaM in SMB-S15 cells might possess its normal biological function.
FIGURE 5.
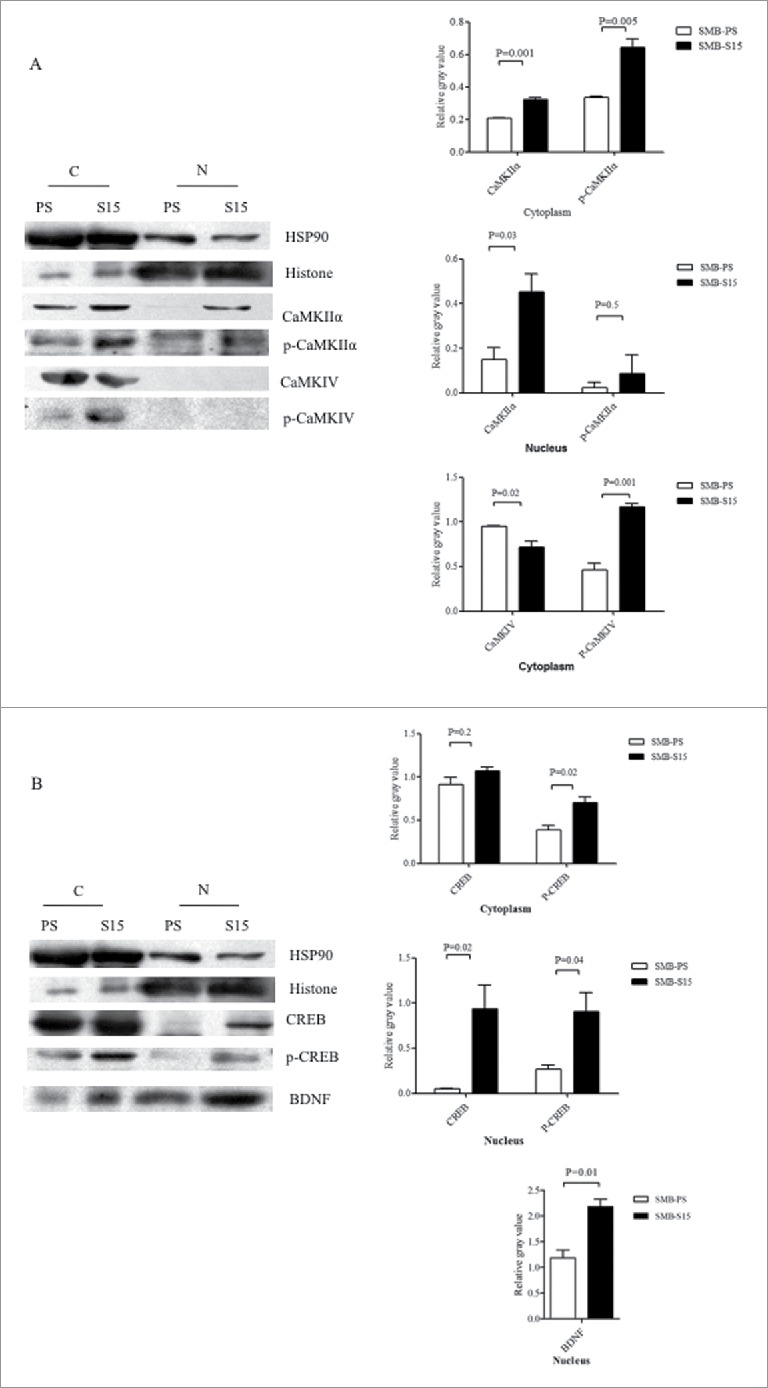
Analysis of the levels of CaMKs, p-CaMKs, CREB, p-CREB and BDNF in the fractions of the nucleus and cytoplasm of SMB cells. Western blots for CaMKIIα, p-CaMKIIα, CaMKIV and p-CaMKIV (A) as well as CREB, p-CREB and BDNF (B) in the fractions of the nucleus and cytoplasm of the cells. N: nucleus, C: cytoplasm. Various blots are shown on the left. Gray value analysis of the signals of CaMKs, p-CaMKs, CREB, p-CREB and BDNF in nucleus and cytosolic fractions are shown on the right. The quantitative results are collected from three independent experiments and are presented as mean+SD.
Upregulation of S-Nitrosylation for Brain CaM and Relative Agents During Prion Infection
S-nitrosylated forms of many proteins are abnormally altered in the brains of patients and animal models with prion infection.16 To see the possible alterations of S-nitrosylated (SNO) CaM during prion infection, the total SNO-proteins were extracted from the brain tissues of scrapie agent 263K infected hamsters at end stage and age-matched normal controls. CaM-specific Western blots for extracts revealed that the levels of SNO-CaM in the brains of 263K-infected hamster were significantly increased compared with these of normal controls, showing statistical difference (Fig. 6A). To test the possible alterations of brain SNO-CaM in the incubation period of prion infection, we prepared the tissue homogenates and whole SNO protein extracts of the brains of 263K-infected hamsters collected at various day post-infection. It demonstrated that the levels of brain SNO-CaM increased in the sample collected at 20 dpi, reached to the peak at middle-late stage (60 dpi) and dropped down but still maintained at high level at terminal stage (80 dpi) (Fig. 6B). It implies that the SNO-CaM abnormally increases and altered in a time-dependent manner during prion infection.
FIGURE 6.
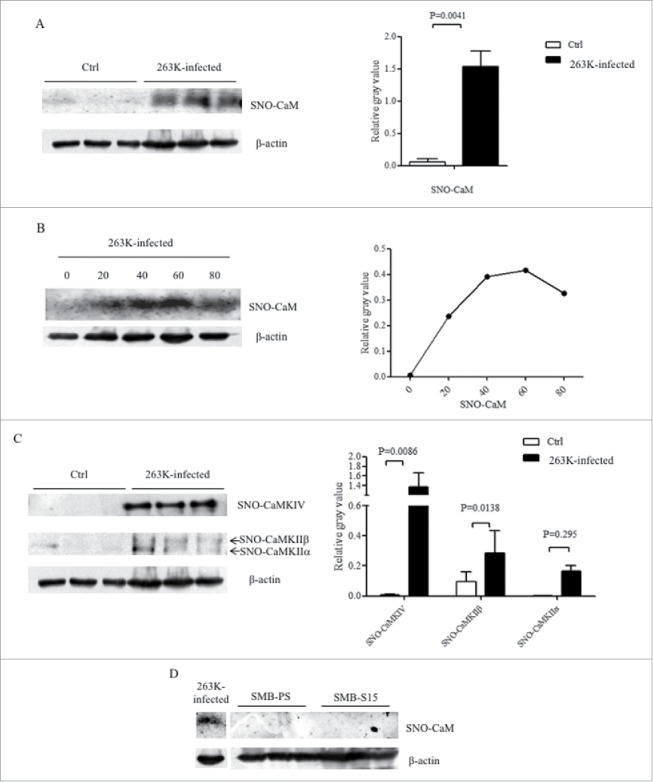
Increases of SNO-CaM and the related kinases in the brains of 263K-infected hamsters. A. The CaM-specific signals in the extracts of brain SNO proteins from three individual 263K-infected hamsters at terminal stage were comparatively evaluated with three normal ones. Graphical data are expressed as mean+SD. B. Dynamic alteration of SNO-CaM in the brains of 263K-infected hamsters during the incubation period. The extracts of brain SNO-proteins were pooled from the brains of three normal hamsters and 263K-infected hamsters collected 20, 40, 60 and 80 dpi. C. Western blots for SNO-CaMKs in the extracts of brain SNO-protein from three individual 263K-infected hamsters at moribund stage were comparatively analyzed with three normal ones. Graphical data are expressed as mean+SD. The relative gray values of CaM were normalized with that of the individual β-actin. D. Western blots for SNO-CaM in the extracts of SNO-protein from SMB-S15 and SMB-PS cells. The blot of SNO-CaM in the extract of brain SNO-protein from a 263K-infected hamster running in the same SDS-PAGE is used as control shown on the left.
To further get more insight into the changes of SNO-CaMKs during prion infection, the extracts of SNO proteins from normal and 263K-infected hamsters at terminal stage were employed into individual CaMKs-specific Western blots. Contrary to the decrease of total brain CaMKII and unchanged CaMKIV in 263K-infected hamsters, both blots of SNO-CaMKIIα and SNO-CaMKIV in the brains of 263K-infected hamsters at terminal stage were remarkably stronger than that of normal control, showing statistical differences (Fig. 6C). It illustrates an obvious upregulation of the nitrosylation forms of CaM and its down-stream kinases in the brain tissues during prion infection. Additionally, the presence of SNO-CaM in the cell lines SMB-S15 and SMB-PS were tested. No CaM-specific signal was detected in the extracts of SNO proteins from those two cell lines (Fig. 6D).
DISCUSSION
CaM is a ubiquitous Ca2+ receptor protein mediating a large number of signaling processes in all eukaryotic cells, meanwhile, it plays a central role in regulating a myriad of cellular functions via interaction with multiple target proteins.8 In the present study, we have firstly investigated the situations of the brain CaM levels in the pathogenesis of prion infection and demonstrated that the brain CaM level is increased in the hamsters infected with scrapie agent 263K with the help of Western blots, IHC, and IFA. However, the expressions and phosphorylations of some downstream factors of CaM, i.e., CaMKII and CaMKIV, are obviously decreased in the brains of 263K-infected hamsters. Those aberrant alterations of the downstream factors of CaM may highlight a malfunction of brain CaM, despite of significantly increased expression, during prion infection. Further analysis has revealed a remarkable increase of SNO-CaM in the brains of 263K-infected hamsters, which may associate with the dysfunction of the brain CaM in prion infection.
As a calcium-binding protein, abnormal changes of CaM level will interrupt Ca2+ homeostasis and further influence the function of many physiological processes in various types of cells. Abnormal increase of CaM level has been repeatedly reported in the specimens of AD patients, AD animal models and the relevant cell lines.8,11,17 Intracellular aberrant Ca2+ homeostasis is also observed in both sporadic and familial forms of AD.18,19 Recently, elevation of CaM level has been described in the brains and CSF of the patients with sCJD.12,13 It seems that increase of CaM is a common phenomenon in neurodegeneration diseases. The morphological data here illustrate that CaM-positive cells colocalize well with NeuN-positive cells, even the damaged cells, but not with GFAP-positive ones, indicating that the increased CaM levels may largely come from the neurons of the infected brains. The higher CaM content is associated with impaired cell survival/death mechanisms.21,22 Studies show that CaM plays an important role in the cell cycle and reaches high level at the G1/S and G2/M transition,23-25 indicating that disease-related cell cycle arrest may influence the brain CaM level. Actually, aberrant expressions of cell cycle regulatory proteins has been identified in the prion-infected experimental animals, which leads to the cell cycle arrest at M phrase.26 One may speculate that large amount of CaM accumulating in neurons during the progression of prion disease may contribute to the abortive cell cycle events observed in the brains during prion infection.
Activation of CaM dependent kinases is one of the most prominent biological functions of CaM, in which CaMKII and CaMKIV are two important ones. However, the increased CaM level in the prion infected brains seems not to be normally functional, since the either the expression or phosphorylation of CaMKs are down-regulated. Study on AD proposes that high level of CaM in brain tissues and lymphoblasts possibly relates with the consequence of impaired proteasomal degradation of the protein.17 Interestingly, we have identified that along with the increase of the total brain CaM level during prion infection, the amounts of SNO forms of CaM and CaMKs are markedly up-regulated. Most of the signal transduction pathways are mediated by protein S-nitrosylation in eukaryotic cells, controls cell processes e.g. metabolism and transcription, and plays an important role in intercellular communication.27 It may mediate either protective or neurotoxic signaling depending on the action of target proteins. A number of S-nitrosylated proteins have been proposed to be able to contribute to the pathogenesis of neurodegenerative diseases.28,29 Although we are still not well understood the precise effectiveness of S-nitrosylation of CaM on its activation for the downstream kinases, it is not hinder us to assume that amounts of SNO-CaM in prion-infected brains will influence its normal functions, which might associate the lack of activities of CaMKs in the late stage of prion disease. Recently, we have confirmed that the levels of nitric oxide (NO) and nitric oxide synthase (NOS) in the brains of several prion infected rodent models, including 263K-infected hamsters, show time-dependent alteration, which started to increase at early stage, reached to the peak in the middle stage and dropped down at late stage.30 Coincidentally, the increase of brain SNO-CaM in this study reveals the same pattern.
Our previous study15 and the data here verify persistent decrease of CaMKII and p-CaMKII in the brain tissues during prion infection. Reduction of the levels of CaMKII and its autophosphorylation form p-CaMKII (T286) has been noticed in hippocampus and frontal cortex of the severe AD brains.31 Although the brain levels of CaMKIV and phosphorylated CaMKIV do not show significant reduction in the scrapie infected hamsters, the ratio of p-CaMKIV/CaMKIV does decrease, which may reflect a down-regulated activity of brain CaMKIV. In addition, increased SNO-forms of both CaMKII and CaMKIV during prion infection in this study may also affect their biological functions. CaMKII is the major post-synaptic protein at excitatory synapses, which is fundamentally important for synaptic plasticity and memory formation.32,33 CaMKIV works as an important player in neuronal cell survival and overexpression of CaMKIV can attenuate neuronal apoptosis in cerebellar granule neurons.34 Both CaMKII and CaMKIV are responsible for phosphorylation of cAMP response element-binding protein (CREB) at Ser 133, which induces a vast amount of gene expressions subsequently.35 One of the regulated genes is Bdnf, encoding BDNF. BDNF affects many processes in neuronal development, such as axonal and dendritic development, synapse formation and maturation, synaptic potentiation, and neuronal survival. More recently, we have reported that BDNF and its signaling components in the brain tissues of scrapie experimental rodents are severely hindered.14 Severe loss of brain BDNF in our previous and present studies may represent one of the consequences of the depression of CaMKs during prion infection.
Compared the dramatically increase of brain CaM in scrapie infected animals, the increase of CaM level in the prion infected cell line SMB-S15 seems to be limited but still significant. However, unlike the observations in the brains of scrapie infected hamsters, the levels of some downstream kinases and effective factors in SMB-S15 cells are also increased, such as CaMKII, p-CaMKII, CREB and BDNF. It seems that the activation of CaM for CaM-dependent kinases in the cultured cells with prion propagation functions well. Moreover, no SNO form of CaM is detected in SMB-S15 cells, which supplies useful evidence for the association between S-nitrosylation of CaM and loss of activation for its downstream kinases. Under our experimental condition, SMB-S15 cells exhibit almost same replicating capacity as its normal partner cell line SMB-PS. However, remarkably more downstream factors of CaM, including CaMKIIα and p-CaMKIIα, CREB and p-CREB, as well as BDNF, distribute in the nucleus fraction of SMB-S15 cells than in that of SMB-PS cells. Based on the results obtained in this study, we may hypothesize that the increased CaM caused by prion infection may present different consequences in vitro and in vivo. In the cultured cells, prion propagation induced the increase of cellular CaM level. The cellular CaM triggers activations of CaM-dependent kinases and other biological components or pathways to overcome the harmful effect of prion accumulation and maintain the cell viability and proliferation. In the brains of prion infected animals, the increased CaM undergoes extensive S-nitrosylation due to the abnormal changes of NO and NOS in CNS and loses its activation for CaM-dependent kinases and other downstream biological modulators. Meanwhile, large amount of CaM accumulates in neurons, possibly due to impaired proteasomal degradation of the protein, may cause turbulence of cell cycle that is arrest at M phase. Further studies to evaluate the situations of other CaM related events during prion infection both in vitro and in vivo will uncover the whole significance of CaM on the pathogenesis of prion disease.
MATERIALS AND METHODS
Ethics Statement
Usage of animal specimens in this study was approved by the Ethical Committee of National Institute for Viral Disease Prevention and Control, China CDC. Animal housing and experimental protocols were in accordance with the Chinese Regulations for the Administration of Affairs Concerning Experimental Animals.
Preparation of Brain Homogenates
The brain samples of Syrian golden hamster inoculated with hamster-adapted scrapie agents 263K were enrolled in this study. The clinical and neuropathological characteristics of these models have been described previously.20,36 The incubation time of 263K-infected hamsters was 70.5±4.93 days. For dynamic study, brain samples of the mice infected with agents 263K were collected on the 20th, 40th, 60th and at terminal stage (80th dpi). The healthy hamsters roughly 3 weeks after weaning were used as pre-infection control.
The brains were removed surgically, immediately dissected, then frozen and stored at -80°C until use. 10% (w/v) brain homogenates were prepared based on the protocol described previously.37 Briefly, brain tissues were homogenized in lysis buffer (100 mM NaCl, 10 mM EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 10 mM Tris, pH 7.5) containing protease inhibitors cocktail set III. The tissue debris was removed with low speed centrifugation at 2000 g for 10 min and the supernatants were collected for further study.
Cell Culture and Cell Lysates
SMB-PS and SMB-S15 cell line derived from mice neurons were obtained from the Roslin Institute (UK) and cultured in Dulbecco's modified Eagle's medium (Gibco, USA) supplemented with 12% fetal bovine serum (FBS) (AusgeneX, Australian). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere. The cell line of SMB-S15 was originally infected with scrapie strain Chandler, in which PrPSc replication was maintained by cell passage. Cell line SMB-PS was SMB-S15 cells that were completely cured of prion by pentosan sulfate (PS) treatment without detectable PrPSc. Cultured cells were washed twice with cold PBS and harvested with cold lysis buffer (100 mM NaCl, 10 mM EDTA, 10 mM Tris, 0.5% sodium deoxycholate, 0.5% Nonidet P-40, pH 7.5) containing protease inhibitors cocktail set III. The lysate was placed on the ice for 30 min and then centrifuged at 5,000 g at 4°C for 5 min. The supernatants were collected and stored at −20°C until needed.
Western Blots
Aliquots of brain homogenates or cell lysates were separated on 12% SDS-PAGE and electroblotted onto NC membrane using a semi-dry blotting system. Membranes were blocked with 5% (w/v) BSA in Tris-buffered saline containing 0.1% Tween 20 (TBST) at room temperature (RT) for 1 h and incubated with various primary antibodies, including anti-CaM polyclonal antibody (pAb, 1:100, Santa Cruz, USA), anti-CaM monoclonal antibody (mAb, 1:1,000, Cell Signaling Technology, USA), anti-CaMKII mAb (1:500, Abcam, USA), anti-p-CaMKII alpha (phospho T286) pAb (1:1,000, Abcam, USA), anti-CaMKIV pAb (1:5,000, Abcam, USA), anti-p-CaMKIV (phospho T196+T200) pAb (1:1,000, Abcam, USA), anti-CREB mAb (1:5,000, Abcam,USA), anti-p-CREB (phospho S133) mAb (1:5,000, Abcam, USA), or anti-BDNF pAb (1:500, Santa Cruz, USA) at 4°C overnight. After washing with TBST, membranes were probed with HRP-conjugated goat-derived anti-mouse or anti-rabbit secondary antibodies at RT for 1 h, and then blots were developed using ECL and visualized on autoradiography films. Images were captured by ChemiDoc™ XRS + Imager and quantified by Image J software.
Immunohistochemical Assays
Brains tissues were fixed in 10% formalin and embedded in paraffin. For IHC, paraffin slides (5-μm thick) were deparaffinised in xylene and ethanol. Antigen retrieval was performed in sodium citrate buffer (pH 6.0) by heating for 30 min followed by enzyme digestion for 1 min. The slides were quenched in 3% H2O2 for 15 min and permeabilised in 0.3% Triton X-100 for 20 min. The sections were blocked with 1% normal goat serum at RT for 15 min and then incubated at 4°C overnight with 1:50-diluted pAb for CaM, the slides were then incubated with 1:200-diluted HRP-conjugated goat-derived anti-rabbit secondary antibody at 37°C for 1 h and visualized by incubation with 3,3′-diaminobenzidine tetrahydrochloride (DAB). The slices were counterstained with hematoxylin, dehydrated and mounted in Permount.
Immunofluorescence Assay
Paraffin embedded brain tissue slices were used in the study. The slices were permeabilizated by 0.3% Triton X-100 in PBS for 30 min and blocked with normal goat serum for 1 h. After blocked, samples were probed with 1:100 diluted pAb anti-CaM (Santa Cruz, USA), 1:200 diluted pAb anti-NeuN (Millipore, USA) or 1:200-diluted pAb anti-GFAP (Cell Signaling Technology, USA) in dilution solution (PBS with 2% BSA and 0.3% Triton X-100) at 4°C overnight. The sections were subsequently probed with 1:400-diluted Alexa Fluor 488-labeled goat anti-rabbit and Alexa Fluor 568-labeled goat-derived anti-mouse secondary antibodies (Invitrogen, USA) for 1 h. After removing secondary antibodies, DAPI (Invitrogen, USA) was used to stain the nucleus at concentration of 1 μg/ml at 37°C for 30 min. The slices were then mounted with permount and viewed using a high content screening system (Operetta Enspire, Perkin Elmer, USA).
Biotin-Switch Assay of Extracting S-Nitrosylated Protein
Brain SNO-proteins were prepared with a commercial detection assay kit (Cayman Chemical, USA) according to the protocol described previously.38 Briefly, brain homogenates of scrapie infected and normal hamsters were incubated with the blocking buffer, gently agitating the mixture at 4°C for 30 min to block free thiol groups. Precipitation of the mixture with ice-cold acetone at -20°C, and the nitrosothiols were reduced to thiols with reducing reagent. The newly formed thiols were labeled with biotinylating reagent. The total biotinylated proteins were immunoprecipitated with streptavidin-agarose beads and various SNO-proteins was detected with the specific Western blots.
Statistical Analysis
In this study, all the experiments were conducted at least three times with consistent results. The mean value and standard error of multiple data points or samples were used to represent the final results. The student's t test was used for statistical analysis and P <0.05 was considered significant. All data were presented as the mean+SD.
Supplementary Material
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No competing interests declared.
ACKNOWLEDGMENT
We thank Mr. Bao-Yun Zhang for his great help in neuropathology.
FUNDING
This work was supported by Chinese National Natural Science Foundation Grants (81401670, 81630062, 81772197), National Key Research and Development Plan (2016YFC1202700), SKLID Development Grant (2012SKLID102, 2016SKLID603), the Young Scholar Scientific Research Foundation of China CDC (2016A101) and Graduate Research and Innovation Projects of Heilongjiang Bayi Agricultural University (YJSCX2016-Y48).
REFERENCES
- 1.Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559. doi: 10.1186/1743-422X-8-559. PMID:22196171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363-83. doi: 10.1073/pnas.95.23.13363. PMID:9811807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;93:337-48. doi: 10.1016/S0092-8674(00)81163-0. PMID:9590169. [DOI] [PubMed] [Google Scholar]
- 4.Chin D, Means AR. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000;10:322-8. doi: 10.1016/S0962-8924(00)01800-6. PMID:10884684. [DOI] [PubMed] [Google Scholar]
- 5.Jurado LA, Chockalingam PS, Jarrett HW. Apocalmodulin. Physiol Rev. 1999;79:661-82. PMID:10390515. [DOI] [PubMed] [Google Scholar]
- 6.Bahler M, Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002;513:107-13. doi: 10.1016/S0014-5793(01)03239-2. PMID:11911888. [DOI] [PubMed] [Google Scholar]
- 7.Hoeflich KP, Ikura M. Calmodulin in action: diversity in target recognition and activation mechanisms. Cell. 2002;108:739-42. doi: 10.1016/S0092-8674(02)00682-7. PMID:11955428. [DOI] [PubMed] [Google Scholar]
- 8.Berchtold MW, Villalobo A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim Biophys Acta. 2014;1843:398-435. doi: 10.1016/j.bbamcr.2013.10.021. PMID:24188867. [DOI] [PubMed] [Google Scholar]
- 9.Bartolome F, de Las Cuevas N, Munoz U, Bermejo F, Martin-Requero A. Impaired apoptosis in lymphoblasts from Alzheimer's disease patients: cross-talk of Ca2+/calmodulin and ERK1/2 signaling pathways. Cell Mol Life Sci. 2007;64:1437-48. doi: 10.1007/s00018-007-7081-3. PMID:17502994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fedida-Metula S, Feldman B, Koshelev V, Levin-Gromiko U, Voronov E, Fishman D. Lipid rafts couple store-operated Ca2+ entry to constitutive activation of PKB/Akt in a Ca2+/calmodulin-, Src- and PP2A-mediated pathway and promote melanoma tumor growth. Carcinogenesis. 2012;33:740-50. doi: 10.1093/carcin/bgs021. PMID:22287561. [DOI] [PubMed] [Google Scholar]
- 11.Esteras N, Alquezar C, de la Encarnacion A, Villarejo A, Bermejo-Pareja F, Martin-Requero A. Calmodulin levels in blood cells as a potential biomarker of Alzheimer's disease. Alzheimers Res Ther. 2013;5:55. doi: 10.1186/alzrt219. PMID:24499616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen C, Xiao D, Zhou W, Shi Q, Zhang HF, Zhang J, Tian C, Zhang JZ, Dong XP. Global protein differential expression profiling of cerebrospinal fluid samples pooled from Chinese sporadic CJD and non-CJD Patients. Mol Neurobiol. 2014;49:290-302. doi: 10.1007/s12035-013-8519-2. PMID:23912784. [DOI] [PubMed] [Google Scholar]
- 13.Shi Q, Chen LN, Zhang BY, Xiao K, Zhou W, Chen C, Zhang XM, Tian C, Gao C, Wang J, et al.. Proteomics analyses for the global proteins in the brain tissues of different human prion diseases. Mol Cell Proteomics. 2015;14:854-69. doi: 10.1074/mcp.M114.038018. PMID:25616867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang TT, Tian C, Sun J, Wang H, Zhang BY, Chen C, Wang J, Xiao K, Chen LN, Lv Y, et al.. Down-regulation of brain-derived neurotrophic factor and its signaling components in the brain tissues of scrapie experimental animals. Int J Biochem Cell Biol. 2016;79:318-26. doi: 10.1016/j.biocel.2016.08.033. PMID:27590859. [DOI] [PubMed] [Google Scholar]
- 15.Fan XY, Tian C, Wang H, Xu Y, Ren K, Zhang BY, Gao C, Shi Q, Meng G, Zhang LB, et al.. Activation of the AMPK-ULK1 pathway plays an important role in autophagy during prion infection. Sci Rep. 2015;5:14728. doi: 10.1038/srep14728. PMID:26423766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen LN, Shi Q, Zhang BY, Zhang XM, Wang J, Xiao K, Lv Y, Sun J, Yang XD, Chen C, et al.. Proteomic analyses for the global S-Nitrosylated proteins in the brain tissues of different human prion diseases. Mol Neurobiol. 2016;53:5079-96. doi: 10.1007/s12035-015-9440-7. PMID:26392294. [DOI] [PubMed] [Google Scholar]
- 17.Esteras N, Munoz U, Alquezar C, Bartolome F, Bermejo-Pareja F, Martin-Requero A. Altered calmodulin degradation and signaling in non-neuronal cells from Alzheimer's disease patients. Curr Alzheimer Res. 2012;9:267-77. doi: 10.2174/156720512800107564. PMID:22044025. [DOI] [PubMed] [Google Scholar]
- 18.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862-72. doi: 10.1038/nrn960. PMID:12415294. [DOI] [PubMed] [Google Scholar]
- 19.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454-63. doi: 10.1016/j.tins.2008.06.005. PMID:18675468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi Q, Zhang BY, Gao C, Zhang J, Jiang HY, Chen C, Han J, Dong XP. Mouse-adapted scrapie strains 139A and ME7 overcome species barrier to induce experimental scrapie in hamsters and changed their pathogenic features. Virol J. 2012;9:63. doi: 10.1186/1743-422X-9-63. PMID:22400710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munoz U, Bartolome F, Bermejo F, Martin-Requero A. Enhanced proteasome-dependent degradation of the CDK inhibitor p27(kip1) in immortalized lymphocytes from Alzheimer's dementia patients. Neurobiol Aging. 2008;29:1474-84. doi: 10.1016/j.neurobiolaging.2007.03.013. PMID:17448572. [DOI] [PubMed] [Google Scholar]
- 22.Esteras N, Alquezar C, Bermejo-Pareja F, Bialopiotrowicz E, Wojda U, Martin-Requero A. Downregulation of extracellular signal-regulated kinase 1/2 activity by calmodulin KII modulates p21Cip1 levels and survival of immortalized lymphocytes from Alzheimer's disease patients. Neurobiol Aging. 2013;34:1090-100. doi: 10.1016/j.neurobiolaging.2012.10.014. PMID:23153928. [DOI] [PubMed] [Google Scholar]
- 23.Chafouleas JG, Bolton WE, Hidaka H, Boyd AE 3rd, Means AR. Calmodulin and the cell cycle: involvement in regulation of cell-cycle progression. Cell. 1982;28:41-50. doi: 10.1016/0092-8674(82)90373-7. PMID:7066986. [DOI] [PubMed] [Google Scholar]
- 24.Sasaki Y, Hidaka H. Calmodulin and cell proliferation. Biochem Biophys Res Commun. 1982;104:451-6. doi: 10.1016/0006-291X(82)90658-1. PMID:7041900. [DOI] [PubMed] [Google Scholar]
- 25.Chafouleas JG, Lagace L, Bolton WE, Boyd AE 3rd, Means AR. Changes in calmodulin and its mRNA accompany reentry of quiescent (G0) cells into the cell cycle. Cell. 1984;36:73-81. doi: 10.1016/0092-8674(84)90075-8. PMID:6692470. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Tian C, Xu Y, Xie WL, Zhang J, Zhang BY, Ren K, Wang K, Chen C, Wang SB, et al.. Abortive cell cycle events in the brains of scrapie-infected hamsters with remarkable decreases of PLK3/Cdc25C and increases of PLK1/Cyclin B1. Mol Neurobiol. 2013;48(3):655-68. doi: 10.1007/s12035-013-8455-1. PMID:23625313. [DOI] [PubMed] [Google Scholar]
- 27.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150-66. doi: 10.1038/nrm1569. PMID:15688001. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596-614. doi: 10.1016/j.neuron.2013.05.005. PMID:23719160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura T, Prikhodko OA, Pirie E, Nagar S, Akhtar MW, Oh CK, McKercher SR, Ambasudhan R, Okamoto S, Lipton SA. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol Dis. 2015;84:99-108. doi: 10.1016/j.nbd.2015.03.017. PMID:25796565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen LN, Sun J, Yang XD, Xiao K, Lv Y, Zhang BY, Zhou W, Chen C, Gao C, Shi Q, et al.. The brain NO levels and NOS activities ascended in the early and middle stages and descended in the terminal stage in scrapie-infected animal models. Mol Neurobiol. 2016;54(3):1786-96. doi: 10.1007/s12035-016-9755-z. PMID:26887380. [DOI] [PubMed] [Google Scholar]
- 31.Amada N, Aihara K, Ravid R, Horie M. Reduction of NR1 and phosphorylated Ca2+/calmodulin-dependent protein kinase II levels in Alzheimer's disease. Neuroreport 2005; 16:1809-13. doi: 10.1097/01.wnr.0000185015.44563.5d. PMID:16237332. [DOI] [PubMed] [Google Scholar]
- 32.Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, Watanabe M, Hatanaka N, Totsuka M, Yagi T, et al.. Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci. 2009;29:7607-18. doi: 10.1523/JNEUROSCI.0707-09.2009. PMID:19515929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hell JW. CaMKII: Claiming center stage in postsynaptic function and organization. Neuron. 2014;81:249-65. doi: 10.1016/j.neuron.2013.12.024. PMID:24462093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.See V, Boutillier AL, Bito H, Loeffler JP. Calcium/calmodulin-dependent protein kinase type IV (CaMKIV) inhibits apoptosis induced by potassium deprivation in cerebellar granule neurons. FASEB J. 2001;15:134-44. doi: 10.1096/fj.00-0106com. PMID:11149901. [DOI] [PubMed] [Google Scholar]
- 35.Yano S, Morioka M, Kuratsu J, Fukunaga K. Functional proteins involved in regulation of intracellular Ca(2+) for drug development: Role of calcium/calmodulin-dependent protein kinases in ischemic neuronal death. J pharmacol Sci. 2005;97:351-4. doi: 10.1254/jphs.FMJ04007X5. PMID:15764842. [DOI] [PubMed] [Google Scholar]
- 36.Chen JM, Gao C, Shi Q, Shan B, Lei YJ, Dong CF, An R, Wang GR, Zhang BY, Han J, et al.. Different expression patterns of CK2 subunits in the brains of experimental animals and patients with transmissible spongiform encephalopathies. Arch Virol. 2008;153:1013-20. doi: 10.1007/s00705-008-0084-z. PMID:18404245. [DOI] [PubMed] [Google Scholar]
- 37.Shi Q, Xiao K, Zhang BY, Zhang XM, Chen LN, Chen C, Gao C, Dong XP. Successive passaging of the scrapie strains, ME7-ha and 139A-ha, generated by the interspecies transmission of mouse-adapted strains into hamsters markedly shortens the incubation times, but maintains their molecular and pathological properties. Int J Mol Med. 2015;35:1138-46. PMID:25683243. [DOI] [PubMed] [Google Scholar]
- 38.Chen LN, Shi Q, Zhang XM, Zhang BY, Lv Y, Chen C, Zhang J, Xiao K, Tian C, Gao C, et al.. Optimization of the isolation and enrichment of S-nitrosylated proteins from brain tissues of rodents and humans with various prion diseases for iTRAQ-based proteomics. Int J Mol Med. 2015;35:125-34. PMID:25339367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.