

Abstract
Giant cell arteritis is an autoimmune disease defined by explicit tissue tropism to the walls of medium and large arteries. Pathognomic inflammatory lesions are granulomatous in nature, emphasizing the functional role of CD4 T cells and macrophages. Evidence for a pathogenic role of antibodies and immune complexes is missing. Analysis of T cell populations in giant cell arteritis, both in the tissue lesions and in the circulation, has supported a model of broad, polyclonal T cell activation, involving an array of functional T cell lineages. The signature of T cell cytokines produced by vasculitic lesions is typically multifunctional, including IL-2, IFN-γ, IL-17, IL-21, and GM-CSF, supportive for a general defect in T cell regulation. Recent data describing the lack of a lymph node-based population of anti-inflammatory T cells in giant cell arteritis patients offers a fresh look at the immunopathology of this vasculitis. Due to defective CD8+NOX2+ regulatory T cells, giant cell arteritis patients appear unable to curtail clonal expansion within the CD4 T cell compartment, resulting in wide-spread CD4 T cell hyperimmunity. Why unopposed expansion of committed CD4 effector T cells would lead to invasion of the walls of medium and large arteries needs to be explored in further investigations.
Keywords: anti-inflammatory T cells, giant cell arteritis, macrophage, pro-inflammatory T cells, CD8+ Treg cells
1. Introduction
Giant cell arteritis (GCA) is a vasculitis of medium and large arteries typically combined with an intense systemic inflammatory syndrome (1–4). Systemic inflammatory syndrome may occur in the absence of frank vasculitis, mostly presenting muscle pain and stiffness known as polymyalgia rheumatica (PMR). GCA is an immune-mediated disease, involving the innate and adaptive branch of the immune system and characterized by granuloma formation in the mural layers of inflamed arteries. By definition, granulomas are organized lymphoid microstructures, composed of two major cell populations: macrophages, sometimes called histiocytes, which may fuse to form giant cells and CD4 T cells. Granulomas have been implicated in containment of intracellular bacterial infections and difficult-to-digest irritants, but convincing data implicating either in the pathogenesis of GCA are missing (5, 6).
While there is no doubt that excess activation of immune cells drives GCA, the original stimulus leading to aberrant immune activation has remained undefined. Granulomatous tissue inflammation is a typical complication of immunodeficiency syndromes (7, 8), exemplifying the coexistence of defective immunity with autoimmune and granulomatous manifestations. In such immune-deficient patients, susceptibility to bacterial and fungal infections is combined with a high risk for excessive inflammation, promoting granuloma formation in essentially any organ system.
Here, we will review current evidence for a fundamental immunodysregulation in GCA, with formation of noninfectious arterial-wall granulomas representing a consequence of insufficient immunosuppression and aberrant threshold setting in CD4 T cell homeostasis.
1.1. GCA – more than one immunopathology
When assessing the immunopathology of GCA it is important to recognize that the disease has several components, which may be partially independent. GCA’s vasculitic component is characterized by granulomatous infiltrates in the wall layers of arteries of sufficient size to have a vasa vasorum network. Data from the last two decades best fit a model in which immune cells enter the target artery through the vasa vasorum network, encounter professional and vessel-specific antigen-presenting cells, are locally stimulated and form granulomatous arrangements of highly-activated macrophages and T cells. In line with this model, the vast majority of tissue-residing lymphocytes are CD4 T cells, which have converted to effector and memory status. A multitude of other immune cells are present in low frequencies, including CD8 T cells, mast cells, NK cells, eosinophils, occasional B cells. Clinical manifestations of vascular inflammation are dominated by vaso-occlusive events that lead to tissue ischemia, most prominently to vison loss due to ischemic optic neuropathy. Recent data suggest that aortic involvement is frequent amongst GCA patients, which may result in dissection or aneurysm formation and rare fatal complications.
GCA’s systemic component has not been localized to a specific tissue site and may occur widespread within lymphoid organs. Fever of unknown origin can be the presenting symptom. Constitutional symptoms, such as weight loss, night sweats, malaise, are not unusual. The muscle pains and stiffness clinically known as PMR can be present early in the disease process and often appear after corticosteroids are reduced during chronic disease management. The underlying immune abnormalities leading to PMR are not understood. Clinically, the intensity of the systemic inflammation is captured by acute phase reactants, measured as elevated C-reactive protein (CRP), acute-phase serum amyloid A protein (A-SAA), erythrocyte sedimentation rate (ESR). Such acute phase reactants may, in turn, have functions in driving disease relevant processes, as supported by a recent study demonstrating that A-SAA induced IL-6 and IL-8 production by temporal artery explants, fostered angiogenic tube formation and promoted myofibroblasts outgrowth (9). Besides hepatocytes, which are also a major producer of CRP, A-SAA can also be released within inflammatory infiltrates, as has been shown for rheumatoid arthritis and psoriatic arthritis (10, 11). Elevation of such acute phase reactants in the circulation gives them widespread access to numerous organ systems, particularly the vascular tree. Accordingly, A-SAA serum levels have been identified as a predictor of coronary artery disease (12) and as an amplifier of atherosclerosis in mice (13). Acute phase reactants respond well to corticosteroid therapy and one of the benefits of this therapy may lie in disrupting nonspecific feed-forward inflammatory amplifier loops. In GCA patients, corticosteroids are excellent suppressors of IL-6, IL-1, IL-17 and IL-23, all products of the innate immune system (14). However, adaptive immunity appears much more resistant to corticosteroids, making GCA a chronic condition, the chronicity of which will require therapeutic interventions counteracting vasculitogenic T cells and their role in granuloma formation.
Accepting the disease model of partially independent disease components has important consequences for the design of explorative studies, the utilization of diagnostic tests (which may only capture one component) and the therapeutic management (15). Ultimately, the immunopathologic processes, vascular inflammation and systemic inflammation, need to be controlled to successfully treat the disease.
1.2. Pro-inflammatory T cells in GCA
Molecular studies of tissue-residing T cells in temporal artery biopsies have demonstrated expansion of individual T cell clones shared amongst disconnected disease lesions; strongly suggestive for antigen-reactive immunity (16). Functional studies in temporal artery tissues support a model of multiple T cell lineages acting as pro-inflammatory effector cells (Fig.1). A multitude of T cell cytokines have been detected in GCA tissue lesions; including IL-2, IFN-γ, IL-17, IL-9, IL-21 and GM-CSF (17–19). Notably, IL-4, the marker cytokine for Th2 cells, has been consistently missing. Either, effector T cells packed into the granulomatous lesions are multifunctional and are capable of producing a variety of cytokines; or the T cell infiltrate is composed of many different specified T cell populations. Both scenarios suggest a fundamental defect in the regulation of the CD4 T cell compartment.
Fig. 1. Multiple T helper cell lineages participate in GCA.
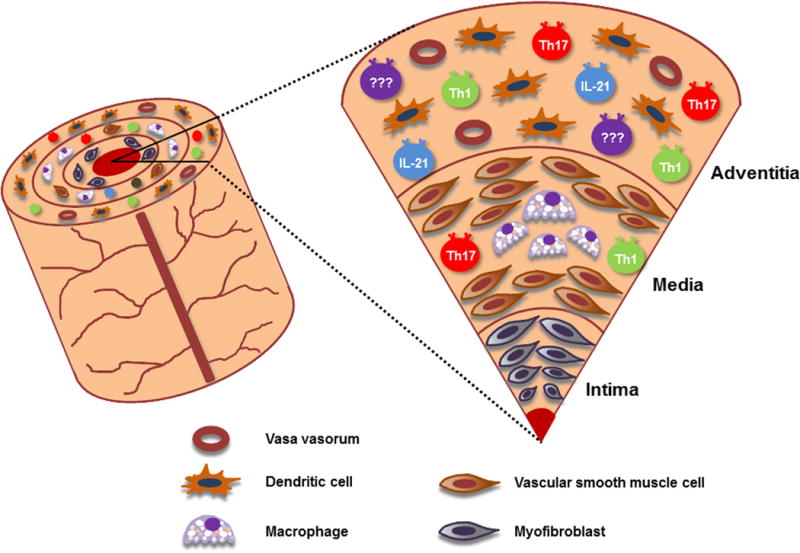
CD4 T cells are the dominant cell population in the vasculitic lesions of GCA, where they partner with macrophages to build granulomatous infiltrates. Based on the production of signature cytokines, CD4 helper cell are subcategorized into distinct effector cell lineages. Vessel wall infiltrates in GCA patients are characterized by a broad array of T helper cell types, suggestive for a broad defect in T cell biology.
Th1 cells are a stable participant in the granulomatous infiltrates of GCA and are also elevated in the circulation (14, 18). IFN-γ has been implicated in a number of pathogenic events in the inflamed vessel wall, predominantly by functioning as a strong activator of endothelial cells, stromal cells, dendritic cells and macrophages (Fig.2). IFN-γ appears critical in inducing vascular endothelial growth factor (VEGF) and promoting neoangiogenesis (20). IFN-γ has been identified as a major chronicity factor in GCA (14). Th1-committed T cells remain the predominant population in both the tissue and the blood (14). Th1 cells are also the most frequent population of differentiated T cells in the blood of healthy individuals, probably a reflection of their critical role in a functional immune system.
Fig. 2. Pro-inflammatory T cells in GCA.
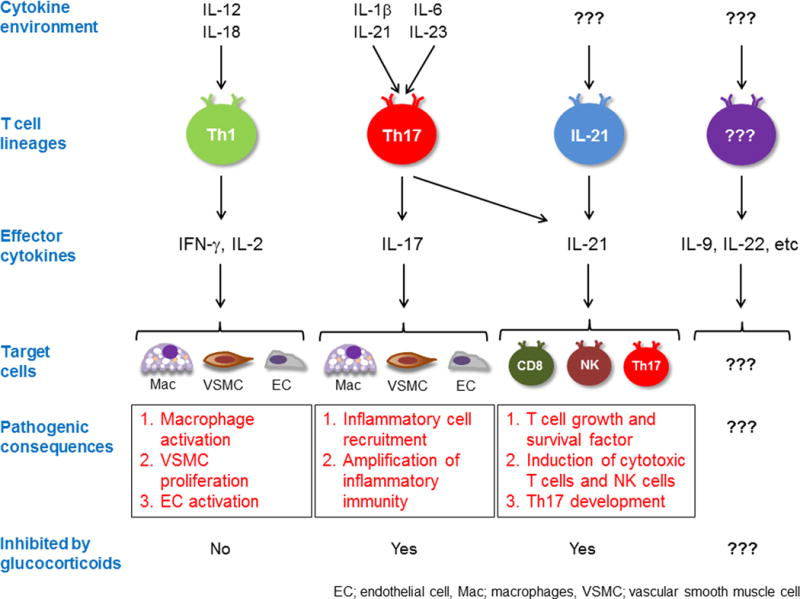
T cells accumulating in the granulomatous infiltrates of GCA are functionally diverse. Based on their cytokine production profile, such lesional T cells are able to interact with selected immune and nonimmune target cells and promote distinct pathogenic pathways. The best understood pathways are outlined. Available data suggest that additional T cell dependent pathogenic cascades are operational in the inflamed arterial wall.
Compared to the dominant role of Th1 cells, all other functional T cell lineages occur at much lower frequencies. Th17 cells are present in the vasculitic infiltrates and in untreated patients are elevated in the peripheral blood. However, the frequencies of Th17 cells, even under conditions of massive expansion, are only around 2% of circulating T cells (14, 18). Th17 cells appear to be easily suppressed with corticosteroid therapy, suggesting dependence of that functional lineage on nonspecific innate amplifiers. Indeed, IL-6, IL-1 and IL-23 are critical drivers of Th17 differentiation and elevated IL-6 levels in GCA patients are promptly responsive to corticosteroids (21). Recent data have emphasized the explicit role of Th17 cells in psoriasis, with prompt and complete resolution of skin lesions when patients are treated with anti-IL-17 reagents (22, 23). In mice, IL-17 cells are critical amplifiers of gut inflammation, but in humans anti-IL-17 therapy is contraindicated in patients with inflammatory bowel disease (24, 25). Thus, the role of Th17-committed T cells may vary with species and organ system. Also, Th17 cells are known to have a high degree of plasticity, able to convert into IL-17–independent effector cells.
IL-9, a typical Th2-related cytokine implicated in asthma (26), has been described in inflamed temporal arteries (17). IL-9 induces the release of VEGF from human mast cells and may have a role in atopic dermatitis (27). Few mast cells have been described as participating in the vessel wall lesions (28). Overall, GCA has no features of an allergic condition and lacks the signature cytokines of Th2-associated inflammation. Further studies will be needed to provide evidence for an active role of IL-9 in affecting the vasculitic response.
Similarly, IL-21, a potent T cell effector cytokine produced mostly by Tfh cells (29), appears abundant in vasculitic lesions and Th21 cells have been described in the circulation (18). Healthy individuals had about 2.5% of IL-21+CD4 T cells in the blood, whereas this frequency was elevated to 8% in patients with active GCA. In ex vivo experiments, recombinant IL-21 shifted the pattern of T cell differentiation, favoring Th1 and Th17 commitment and reducing FoxP3 expression (18). CD4+IL-21+ T cell frequencies correlated closely with IFN-γ–producing Th1 and IL-17-producing Th17 cells and frequencies of all T cell populations responded to therapy. Overall, the T cell compartment of patients with active GCA appeared to be in a state of broad and polyclonal stimulation, with the expression of the T ell activation marker HLA-DR doubled in active versus inactive cases. Terrier et al. have proposed that IL-21 is hierarchically placed at the top of a broad and nonselective T cell activation and that corticosteroid therapy is sufficient to correct this fundamental defect in the immune system of affected individuals (18). This appealing concept opens numerous new questions. What is the cellular source of IL-21? Is the IL-21 produced in the vasculitic lesions sufficient to sustain a system-wide activation status? Therapy seems to normalize T cell activation. Why do vasculitic lesions persist? And, finally, why does GCA lack features of a Tfh disease? Granulomatous lesions, by definition, contain few B cells and have no resemblance to germinal centers, the “playing ground” of Tfh cells. Also, production of autoantibodies or hypergammaglobulinemia has not been identified as a feature of GCA, complicating the design of studies exploring the precise role of IL-21–producing T cells in this vasculitis.
Little is known about a potential role of IL-2 in GCA. A study comparing T cell cytokine signatures in temporal arteries of patients with and without large vessel involvement revealed higher levels of IL-2 gene transcripts in cases with subclavian and aortic GCA (30), suggesting a possible contribution for this T cell growth factor in shaping disease patterning.
Recent elegant work on CD8 T cells in GCA has provided valuable information on long-standing debates in whether this T cell subpopulation participates in the disease process. In general, <5% of tissue-infiltrating cells are CD8+ (31, 32). Reports more than 25 years ago had alerted the community to a selective loss of circulating CD8 T cells, both in percentage and absolute numbers (33–36), with activation markers expressed on remaining CD8 T cells (37). The recent study by Samson et al. confirms the polyclonal stimulation of CD8 T cells and points towards higher representation of tissue CD8 T cells in cases of more severe vasculitis (31). Frequencies and repertoire diversity of CD8 T cells in older individuals is strongly determined by their cytomegalo virus (CMV) infection status (38–41), as CMV is now emerging as a commensurable pathogen in humans, with a strong impact on the fate of CD8 T cells. CMV infections rates and viral loads in GCA patients have not been reported, but may be different in different geographic regions and social strata and immunosuppression may reactivate CMV viral replication. Exploring the interplay of CMV infection and pathogenicity of CD8 T cells may be a fruitful field of exploration.
1.3. Anti-inflammatory T cells in GCA
The intensity and duration of adaptive immune responses depends upon antigen amount, microenvironment in which the antigen is encountered, cytokine signals received by T cells, propensity of T cells to survive or undergo apoptosis, ability of T cells to convert into effector T cells and long-lived memory T cells; and finally, the functional activity of regulatory T cells (Treg), which can modulate each single step of this complex chain of events. Tregs are powerful regulators of immunity, their deficiency linked to hyperimmune syndromes and their surplus linked to immune failure, such as in anti-tumor responses (42, 43). Treg function is therefore a prime concern when considering the immunopathology in GCA; a simple hypothesis being that insufficient Treg function is mechanistically involved in excess immune reactivity in GCA.
Data by Terrier et al. have supported the model that GCA patients may lack sufficient anti-inflammatory T cell function (18). In the blood of patients, the authors found decreased frequencies of CD4+FoxP3+CD25highCD127− Tregs (3% versus 4.4% in age-matched controls), with no improvement upon successful immunosuppressive therapy. In inflamed temporal arteries, no FoxP3 expression was encountered. In further analysis, both resting and activated memory Treg cells were affected by the downregulation; possibly caused by the exuberant Il-21–dependent immune activation.
A recent study has reported a defect in CD8+ anti-inflammatory T cells in GCA and has shifted interest towards a different disease model for GCA (44, 45). In essence, a newly described population of CD8 Tregs has been implicated in controlling clonal expansion and activation threshold setting in the CD4 T cell pool (Fig.3). Localized in the T cell zones of lymphoid organs such CD8 Tregs exert control over CD4 T cells, with no evidence of specific antigen driving the process. Instead, when activated in the presence of IL-15 and distinctly low antigen dose, CD8+ T cells develop into CD8+CCR7+FoxP3+ T cells, that upregulate the enzyme NADPH oxidase 2 (NOX2). NOX2 packaged into membrane microvesicles is released and absorbed by neighboring CD4 T cells, where the oxidase disrupts membrane-proximal signaling pathways and suppresses activation, proliferation and long-term survival of CD4 T cells. CD8 T cells from GCA patients, whether treated or untreated, failed to acquire NOX2 expression, rendering them incapable for lymphoid-tissue-based tolerance induction. The functional consequences should include a robust and polyclonal expansion of CD4 T cells and a generalized bias towards pro-inflammatory effector functions. The loss of CD8+NOX2+ Tregs was explored in other inflammatory conditions. Patients with small vessel vasculitis or with the inflammatory syndrome psoriatic arthritis were capable of inducing this immunoregulatory CD8 T cell population. Corticosteroids could not easily correct the defect, pointing towards a fundamental immunopathology in how GCA patients control the size and the function of their CD4 T cell compartment (44).
Fig. 3. CD8+ CCR7+ FoxP3+ Treg as the missing link in GCA.
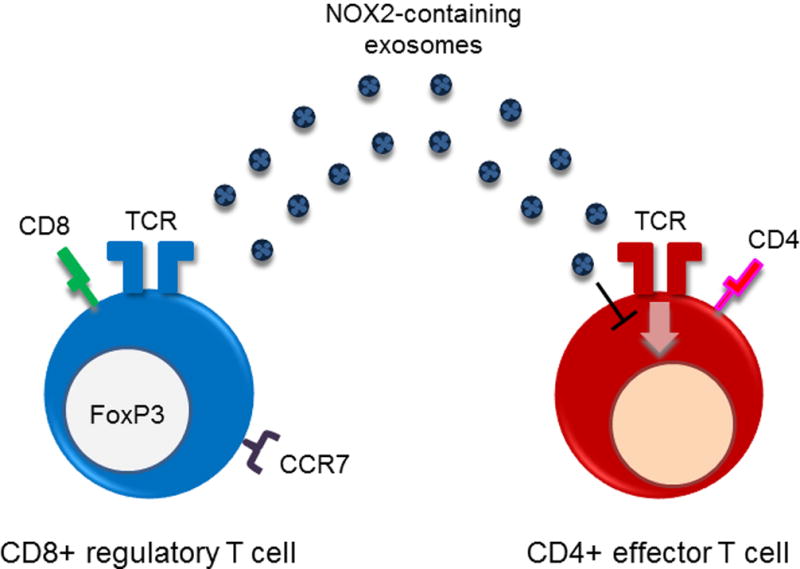
Recently described CD8+ Treg cells suppress CD4 T cell responses by releasing microvesicles packaged with the NADPH oxidase NOX2, thereby controlling the activation of nearby CD4 T cells. NOX2-containing microvesicles are absorbed by CD4 T cells, where they inhibit membrane-proximal T cell receptor signaling events and effectively suppress CD4 T cell expansion and survival. Patients with GCA essentially have lost the ability to induce CD8+NOX2+ Tregs, disrupting control of the CD4 T cell compartment.
Considering the function of Treg cells as overall regulators of the immune system, instead of analyzing their particular role within the inflammatory lesion, opens new opportunities to elucidate the mistakes that the GCA patient’s immune system makes. CD8+Nox2+ Treg cells are typically not present in peripheral lesions, because that is not where they function. Whether Treg cells participate in the regulation of inflammatory intensity in vasculitic lesions is insufficiently understood. Available studies suggest that their number in the lesions may be distinctly low (44). Instead of focusing on what happens in the vasculitic lesions, assessing the overall threshold setting of the immune system may be a more fruitful approach to understand the immunopathology of GCA (Fig.4).
Fig. 4. Defects in central and peripheral immunoregulation in GCA.
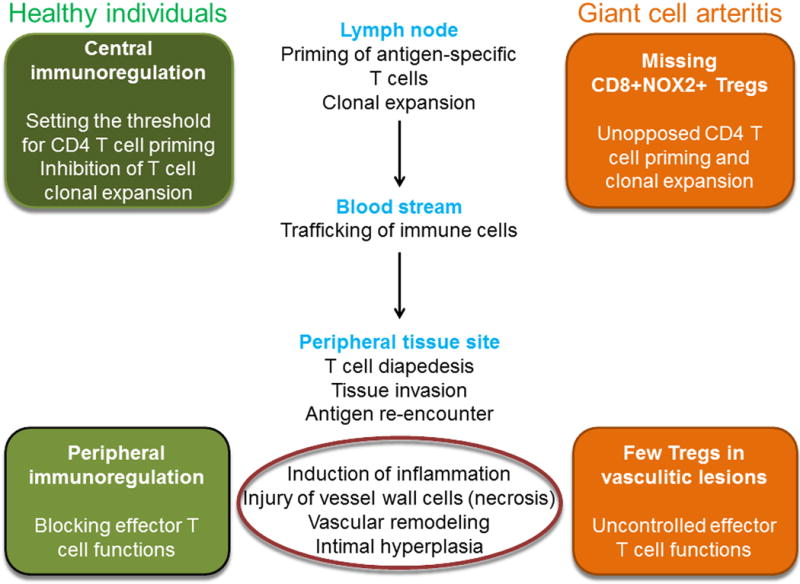
To prevent uncontrolled inflammation, antigen-specific T cells are subject to tolerance-inducing mechanisms both in central lymphoid organs as well as in peripheral tissue sites. Both sites need to be integrated into pathogenic models for GCA. Available data suggest that GCA vascular lesions are poor in anti-inflammatory T cells. Recent studies have revealed a defect in CD8 Tregs, which function in central lymphoid organs and are essentially absent in GCA patients.
Conclusions and missing links
Deviating the focus away from the vasculitic lesions towards broader immune-regulation in the patient’s T cell compartment offers new opportunities to study the immunopathology of GCA. Granuloma formation in the mural layers of vasa-vasorum–containing arteries may simply be the tip of the iceberg of an underlying problem in containing immunity. An insufficiently considered component of disease risk is the strict age association of GCA. Why do patients have to be older than 50 years before they become susceptible? And does this indicate a fundamental difference in immunopathology in GCA and Takayasu’s arteritis, the two closely related subtypes of granulomatous vasculitis.
The immune system changes fundamentally with progressive age (46–48). A reduction in adaptive immunity is often combined with increasing risk for inflammation. Here, failure of anti-inflammatory CD8 Tregs offers an excellent explanation, why CD4 T cells can display strong immune reactivity and produce chronic inflammation. Notably, the discovery of the CD8+NOX2+ Treg defect in GCA was prompted by the observation that inducibility and functional competence of this T cell population is age-dependent. Even in healthy individuals, CD8+NOX2+ Tregs progressively fail with aging. In GCA patients, this process is accelerated. These data are the first evidence that GCA patients have a specific pattern of immunosenescence.
If GCA patients have a global defect in how to control the size and the activation threshold of their CD4 T cell compartment, why would granulomas be formed in a specific tissue niche? Experimental evidence explaining the strict tissue tropism of GCA has been slow in forthcoming. Local amplification mechanisms, such as expression of receptor and ligands on vascular smooth muscle cells fostering immune-stromal interactions, have been described (49). Localization of vessel-specific dendritic cells positioned in close vicinity of the vasa vasorum network has provided insights into the vessel-targeting process (50). Recent data suggesting the local expression of varicella zoster virus in inflamed temporal arteries has added further building blocks to explain a localized persistent immune response (51, 52).
A missing link in understanding the immunopathology of GCA lies in the special relationship of selected arteries with an aging and restructured immune system. Understanding how the process of vascular aging impacts such relationship should be a priority for further research studies.
Finally, the question is raised how the changing concept of GCA pathology affects our approach to diagnosis and therapy. Acute phase reactants, e.g. ESR and CRP, have bene very helpful tools in assessing the activation state of the immune system, as they can be measured reliably and fast. It remains unclear whether there is a biomarker of immune activation that would supplant ESR and CRP measurements. Therapeutic management requires broad-based immunosuppression, given the participation of so many arms of the immune system in the disease process. There is beginning evidence that the vascular and the systemic competent of GCA respond differentially to therapy (53). Ultimately, we will need to deplete the vasculitic infiltrates from the vessel wall as suppression of the systemic inflammation represents only half of the therapeutic goal.
Acknowledgments
This work was supported by the National Institutes of Health (R01 AR042527, R01 AI044142, HL 117913, R01 AI108906, R01 AI108891, R01 AG045779, U19 AI057229, U19 AI057266, I01 BX001669) and the Govenard Discovery Fund.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.”
Footnotes
DISCLOSURE OF INTERESTS
The authors declare no competing financial interests.
References
- 1.Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. Lancet. 2008;372(9634):234–45. doi: 10.1016/S0140-6736(08)61077-6. [DOI] [PubMed] [Google Scholar]
- 2.Weyand C, Goronzy J. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349:160–9. doi: 10.1056/NEJMra022694. [DOI] [PubMed] [Google Scholar]
- 3.Weyand C, Goronzy J. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9:731–40. doi: 10.1038/nrrheum.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weyand C, Goronzy J. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. 2014;371:50–7. doi: 10.1056/NEJMcp1214825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen HJ, Smith AM. The role of the innate immune system in granulomatous disorders. Front Immunol. 2013;4:120. doi: 10.3389/fimmu.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hilhorst M, Shirai T, Berry GJ, Goronzy J, Weyand C. T-cell macrophage interactions in vasculitis and granuloma formation. Front Immunol. 2014;5:432. doi: 10.3389/fimmu.2014.00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prasse A, Kayser G, Warnatz K. Common variable immunodeficiency-associated granulomatous and interstitial lung disease. Curr Opin Pulm Med. 2013;19(5):503–9. doi: 10.1097/MCP.0b013e3283642c47. [DOI] [PubMed] [Google Scholar]
- 8.Rose CD, Neven B, Wouters C. Granulomatous inflammation: The overlap of immune deficiency and inflammation. Best practice & research. 2014;28(2):191–212. doi: 10.1016/j.berh.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 9.O’Neill L, Rooney P, Molloy D, Connolly M, McCormick J, McCarthy G, et al. Regulation of Inflammation and Angiogenesis in Giant Cell Arteritis by Acute-Phase Serum Amyloid A. Arthritis Rheumatol. 2015;67(9):2447–56. doi: 10.1002/art.39217. [DOI] [PubMed] [Google Scholar]
- 10.Mullan RH, McCormick J, Connolly M, Bresnihan B, Veale DJ, Fearon U. A role for the high-density lipoprotein receptor SR-B1 in synovial inflammation via serum amyloid-A. Am J Pathol. 2010;176(4):1999–2008. doi: 10.2353/ajpath.2010.090014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Connolly M, Marrelli A, Blades M, McCormick J, Maderna P, Godson C, et al. Acute serum amyloid A induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/SCID mouse chimera model. J Immunol. 2010;184(11):6427–37. doi: 10.4049/jimmunol.0902941. [DOI] [PubMed] [Google Scholar]
- 12.Johnson BD, Kip KE, Marroquin OC, Ridker PM, Kelsey SF, Shaw LJ, et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE) Circulation. 2004;109(6):726–32. doi: 10.1161/01.CIR.0000115516.54550.B1. [DOI] [PubMed] [Google Scholar]
- 13.Lewis KE, Kirk EA, McDonald TO, Wang S, Wight TN, O’Brien KD, et al. Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation. 2004;110(5):540–5. doi: 10.1161/01.CIR.0000136819.93989.E1. [DOI] [PubMed] [Google Scholar]
- 14.Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121(7):906–15. doi: 10.1161/CIRCULATIONAHA.109.872903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia Rheumatica and Giant Cell Arteritis: A Systematic Review. JAMA. 2016;315(22):2442–58. doi: 10.1001/jama.2016.5444. [DOI] [PubMed] [Google Scholar]
- 16.Weyand CM, Schonberger J, Oppitz U, Hunder NN, Hicok KC, Goronzy JJ. Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J Exp Med. 1994;179(3):951–60. doi: 10.1084/jem.179.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology (Oxford) 2015;54(9):1596–604. doi: 10.1093/rheumatology/kev102. [DOI] [PubMed] [Google Scholar]
- 18.Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. 2012;64(6):2001–11. doi: 10.1002/art.34327. [DOI] [PubMed] [Google Scholar]
- 19.Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Annals of internal medicine. 1994;121(7):484–91. doi: 10.7326/0003-4819-121-7-199410010-00003. [DOI] [PubMed] [Google Scholar]
- 20.Kaiser M, Younge B, Bjornsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am J Pathol. 1999;155(3):765–74. doi: 10.1016/S0002-9440(10)65175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum. 1993;36(9):1286–94. doi: 10.1002/art.1780360913. [DOI] [PubMed] [Google Scholar]
- 22.Fragoulis GE, Siebert S, McInnes IB. Therapeutic Targeting of IL-17 and IL-23 Cytokines in Immune-Mediated Diseases. Annu Rev Med. 2016;67:337–53. doi: 10.1146/annurev-med-051914-021944. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Qian T, Zhang D, Yan H, Hao F. Clinical efficacy and safety of anti-IL-17 agents for the treatment of patients with psoriasis. Immunotherapy. 2015;7(9):1023–37. doi: 10.2217/imt.15.50. [DOI] [PubMed] [Google Scholar]
- 24.Ueno A, Ghosh A, Hung D, Li J, Jijon H. Th17 plasticity and its changes associated with inflammatory bowel disease. World J Gastroenterol. 2015;21(43):12283–95. doi: 10.3748/wjg.v21.i43.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzpatrick LR. Inhibition of IL-17 as a pharmacological approach for IBD. Int Rev Immunol. 2013;32(5–6):544–55. doi: 10.3109/08830185.2013.821118. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol. 2015;15(5):295–307. doi: 10.1038/nri3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sismanopoulos N, Delivanis DA, Alysandratos KD, Angelidou A, Vasiadi M, Therianou A, et al. IL-9 induces VEGF secretion from human mast cells and IL-9/IL-9 receptor genes are overexpressed in atopic dermatitis. PLoS One. 2012;7(3):e33271. doi: 10.1371/journal.pone.0033271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cavazza A, Muratore F, Boiardi L, Restuccia G, Pipitone N, Pazzola G, et al. Inflamed temporal artery: histologic findings in 354 biopsies, with clinical correlations. Am J Surg Pathol. 2014;38(10):1360–70. doi: 10.1097/PAS.0000000000000244. [DOI] [PubMed] [Google Scholar]
- 29.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–63. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 30.Brack A, Martinez-Taboada V, Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum. 1999;42(2):311–7. doi: 10.1002/1529-0131(199902)42:2<311::AID-ANR14>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 31.Samson M, Ly KH, Tournier B, Janikashvili N, Trad M, Ciudad M, et al. Involvement and prognosis value of CD8 T cells in giant cell arteritis. J Autoimmun. 2016 doi: 10.1016/j.jaut.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Braun N, Fritz P, Rieth A, Schroth W, Kimmel M, Biegger D, et al. Predictors for treatment success and expression of glucocorticoid receptor in giant cell arteritis and polymyalgia rheumatica. J Rheumatol. 2009;36(10):2269–76. doi: 10.3899/jrheum.090075. [DOI] [PubMed] [Google Scholar]
- 33.Salvarani C, Boiardi L, Macchioni P, Rossi F, Tartoni P, Casadei Maldini M, et al. Role of peripheral CD8 lymphocytes and soluble IL-2 receptor in predicting the duration of corticosteroid treatment in polymyalgia rheumatica and giant cell arteritis. Ann Rheum Dis. 1995;54(8):640–4. doi: 10.1136/ard.54.8.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnold MH, Corrigall VM, Pitzalis C, Panayi GS. The sensitivity and specificity of reduced CD8 lymphocyte levels in the diagnosis of polymyalgia rheumatica/giant cell arteritis. Clin Exp Rheumatol. 1993;11(6):629–34. [PubMed] [Google Scholar]
- 35.Elling P, Olsson A, Elling H. CD8+ T lymphocyte subset in giant cell arteritis and related disorders. J Rheumatol. 1990;17(2):225–7. [PubMed] [Google Scholar]
- 36.Elling H, Elling P, Olsson A. CD8+ lymphocyte subset in polymyalgia rheumatica and arteritis temporalis. Inverse relationship between the acute hepatic phase reactants and the CD8+ T-cell subset. Clin Exp Rheumatol. 1989;7(6):627–30. [PubMed] [Google Scholar]
- 37.Dasgupta B, Duke O, Timms AM, Pitzalis C, Panayi GS. Selective depletion and activation of CD8+ lymphocytes from peripheral blood of patients with polymyalgia rheumatica and giant cell arteritis. Ann Rheum Dis. 1989;48(4):307–11. doi: 10.1136/ard.48.4.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hadrup SR, Strindhall J, Kollgaard T, Seremet T, Johansson B, Pawelec G, et al. Longitudinal studies of clonally expanded CD8 T cells reveal a repertoire shrinkage predicting mortality and an increased number of dysfunctional cytomegalovirus-specific T cells in the very elderly. J Immunol. 2006;176(4):2645–53. doi: 10.4049/jimmunol.176.4.2645. [DOI] [PubMed] [Google Scholar]
- 39.Leng SX, Qu T, Semba RD, Li H, Yao X, Nilles T, et al. Relationship between cytomegalovirus (CMV) IgG serology, detectable CMV DNA in peripheral monocytes, and CMV pp65(495-503)-specific CD8+ T cells in older adults. Age (Dordr) 2011;33(4):607–14. doi: 10.1007/s11357-011-9205-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202(5):673–85. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whiting CC, Siebert J, Newman AM, Du HW, Alizadeh AA, Goronzy J, et al. Large-Scale and Comprehensive Immune Profiling and Functional Analysis of Normal Human Aging. PLoS One. 2015;10(7):e0133627. doi: 10.1371/journal.pone.0133627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. doi: 10.1016/j.coi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 43.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 44.Wen Z, Shimojima Y, Shirai T, Li Y, Ju J, Yang Z, et al. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest. 2016;126(5):1953–67. doi: 10.1172/JCI84181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collison J. Vasculitis syndromes: Dysfunctional CD8 TREG cells implicated in GCA. Nat Rev Rheumatol. 2016;12(6):314. doi: 10.1038/nrrheum.2016.72. [DOI] [PubMed] [Google Scholar]
- 46.Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol. 2009;9(1):57–62. doi: 10.1038/nri2471. [DOI] [PubMed] [Google Scholar]
- 47.Goronzy JJ, Li G, Yang Z, Weyand CM. The janus head of T cell aging - autoimmunity and immunodeficiency. Front Immunol. 2013;4:131. doi: 10.3389/fimmu.2013.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goronzy JJ, Weyand CM. Immune aging and autoimmunity. Cell Mol Life Sci. 2012;69(10):1615–23. doi: 10.1007/s00018-012-0970-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, et al. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. 2011;123(3):309–18. doi: 10.1161/CIRCULATIONAHA.110.936203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han JW, Shimada K, Ma-Krupa W, Johnson TL, Nerem RM, Goronzy JJ, et al. Vessel wall-embedded dendritic cells induce T-cell autoreactivity and initiate vascular inflammation. Circ Res. 2008;102(5):546–53. doi: 10.1161/CIRCRESAHA.107.161653. [DOI] [PubMed] [Google Scholar]
- 51.Gilden D, Nagel MA. Varicella zoster virus triggers the immunopathology of giant cell arteritis. Curr Opin Rheumatol. 2016;28(4):376–82. doi: 10.1097/BOR.0000000000000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilden D, White T, Khmeleva N, Heintzman A, Choe A, Boyer PJ, et al. Prevalence and distribution of VZV in temporal arteries of patients with giant cell arteritis. Neurology. 2015;84(19):1948–55. doi: 10.1212/WNL.0000000000001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watanabe R, Goronzy JJ, Berry G, Liao YJ, Weyand CM. Giant Cell Arteritis: From Pathogenesis to Therapeutic Management. Curr Treatm Opt Rheumatol. 2016;2(2):126–37. doi: 10.1007/s40674-016-0043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]