

Abstract
Recently, bi-allelic mutations in cytosolic isoleucyl-tRNA synthetase (IARS) have been described in three individuals with growth delay, hepatic dysfunction, and neurodevelopmental disabilities. Here we report an additional subject with this condition identified by whole-exome sequencing. Our findings support the association between this disorder and neonatal cholestasis with distinct liver pathology. Furthermore, we provide functional data on two novel missense substitutions and expand the phenotype to include mild developmental delay, skin hyper-elasticity, and hypervitaminosis D.
Keywords: cholestasis, connective tissue, development, growth, IARS, vitamin D
Graphical abstract
Introduction
Aminoacyl tRNA synthetases (ARSs) are ubiquitously expressed, highly conserved enzymes involved in attaching the appropriate amino acid to tRNA to enable protein synthesis [1]. All ARSs contain catalytic and anticodon recognition domains to catalyse the aminoacylation reactions specific for their cognate amino acids [2]. Some ARSs harbor non-canonical domains with various activities including regulation of gene transcription and expression. In the past decade mutations in a growing number of ARSs were linked to human disease [3, 4]. Four human cytosolic ARSs have been implicated in autosomal dominant Charcot-Marie-Tooth disease [2, 5–7] and mutations in mitochondrial ARSs cause a spectrum of mitochondrial disorders [8]. Recently, Kopajtich et al reported three individuals harboring bi-allelic mutations in cytosolic isoleucyl-tRNA synthetase (IARS) (MIM: 600709). These individuals had a distinct syndrome of prenatal onset growth retardation, intellectual disability, and liver dysfunction [9]. Here, we report an additional subject with bi-allelic missense substitutions in IARS and overlapping clinical findings. This report confirms the association with neonatal cholestasis, expands the IARS-related phenotype and provides functional data on two pathogenic variants detected in a family of Arab descent.
Materials and methods
Genetic testing
The family consented for whole exome sequencing, biological assays and publication of photos according to our IRB approved protocol (clinicaltrials.gov: NCT01294345). Whole-exome sequencing was performed at the NIH Intramural Sequencing Center (NISC) using the SeqCap EZ Exome version 3.0 capture kit (Roche NimbleGen) and the Illumina HiSeq2500 platform. Sequencing data were aligned to the human reference genome using Novoalign (Novocraft Technologies). Variants were called using the in house MPG genotype caller. Detected variants were annotated and filtered using PhenoDB [10].
Yeast complementation assays
To evaluate the functional consequence of the IARS variants identified in the patient described here we performed yeast complementation assays utilizing a pYY1 expression construct. Similar studies have successfully revealed the functional consequences of many disease-associated aminoacyl-tRNA synthetase mutations [4] and were recently employed to study the functional consequences of other IARS variants [9]. Here, the endogenous yeast gene is deleted and the ability of wild-type and mutant human IARS alleles to support yeast cell growth is used as a proxy for enzyme function. Detailed methods are described in the ‘supplemental note’.
Results
Case description
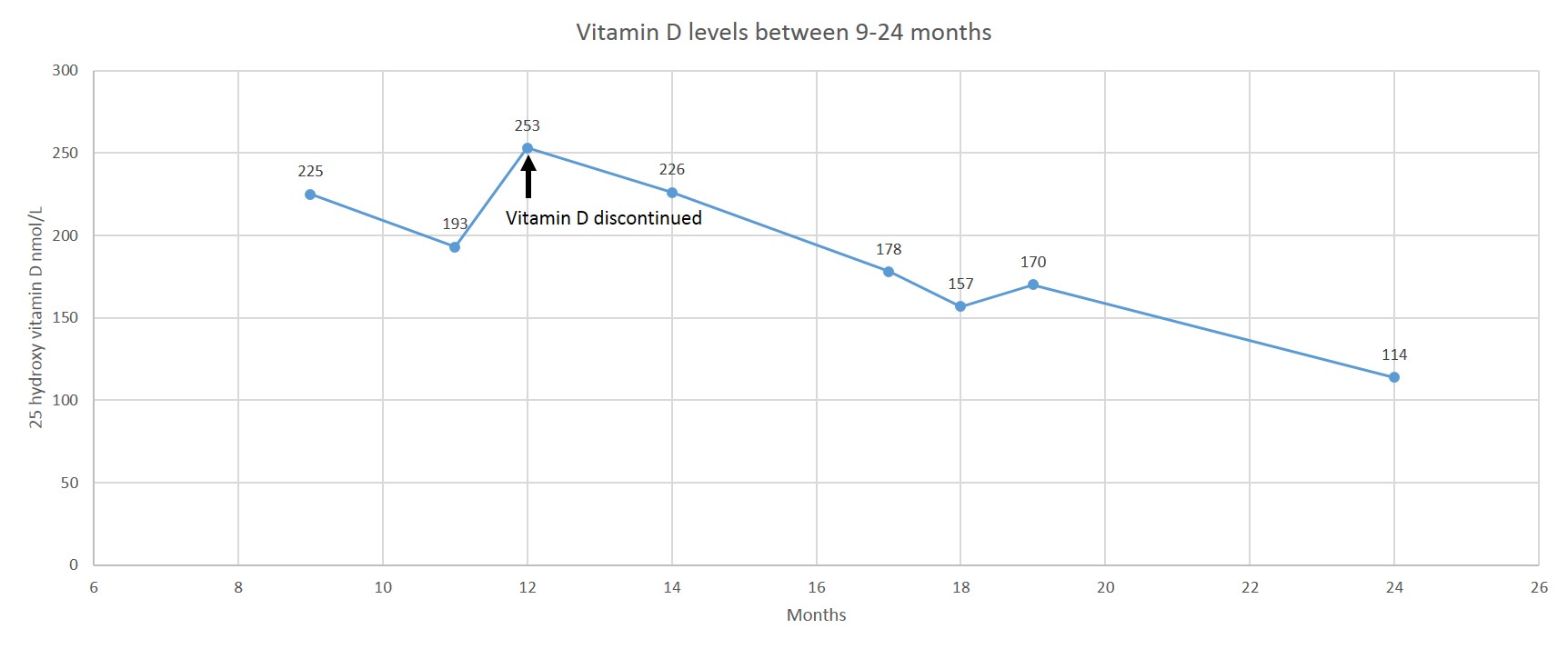
The proband is the third son of non-consanguineous parents of Arab descent. The pregnancy was complicated by abnormal first trimester screening with 1:35 risk for Down syndrome due to increased βHCG. Anatomic ultrasound at 18 weeks was normal, but at 24 weeks decreased growth was noted. During the third trimester there was growth delay and oligohydramnios. He was delivered via spontaneous vaginal delivery at 37 weeks gestation. The birth weight was 1840 grams (Z= −2.52) and the head circumference (HC) was 29.7 cm (Z= −2.6). The Apgar scores were 9 and 10 at 1 and 5 minutes respectively. He was admitted to the NICU for low birth weight and hypoglycemia (27mg/dL) that resolved with feeding. During his admission he developed direct hyperbilirubinemia up to 7.5mg/dL, increased alkaline phosphatase 1450 (U/L) and GGT 311 (U/L). He had abnormal liver enzymes GOT 200 (U/L), GPT 108 (U/L), hypoalbuminemia and hyperammonemia (139). He was treated with vitamin K and fresh frozen plasma because his blood did not clot and INR was not measurable, with improvement in the INR (1.5) that later on normalized. Treatment with Ursodeoxycholic acid was initiated and he received Pregestimil per nasogastric (NG) tube. He had a normal echocardiogram. On abdominal US the liver was normal in size and texture. There was a duplicated collecting system on the left and mild bilateral hydronephrosis. A HIDA scan was normal with no evidence of obstruction. A core needle biopsy of the liver was performed at the age of one month (Fig. 1A). The lobular architecture was preserved, with bile stasis in hepatocytes. Fibrosis with new vessel formation was seen in portal spaces. No bile duct was traced in 8 portal spaces. Bile ductular proliferation was seen and demonstrated by immunostain for Cytokeratin (CK). Gradually the jaundice resolved and the liver enzymes normalized. He had increased levels of 25-hydroxy vitamin D (up tp 253 nmol/L)with normal PTH levels of unknown cause. He received routine supplements until 12 months of age (400 units daily). After discontinuation of supplements levels decreased to the high normal range(Fig. 1S). Repeated zinc levels were within normal range. During his NICU admission there was one episode of a urinary tract infection with E.coli. At 1 year his length, weight and HC measured at −5 SD. On exam he had abnormal fat distribution with a round face, full cheeks and increased fat in the genitalia area. He had joint hyperlaxity and his skin was hyper-elastic, doughy like and transparent (Fig. 1B). He received formula supplements via NG tube until the age of 4 years. He is treated with Growth hormone (GH) since the age of 19 months for GH resistance (abnormal IGF1 generation test), potentially secondary to his liver dysfunction and poor nutrition status. At the age of 4 years the height and length were within normal range (−0.5 SD) and the HC was at −2.5 SD. In terms of his development he walked at 18 months and his first words were at 18 months. Currently he attends a regular preschool but a neuro-psychological evaluation demonstrated a 4–5 months delay on the Cognitive Adaptive Test/Clinical Linguistic and Auditory Milestone Scale (CAT CLAMS) and he is now receiving occupational and physical therapy.
Figure 1. Liver biopsy and photographs of the hands and arms.
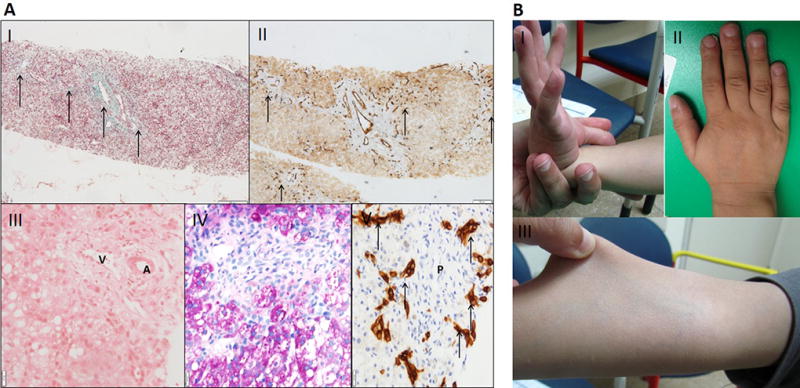
A: Liver biopsy at the age of 1 month. I- Fibrosis in and between portal spaces (arrows) (Masson trichrome stain, X100). II- Multiple new vessels in periphery of portal fibrosis (arrows) (CD34 immunostain, X100). III- Portal space with a vein (V), artery (A). no bile duct is seen (H&E, X200). IV- Portal space with no discernible bile duct (H&E, X200). V- Bile ductular proliferation around portal space (P) (CK19 immunostain, X200).
B: Photographs demonstrating joint laxity and skin hyper-elasticity. I- passive dorsiflexion of the right thumb beyond the flexor aspect of the forearm consistent with joint hypermobility. II- Short hands squared distal fingers and 5th finger clinodactyly. III- The skin of the lower arm is easily stretched indicating abnormal skin elasticity.
Identification of pathogenic IARS variants
The subject underwent a basic metabolic evaluation that did not reveal a diagnosis. . WES identified compound heterozygous variants in IARS: c.2215C>T, p.Arg739Cys and c.1667T>C, p.Phe556Ser (NM_002161) with a low mean allele frequency in the ExAC database of ~60,000 exomes (2.47−5 and 0 respectively). The unaffected sisters did not carry these variants (Fig. 2A). Complementation studies in yeast demonstrated the IARS variants cause a loss-of-function effect on yeast viability (Fig. 3). Growth associated with the p.Phe556Ser variant was only visible after 4 days of incubation at 30°C, while growth associated with wild-type IARS was visible after 2 days of incubation. Furthermore, the number and size of colonies associated with p.Phe556Ser IARS were visibly reduced even after 4 days of incubation. This indicates that p.F556S has impaired function in vivo and is thus a hypomorphic allele. The p.Arg739Cys variant was unable to support any yeast cell growth at both 2 and 4 days of incubation consistent with p.Arg739Cys resulting in a functional null allele.
Figure 2. Segregation, protein location and conservation of IARS variants.
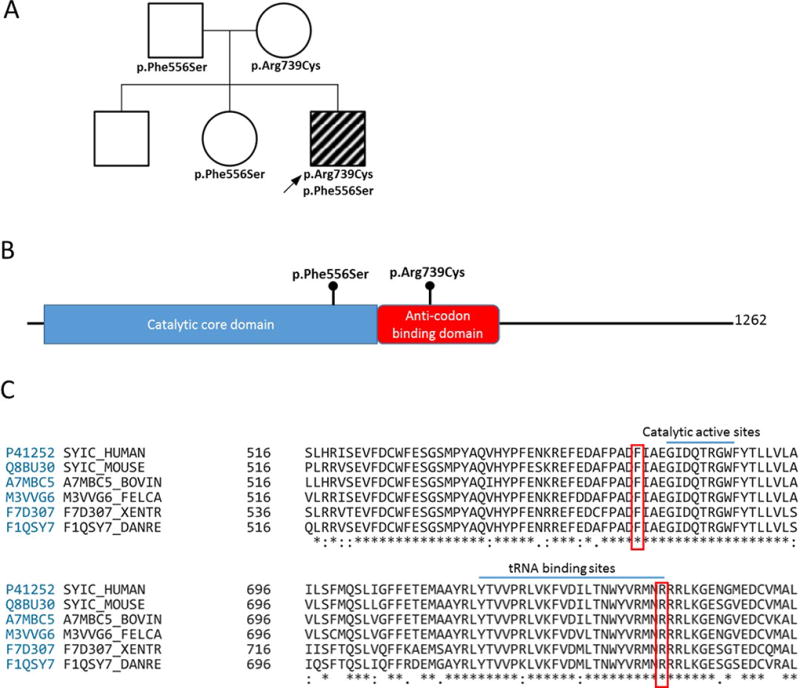
A: Pedigree showing the segregation of IARS variants detected. The two unaffected sisters did not carry both variants. B: IARS protein domains and location of the missense substitutions. C: Conservation down to yeast and previously described catalytic active site [13] and tRNA binding site [12].
Figure 3. Effects of IARS variants on cell growth in yeast complementation assays.
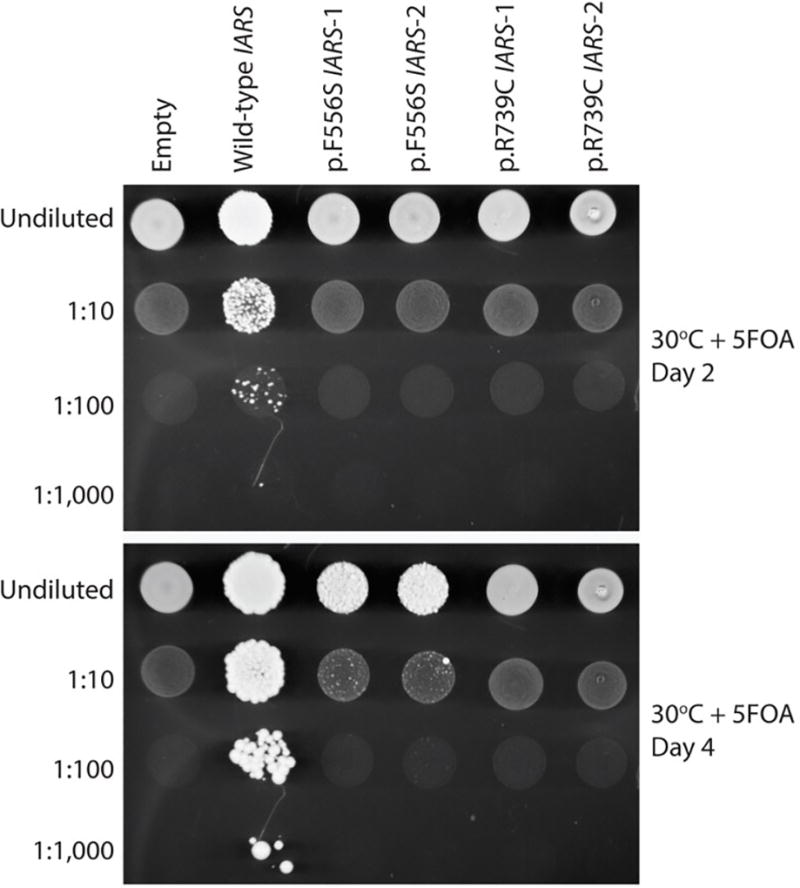
The p.Phe556Ser and p.Arg739Cys IARS variants cause severely depleted cell growth in yeast complementation assays. A haploid yeast strain deleted for endogenous ISL1 (with viability maintained by a URA3-bearing plasmid harboring wild-type ISL1) was transformed with a pYY1 plasmid containing wild-type IARS, the indicated mutant form of IARS, or no insert (“Empty”). Cultures for each strain (labeled along the top) were grown for 2 days in liquid medium, diluted or left saturated (indicated along the left), spotted on solid medium containing 5-FOA to select for spontaneous loss of the URA3-bearing maintenance vector, and grown at 30°C for either two days (top panel) or four days (bottom panel). The ‘1’ and ‘2’ annotations indicate the use of two independently generated mutant IARS expression constructs. Please note the weak growth of cells expressing p.Ph556Ser IARS after four days (compare the 1:10 dilutions of wild-type and p.Phe556Ser IARS in the bottom panel).
Discussion
This report supports the association between bi-allelic pathogenic variants in IARS and a distinct syndrome characterized by intra-uterine-growth delay, liver dysfunction, and neurodevelopmental disabilities. Significant growth delay is a consistent finding in all the reported subjects as well as the potentially equivalent disorder seen in cattle [11]. The cause for growth delay is unknown and might improve with age and / or GH therapy. One subject was reported to have GH deficiency, however here we report GH resistance. Liver dysfunction and abnormal liver pathology was seen in two previously reported patients and only one presented with neonatal cholestasis. That individual also carried a variant of uncertain significance in ABCB11 (MIM: 603201). The subject reported here had a similar presentation with neonatal liver failure and cholestasis. We did not identify any rare variants in genes related to neonatal cholestasis, supporting the role of IARS in this presentation. Here, the liver biopsy was performed at the age of 1 month compared to 1–2 years in previously reported cases. Interestingly, portal tract fibrosis and bile ductular proliferation was already observed. Steatosis was not seen and might be a late manifestation. Bile ducts paucity is seen in very few conditions such as congenital sclerosing cholangitis, Alagille syndrome [MIM: 118450], drug induced liver injury and the vanishing bile duct syndrome. Here, we add an additional condition that may present this way and also associated with cholangiolar proliferation. In contrast to the previously reported cases, development was normal up to the age of 3 years. At 4 years there is evidence of mild developmental delay based on CAT/CLAMS scores but he attends a regular preschool. This supports a variable neurodevelopmental outcome in individuals with this disorder and potentially typical development and IQ in some of the cases. This may depend on the severity of the variants and enzyme residual activity. Recurrent infections, low zinc levels and clinical improvement with zinc supplements were reported in the previous cases. However, in this case zinc levels were normal on repeated tests and there was a single bacterial infection. Interestingly, 25-hydroxy vitamin D levels were high in spite of liver failure, however the levels decreased after discontinuation of routine supplements and the etiology is not clear. . On physical exam there was a strikingly round face compared to trunk, increased fat in the genitalia area, as well as soft, translucent and hyper-elastic skin with joint laxity (Fig. 1B). These findings may have been overlooked previously, or more prominent at a younger age. Reports on additional individuals with this condition will help determine whether this is a common feature that may assist in clinical diagnosis and point to a role of IARS in connective tissue development.
The subject had two novel IARS variants in-trans. The variants were located within the catalytic and anticodon recognition domains and are conserved down to yeast (Fig. 2CD). The c.2215C>T, p.Arg739Cys variant is in a previously described tRNA binding site [12] and the c.1667T>C, p.Phe556Ser variant is 4 residues upstream of a catalytic active site [13]. We performed complementation studies in yeast using the human IARS gene. The IARS variants assessed affected yeast viability to a varying extent. The p.Phe556Ser variant had impaired function but did enable growth, however p.Arg739Cys was unable to support any yeast cell growth (Fig. 3). Therefore, similar to the previously reported cases, the proband has one null allele and one hypomorphic allele. There are currently no reports of individuals with two null alleles, potentially because these will result in early lethality. Similarly, there are no reports on two hypomorphic alleles (compound heterozygous or homozygous). It is possible that such individuals will have a milder and less recognizable phenotype.
In summary, this report supports the association between bi-allelic pathogenic variants in IARS and intra uterine growth delay and liver dysfunction with neonatal cholestasis. We expand the phenotype to include mild developmental delay, skin and joint laxity and hypervitaminosis D. Further reports on individuals with this condition will help define the clinical spectrum.
Supplementary Material
25-hydroxy vitamin D levels were elevated on a few measurements and decreased after routine vitamin D supplements (400 U daily) were discontinued at 12 months.
{kind=link}
Acknowledgments
We are thankful to the family for consenting to participate in this publication. The pYY1 expression construct was a gift from Chin-l Chien and Chien-Chia Wang (National Central University, Taiwan). A.A. is supported by a grant from the National Institute of General Medical Sciences (GM118647). The NIH Intramural Sequencing Center (NISC) conducted the sequencing experiments in this project. This work was supported by the intramural program of the National Human Genome Research Institute, NIH.
Footnotes
All contributors have read and approved the submission to the journal. We are not aware of any conflict of interest.
References
- 1.Schimmel P. Aminoacyl tRNA synthetases: general scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annu Rev Biochem. 1987;56:125–58. doi: 10.1146/annurev.bi.56.070187.001013. [DOI] [PubMed] [Google Scholar]
- 2.Yao P, Fox PL. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med. 2013;5(3):332–43. doi: 10.1002/emmm.201100626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo M, Yang XL, Schimmel P. New functions of aminoacyl-tRNA synthetases beyond translation. Nature Reviews Molecular Cell Biology. 2010;11(9):668–674. doi: 10.1038/nrm2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallen RC, Antonellis A. To charge or not to charge: mechanistic insights into neuropathy-associated tRNA synthetase mutations. Curr Opin Genet Dev. 2013;23(3):302–9. doi: 10.1016/j.gde.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonellis A, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72(5):1293–9. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jordanova A, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38(2):197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 7.Vester A, et al. A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum Mutat. 2013;34(1):191–9. doi: 10.1002/humu.22210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konovalova S, Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol Genet Metab. 2013;108(4):206–11. doi: 10.1016/j.ymgme.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Kopajtich R, et al. Biallelic IARS Mutations Cause Growth Retardation with Prenatal Onset, Intellectual Disability, Muscular Hypotonia, and Infantile Hepatopathy. Am J Hum Genet. 2016;99(2):414–22. doi: 10.1016/j.ajhg.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamosh A, et al. PhenoDB: a new web-based tool for the collection, storage, and analysis of phenotypic features. Hum Mutat. 2013;34(4):566–71. doi: 10.1002/humu.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirano T, et al. IARS mutation causes prenatal death in Japanese Black cattle. Anim Sci J. 2016 doi: 10.1111/asj.12639. [DOI] [PubMed] [Google Scholar]
- 12.Silvian LF, Wang J, Steitz TA. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science. 1999;285(5430):1074–7. [PubMed] [Google Scholar]
- 13.Nakama T, Nureki O, Yokoyama S. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J Biol Chem. 2001;276(50):47387–93. doi: 10.1074/jbc.M109089200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
25-hydroxy vitamin D levels were elevated on a few measurements and decreased after routine vitamin D supplements (400 U daily) were discontinued at 12 months.