

Abstract
Cytosine methylation at CpG dinucleotides is a chief mechanism in epigenetic modification of gene expression patterns. Previous studies demonstrated that increased CpG methylation of Sp1 sites at -268 and -346 of protein kinase C ε promoter repressed the gene expression. The present study investigated the impact of CpG methylation on the Sp1 binding via molecular modeling and electrophoretic mobility shift assay. Each of the Sp1 sites contain two CpGs. Methylation of either CpG lowered the binding affinity of Sp1, whereas methylation of both CpGs produced a greater decrease in the binding affinity. Computation of van der Waals (VDW) energy of Sp1 in complex with the Sp1 sites demonstrated increased VDW values from one to two sites of CpG methylation. Molecular modeling indicated that single CpG methylation caused underwinding of the DNA fragment, with the phosphate groups at C1, C4 and C5 reoriented from their original positions. Methylation of both CpGs pinched the minor groove and increased the helical twist concomitant with a shallow, hydrophobic major groove. Additionally, double methylation eliminated hydrogen bonds on recognition helix residues located at positions -1 and 1, which were essential for interaction with O6/N7 of G-bases. Bonding from linker residues Arg565, Lys595 and Lys596 were also reduced. Methylation of single or both CpGs significantly affected hydrogen bonding from all three Sp1 DNA binding domains, demonstrating that the consequences of cytosine modification extend beyond the neighboring nucleotides. The results indicate that cytosine methylation causes subtle structural alterations in Sp1 binding sites consequently resulting in inhibition of side chain interactions critical for specific base recognition and reduction of the binding affinity of Sp1.
Keywords: DNA methylation, Sp1, protein-DNA recognition, zinc finger, molecular modeling
1. Introduction
Epigenetic mechanisms are essential for development and differentiation and allow an organism to respond to the environment through changes in gene expression patterns. DNA methylation is a chief mechanism in epigenetic modification of gene expression patterns, and occurs at cytosine of the dinucleotide sequence CpG [1-3]. Although cytosine is methylated in 70% of CpGs of mammalian DNA, CpGs in the promoter/enhancer regions of many mammalian genes are not methylated. Methylation in promoter regions is generally associated with transcription repression of the associated genes [1,2,4-6]. Although the molecular mechanisms behind gene repression remain uncertain, studies have indicated that cytosine methylation has a distinct structural impact on DNA. Molecular dynamics and spectral studies demonstrate that subtle changes in nucleotide, sugar pucker and phosphodiester linkage conformation may play a critical role [7-9]. The addition of alkyl groups has also been shown to increase the rigidity of the DNA backbone and adversely affect protein binding [10].
Recent study demonstrated that increased CpG methylation of the Sp1 sites at -268 and -346 of protein kinase C ε (PKCε) promoter caused the gene repression in the developing heart, resulting in heightened susceptibility of ischemia and reperfusion injury [11]. Yet, a molecular explanation for the presumed decreased Sp1 binding affinity as a result of cytosine methylation has not been described. Sp1 is a zinc finger protein consisting of 3 domains each with an alpha helix and an antiparallel beta sheet coordinated to a Zn2+ ion with a Cys2His2 motif [12]. Because the only structural data available is that of the solution structure of the DNA binding domain in its unbound state, its exact mode of recognition remains uncertain. Herein, we present results of a combined experimental and modeling study of Sp1 in complex with the binding sites at -268 and -346 of the PKCε promoter. Using Electrophoretic Mobility Shift Assay (EMSA) and molecular modeling we provide models for cytosine methylation of Sp1 binding sequences causing distinct structural alterations of DNA that inhibit its interaction with Sp1 binding domain.
2. Materials and Methods
2.1. Molecular Modeling
All computations were performed using the software Molecular Operating Environment (MOE) 2008.10 [13]. The 3D coordinates of the molecular model of Sp1 in complex with the sequence AGGGCGGGGCCT was obtained from Dr. Takuya Yoshida from the Graduate School of Pharmaceutical Sciences, Osaka University, Japan [14]. Individual nucleotide bases were manually modified using the Sequence Editor to reflect the -268 and -346 Sp1 binding sites (Table 1). Methyl groups were added manually using the Molecule Builder. All lone pair charges and hydrogen atoms were added with the CHARMM27 [15] force-field loaded. The system was energy minimized using the Generalized Born model of implicit solvation to a 0.05 kcal/mol gradient with Current Geometry selected under Chiral Constraints, to reach a local minimum. Following energy optimization, the van der Waals (VDW) energy of the system was calculated using CHARMM27. VDW potential maps were computed by isolating the DNA sequence and drawing a 4.5Å potential surface [16,17]. DNA-Protein hydrogen interactions were scored as probabilistic potentials of bond formation and were obtained using the Ligand Interactions feature of MOE [18].
Table 1.
Sp1 binding sites.
| -268 | -346 | |
|---|---|---|
| Unmethylated | 5′- CGGGGCGGGGACG- 3′ 3′- GCCCCGCCCCTGC- 5′ |
5′- CGGGGCGGAGGGT- 3′ 3′- GCCCCGCCTCCCA – 5′ |
| C1 methylation | 5′- mCGGGGCGGGGACG-3′ 3′- GmCCCCGCCCCTGC- 5′ |
5′- mCGGGGCGGAGGGT- 3′ 3′- GmCCCCGCCTCCCA - 5′ |
| C6 methylation | 5′- CGGGG mCGGGGACG-3′ 3′- GCCCCG mCCCCTGC- 5′ |
5′- CGGGG mCGGAGGGT – 3′ 3′- GCCCCGmCCTCCCA – 5′ |
| Full methylation | 5′- mCGGGGmCGGGGACG-3′ 3′- GmCCCCGmCCCCTGC- 5′ |
5′- mCGGGGmCGGAGGGT -3′ 3′- GmCCCCGmCCTCCCA - 5′ |
2.2. Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed using oligonucleotides of the -268 and -346 Sp1 binding sites with the recombinant Sp1 protein (Promega), as previously described [19]. Unmethylated and methylated Sp1 oligonucleotides at each CpG site or both CpG sites were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). Probe labeling and EMSA were performed using a biotin 3′ end-labeling kit and a LightShift chemiluminescent EMSA Kit per the manufacture's protocol (Pierce Biotechnology, Rockford, IL). Dot blot and series of controls were performed to ensure sufficient labeling and successful shift. Binding reactions were performed in 20 μl containing 50 fmol of the labeled double-stranded oligonucleotide probes, 1× binding buffer, 1 μg of polydeoxyinosinic acid and polydexycytidylic acid, and 0.2 μg of purified Sp1, in the presence of increasing concentrations of the unlabeled oligonucleotides. DNA-protein complexes were resolved with 6% nondenaturing polyacrylamide minigels (29:1 cross-linking ratio). Nylon membranes were used for transferring DNA-protein complex, followed by UV cross-linking to the membrane. After applying chemiluminescent substrate, the membranes were exposed to Kodak x-ray film (Eastman Kodak) for autoradiography.
3. Results
Two Sp1 binding sites were identified at -346 and -268 in the rat PKCε gene promoter [19]. Each Sp1 binding element contains two CpG dinucleotide sequences at C1 and C6 positions. The eight Sp1 binding sequences studied, with all combinations of unmethylated or methylated cytosine at C1 and C6 are shown in Table 1. EMSA demonstrated that single CpG methylation at either C1 or C6 position decreased the binding affinity of recombinant Sp1 to both Sp1-346 and Sp1-268 binding elements, with the C6 methylation showing a larger decrease at -268 site (Fig. 1). Methylation of both CpG dinucleotide sequences at C1 and C6 positions caused an even greater inhibition of Sp1 binding (Fig. 1).
Fig. 1. Effect of CpG methylation on Sp1 binding.
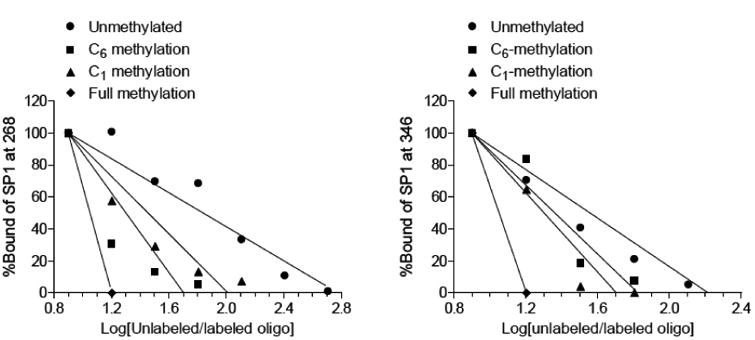
The binding affinity of Sp1 protein to the Sp1 binding sites at -268 and -346 was determined in competition EMSA assays as described in Methods. Recombinant Sp1 was incubated with labeled double-stranded oligonucleotide probes containing the consensus Sp1−346 and Sp1−268 binding motifs with unmethylated, methylated at each of the two CpG sites or both CpG sites. The competition EMSA was performed in the presence of increasing concentrations of the unlabeled oligonucleotides.
To determine the underlying mechanisms of cytosine methylation inhibiting Sp1 binding, a molecular model was built for Sp1 in complex with each of the eight sequences. For each system, the van der Waals (VDW) energy was computed (Fig. 2). VDW values increased progressively with the number of cytosine methylated at both the -268 and -346 site (Fig. 2). In both binding sites, the C6 methylation showed a greater increase in VDW energy than the C1 methylation, and the increases of VDW energy for corresponding systems at -346 were less drastic than at the -268 site (Fig. 2). The higher VDW energy increase by C6 methylation versus C1 methylation partially explains the greater inhibiting effect of C6 methylation on Sp1 binding demonstrated in EMSA.
Fig. 2. VDW Energies of Sp1 bound to -268 and -346 binding sequences.
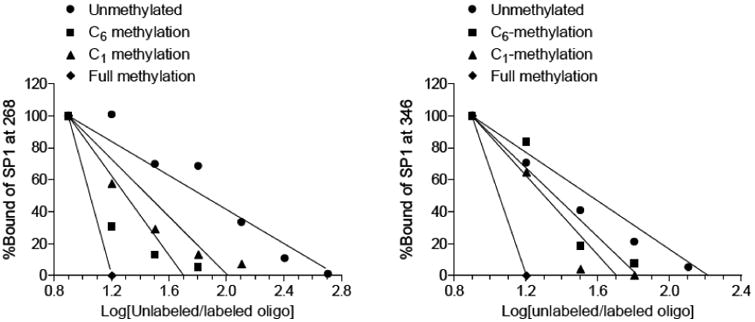
Cytosine methylation of -268 and -346 binding sequences significantly increased the energy of both strands. In either case, C6 methylation had a more drastic effect than that of C1 while methylation of both C1 and C6 was impacted the most.
A molecular interaction (binding) model was developed to ascertain the exact mode of recognition of Sp1 to the -268 and -346 sequences (Table 2, hydrogen bond scores; Figs. (3 and 4), strongest predicated intermolecular interactions). Intermolecular interaction mapping revealed that Sp1 binds to the DNA sequences utilizing residues along the finger 1 beta sheet (Tyr548 and Gly549) as well as positions -1, 1, 3, 5, 6, and 9 on its recognition helices. Also essential to the specificity of Sp1 is Lys535 and the linker residues Arg565 and Lys595, which all reside in the same position relative to the Cys2His2 motif [20].
Table 2.
Intermolecular interactions between Sp1 and -268, -346. Hydrogen bond scores are presented as the percent likelihood of an ideal hydrogen bond formation based on a 4,000 structure PDB training set.25
| Structure | Position | Sp1(-268) Residue | Nucleotide | Atom | Control (%) | meC1 (%) | meC6 (%) | Full (%) |
|---|---|---|---|---|---|---|---|---|
| Turn | Lys535 (NZ) | G8 | O2(Pi) | 62.6 | 77.2 | 0 | 90.4 | |
| β-Sheet | Tyr548 (OH) | G7 | O1(Pi) | 10.2 | 0 | 99.1 | 0 | |
| Turn | Gly549 (N) | G8 | O1(Pi) | 52.7 | 0 | 0 | 0 | |
| Helix 1 | -1 | Lys550 (NZ) | G10 | N7 | 23.1 | 39.1 | 12.9 | 27.3 |
| -1 | Lys550 (NZ) | G10 | O6 | 36.7 | 61 | 82.1 | 85.5 | |
| -1 | Lys550 (NZ) | T11′ | O4 | 10.1 | 0 | 0 | 0 | |
| 1 | Thr551 (OG1) | G12′ | O2(Pi) | 76.2 | 73 | 59.4 | 62.2 | |
| 3 | His553 (NE2) | G9 | N7 | 11.7 | 13.8 | 28.1 | 19.3 | |
| 5 | Arg555 (NH2) | G12′ | O1(Pi) | 62.6 | 56.5 | 48 | 56.4 | |
| Linker | Arg565 (NH2) | G5 | O2(Pi) | 73.4 | 0 | 71.3 | 44.7 | |
| Helix 2 | -1 | Arg580 (NH1) | G7 | N7 | 53 | 0 | 58 | 0 |
| 5 | Gln585 (NE2) | C9′ | O1(Pi) | 31 | 0 | 44.1 | 0 | |
| Linker | Lys595 (NZ) | G2 | O2(Pi) | 84.9 | 91.9 | 60 | 53.1 | |
| Helix 3 | -1 | Arg608 (N) | G3 | O1(Pi) | 12.2 | 0 | 0 | 0 |
| -1 | Arg608 (NH1) | G5 | O6 | 38.7 | 0 | 0 | 0 | |
| 1 | Ser609 (OG) | C7′ | O5(Pi) | 12.1 | 0 | 0 | 0 | |
| 5 | Ser613 (OG) | G6′ | O1(Pi) | 82.7 | 0 | 41.5 | 87.2 | |
| 6 | Lys614 (NZ) | G2 | N7 | 24.7 | 33.7 | 31.3 | 19.1 | |
| 9 | Lys617 (NZ) | C5′ | O1(Pi) | 45.4 | 0 | 49.1 | 44.3 | |
| Structure | Position | Sp1(-346) Residue | Nucleotide | Atom | Control (%) | meC1 (%) | meC6 (%) | Full (%) |
| Turn | Lys535 (NZ) | G8 | O2(Pi) | 54.1 | 68.6 | 39.8 | 48.6 | |
| β-Sheet | His537 (NE2) | A13′ | O2(Pi) | 15.4 | 0 | 19.1 | 20.2 | |
| β-Sheet | Lys546 (NZ) | G7 | O2(Pi) | 55.4 | 62 | 61 | 38.1 | |
| Helix 1 | -1 | Lys550 (NZ) | G10 | O6 | 75.1 | 26.3 | 42.1 | 63.9 |
| 3 | His553 (NE2) | A9 | N7 | 15.4 | 0 | 16.9 | 0 | |
| 5 | Arg555 (NH1) | C11′ | O1(Pi) | 70.7 | 0 | 65.4 | 72.3 | |
| 9 | Arg559 (NH2) | C11′ | O1(Pi) | 51.9 | 71.7 | 62.5 | 96.9 | |
| Linker | Arg565 (NH1) | G5 | O2(Pi) | 87.2 | 11.3 | 52.9 | 0 | |
| Helix 2 | -1 | Arg580 (NH1) | G8 | N7 | 64.3 | 0 | 0 | 0 |
| -1 | Arg580 (NH2) | G8 | O6 | 12.3 | 0 | 0 | 0 | |
| 1 | Ser581 (OG) | C10′ | O1(Pi) | 53.8 | 0 | 72.2 | 0 | |
| Linker | Lys595 (NZ) | G2 | O2(Pi) | 88.6 | 62 | 85.4 | 0 | |
| Linker | Lys596 (NZ) | C8′ | O5(Pi) | 41.5 | 13.1 | 58.9 | 0 | |
| Helix 3 | -1 | Arg608 (NH1) | G4 | O6 | 46.9 | 41.3 | 56.9 | 0 |
| 1 | Ser609 (OG) | C7′ | O5(Pi) | 16 | 10.9 | 14.1 | 0 | |
| 5 | Ser613 (OG) | G6′ | O1(Pi) | 72.3 | 88.6 | 67.3 | 95.3 | |
| 6 | Lys614 (NZ) | G2 | N7 | 33.9 | 42.3 | 49 | 61.2 | |
| 9 | Lys617 (NZ) | C5′ | O1(Pi) | 49.3 | 38 | 46.9 | 42.4 |
Fig. 3. Schematic of major Sp1 H-bonding interactions with the promoter sequence 268.
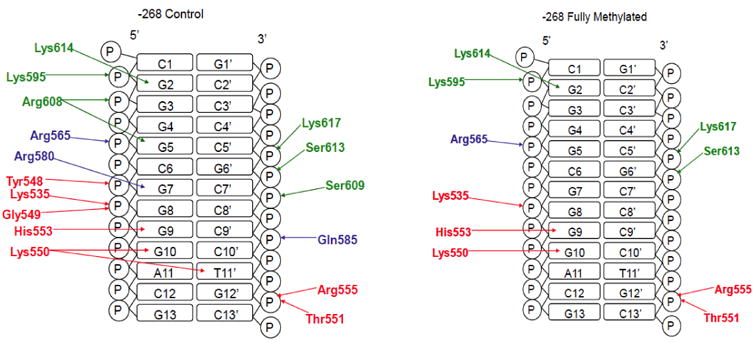
Finger 1 residues are colored red, finger 2 blue, and finger 3 green.
Fig. 4. Schematic of major Sp1 H-bonding interactions with the promoter sequence 346.
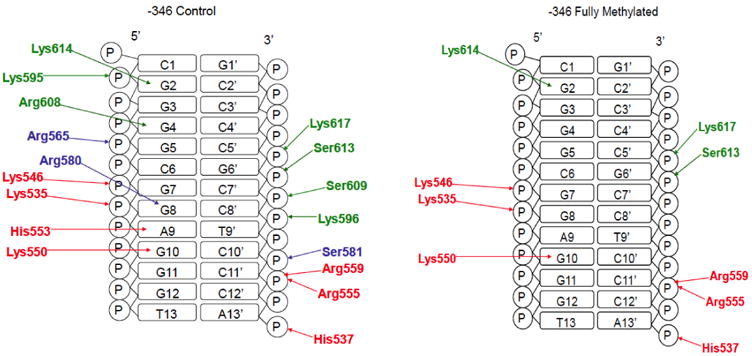
Finger 1 residues are colored red, finger 2 blue, and finger 3 green.
When bound to the non-methylated -268 sequence, several amino acids from zinc finger 3 interact with the phosphate backbone and nucleotide bases (Fig. 3, Fig. 5A). Along the finger 3 recognition helix, a side chain NH3+ hydrogen on Lys617 interacts with the phosphate group of C5′, while another from Lys614 bonds with the N7 at the G2 base. Hydrogen bonding also occurs between the hydroxyl groups of Ser613 and Ser609, and the phosphate groups of G6′ and C7′, respectively. Arg608 interacts with two separate nucleotides: the O6 from the G5 base interacts with the NH proton, and the O1 of the G3 phosphate hydrogen bonds with the backbone nitrogen. Lys595 exists on the finger 2-finger 3 linker with a proton from its NH3+ donated to the O2 on the phosphate group of G2 (Fig. 5B).
Fig. 5. Predicted binding mode of Sp1 to -268.
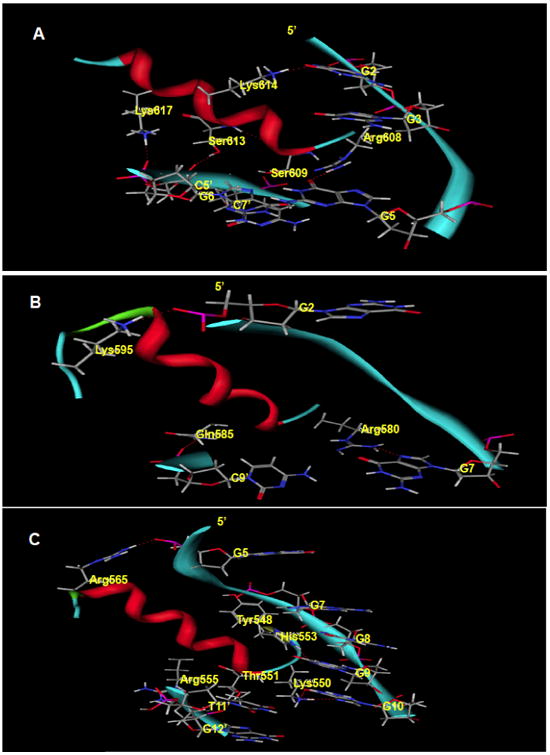
Hydrogen bonding is displayed in dotted red lines. A. The finger 3 helix binding mode involves recognition of the G5 base by Arg608 (-1), bonding between Ser609 (1) and C7′ O5, and interchange between Lys614 and the G2 N7. Ser613 and Lys617 make contacts at phosphate oxygen atoms from G6′ and C5′ respectively. Sp1 binds to the -346 sequence in essentially the same manner only using Arg559 to contact C11′ O1 instead of His553 N7 in finger 1 and Ser581 bonds with C10′ O1 as opposed to Gln585 with C9′ O1. Amino acid backbones have been omitted for clarity. B. At the finger 2 domain, a guanidinyl nitrogen atom from Arg580 (-1) bonds with the N7 of G7 while Gln585 and Lys595 interact with phosphate groups from C9′ and G2 respectively. C. Finger 1 interacts with the phosphate backbone using components of the antiparallel beta sheet: Lys535 (not shown), Tyr548, Gly549 (not shown). Lys550 (-1) recognizes N7 and O6 atoms from the G10 base along with His553 (3). Thr551 (1), Arg555 (5) and the linker residue Arg565 interact with phosphate backbones from Guanine nucleotides.
Along the finger 2 helix, Gln585 interacts with the O1 of the C9′ phosphate via its side chain amide group (Fig. 3, Fig. 5B). Also in the finger 2 domain, the NH of the Arg580 side chain bonds with the N7 of the G7 base (Fig. 3, Fig. 5B). The finger 1-finger 2 linker provides an additional interaction from Arg565, where one of its guanidinyl amines interacts with the O2 located on the phosphate backbone of G5 (Fig. 3, Fig. 5C).
Similarly, along the finger 1 helix, an Arg555 amine interacts with the O1 of G12′ (Fig. 2, Fig. 5C). Further down the helix, the NE2 of His553 forms a hydrogen bond with the N7 of G9. At the N-terminus of the recognition helix, Lys550 forms three hydrogen bonds on the bases of two nucleotides. The side chain amino group contacts the G10 base at O6 (carbonyl oxygen) and N7, while also interacting with the T11′ base at O4. At the N-terminus of finger 1, a phosphate contact is made between the side chain NH3+ of Lys535 and the O2 of G8. Tyr548, a component of the antiparallel beta sheet (His537-Cys539 and Lys546-Tyr548), engages in hydrogen bonding with the O2 at the phosphate of G7 via its hydroxyl group. Additional phosphate interactions occur through the backbone nitrogen of Gly549, which donates a proton to the O1 of G8, and the Thr551 hydroxyl that bonds with the O2 of G12′.
Sp1 binds to the -346 site in a nearly identical manner (Table 2 and Fig. 4). At finger 3, Lys617, Lys614, Ser613, Ser609 and Lys595 all interact with the same functional groups as in the -268 site. However, what is distinctly different from the -268 recognition mode is that Arg608 recognizes the G4 base rather than G5, and that the Lys596 NZ contacts a C8′ phosphate oxygen. At the finger 2 domain, Arg580 contacts the N7 of G8 and the hydroxyl of Ser581 undergoes an exchange with the phosphate O1 of C10′. Arg565 contacts the G5 phosphate as in the -268 pattern. The finger 1 recognition pattern utilizes Arg559 and Arg555 to recognize oxygen atoms on the C11′ phosphate while His553 and Lys550 recognize the A9 and G10 bases, respectively. Phosphate oxygen atoms of G7, G8 and A13′ are bonded to side chain atoms of Lys546, Lys535 and His537, respectively.
Probabilistic hydrogen bond scoring (Table 2) indicates that cytosine methylation significantly reduces or eliminates the percent likelihood of hydrogen bond formation between most of the base/phosphate atoms and Sp1 residues critical for recognition (Figs. 3 and 4). This is particularly true for residues on positions -1 and 1 of the recognition helices. At the -268 sequence, Lys550 (-1) had its predicted interaction with the T11′ O4 eliminated in response to single site or both CpG methylation. Thr551, the first residue of the finger 1 helix, had a reduced score with the G12′ O2 in singly or fully methylated scenarios. Along the finger 2 helix, the interaction from the NH1 nitrogen of Arg580 (-1) to the N7 of G7 was disrupted in the cases of C1 and full methylation. Similarly, at the finger 3 helix, Arg608 (-1) and Ser609 (1) no longer interacted with the oxygen atom at the phosphate and cytosine base when -268 was singly or fully methylated. Likewise, the methylation of the -346 region reduced hydrogen bond scoring between the residues located at -1 and 1 of the recognition helix and the nucleotides of DNA (Table 2). In either single site methylation or full methylation, the interaction probability between Lys550 (-1) of the finger 1 helix and the G10 base was significantly reduced while the bonds from the guanidinyl group of Arg580 (-1) of the finger 2 helix to the N7 and O6 base atoms of G8 were not projected at all (0%). Full methylation of -346 also completely eliminated projected hydrogen bonds from Ser581 (1), Arg608 (-1) and Ser609 (1). In addition to the loss of the predicted H-bonding from the residues at -1 and 1 of the recognition helices, the H-bonding from linker residues Arg565, Lys595, Lys596 (-346 only) and Gly549 (-268 only) were also reduced or abolished in response to the full methylation at -268 site and -346 site.
Van der Waals (VDW) contact mapping of -268 and -346 sequences revealed that CpG methylation causes distinct structural alterations at the major and minor grooves (Figs. 6 and 7). The unmethylated sequence of -268 has a polar major groove (Fig. 6A). Methylation of C1 caused an unwinding of the first three base pairs in the -268 sequence (Fig. 6B), and this was considerably more pronounced in the case of C6 methylation (Fig. 6C). This change in structure had the effect of widening the minor groove opposite to the finger 3 helix site. C6 methylation caused a pinching of the minor groove opposite the finger 1 site as well as an increasing of the hydrophobicity of the major groove (Fig. 6C). Methylation at both C1 and C6 caused further decrease in major groove polarity, while maintaining the unwinding and the minor groove pinching seen for the individual methylations (Fig. 6D).
Fig. 6. VDW Potential Map of -268 DNA.
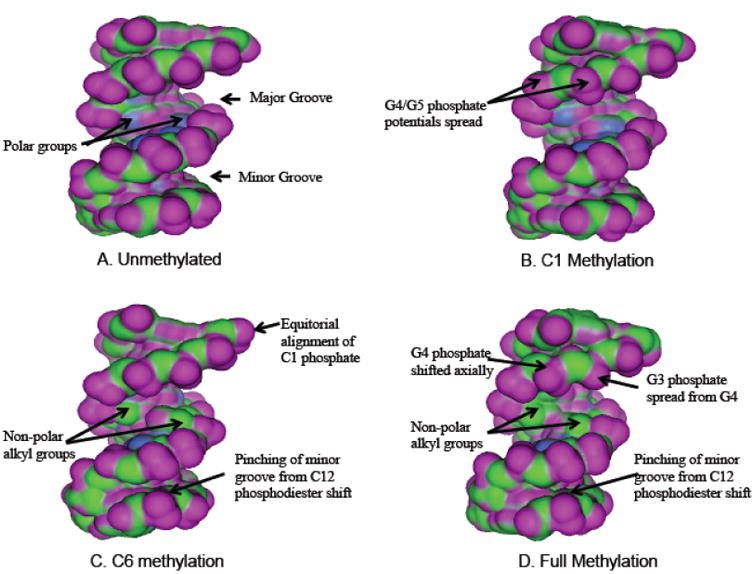
A unmethylated. B. C1 methylation. C. C6 methylation. D: full methylation. Purple represents regions of Hydrogen bonding potential, blue is polar and green is hydrophobic. Realignments of the phosphate potentials in response to cytosine methylation are labeled with arrows and are described in the text.
Fig. 7. VDW Potential Map of -346 DNA.
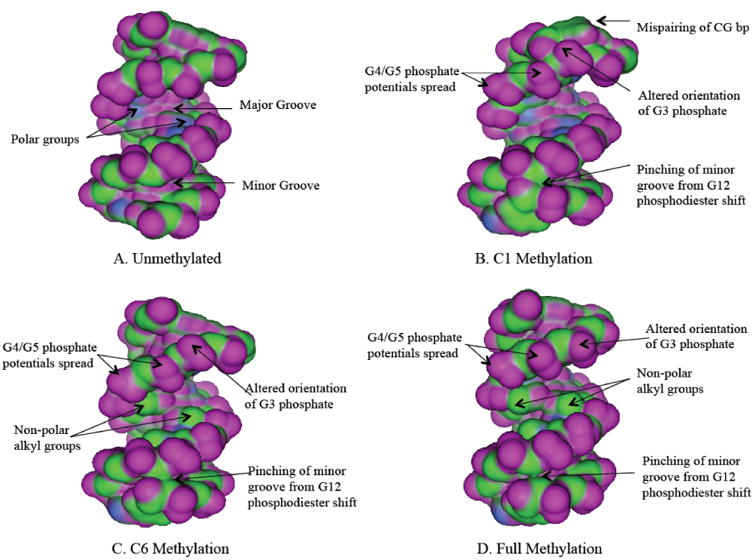
A unmethylated. B. C1 methylation. C. C6 methylation. D: full methylation. Color coding of potentials is explained for Fig. (6).
Methylation of -346 resulted in conformational changes similar to that of -268. Pinching of the minor groove was observed in single pair methylations as well as when the sequence had both pairs methylated (Fig. 7). Methyl groups added to C1 caused C1-G1′ mispairing, thus drastically altering the backbone conformation of both the 5′ and 3′ strands. The G2 phosphate oriented downward while the G3 phosphate became flipped away from the major groove. Moreover, the T13-A13′ positions also mis-paired, leading to pinching of the minor groove. C6 methylation produced a similar effect at the minor groove near finger 1. When both cytosine pairs were methylated in the -346 sequence, the entire minor groove became narrower while the major groove showed a significant decrease in polarity.
The local conformational changes of the DNA sequence by methylation led to an increase in the helical twisting of its overall structure, resulting in an upward shift in the -268 sequence and a downward shift in the -346 sequence (Fig. 8). The shifting of the sequences may be one of the major reasons for the loss of many of side chain interactions with Sp1.
Fig. 8. Impact of cytosine methylation on the structure of sp1 binding sites.
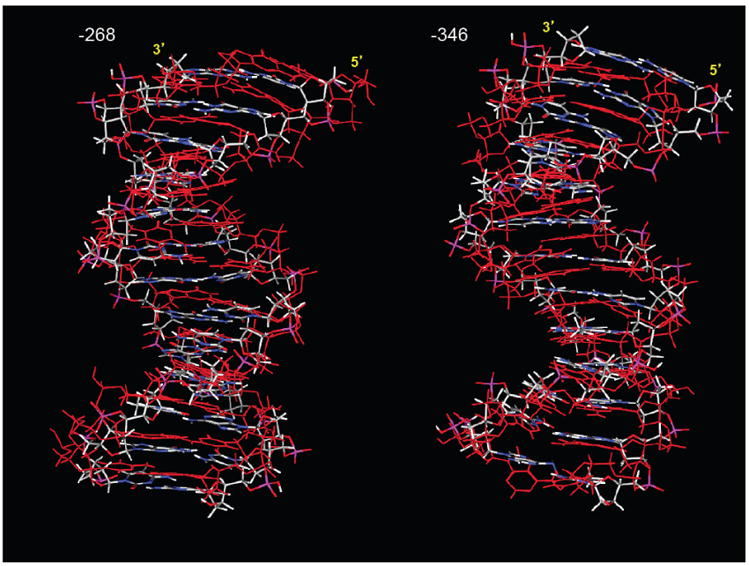
Fully methylated sequences are displayed in red, overlayed on control (unmethylated) sequences colored in grey (carbon)-blue (nitrogen)-white (hydrogen)-red (oxygen).
Discussion
The computed models were based on DNA binding model structure of Sp1 built by Dr. Takuya Yoshida' group [14], modified in silico and minimized to create the eight bound species. Despite the simplicity of the approach, the models show remarkable agreement with the experimental results; the VDW energies, used as a proxy for binding energies largely parallel the observed binding affinities, and the models show distinctive conformational change of the DNA fragment with corresponding reduced binding interactions, upon increasing methylation.
Reorientation of the phosphate groups from steric crowding of methylated cytosine bases appears to be a key facilitator of the observed structural alterations. When cytosine was methylated at C6 position, the major groove at the finger 2 active site became shallow and sterically crowded. The 5′ strand of -268 binding sequences appeared more extended accommodate changes in orientation of C1 and G2 phosphates upon C6 cytosine methylation, specifically these two phosphates which were pointed toward the major groove before methylation, oriented equatorially after methylation. Contraction of the minor grooves appears to occur as a function of the phosphodiester linkages on opposing strands coming into closer contact with one another. Specifically, this was observed in -268 binding sequences where the O5 of G5 shifted toward the corresponding linkage at G1′ while the O5 of C12 moved closer to the C9′ backbone. In both cases, the minor groove was tightened and the aforementioned 5′ phosphate groups were realigned. Likewise, at -346 binding sequences the O5 bonds of G5 and G12 underwent comparable translations.
The impact of cytosine methylation-induced structural alterations on Sp1 binding was evaluated. Overall, methylation of both C1 and C6 cytosine causes an increase in the helical twisting of the Sp1 binding sites (Fig. 8). This causes a significant change in the conformation of the major and minor grooves and, thereby, impairs the extent to which Sp1 can bind. Alteration of the major groove inhibits side chain interactions from residues at positions -1 and 1 of the zinc-finger helices, which have been documented to be critical for recognition of G and C bases [21, 22]. Moreover, it has been suggested that the backbones of linker residues become more rigid and ordered upon binding, thus, indicating their critical role in molecular stabilization [14]. Our results indicate that cytosine methylation abolishes contacts between the phosphate backbone and the linkers Arg565, Lys595, Lys596 at -346 binding sequences, as well as the turn residue Gly549 at -268 binding sequences, thereby, providing an additional explanation for the observed reduction in Sp1 binding affinity.
Methylation of single or both cytosine at C1 and C6 caused conformational changes throughout each oligonucleotide binding sequences and affected hydrogen bonding from all three Sp1-DNA binding domains, demonstrating that the consequences of cytosine methylation extend beyond the adjacent nucleotides. Also noteworthy is the markedly greater impact of cytosine methylation on -268 binding sequences as opposed to -346 binding sequences exhibited in van der Waals energy computations, which implies that the consequences of cytosine methylation are sequence specific. A possible explanation for the relative stability of -346 binding sequences is the presence of an additional A-T base pair that may make the sequence less susceptible to structural shifts due to cytosine methylation. Furthermore, the inhibiting effect of C6 methylation was shown to be greater than that of C1 based on VDW energy calculations, which is consistent with the result of EMSA in the -268 sequence, but not in -346. It is possible that Sp1 affinity is not only determined by the value of VDW energy, but also by the steric factor. As we can see from Figs. (6 and 7), C6 methylation changed the major groove from polar to nonpolar while C1 methylation did not. The loss of many strong polar interactions at the major groove probably led to the greater increase of VDW energy for C6 methylation in both -268 and -346. The enigma that C1 methylation had a slightly greater inhibiting effect than C6 methylation in -346 could be answered by its drastic structural changes due to C1 methylation (Fig. 7B). As demonstrated in Fig. (7B), the C1 methylation in -346 binding sequences totally changed its local conformation and destroyed the initial GC pairing, which is still partially preserved in C6 methylation.
In conclusion, the present study has demonstrated the structural impact of combinations of cytosine methylation on Sp1 binding sites in gene promoter as well as the resulting changes in Sp1 hydrogen bonding and VDW interactions that consequently reduce Sp1 binding affinity. Steric crowding of the methyl groups appears to be a primary contributor to the reorganization of the DNA backbone by altering the orientation of phosphate groups and sugar-phosphate bonds. Changes in phosphate position reported previously [8] using 31P NMR spectroscopy indicated that cytosine methylation impacts the phosphate of the altered base and those of its flanking nucleotides. This is consistent with our data, which show changes both in orientation at the sites of methylation and in hydrogen bonding contacts at nearby nucleotides. Furthermore, several studies have also reported similar modifications in the helical twist and major/minor grooves as a direct consequence of DNA methylation [8,23,24]. Using NMR and MD simulations, Derreumaux and coauthors also reported similar findings of the methylated consensus sequence of the cAMP response element, which they attributed to steric crowding and changes in the solvation energy [9]. It is also plausible that such structural shifts, particularly along the sugars and phosphate groups, may be responsible for the reduced flexibility of the DNA backbone that has been observed in response to cytosine methylation [7,10,25]. Given that Sp1 is an important transcription factor that participates in the regulation of expression of many genes, the present study provides molecular insight into the role of cytosine methylation in regulating the transcription factor binding that contributes to the understanding of epigenetic modification of gene expression patterns.
Acknowledgments
We gratefully acknowledge Dr. Takuya Yoshida from the Graduate School of Pharmaceutical Sciences at Osaka University, Japan for generously providing the 3-D coordinates of the DNA binding model structure of Sp1. We also thank Haitao Zhang for his technical support. This work was supported by National Institutes of Health Grants S06GM073842 (to S.Y.) and HL118861 (to L.Z.). John Tenayuca is a NIH MIDARP Scholar.
List of Abbreviations
- EMSA
Electrophoretic Mobility Shift Assay
- Prkcε
Protein kinase c epsilon
- VDW
Van Der Waals
Footnotes
Conflict of Interest: The authors confirm that this article content has no conflict of interest.
Disclaimer: The above article has been published in Epub (ahead of print) on the basis of the materials provided by the author. The Editorial Department reserves the right to make minor modifications for further improvement of the manuscript.
References
- 1.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinisic and environmental signals. Nat Gen. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Takei D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 3.Reik W, Dean W. DNA methylation and mammalian epigenetics. Electrophoresis. 2001;22:2838–2843. doi: 10.1002/1522-2683(200108)22:14<2838::AID-ELPS2838>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 4.Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM. Epi-genetic regulation of 11 beta-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest. 2004;114:1146–1157. doi: 10.1172/JCI21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antequera F, Boyes J, Bird A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. 1990;62:503–514. doi: 10.1016/0092-8674(90)90015-7. [DOI] [PubMed] [Google Scholar]
- 6.Jones PL, Wolffe AP. Relationships between chromatin organization and DNA methylation in determining gene expression. Semin Cancer Biol. 1999;9:339–347. doi: 10.1006/scbi.1999.0134. [DOI] [PubMed] [Google Scholar]
- 7.Banyay M, Gräslund A. Structural effects of cytosine methylation on DNA sugar pucker studied by FTIR. J Mol Biol. 2002;324:667–676. doi: 10.1016/s0022-2836(02)01104-x. [DOI] [PubMed] [Google Scholar]
- 8.Lefebvre A, Mauffret O, Saïd EA, Monnot M, Lescot E, Fermandjian S. Sequence dependent effects of CpG cytosine methylation A joint 1H-NMR and 31P-NMR study. Eur J Bio-chem. 1995;229:445–454. doi: 10.1111/j.1432-1033.1995.0445k.x. [DOI] [PubMed] [Google Scholar]
- 9.Derreumaux S, Chaoui M, Tevanian G, Fermandjian S. Impact of CpG methylation on structure, dynamics and salvation of cAMP DNA responsive element. Nucleic Acids Res. 2001;29:2314–2326. doi: 10.1093/nar/29.11.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nathan D, Crothers DM. Bending and flexibility of methylated and unmethylated EcoRI DNA. J Mol Biol. 2002;316:7–17. doi: 10.1006/jmbi.2001.5247. [DOI] [PubMed] [Google Scholar]
- 11.Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKCε gene repression in rat hearts. Circ Res. 2010;107:365–373. doi: 10.1161/CIRCRESAHA.110.221259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narayan VA, Kriwacki RW, Caradonna JP. Structures of zinc finger domains from transcription factor sp1. J Biol Chem. 1997;272:7801–7809. doi: 10.1074/jbc.272.12.7801. [DOI] [PubMed] [Google Scholar]
- 13.Chemical Computing Group Inc.; Montreal Quebec, Canada: Molecular Operating Environment 2008.10 (MOE) http://www.chemcomp.com. [Google Scholar]
- 14.Oka S, Shiraishi Y, Yoshida T, Ohkubo T, Sugiura Y, Kobayashi Y. NMR structure of transcription factor Sp1 DNA binding domain. Biochemistry. 2004;43:16027–16035. doi: 10.1021/bi048438p. [DOI] [PubMed] [Google Scholar]
- 15.Feller SE, MacKerell AD. An improved empirical potential energy function for molecular simulations of phospholipids. J Phys Chem. 2000;104:7510–7515. [Google Scholar]
- 16.Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macro-molecules. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- 17.Bobbyer DNA, Goodford PJ, McWhinnie PM, Wade RC. New hydrogen-bond potentials for use in determining energetically favorable binding sites on molecules of known structure. J Med Chem. 1989;32:1083–1094. doi: 10.1021/jm00125a025. [DOI] [PubMed] [Google Scholar]
- 18.Labute P. Probabilistic Receptor Potentials. Journal of the Chemical Computing Group. 2001 http://www.chemcomp.com/journal/cstat.htm.
- 19.Zhang H, Meyer KD, Zhang L. Fetal exposure to cocaine causes programming of Prkce gene repression in the left ventricle of adult rat offspring. Biol Reprod. 2009;80:440–448. doi: 10.1095/biolreprod.108.072983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marco E, Garcίa-Nieto R, Gago F. Assesment by molecular dynamics simulations of the structural determinants of DNA-binding specificity for transcription factor Sp1. J Mol Biol. 2003;328:9–32. doi: 10.1016/s0022-2836(03)00243-2. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki M, Gerstein M, Yagi N. Stereochemical basis of DNA recognition by Zn fingers. Nucleic Acids Res. 1994;22:3397–3405. doi: 10.1093/nar/22.16.3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elrod-Erickson M, Rould MA, Nekludova L, Pabo CO. Zif268 protein-DNA complex refined at 1.6Å: a model system for understanding zinc finger-DNA interactions. Structure. 1996;4:1171–1180. doi: 10.1016/s0969-2126(96)00125-6. [DOI] [PubMed] [Google Scholar]
- 23.Marcourt L, Cordier C, Couesnon T, Dodin G. Impact of C5-cytosine methylation on the solution structure of d(GAAAACGTTTTC)2 An NMR and molecular modeling investigation. Eur J Biochem. 1999;265:1032–1042. doi: 10.1046/j.1432-1327.1999.00819.x. [DOI] [PubMed] [Google Scholar]
- 24.Heinemann U, Hahn M. CCAGGCm5CTGG helical fine structure, hydration, and comparison with CCAGGCCTGG. J Biol Chem. 1992;267:7332–7341. [PubMed] [Google Scholar]
- 25.Geahigan KB, Meints GA, Hatcher ME, Orban J, Drobny GP. The dynamic impact of CpG methylation in DNA. Biochemistry. 2000;39:4939–4946. doi: 10.1021/bi9917636. [DOI] [PubMed] [Google Scholar]