

Abstract
Maintenance of genome stability depends on efficient, accurate repair of DNA damage. DNA double-strand breaks (DSBs) are among the most lethal types of DNA damage, with the potential to cause mutation, chromosomal rearrangement, and genomic instability that could contribute to cancer. DSB damage can be repaired by various pathways including nonhomologous end-joining (NHEJ) and homologous recombination (HR). However, the cellular mechanisms that regulate the choice of repair pathway are not well understood. Recent studies suggest that the tumor suppressor protein CtIP controls the decision to repair DSB damage by HR. It does so by regulating the initiation of DSB end resection after integrating signals from the DNA damage checkpoint response and cell cycle cues.
CtIP links cell cycle control, DNA damage checkpoints and repair
CtIP, the CtBP (carboxy-terminal binding protein) interacting protein, was initially characterized for its role in transcription – first, as a cofactor for the transcriptional repressor CtBP, and also as a binding partner for other proteins including the cell cycle regulators retinoblastoma protein (Rb) and breast cancer 1 (BRCA1) [1–4]. In the G1 phase of the cell cycle, CtIP associates with Rb, allowing CtIP to bind its own promoter as well as the promoters of other E2F target genes such as Cyclin D1. This releases Rb-mediated transcriptional repression and increases expression of genes required for S phase entry [5]. CtIP and Rb could also directly regulate the initiation of DNA synthesis by interaction with MCM7, a component of the replicative helicase [6]. CtIP therefore has both transcription-dependent and -independent roles in cell cycle progression [7,8]. CtIP also plays a central role in the cell cycle checkpoint response to DNA DSBs, with new evidence demonstrating that CtIP controls the choice of DSB repair pathway (Figure 1).
Figure 1.

Overview of the role of CtIP in the cellular response to DSB damage. DSB damage induces checkpoint activation at the G1/S transition, within the S phase, or at the G2/M phase transition. The G1/S and G2/M checkpoints block entry to the S and M phases, respectively, whereas the intra-S phase checkpoint inhibits late origin firing and slows down DNA replication elongation. CtIP is required for activation of the G2/M checkpoint and could also act in the intra-S phase checkpoint. The phase of the cell cycle in which DSB damage occurs influences the choice of repair pathway. In the G1 phase, DNA DSBs are primarily repaired by NHEJ. In the S phase and G2 phases, DSBs can be repaired by HR as well as by NHEJ. The HR pathway requires CtIP, together with CDK, BRCA1, ATM and MRN, to initiate DSB end resection. CtIP is also involved in MMEJ, a form of NHEJ that involves limited DSB end resection.
DNA DSBs can be induced in cells by ionizing radiation, treatment with radiomimetic chemicals and perturbations in DNA replication. DSBs induce activation of the G1/S checkpoint, intra-S checkpoint or G2/M checkpoint, depending on when the DNA damage occurs (Figure 1). In the S and G2 phases of the cell cycle, CtIP undergoes CDK-dependent phosphorylation, which promotes its binding to the tumor suppressor protein BRCA1 and the Mre11–Rad50–NBS1 (MRN) complex [9–11]. CtIP undergoes additional phosphorylation by the checkpoint protein kinase ATM in response to DSB damage [12]. Although the underlying mechanism remains controversial, ATM phosphorylation of CtIP leads to transcription of cell cycle inhibitor genes, such as p21 and Gadd45 [12,13]. CtIP also promotes checkpoint signaling and subsequent cell cycle arrest by the Chk1 protein kinase [9,11]. Chk1 activation requires phosphorylation by the ATM-related checkpoint protein kinase ATR, which is activated on single-stranded DNA (ssDNA) coated with RPA protein [14,15]. The role of CtIP in Chk1 activation could be indirect, however; CtIP promotes formation of ssDNA and subsequent ATR activation through its role in DSB resection, a process that digests 5′ termini of DNA ends (see below) [10,16–18]. This model also explains why CtIP is required for Chk1 phosphorylation and checkpoint activation in response to DSBs, but not to stalled replication forks [11]. At stalled replication forks, the ssDNA structure that activates ATR is generated through the uncoupling of the DNA helicase activity from the DNA polymerase activity [19], a mechanism that is distinct from DSB resection.
Recent studies have revealed that DSB repair is a key function of CtIP. DSB damage can be repaired by different pathways, determined in part by the cell cycle stage in which the damage occurs [20]. In G1 phase of the cell cycle, DSBs are mainly repaired by the nonhomologous end-joining (NHEJ) pathway, which re-ligates broken DNA ends. In the S and G2 phases, owing to the presence of sister chromatids, DSBs can also be repaired by homologous recombination (HR) [20]. A fundamental difference between HR and NHEJ is that HR-mediated repair requires substantial DSB resection (approx. 100–200 nucleotides), which generates ssDNA required for homology searching and strand invasion [21]. CtIP is required for DSB resection, and therefore is required for HR [10,16–18,22]. Interestingly, CtIP can also participate in DSB repair by microhomology-mediated end-joining (MMEJ) during the G1 phase [22]. In contrast to NHEJ, MMEJ requires limited resection of DSB ends (four to six nucleotides) to provide short, complementary sequences that stabilize DNA end ligation. CtIP might promote this limited DSB resection, thereby allowing cells to repair DSB damage by MMEJ [22] (Figure 1).
Despite limited sequence homology, evidence strongly suggests that mammalian CtIP is the functional equivalent of S. cerevisiae Sae2 and S. pombe Ctp1 [17,23,24]. CtIP orthologs have also been identified in C. elegans (COM-1), Xenopus (xCtIP), chicken, mouse and A. thaliana (AtGR1) [18,22,25–27]. The main regions of similarity of the proteins are in the carboxyl terminus, which includes a conserved CDK phosphorylation site, T847 [17,23,26–28], and in the amino terminus, which is believed to mediate dimerization or oligomerization of these proteins [29,30]. Studies on these CtIP orthologs all support the idea that these proteins are required for DSB resection (Table 1).
Table 1.
Functions and regulation of CtIP orthologs
| CtIP ortholog | Organism | Protein | Function | Regulation |
|---|---|---|---|---|
| Sae2 | S. cerevisiae | 235 a.a. | DSB resection and HR in meiotic and mitotic cells [28,57–59,68,69,94–98] Acts in the same epistasis pathway as MRX in DSB resection [58,97] Recombinant protein has intrinsic endonuclease activity [56] Cooperates with MRX in removing hairpin structure [56,96,98] |
S267 phosphorylation by CDK1 is required for DSB resection [28] Phosphorylation by Tel1/Mec1 is required for meiosis and cell survival after DNA damage [99,100] |
| Ctp1 | S. pombe | 294 a.a. | DSB resection and HR in meiotic and mitotic cells Acts in the same epistasis pathway as MRN in DSB resection [23,24] |
Ctp1 expression is regulated by MBF [23] Ctp1 phosphorylation at CK2 SXT sites is required for Nbs1 binding [48,49] |
| COM1 | C. elegans | 525 a.a. | DSB processing in meiosis [26] | Not determined |
| AtGR1 | A. thaliana | 588 a.a. | DSB processing in meiosis [27] | Not determined |
| CtIP | X. laevis | 856 a.a. | DSB resection in Xenopus egg extracts [18] | CtIP associated with damaged chromatin is highly modified [18] CtIP damage recruitment is regulated by MRN and ATM [18] |
| CtIP | Chicken | 912 a.a. | HR in S and G2 phases and MMEJ in G1 phase [22,77] | CtIP S332-phosphorylation is not required for DSB resection and HR [77] |
| CtIP | Mouse | 893 a.a. | Required for embryonic development; haploinsufficient [25] Promotes G1/S transition by activating E2F/Rb-regulated promoters [5] Transcriptional corepressor together with BRCA1 and ZBRK1 [101] |
Not determined |
| CtIP | Human | 897 a.a. | DSB resection, ATR activation and HR [9–11,16–18,73] Transcription corepressor together with CtBP, RBP-Jκ/SHARP, Ikaros and LOM4 [102,103] |
Phosphorylation at S664 and S745 by ATM regulates transcription of cell cycle inhibitor genes [12] Phosphorylation at S327 and T847 by CDK, and ubiquitination by BRCA1 are required for DSB resection [73,76] CtIP protein levels are cell cycle-regulated [9,82] SIAH promotes CtIP degradation through the ubiquitin-proteosome pathway [81] |
CtIP promotes DSB resection together with MRN
CtIP is required, but not sufficient, for DSB end resection – this process also requires the MRN complex [31–33]. The MRN complex is highly conserved and is involved in nearly every aspect of the DSB damage response, including DNA damage sensing, signaling and repair [34]. In response to DSBs, MRN acts to recognize damaged chromatin, and binds broken DNA ends [35–37]. Subsequently, MRN initiates checkpoint signaling through recruitment and activation of ATM on damaged chromatin [38,39]. MRN together with activated ATM promotes DSB end resection, and subsequent activation of ATR and repair by HR [40–43]. The Mre11 subunit of MRN harbors DNA-binding, exonuclease and endonuclease activities [34]. It also displays limited DNA unwinding capability in the presence of Rad50 and NBS1 [44,45]. The endonuclease activity of Mre11 is apparently directly involved in DSB resection; however, this activity is not required for MRN function in DSB sensing or for recruitment and activation of ATM [46,47]. These findings suggest the possibility that association of additional proteins with MRN at DSB sites redirects its function from damage sensing and checkpoint initiation to DNA end resection and repair.
CtIP physically interacts with MRN, and both the N-terminus and C-terminus of CtIP have been shown to interact with the NBS1 subunit of MRN [10,16,17]. CtIP also colocalizes with MRN at DSB ends, suggesting that CtIP could influence MRN function [10,16–18]. The molecular nature of the CtIP–MRN interaction has been best characterized in fission yeast. Ctp1 is phosphorylated at SXT/SDT repeats (potential CK2 phosphorylation sites) which mediate binding to the FHA motif of NBS1. This interaction is important for Ctp1 localization to DSBs and Ctp1-dependent resistance to DNA damaging agents [48,49]. It has not been determined whether these CK2 phosphorylation sites are conserved in CtIP proteins in metazoans. However, a similar mechanism mediates the association of the mammalian checkpoint mediator protein MDC1 with NBS1, leading to the retention of the MRN complex on chromatin [50–53].
Recruitment of metazoan CtIP to DSBs also requires MRN [18]. However, CtIP damage recruitment is significantly delayed (by 5–15 min) compared with that of NBS1, suggesting that CtIP is not brought to DSBs passively through its interaction with MRN [18]. Given that CtIP damage recruitment also requires ATM kinase activity, and that ATM activation depends on MRN, MRN could play an indirect role in the initial damage recruitment of CtIP by activating ATM [18]. MRN could also facilitate CtIP damage relocation by establishing a specific chromatin structure at the DSB ends. Consistent with this idea, S. cerevisiae MRX (Mre11-Rad50-Xrs2, the counterpart of MRN in metazoans) interacts with components of the chromatin remodeling complexes RSC and INO80 to promote histone removal at DSB ends [54,55]. However, it is formally possible that the CtIP–MRN interaction directly mediates CtIP damage recruitment at a later stage. MRN might also be required to retain CtIP at DSBs after its damage recruitment.
Emerging evidence suggests the hypothesis that CtIP regulates MRN function at DSBs, and redirects MRN function from DNA damage sensing to end resection. Consistent with this idea, purified recombinant human CtIP protein stimulates the nuclease activity of a Mre11–Rad50 complex towards a closed ssDNA substrate in vitro [17]. This finding is also consistent with the observation that purified CtIP can interact with Mre11 and Rad50 [16]. Interestingly, yeast Sae2 has been shown to possess endonuclease activity itself in vitro [56]. This raises the intriguing possibility that both Sae2/CtIP and MRX/N directly participate in DSB end processing.
Recently, a two-step model has been proposed for DSB end resection, based on studies in budding yeast [32,57–59]. In this model, Sae2 and MRX initiate the DSB resection process to remove 50–100 nucleotides from the 5′ termini of the DNA. Following this first step, the Exo1 exonuclease and Sgs1-DNA2 helicase/endonuclease function redundantly to carry out further resection to generate long 3′ ssDNA tails [32,57–59]. Notably, functional homologs of all of these proteins are present in fission yeast and metazoans, suggesting that the model for DSB end resection could be conserved in higher organisms (Figure 2). Consistent with this idea, human Exo1 and the Sgs1 ortholog BLM have been shown to be involved in DSB resection [60–62]. Another Sgs1 ortholog, the WRN helicase, might promote the initial unwinding of DNA ends at the DSB, together with the activity of the MRN complex [45,63]. CtIP together with MRN might then cleave the 5′ ssDNA flap at the ssDNA–dsDNA junction to initiate DSB resection. This endonucleolytic cleavage of 5′ termini is absolutely essential for resection of DNA ends that are blocked to processing by a covalently attached protein, such as the DSB ends generated during meiosis that are linked to the Spo11 protein, or DSB ends bound to topoisomerases, which remain after treatment with topoisomerase inhibitors [64–70]. This endo-cleavage step might occur repeatedly, or could be switched to resection by Exo1 or by BLM-DNA2 (Figure 2).
Figure 2.
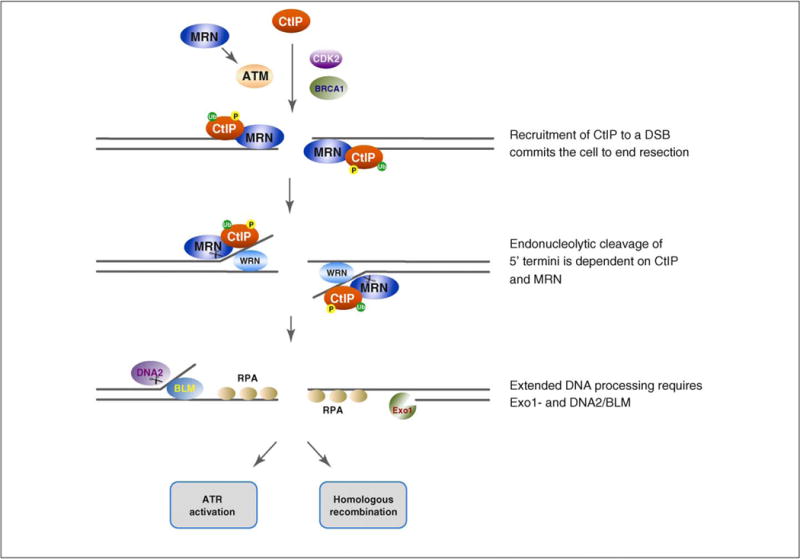
A model for the role of CtIP in DSB end resection. After DSB damage, CtIP is phosphorylated by CDK and ATM, and ubiquitinated by BRCA1, leading to its recruitment to the DNA ends. CtIP damage recruitment commits cells to DNA end resection and repair by HR. CtIP together with MRN initiate DSB resection by promoting endonucleolytic cleavage of the 5′-DNA termini. The WRN helicase might promote this initial DSB resection by unwinding the DNA ends. The 5′ termini are then further resected by the overlapping activities of Exo1 and DNA2/BLM. The ssDNA generated from the DSB end processing is bound by RPA, and the RPA-coated ssDNA structure triggers activation of the ATR checkpoint protein kinase and DNA repair by HR.
Regulation of CtIP by CDK, BRCA1 and ATM
The function of CtIP is highly regulated to ensure that DSB resection and HR occur in appropriate stages of the cell cycle (Figure 3). The kinase activity of CDK (CDK1 in yeast and CDK2 in mammals) in the S and G2 phases is required for DSB resection [42,71,72]. In addition, Sae2/CtIP is a target of CDK [9,28,73]. These findings provide a mechanistic explanation for why HR primarily operates in the S and G2 phases of the cell cycle. Two CDK consensus phosphorylation sites, S327 and T847, have been identified in CtIP (Figure 4) [9,73]. CDK-dependent phosphorylation of S327 mediates CtIP association with the C-terminal BRCT domains of BRCA1 as well as with MRN [9,10,74,75]. This interaction promotes the ubiquitination of CtIP by the N-terminal ubiquitin-ligase activity of BRCA1 [76]. CtIP phosphorylation at S327 and ubiquitination are required for the damage localization of CtIP and therefore also needed for its function in DSB end resection [76]. Consistent with this idea, CtIP is reported to be an essential gene in chicken DT40 cells, and the human CtIP(S327A) mutant introduced into CtIP-knockout DT40 cells is deficient in supporting HR [22]. However, a more recent study reported that phosphorylation of chicken CtIP at S332 (equivalent to S327 in human CtIP) is not required for DSB resection or HR, and that CtIP-knockout chicken DT40 cells are viable [77]. Further work is needed to resolve these discrepancies. It is possible that phosphorylation of chick CtIP by CDK at other sites plays a redundant role in supporting CtIP damage recruitment and its subsequent role in DSB resection. Interestingly, the human CtIP(S327A) mutant introduced into CtIP-knockout chicken DT40 cells is proficient in supporting MMEJ in the G1 phase [22]. This suggests that human CtIP can translocate to DSBs and promote limited DSB resection in the absence of S327-phosphorylation and BRCA1-dependent ubiquitination, at least during the G1 phase. To date, it is not clear how BRCA1-mediated ubiquitination facilitates association of human CtIP with DNA damage sites. One possibility is that the polyubiquitin chains on CtIP interact with other DSB-associated proteins such as RAP80/Abraxas that contain an ubiquitin-interacting motif [78–80]. It will be important to understand how CtIP activity in DSB end resection is regulated in the G1 phase, and whether CDK and BRCA1 control the extent of CtIP-dependent DNA end resection in the S and G2 phases.
Figure 3.
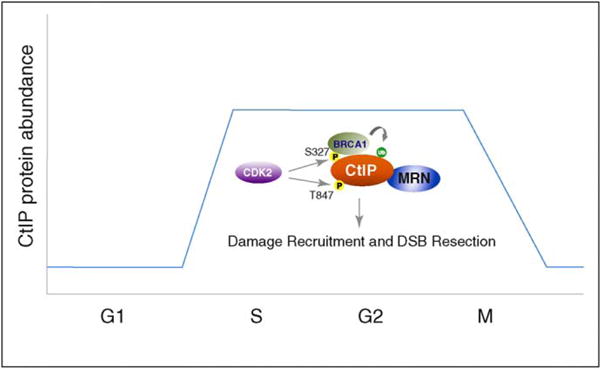
Cell cycle regulation of CtIP. CtIP function is regulated both at the level of protein expression and post-translational modifications. CtIP protein levels are low in G1 phase and high in the S, G2 and M phases, although the gene encoding CtIP is transcribed throughout the cell cycle [82]. Modification of CtIP by CDK2 and BRCA1 occurs in the S/G2 phase and is required for CtIP function in HR. CDK2-mediated phosphorylation of CtIP at S327 promotes its association with BRCA1 and the MRN complex [9,10,74,75]. CtIP also is ubiquitinated by BRCA1 [76]. CtIP phosphorylation and association with BRCA1 are required for CtIP localization to damaged chromatin [76]. Additional phosphorylation of CtIP at T847 by CDK2 could facilitate CtIP function in DSB resection [73]. Although the mechanism of CtIP function in DNA resection remains to be determined, CtIP modification or binding to MRN could influence MRN enzymatic activity. CtIP might also promote limited DNA resection in G1 in the absence of CDK2 or BRCA1.
Figure 4.
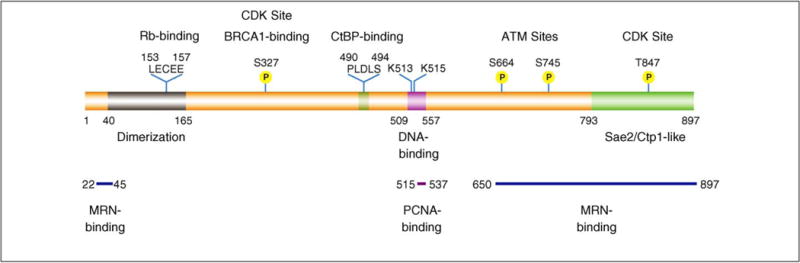
Protein domains of human CtIP important for its function and regulation. Human CtIP contains a dimerization domain (amino acids 40–165) [29] and a Rb consensus binding site (LECEE) [2] in the N-terminus, and a CtBP-binding motif (PLDLS) [3] in the middle region. The C-terminal Sae2/Ctp1-like region (amino acids 793–897) is conserved from yeast to humans [17,23,26,27]. CDK-dependent phosphorylation of CtIP at S327 mediates its association with BRCA1, CtIP ubiquitination by BRCA1 and CtIP recruitment to damaged chromatin [76,82]. The 509–557 region of CtIP has DNA-binding activity in vitro and might mediate CtIP damage recruitment through direct binding to the DNA at DSBs [18]. Two key lysine residues conserved in vertebrates within this region, K513 and K515, are important for both its DNA binding activity in vitro and damage recruitment in cells [18]. A PCNA-binding motif was also identified in this DNA binding region [104]. Both the N-terminal region (amino acids 22–45) and the C-terminal region (amino acids 650–897) of CtIP are reported to bind the MRN complex [10,16,17]. Two ATM phosphorylation sites (S664 and S745) are conserved in vertebrates [12]. CDK-mediated phosphorylation of T847 is important for CtIP function in DNA end resection, but is not required for CtIP damage recruitment [73].
CDK-dependent phosphorylation of CtIP at T847 is also required for DSB resection and subsequent HR in the S and G2 phases [73]. The non-phosphorylatable CtIP(T847A) mutant is still recruited to DNA damage sites, suggesting that CDK2 phosphorylation at T847 regulates the function of CtIP in DSB resection after damage recruitment. The corresponding CDK phosphorylation site in S. cerevisiae Sae2, S267, is also required for DSB resection and HR, indicating a conserved mechanism of regulation [28]. However, this site is not present in S. pombe Ctp1. Unlike the other CtIP orthologs, Ctp1 is regulated at the level of transcription by the MBF transcription factor that controls S phase gene expression. The absence of Ctp1 gene expression in the G1 phase prevents DSB resection and HR in the G1 phase of the cell cycle in fission yeast [23]. In budding yeast, where Sae2 is expressed throughout the cell cycle, CDK-dependent, post-translational regulation is required to restrict Sae2 function in DSB resection to the S and G2 phases. The activity of human CtIP in DSB resection is regulated by both CDK activity and the cell cycle-dependent oscillation of CtIP protein abundance (Figure 3) [9,73,81,82].
In addition to CDK, efficient DSB resection and subsequent ATR activation also require ATM kinase activity [40–43]. Although the contribution of ATM kinase activity to DSB resection has not been clear, our recent study indicates that ATM promotes CtIP recruitment to DSBs, suggesting that CtIP is a target of ATM in DNA end resection [18]. Thus, CtIP plays a crucial role in linking ATM activation to ATR activation after DNA damage [18,83]. Two ATM phosphorylation sites, S664 and S745, have been identified in human CtIP (Figure 4) [12]. However, these sites apparently are dispensable for CtIP recruitment to DSBs [18], suggesting that phosphorylation at other sites by ATM or another protein downstream of ATM facilitates CtIP damage relocation. One possible role of ATM kinase activity in CtIP damage recruitment is to promote conformational changes in CtIP that lead to exposure of a DNA-binding motif in a central region of CtIP (Figure 4) [18]. The CtIP DNA binding motif is required for its recruitment to DSBs, probably by directly binding to DNA at the damage sites [18]. Another possibility is that CtIP phosphorylation by ATM or a kinase downstream of ATM such as Chk2 primes CtIP for CK2-dependent phosphorylation, which facilitates CtIP interaction with the FHA domain of NBS1, and association with DNA damage sites. Future studies are needed to define the functional relationship between CDK, BRCA1, ATM and the DNA-binding activity in CtIP in the regulation of CtIP damage recruitment and its function in DNA end resection after damage recruitment.
CtIP and cancer
CtIP is an essential gene in mammalian cells, with homozygous inactivation causing embryonic lethality in mice [25]. Inactivation of just one CtIP allele predisposes mice to multiple types of cancers, particularly lymphomas, suggesting that CtIP functions as a tumor suppressor protein [25]. Mutations in CtIP have also been identified in human cancer cell lines [4]. In colorectal cancer cells with mutations in the mismatch repair (MMR) pathway, the A9 repeat in the middle region of the gene encoding CtIP is frequently mutated as a result of microsatellite instability, resulting in translation of a truncated CtIP protein of 357 amino acids [84]. Although no biallelic mutations of CtIP have been identified in cancers, the haploinsufficiency observed in mice suggests that mutation of a single allele of CtIP might promote genome instability and tumorigenesis. Interestingly, epigenetic inactivation of CtIP has been reported in breast cancer, and is associated with increased resistance to tamoxifen treatment [85].
Disruption of CtIP function could also be the underlying mechanism for cancer susceptibility in cells with certain BRCA1 mutations. BRCA1 mutations are associated with hereditary predisposition to breast and ovarian cancer, with the most common mutations occurring in the C-terminal BRCT tandem repeats [86]. BRCA1 forms distinct complexes in cells, owing to the mutually exclusive binding of its BRCT domains with phosphorylated proteins including CtIP, BACH1/BRIP1/FancJ and RAP80/Abraxas [74,79,80,87]. Structural studies have demonstrated that two cancer-associated mutations in the BRCT domains of BRCA1, M1775R and R1699W, directly interfere with the hydrophobic pocket to which phosphorylated CtIP binds [75,88]. Defects in the BRCA1–CtIP interaction and the DNA damage response have also been observed in HCC1937 breast cancer cells that harbor a BRCA1 mutant protein lacking a BRCT repeat, consistent with the notion that a functional CtIP–BRCA1 complex is important for tumor suppression [4,74].
Although inactivation and mutation of CtIP and BRCA1 promote cancer formation and development, these might also be exploited for targeted cancer therapy on the basis of synthetic lethality [89]. Recently, inhibitors of the base excision repair enzyme PARP1 have been used to preferentially kill BRCA1-deficient cancer cells [90,91]. This selective killing effect is believed to occur as a result of the defects in HR-mediated repair of the DSBs that are converted from the unrepaired single-strand breaks induced by PARP1 inhibition in the BRCA1-deficient cells [90,91]. Synthetic lethality also was observed between PARP1 inhibitors and mutation in other genes in the HR pathway [92], or mutation of genes such as PTEN that influence HR gene expression [93]. Given the crucial role of CtIP in HR and its intimate relationship with BRCA1, PARP1 inhibitors could be also efficacious for treatment of cancers where CtIP is mutated or epigenetically inactivated.
Concluding remarks
Recent studies on CtIP place this protein at the intersection of cell cycle control, checkpoint signaling and DNA repair. After DNA damage, CtIP receives signals from both the cell cycle kinase CDK and checkpoint kinase ATM that activate its function in DNA damage signaling and repair. CtIP is then recruited to DSB damage sites, where it interacts with the MRN complex to mediate the initiation step of DNA end resection. In so doing, CtIP drives the transition from ATM-dependent checkpoint signaling to ATR-dependent checkpoint signaling. Importantly, the engagement of CtIP-dependent DNA end resection commits cells irreversibly to repair by the HR pathway. Although much has been learned about what regulates CtIP relocalization to DNA damage sites, many questions remain as to how CtIP facilitates DSB resection after damage recruitment (Box 1). In particular, it will be important to determine whether CtIP, as with yeast Sae2, functions as an endonuclease, and if so, whether and how this activity is regulated by CDK, BRCA1 and ATM. The mechanism of cooperation between CtIP and MRN, and the functional relationship between the potential nuclease activity of CtIP and that of MRN in DSB resection also remain to be delineated. Furthermore, it remains to be investigated whether the two-step model for DSB resection proposed in budding yeast applies to higher organisms, and if it does, how the switch is made from the initial MRN-CtIP dependent endonucleolytic cleavage step to the second resection step that is potentially carried out by Exo1 and BLM-DNA2. Finally, it will be interesting to find out whether the DNA binding motif identified in CtIP is required for the transcription function of CtIP, and whether CtIP promotes transcription of additional genes that are involved in DNA damage signaling and repair. Future studies on CtIP function and regulation will provide insights into how defects in the DSB damage response predispose patients to cancer, and might help identify and treat patients that could best respond to cancer therapies that cause DNA damage or target the DNA damage checkpoint response pathway.
Box 1. Outstanding questions on CtIP function in the DNA damage response.
How do CtIP and MRN cooperate in DSB end resection? Does CtIP have intrinsic nuclease activity? If so, what regulates this activity in resection, and what is the molecular nature of the relationship between CtIP nuclease activity and Mre11 nuclease activity?
How is the transition made from CtIP/MRN-dependent DSB end processing to resection by Exo1, BLM/Dna2 or other enzymes?
How is CtIP retained at DSBs? Is the CtIP-NBS1 interaction mediated by phosphorylated SXT repeats in CtIP and the FHA phosphopeptide domain of NBS1, analogous to the Ctp1-Nbs1 interaction in fission yeast?
Do CtIP phosphorylation by ATM and CDK and ubiquitination by BRCA1 regulate the function of CtIP in DSB resection?
What is the function of CtIP in MMEJ in G1 phase? Does CtIP cooperate with MRN in MMEJ?
How does CtIP dimerization contribute to its association with other proteins, and its function in DSB resection and in transcription?
Is the DNA-binding motif of CtIP required for its function in transcription?
Is CtIP a tumor suppressor because of its function in DNA end resection, its regulation of BRCA1, or its function in transcription?
Acknowledgments
We apologize to colleagues whose work we were not able to cite in this review owing to space limitations. We thank Drs. Tony Hunter, Helen Piwnica-Worms, Tony Polverino and Susana Gonzalo for critical reading of the manuscript and their insightful input. Research in Z.Y.’s lab is supported by startup funds from the Department of Cell Biology and Physiology at Washington University School of Medicine, a grant from the American Cancer Society (IRG-58-010-52) and a Washington University Molecular Imaging Center Pilot Research Project grant.
References
- 1.Fusco C, et al. Molecular cloning and characterization of a novel retinoblastoma-binding protein. Genomics. 1998;51:351–358. doi: 10.1006/geno.1998.5368. [DOI] [PubMed] [Google Scholar]
- 2.Meloni AR, et al. A mechanism for Rb/p130-mediated transcription repression involving recruitment of the CtBP corepressor. Proc Natl Acad Sci U S A. 1999;96:9574–9579. doi: 10.1073/pnas.96.17.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaeper U, et al. Interaction between a cellular protein that binds to the C-terminal region of adenovirus E1A (CtBP) and a novel cellular protein is disrupted by E1A through a conserved PLDLS motif. J Biol Chem. 1998;273:8549–8552. doi: 10.1074/jbc.273.15.8549. [DOI] [PubMed] [Google Scholar]
- 4.Wong AK, et al. Characterization of a carboxy-terminal BRCA1 interacting protein. Oncogene. 1998;17:2279–2285. doi: 10.1038/sj.onc.1202150. [DOI] [PubMed] [Google Scholar]
- 5.Liu F, Lee WH. CtIP activates its own and cyclin D1 promoters via the E2F/RB pathway during G1/S progression. Mol Cell Biol. 2006;26:3124–3134. doi: 10.1128/MCB.26.8.3124-3134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sterner JM, et al. Negative regulation of DNA replication by the retinoblastoma protein is mediated by its association with MCM7. Mol Cell Biol. 1998;18:2748–2757. doi: 10.1128/mcb.18.5.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chinnadurai G. CtIP, a candidate tumor susceptibility gene is a team player with luminaries. Biochim Biophys Acta. 2006;1765:67–73. doi: 10.1016/j.bbcan.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 8.Wu G, Lee WH. CtIP, a multivalent adaptor connecting transcriptional regulation, checkpoint control and tumor suppression. Cell Cycle. 2006;5:1592–1596. doi: 10.4161/cc.5.15.3127. [DOI] [PubMed] [Google Scholar]
- 9.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, et al. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg RA, et al. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li S, et al. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature. 2000;406:210–215. doi: 10.1038/35018134. [DOI] [PubMed] [Google Scholar]
- 13.Wu-Baer F, Baer R. Effect of DNA damage on a BRCA1 complex. Nature. 2001;414:36. doi: 10.1038/35102118. [DOI] [PubMed] [Google Scholar]
- 14.You Z, et al. The role of single-stranded DNA and polymerase alpha in establishing the ATR, Hus1 DNA replication checkpoint. J Biol Chem. 2002;277:27088–27093. doi: 10.1074/jbc.M204120200. [DOI] [PubMed] [Google Scholar]
- 15.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 16.Yuan J, Chen J. N terminus of CtIP is critical for homologous recombination-mediated double-strand break repair. J Biol Chem. 2009;284:31746–31752. doi: 10.1074/jbc.M109.023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.You Z, et al. CtIP links DNA double-strand break sensing to resection. Mol Cell. 2009;36:954–969. doi: 10.1016/j.molcel.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byun TS, et al. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 21.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 22.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Limbo O, et al. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell. 2007;28:134–146. doi: 10.1016/j.molcel.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akamatsu Y, et al. Molecular characterization of the role of the Schizosaccharomyces pombe nip1+/ctp1+ gene in DNA double-strand break repair in association with the Mre11-Rad50-Nbs1 complex. Mol Cell Biol. 2008;28:3639–3651. doi: 10.1128/MCB.01828-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen PL, et al. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol Cell Biol. 2005;25:3535–3542. doi: 10.1128/MCB.25.9.3535-3542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penkner A, et al. A conserved function for a Caenorhabditis elegans Com1/Sae2/CtIP protein homolog in meiotic recombination. EMBO J. 2007;26:5071–5082. doi: 10.1038/sj.emboj.7601916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uanschou C, et al. A novel plant gene essential for meiosis is related to the human CtIP and the yeast COM1/SAE2 gene. EMBO J. 2007;26:5061–5070. doi: 10.1038/sj.emboj.7601913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huertas P, et al. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dubin MJ, et al. Dimerization of CtIP, a BRCA1- and CtBP-interacting protein, is mediated by an N-terminal coiled-coil motif. J Biol Chem. 2004;279:26932–26938. doi: 10.1074/jbc.M313974200. [DOI] [PubMed] [Google Scholar]
- 30.Kim HS, et al. Functional interactions between Sae2 and the Mre11 complex. Genetics. 2008;178:711–723. doi: 10.1534/genetics.107.081331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernstein KA, Rothstein R. At loose ends: resecting a double-strand break. Cell. 2009;137:807–810. doi: 10.1016/j.cell.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mimitou EP, Symington LS. DNA end resection: many nucleases make light work. DNA Repair (Amst) 2009;8:983–995. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda S, et al. Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Mol Cell. 2007;28:351–352. doi: 10.1016/j.molcel.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 34.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 35.Hopfner KP. DNA double-strand breaks come into focus. Cell. 2009;139:25–27. doi: 10.1016/j.cell.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 36.Hopfner KP, et al. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001;105:473–485. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 37.Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13:458–462. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 38.Difilippantonio S, Nussenzweig A. The NBS1-ATM connection revisited. Cell Cycle. 2007;6:2366–2370. doi: 10.4161/cc.6.19.4758. [DOI] [PubMed] [Google Scholar]
- 39.You Z, et al. Rapid activation of ATM on DNA flanking double-strand breaks. Nat Cell Biol. 2007;9:1311–1318. doi: 10.1038/ncb1651. [DOI] [PubMed] [Google Scholar]
- 40.Adams KE, et al. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene. 2006;25:3894–3904. doi: 10.1038/sj.onc.1209426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cuadrado M, et al. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J Exp Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jazayeri A, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 43.Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem. 2006;281:9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 46.Buis J, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams RS, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lloyd J, et al. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell. 2009;139:100–111. doi: 10.1016/j.cell.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams RS, et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chapman JR, Jackson SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008;9:795–801. doi: 10.1038/embor.2008.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Melander F, et al. Phosphorylation of SDT repeats in the MDC1N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol. 2008;181:213–226. doi: 10.1083/jcb.200708210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spycher C, et al. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol. 2008;181:227–240. doi: 10.1083/jcb.200709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu L, et al. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci U S A. 2008;105:11200–11205. doi: 10.1073/pnas.0802885105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsukuda T, et al. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shim EY, et al. The yeast chromatin remodeler RSC complex facilitates end joining repair of DNA double-strand breaks. Mol Cell Biol. 2005;25:3934–3944. doi: 10.1128/MCB.25.10.3934-3944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lengsfeld BM, et al. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007;28:638–651. doi: 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Budd ME, Campbell JL. Interplay of Mre11 nuclease with Dna2 plus Sgs1 in Rad51-dependent recombinational repair. PLoS ONE. 2009;4:e4267. doi: 10.1371/journal.pone.0004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu Z, et al. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nimonkar AV, et al. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci U S A. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schaetzlein S, et al. Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell. 2007;130:863–877. doi: 10.1016/j.cell.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gravel S, et al. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toczylowski T, Yan H. Mechanistic analysis of a DNA end processing pathway mediated by the Xenopus Werner syndrome protein. J Biol Chem. 2006;281:33198–33205. doi: 10.1074/jbc.M605044200. [DOI] [PubMed] [Google Scholar]
- 64.Milman N, et al. Meiotic DNA double-strand break repair requires two nucleases, MRN and Ctp1, to produce a single size class of Rec12 (Spo11)-oligonucleotide complexes. Mol Cell Biol. 2009;29:5998–6005. doi: 10.1128/MCB.01127-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farah JA, et al. Ctp1 and Exonuclease 1, alternative nucleases regulated by the MRN complex, are required for efficient meiotic recombination. Proc Natl Acad Sci U S A. 2009;106:9356–9361. doi: 10.1073/pnas.0902793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rothenberg M, et al. Ctp1 and the MRN-complex are required for endonucleolytic Rec12 removal with release of a single class of oligonucleotides in fission yeast. PLoS Genet. 2009;5:e1000722. doi: 10.1371/journal.pgen.1000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hartsuiker E, et al. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol Cell. 2009;33:117–123. doi: 10.1016/j.molcel.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keeney S, Kleckner N. Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci U S A. 1995;92:11274–11278. doi: 10.1073/pnas.92.24.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neale MJ, et al. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–1057. doi: 10.1038/nature03872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartsuiker E, et al. Ctp1CtIP and Rad32Mre11 nuclease activity are required for Rec12Spo11 removal, but Rec12Spo11 removal is dispensable for other MRN-dependent meiotic functions. Mol Cell Biol. 2009;29:1671–1681. doi: 10.1128/MCB.01182-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aylon Y, et al. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004;23:4868–4875. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ira G, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu X, et al. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998;273:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 75.Varma AK, et al. Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry. 2005;44:10941–10946. doi: 10.1021/bi0509651. [DOI] [PubMed] [Google Scholar]
- 76.Yu X, et al. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006;20:1721–1726. doi: 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakamura K, et al. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 6:e1000828. doi: 10.1371/journal.pgen.1000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim H, et al. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- 79.Sobhian B, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang B, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Germani A, et al. SIAH-1 interacts with CtIP and promotes its degradation by the proteasome pathway. Oncogene. 2003;22:8845–8851. doi: 10.1038/sj.onc.1206994. [DOI] [PubMed] [Google Scholar]
- 82.Yu X, Baer R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J Biol Chem. 2000;275:18541–18549. doi: 10.1074/jbc.M909494199. [DOI] [PubMed] [Google Scholar]
- 83.Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33:547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vilkki S, et al. Screening for microsatellite instability target genes in colorectal cancers. J Med Genet. 2002;39:785–789. doi: 10.1136/jmg.39.11.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu M, et al. CtIP silencing as a novel mechanism of tamoxifen resistance in breast cancer. Mol Cancer Res. 2007;5:1285–1295. doi: 10.1158/1541-7786.MCR-07-0126. [DOI] [PubMed] [Google Scholar]
- 86.Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer. 2007;7:937–948. doi: 10.1038/nrc2054. [DOI] [PubMed] [Google Scholar]
- 87.Cantor SB, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 88.Drikos I, et al. Characterization of cancer-linked BRCA1-BRCT missense variants and their interaction with phosphoprotein targets. Proteins. 2009;77:464–476. doi: 10.1002/prot.22460. [DOI] [PubMed] [Google Scholar]
- 89.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 90.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 91.Bryant HE, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 92.McCabe N, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 93.Mendes-Pereira AM, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–322. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Clerici M, et al. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J Biol Chem. 2005;280:38631–38638. doi: 10.1074/jbc.M508339200. [DOI] [PubMed] [Google Scholar]
- 95.McKee AH, Kleckner N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics. 1997;146:797–816. doi: 10.1093/genetics/146.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lobachev KS, et al. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 97.Prinz S, et al. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics. 1997;146:781–795. doi: 10.1093/genetics/146.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rattray AJ, et al. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics. 2001;158:109–122. doi: 10.1093/genetics/158.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cartagena-Lirola H, et al. Budding yeast Sae2 is an in vivo target of the Mec1 and Tel1 checkpoint kinases during meiosis. Cell Cycle. 2006;5:1549–1559. doi: 10.4161/cc.5.14.2916. [DOI] [PubMed] [Google Scholar]
- 100.Baroni E, et al. The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol Cell Biol. 2004;24:4151–4165. doi: 10.1128/MCB.24.10.4151-4165.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ahmed KM, et al. Derepression of HMGA2 via removal of ZBRK1/BRCA1/CTIP complex enhances mammary tumorigenesis. J Biol Chem. 2009;285:4464–4471. doi: 10.1074/jbc.M109.062265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koipally J, Georgopoulos K. Ikaros-CtIP interactions do not require C-terminal binding protein and participate in a deacetylase-independent mode of repression. J Biol Chem. 2002;277:23143–23149. doi: 10.1074/jbc.M202079200. [DOI] [PubMed] [Google Scholar]
- 103.Sum EY, et al. The LIM domain protein LMO4 interacts with the cofactor CtIP and the tumor suppressor BRCA1 and inhibits BRCA1 activity. J Biol Chem. 2002;277:7849–7856. doi: 10.1074/jbc.M110603200. [DOI] [PubMed] [Google Scholar]
- 104.Gu B, Chen PL. Expression of PCNA-binding domain of CtIP, a motif required for CtIP localization at DNA replication foci, causes DNA damage and activation of DNA damage checkpoint. Cell Cycle. 2009;8:1409–1420. doi: 10.4161/cc.8.9.8322. [DOI] [PubMed] [Google Scholar]