

ABSTRACT
An increasing number of studies have demonstrated that macroautophagy/autophagy plays an important role in the infectious processes of diverse pathogens. However, it remains unknown whether autophagy is induced in avian metapneumovirus (aMPV)-infected host cells, and, if so, how this occurs. Here, we report that aMPV subgroup C (aMPV/C) induces autophagy in cultured cells. We demonstrated this relationship by detecting classical autophagic features, including the formation of autophagsomes, the presence of GFP-LC3 puncta and the conversation of LC3-I into LC3-II. Also, we used pharmacological regulators and siRNAs targeting ATG7 or LC3 to examine the role of autophagy in aMPV/C replication. The results showed that autophagy is required for efficient replication of aMPV/C. Moreover, infection with aMPV/C promotes autophagosome maturation and induces a complete autophagic process. Finally, the ATF6 pathway, of which one component is the unfolded protein response (UPR), becomes activated in aMPV/C-infected cells. Knockdown of ATF6 inhibited aMPV/C-induced autophagy and viral replication. Collectively, these results not only show that autophagy promotes aMPV/C replication in the cultured cells, but also reveal that the molecular mechanisms underlying aMPV/C-induced autophagy depends on regulation of the ER stress-related UPR pathway.
KEYWORDS: aMPV/C replication, autophagy, avian metapneumovirus, ER stress, UPR
Introduction
Avian metapneumovirus (aMPV) causes acute upper respiratory tract infection in turkeys and chickens. These infections are mainly characterized by nasal discharge, tracheal and facial congestion, and swollen infraorbital sinuses.1 In laying birds, infection results in egg drops and poor egg quality.2 aMPV belongs to the genus Metapneumovirus within the subfamily Pneumovirinae of family Paramyxoviridae and has a nonsegmented, single-stranded, negative-sense RNA genome.3 aMPV has first been described in South Africa in 1978,4 and subsequently reported in many other countries.5 Four subgroups of aMPV (A, B, C, and D) have been recognized based on genetic characterization and antigenic differences.6 Subgroups C aMPV (aMPV/C) have first been identified in turkeys in the USA in 1996 and subsequently isolated from farmed ducks in France.7,8 This virus has also spread to Asia, found in pheasants in Korea and in chickens in China.9,10 There is low sequence identity between aMPV/C and subgroups A and B, which possess weak cross-reactivity in neutralization and enzyme-linked immunosorbent assay. However, aMPV/C has closer genetic and antigenic relatedness to human metapneumovirus (hMPV) than other aMPV subgroups.11-13
Autophagy is a dynamic and conserved eukaryotic process that delivers protein aggregates and obsolete or damaged organelles into lysosomes for degradation through autophagosomes, which are single- or double-membrane structures.14-18 The autophagic process is completed after an autophagosome fuses to a lysosome, substrates contained inside are digested, and breakdown products are released back into the cytosol. Many autophagy-related and regulatory genes have been identified.19 During autophagy, MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) is conjugated to phosphatidylethanolamine to form lipidated LC3-II, which can be used as an autophagosomal marker in host cells. The multifunctional polyubiquitin-binding protein SQSTM1/p62 (sequestosome 1) serves as a substrate for autophagic degradation and can be used to assess autophagic flux.20,21 Autophagy plays an important role not only in cellular homeostasis but also in response to cellular stressors, such as nutrient starvation or pathogen infection.20,22 Some viruses inhibit and block autophagosome maturation through different strategies,23-25 whereas other viruses exploit autophagy to benefit their own replication.26,27
The endoplasmic reticulum (ER) is a multifunctional organelle in eukaryotic cells that is involved in the post-translational modification, folding and oligomerization of newly synthesized intracellular proteins. In particular, the ER may serve as one of the origins of the autophagosomal membrane.28 However, ER stress occurs in response to endogenous imbalances and can result in ER malfunction.29,30 In response to ER stress, cells activate the unfolded protein response (UPR) to maintain ER homeostasis by minimizing the accumulation of unfolded or misfolded proteins. Three UPR pathways that respond to ER stress have been reported to maintain intracellular homeostasis; these include the EIF2AK3/PERK (eukaryotic translation initiation factor 2 α kinase 3) pathway, the ATF6 (activating transcription factor 6) pathway and the ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1) pathway. ER stress and the activation of the UPR pathway occur during viral infection. Additionally, ER stress can trigger autophagy through activation of UPR components,31-33 and several viruses of the family Paramyxoviridae have been reported to activate autophagy, which is involved in viral replication.34-36 These findings motivated us to investigate the interplay and molecular mechanisms that exist between aMPV infection and the activation of autophagy.
In this study, we showed that complete autophagy is induced in aMPV/C-infected cells and that knockdown of genes crucial for autophagosome formation significantly decreases viral yield. Furthermore, we found that aMPV/C infection induces autophagy via ER stress, specifically via regulation of the ATF6 UPR pathway, and that silencing the ATF6 gene suppresses aMPV/C replication in cultured cells.
Results
Infection with aMPV/C activates autophagy in cultured cells
Transmission electron microscopy (TEM) is an accepted standard method for observing the formation of single- or double-membrane autophagic compartments around the perinuclear region and evaluating the morphology of autophagic compartments.20,22 Thus, to determine whether autophagy is triggered upon aMPV/C infection, TEM was used to perform ultrastructural analysis of aMPV/C-infected Vero cells. Our results showed that aMPV/C-infected cells had significantly increased numbers of single- or double-membrane vesicles around the perinuclear region and that recognizable cytoplasmic contents or degraded organelles were sequestered in homogeneously-sized vesicles with morphologically typical characteristics of autophagic vacuoles. In contrast, similar vesicles were rarely observed in uninfected (mock-infected) cells; rather, these cells exhibited extremely dense cytoplasm and contained many morphologically normal organelles (Fig. 1A, panels i and ii). Immunoelectron microscopy (IEM) was further used to observe whether aMPV/C replication was located in the vesicles. As shown in Fig. 1A (panel iii), many viral fusion (F) protein immunogold labels were localized on the membranes of autophagosome-like vesicles (arrowheads in panel iii), which likely represented synthesized viral proteins or virions. In contrast, no immunogold labels were observed in mock-infected Vero cells that were incubated with anti-F protein antibody (data not shown). For negative controls, normal mouse immunoglobulin G were gold labeled without primary antibody incubation, showed no positive labels (data not shown). Taken together, these data strongly demonstrate that aMPV/C infection could induce autophagosome-like vesicle generation in Vero cells and aMPV/C replication mainly localized on the membranes of these vesicles.
Figure 1.

Infection with aMPV/C induces autophagy in cultured cells. (A) Vero cells were mock-infected or infected with aMPV/C for 72 h and then processed for and observed using transmission electron microscopy (TEM) (panels i and ii). aMPV/C-infected Vero cells were incubated with anti-F protein antibody followed by incubation with a secondary antibody conjugated to 10-nm colloidal gold particles, and then observed by immunoelectron microscopy (IEM) (panel iii). Black arrows and arrowheads indicate characteristic single- or double-membrane vesicles and colloidal gold particles, respectively. Scale bars: 2 μm. (B and C) Vero cells and DF-1 cells were mock-infected or infected with aMPV/C at the indicated time points (0, 24, 48, 72, 96, and 120 h) postinfection. Cell samples were harvested and processed, and their proteins were separated by SDS-PAGE and were analyzed by western blotting with anti-LC3 and anti-viral F antibodies. ACTB was used as a protein loading control. Representative results are displayed with graphs corresponding to the ratios of LC3-II:ACTB bands normalized to the control conditions. (D) Vero cells transfected with pEGFP-LC3 plasmids for 18 h were infected with aMPV/C, and then subjected to confocal immunofluorescence analysis at 72 hpi. The GFP-LC3 expression signal (green) and viral N or F protein staining (red) are shown. Scale bars: 10 μm. The GFP-LC3 puncta were counted in stained cells for viral N protein. (E) Vero cells were cultured with replication-competent aMPV/C for 72 h or heat-inactivated aMPV/C for 72 and 120 h. Subsequently, cell samples were processed and analyzed as described in B and C. Error bars, mean ± SD of 3 independent tests. Two-way ANOVA, #P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001, compared with the control group.
To further verify that the observed vesicles were related to autophagy in the aMPV/C-infected cells, we used western blotting to examine the conversion of endogenous LC3 protein, which is an important hallmark of autophagy.20,21 The results showed that the amounts of LC3-II increased during the first 0 to 72 h and then decreased at 96 and 120 h after aMPV/C infection, whereas the amounts of LC3-II did not substantially change in mock-infected Vero cells (Fig. 1B). In addition, viral F protein was used to track the progression of aMPV/C infection (Fig. 1B). DF-1 cells, an avian cell line, which supports aMPV growth,37 was further used to evaluate autophagy induced by aMPV/C infection. Similar results were also displayed in aMPV/C-infected DF-1 cells on a western blotting. Furthermore, the relative amount of LC3-II reached a peak at 72 h postinfection (hpi) (Fig. 1C). Because LC3-II level is well correlated with the number of autophagosomes,38 the ratios of LC3-II to ACTB levels in cells are known as an accurate indicator of autophagy activity.20,36 As illustrated in Fig. 1B and C, from 72 hpi onward, the densitometry ratios of LC3-II to ACTB bands in the aMPV/C-infected Vero or DF-1 cell lysates were much higher than those in the mock-infected cells, indicating that there was a cumulative increase in autophagosome formation occurred during the first 0 to 72 h after viral infection (P < 0.01). Importantly, it was found that LC3-II levels increased from 24 to 72 h, in agreement with F protein expression patterns (Fig. 1B and C). These results suggest that autophagy is obviously induced in Vero cells and DF-1 cells during the first 0 to 72 h after aMPV/C infection.
Another good marker of autophagic vesicle formation is the punctate accumulation of LC3, which represents the recruitment of LC3-II to phagophores, the precursors to autophagosomes.20,22 To facilitate the observation of autophagic vesicles in aMPV/C-infected cells by confocal immunofluorescence microscopy, Vero cells were transfected with a green fluorescent protein-tagged LC3 plasmid (GFP-LC3). The green fluorescent pattern exhibited obvious punctate morphology upon aMPV/C infection, resembling the pattern of autophagosome-like vesicles, as evidenced by positive red aMPV/C antigen (viral N and F proteins) staining. In contrast, the patterns produced by GFP-LC3 in the mock-infected cells were diffuse (Fig. 1D, P < 0.01). These results confirmed that aMPV/C infection could induce the formation of autophagosomes.
A recent report shows that binding of a pathogen receptor of infectious bursal disease virus (IBDV) to host cells induced autophagy.39 Heat-inactivated viruses are considered to lose their abilities to replicate in host cells. Thus, to examine whether aMPV/C active replication is required for the induction of autophagy, we used western blotting to evaluate the amounts of LC3-II in cultured Vero cells after inoculation with a heat-inactivated virus sample (corresponding to a multiplicity of infection of 0.1). The complete eradication of viral infectivity by heat inactivation was confirmed by assaying virus titer with the activated undiluted viral suspension (data not shown). As shown in Fig. 1E, LC3-II expression decreased to marginal levels similar to those observed in mock-infected cells and in cells infected with heat-inactivated aMPV/C at 72 and 120 hpi. Meanwhile, no F protein synthesis was detected in the cultured cells inoculated with heat-inactivated aMPV/C, suggesting that aMPV/C active replication is indispensable for the induction of autophagy.
Induction of autophagy regulates aMPV/C replication
To further analyze the relationship between autophagy and aMPV/C viral replication, we used rapamycin (Rap), a widely used inducer of autophagy, or 3-methyladenine (3-MA), a widely used inhibitor of autophagy,40 to treat aMPV/C-infected cells for evaluation of viral protein expression and virus production. As illustrated in Fig. 2, both the intensity of the LC3-II band (P < 0.001) and the expression of the viral F protein (P < 0.01) were significantly increased upon Rap treatment compared with the untreated groups, and Rap-treated cells resulted in a notable raise in viral yields at 72 hpi (Fig. 2B, P < 0.05). In contrast, 3-MA treatment significantly inhibited the conversion of LC3-II protein and reduced the expression levels of viral F protein compared with those observed in untreated aMPV/C-infected groups (Fig. 2C, P < 0.001), and the viral titers were obviously decreased in 3-MA-treated cells at 72 hpi (Fig. 2D, P < 0.01). In addition, the MTT assay demonstrated that the viability of Vero cells was not obviously affected by Rap or 3-MA treatment (data not shown). These results further confirmed that autophagy plays a critical role in aMPV/C replication.
Figure 2.
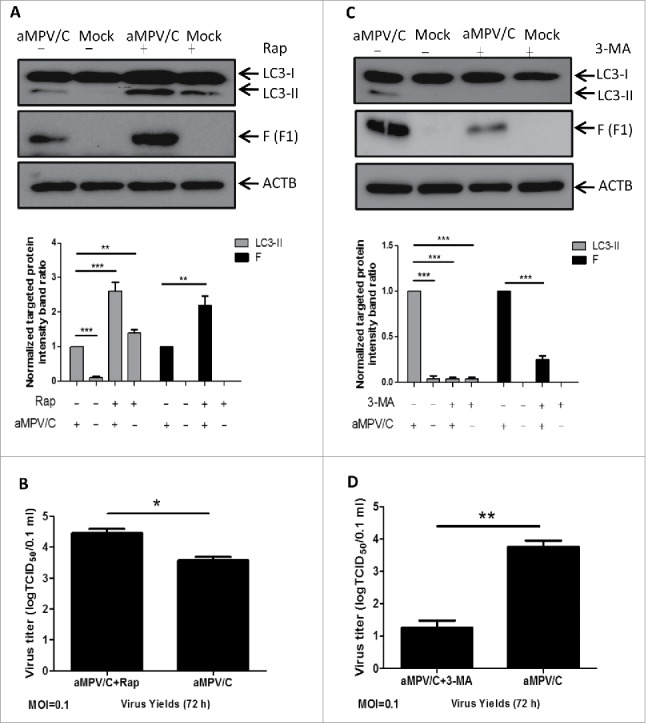
The effect of autophagy on aMPV/C replication in cells treated with different pharmacological compounds. (A) Vero cells were mock-pretreated or pretreated with complete medium containing 500 nM rapamycin (Rap) for 4 h and then subjected to aMPV absorption for 1.5 h and further cultured in fresh medium with the presence or absence of 500 nM Rap for 72 h. Cell samples were then processed and analyzed using anti-LC3 and -viral F antibodies as described in the legend to Figure 1B. The presence of Rap is indicated with “+.” Representative results are displayed with graphs corresponding to the ratios of LC3-II:ACTB or viral F:ACTB normalized to the control conditions. (B) Vero cells were pretreated and infected as described in A. Progeny virus yields were examined based on 50% tissue culture infective dose (TCID50) per 0.1 mL at 72 hpi in Vero cells. MOI, multiplicity of infection. (C) Vero cells were mock-pretreated or pretreated with complete medium containing 2 mM 3-methyladenine (3-MA) for 4 h and then subjected to aMPV/C absorption for 1.5 h and further cultured in fresh medium with the presence or absence of 2 mM 3-MA for 72 h. Cell samples were then processed and analyzed as described in the legend to Figure 1B. The presence of 3-MA is indicated with “+.” Representative results are displayed with graphs corresponding to the ratios of LC3-II:ACTB or viral F:ACTB normalized to the control conditions. (D) Progeny virus yields were examined as described in B. Error bars, mean ± SD of 3 independent experiments. Two-way ANOVA, *P < 0.05; **P < 0.01; ***P < 0.001, compared with the control group.
Knockdown of the endogenous ATG7 or LC3 genes reduces aMPV/C replication
To exclude the effects of pharmacological treatment on viral replication, we extended the above studies by further examining the effects of knockdown of the intracellular autophagy proteins ATG7 or LC3 on the replication of aMPV/C using target-specific RNA interference. The ATG7 protein and the LC3 protein are both essential for inducing autophagosome formation.26,41 Therefore, we used specific siRNAs to silence the ATG7 gene or the LC3 gene in Vero cells. As shown in Fig. 3, the cells transfected with ATG7 siRNA or LC3 siRNA showed dose-dependent reductions in ATG7 protein or LC3 protein levels. In particular, 30 pmol of siRNA treatment noticeably reduced the expression levels of endogenous ATG7 protein or LC3 protein, and this dose was therefore used for subsequent studies (Fig. 3A). Fig. 3B shows that the increased levels of viral F protein were significantly inhibited in the ATG7-silenced group compared with the scrambled siRNA group and control group (P < 0.001), and the reduction in ATG7 protein expression resulted in a notable decrease in viral yields at 72 hpi (Fig. 3C, P < 0.05). Similarly, Vero cells transfected with LC3 siRNA also showed an obvious decrease in the synthesis of viral F protein (Fig. 3D, P < 0.01) and in viral yields at 72 hpi (Fig. 3E, P < 0.05). Taken together, these results further demonstrate that autophagy is required for efficient replication of aMPV/C.
Figure 3.

Knockdown of the endogenous ATG7 or LC3 genes inhibits the replication of aMPV/C. (A) Vero cells were mock-transfected (mock) or transfected with ATG7-siRNA or LC3-siRNA at different concentrations (5, 10, 20 or 30 pmol), or 30 pmol scrambled-siRNA (Scra). ATG7 and LC3 proteins were detected by western blotting at 48 h. (B) Vero cells were transfected with ATG7-siRNA, Scra and Con, and then infected with aMPV/C. At 72 hpi, the cells were harvested and analyzed by western blotting with anti-ATG7, anti-viral F, and anti-ACTB antibodies. Representative results are displayed with graphs corresponding to the ratios of ATG7:ACTB or viral F:ACTB normalized to the control conditions. (C) Vero cells transfected with ATG7-siRNA or Scra were infected with aMPV/C for 72 hpi. Progeny virus yields were examined by TCID50 assay in Vero cells. (D) Vero cells were transfected with LC3-siRNA, Scra and Con and then infected, processed and analyzed with anti-LC3, anti-viral F and anti-ACTB antibodies. Representative results are displayed with graphs corresponding to the ratios of LC3-II:ACTB or viral F:ACTB normalized to the control conditions. (E) Vero cells transfected with LC3-siRNA or Scra were infected, processed and examined as described in C. Error bars, mean ± SD of 3 independent experiments. Two-way ANOVA test, *P < 0.05; **P < 0.01; ***P < 0.001, compared with the control group.
Induction of complete autophagy by aMPV/C is beneficial to viral replication
The SQSTM1/p62 protein is a selective autophagic molecule that is incorporated into autophagosomes and degraded along with other cell substrates by lysosomal hydrolases.20,22,42 These events represent a complete autophagic process, which induces autophagic flux and autophagosome maturation. To determine whether aMPV/C-induced autophagy is a complete process, we first detected intracellular SQSTM1 levels in aMPV/C-infected Vero cells by western blotting analysis (Fig. 4A). We found that the expression of intracellular SQSTM1 gradually decreased in the aMPV/C-infected cells during the first 0 to 72 h after viral infection, and this degradation was weakly recovered at 96 and 120 h. In contrast, no obvious change in SQSTM1 expression was found in mock-infected cells. Moreover, the densitometry ratios of SQSTM1 to ACTB bands in the aMPV/C-infected cells were much lower than those in the mock-infected cells from 48 to 96 h (P < 0.01), indicating that autophagic flux occurred in the aMPV/C-infected cells.
Figure 4.

aMPV/C-induced complete autophagy is beneficial to viral replication. (A) Vero cells were mock-infected or infected with aMPV/C at the indicated time points (0, 24, 48, 72, 96, and 120 h) postinfection and then lysed and subjected to western blotting with anti-SQSTM1 antibody. ACTB was used as a protein loading control. The ratios of SQSTM1:ACTB were normalized to the control conditions. (B) Vero cells were mock-pretreated or pretreated with complete medium containing 20 μM chloroquine (CQ) for 1 h and then subjected to aMPV/C absorption for 1.5 h and further cultured in fresh medium with the presence or absence of 20 μM CQ for 72 hpi. Cell samples were then processed and analyzed using anti-LC3, anti-SQSTM1, and anti-viral F antibodies as described in (A). The presence of CQ is indicated with “+.” Representative results are displayed with graphs corresponding to the ratios of LC3-II:ACTB or viral F:ACTB normalized to the control conditions. (C) Vero cells were pretreated and infected as described in B. Progeny virus yields were examined by TCID50 at 72 hpi in Vero cells. Error bars, mean ± SD of 3 independent experiments. Two-way ANOVA test, #P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001, compared with the control group.
To further characterize whether aMPV/C infection induced autophagosome maturation, we measured LC3-II accumulation and autophagic flux in aMPV/C-infected and mock-infected cells treated with or without chloroquine (CQ). CQ is a nonspecific inhibitor that increases the pH in lysosomes to prevent autophagosome-lysosome fusion.20,43,44 As shown in Fig. 4B, following CQ treatment, there was an obvious increase in LC3-II accumulation and significant inhibition of SQSTM1 degradation in the aMPV/C-infected Vero cells compared with the mock-infected cells (P < 0.05). Moreover, viral F protein expression was significantly decreased following CQ treatment (Fig. 4B, P < 0.001). As expected, the viral titers in CQ-treated cells were noticeably decreased compared with those in the aMPV/C-infected cells at 72 hpi (Fig. 4C, P < 0.01). In addition, the MTT assay demonstrated that the viability of Vero cells was not perceptibly affected by CQ treatment (data not shown). Taken together, these results demonstrated that aMPV/C infection enhances autophagosome maturation and induces a complete autophagic process, which is beneficial to viral production.
aMPV/C infection induces ER stress via activating UPR signaling through the ATF6 pathway, but not the EIF2AK3 and ERN1 pathways
To elucidate how aMPV/C infection induces autophagy, we investigated whether ER stress was present and UPR pathways were activated in aMPV/C-infected Vero cells, as these factors have been reported to induce autophagy.27,29,45 Vero cells treated with thapsigargin (Tg), a known ER stress inducer,46 for 48 h served as a positive control. HSPA5/GRP78 (heat shock protein family A [Hsp70] member 5) is an ER chaperone protein that mainly regulates 3 proximal ER stress transducers, including EIF2AK3, ATF6 and ERN1, by interacting with a lumenal domain. As expected, western blotting analysis showed that aMPV/C infection and Tg treatment could enhance levels of the ER stress-marker protein HSPA5 compared with control cells (Fig. 5A). Moreover, we analyzed autophagy in Vero cells infected with aMPV/C or treated with Tg. The conversion of LC3-II was significantly increased by aMPV/C or Tg treatment in comparison with mock-infected cells, indicating that ER stress can induce autophagic signaling in aMPV/C-infected Vero cells.
Figure 5.

aMPV/C infection activates the ATF6 pathway of the UPR. (A and B) After infection with aMPV/C for 72 h or NDV for 24 h, Vero cells were processed and subjected to western blotting analysis using the indicated antibodies (HSPA5, EIF2AK3, p-EIF2AK3, EIF2S1, p-EIF2S1, precursor ATF6, cleaved ATF6, LC3, and ACTB). ACTB was used as a protein loading control. (C) Schematic showing the analysis of XBP1 mRNA splicing. The PstI restriction site and the relative location of the 26-nucleotide (nt) intron are shown. The sizes of PCR-amplified fragments from spliced XBP1 (XBP1s) and unspliced XBP1 (XBP1u) with or without PstI cleavage are also listed. (D and E) Vero cells were treated with Tg (400 nM) for 48 h as a positive control and were infected with aMPV/C for 72 h or NDV for 24 h. Total cellular RNAs were prepared, and RT-PCR was performed by using specific primers to detect XBP1s levels. To determine XBP1s levels, the PCR products were digested with PstI and then electrophoresed. XBP1u (283/315 base pairs) and XBP1s (572 base pairs) bands are indicated.
In response to ER stress, the UPR pathway becomes activated to minimize ER malfunction. To dissect the mechanism underlying how aMPV/C infection induces autophagy, we first examined whether the EIF2AK3 pathway became activated in aMPV/C-infected Vero cells by analyzing the phosphorylation of EIF2AK3 and its downstream effector EIF2S1/EIF2A (eukaryotic translation initiation factor 2 subunit α) by western blotting. As shown in Fig. 5A, the cells infected with aMPV/C exhibited the same expression levels of EIF2AK3, phosphorylated (p)-EIF2AK3, EIF2S1, and p-EIF2S1 compared with mock-infected cells. Higher levels of EIF2AK3 and EIF2S1 phosphorylation were observed in Tg-treated cells than in the mock-infected cells. ATF6, a second sensor of ER stress, is cleaved by proteases and is translocated to the nucleus, where it activates many genes responsible for the ER stress response.47,48 Western blotting analysis showed that Tg-treatment or aMPV/C infection significantly increased the degradation of the 90-kDa ATF6 precursor, yielding 50-kDa cleaved products. These results suggested that the ATF6 pathway becomes activated in aMPV/C-infected Vero cells.
Activation of ERN1 in response to ER stress could cause alternative splicing of XBP1 mRNA. XBP1 cDNA was amplified by RT-PCR and digested with PstI; the PstI restriction site is located in a 26-nucleotide region of XBP1 cDNA that is removed by ERN1-mediated splicing, as described previously (Fig. 5C).49,50 Tg served as a positive control for inducing XBP1 splicing (to generate XBP1s).27 As shown in Fig. 5D, high levels of XBP1s were present in Tg-treated cells, as shown by the resistance of the spliced products to PstI digestion. Moreover, when compared with mock-infected cells and aMPV/C-infected cells, no significant differences were observed in the levels of XBP1 expression, which was mostly not spliced. These results suggest that aMPV/C infection does not robustly activate the ERN1 pathway through the splicing of XBP1.
Recent studies report that Newcastle disease virus (NDV), another member of Paramyxoviridae family, can induce autophagy through activation of the EIF2AK3 and ATF6 pathways in A549 cells.51 As a virus control, we further used NDV strain LaSota to inoculate into Vero cells for determination of autophagy induction. Vero cells treated with Tg for 48 h or infected with NDV for 24 h were analyzed by western blotting and RT-PCR. As shown in Fig. 5B, besides the notable increases in HSPA5 expression and LC3 conversion, both EIF2AK3 and EIF2S1 were significantly phosphorylated, while the total EIF2AK3 and EIF2S1 levels remained unchanged in NDV-infected or Tg-treated cells compared with mock-infected cells. As Tg-treatment, NDV infection significantly increased the degradation of the ATF6 precursor, yielding the cleaved products (Fig. 5B). Also, the RT-PCR results showed that high levels of XBP1s were present in Tg-treated Vero cells, whereas XBP1s was rarely observed in NDV-infected or mock-infected Vero cells, which were similar to that of aMPV/C infection (Fig. 5E). These results further demonstrated that NDV infection induced authphagy through the EIF2AK3 and ATF6 pathways in the cultured cells, regardless of different cell types. Unlike NDV, our results suggest that aMPV/C infection induces autophagy through the ATF6 pathway but not the EIF2AK3 and ERN1 pathways in Vero cells.
Knockdown of the endogenous ATF6 gene inhibits aMPV/C-induced autophagy and virus production
To verify if autophagy occurred through the ATF6 UPR pathway in aMPV/C-infected Vero cells, we examined whether knockdown of the endogenous ATF6 gene would inhibit aMPV/C-induced autophagy and viral yields. First, we used a specific siRNA to silence the ATF6 gene in Vero cells. Cells transfected with the ATF6 siRNA showed a dose-dependent decrease in ATF6 expression (Fig. 6A), with 40 pmol of siRNA being chosen for further study. Next, we assessed the effect of ATF6 knockdown on aMPV/C-induced autophagy. Compared to control cells (siRNA-untransfected cells) and scrambled siRNA-transfected cells, knockdown of the ATF6 gene reduced aMPV/C-induced accumulation of LC3-II and degradation of SQSTM1 (Fig. 6B). This result suggests that autophagy was activated via the ATF6 pathway in the aMPV/C-infected Vero cells. In addition, we detected the effect of silencing ATF6 expression on virus production. As shown in Fig. 6B, there was significantly decreased viral F protein expression in Vero cells (P < 0.001) compared with control cells and scrambled siRNA-transfected cells. Likewise, virus titer assays showed that downregulation of ATF6 expression noticeably decreased viral yields at 72 hpi in Vero cells (Fig. 6C, P < 0.05). These results suggest that silencing ATF6 by siRNA can inhibit aMPV/C-induced autophagy and viral yields. In addition, we examined the effect of ATF6 overexpression on aMPV/C replication in Vero cells. Vero cells were transfected with pCMV-HA-ATF6 plasmid and then infected with aMPV/C. As shown in Fig. 6D, the accumulation of LC3-II or expression of SQSTM1 and F proteins were not significantly changed in the pCMV-HA-ATF6 plasmid-transfected cells compared with pCMV-HA plasmid-transfected cells or mock-treated cells (P > 0.05). Moreover, differences of viral yields were not found between ATF6 overexpression group and control group (Fig. 6E, P > 0.05). These results suggest that overexpression of ATF6 cannot enhance aMPV/C replication. Overall, these data indicated that aMPV/C-infection induces autophagy by activating the ATF6 UPR pathway.
Figure 6.

Knockdown of the endogenous ATF6 gene inhibits aMPV/C-induced autophagy and virus production. (A) Vero cells were mock-transfected (mock) or transfected with ATF6-siRNA at different concentrations (10, 20, 30 or 40 pmol), or 40 pmol scrambled-siRNA (Scra). ATF6 protein was detected by western blotting at 48 h. (B) Vero cells were transfected with ATF6-siRNA, Scra and Con (siRNA-untransfected) and then infected with aMPV/C for 72 h. The cells were harvested and analyzed by western blotting with anti-ATF6, anti-SQSTM1, anti-LC3, anti-viral F, and anti-ACTB antibodies. Representative results are displayed with graphs corresponding to the ratios of ATF6:ACTB or viral F:ACTB normalized to the control conditions. (C) Vero cells were transfected and infected as described in B. Progeny virus yields were examined by TCID50 at 72 hpi in Vero cells. (D) Vero cells were transfected with pCMV-HA (HA) or pCMV-HA-ATF6 (HA-ATF6) plasmid and then infected with aMPV/C for 72 h. The cells were harvested and analyzed by western blotting with anti-ATF6, anti-SQSTM1, anti-LC3, anti-viral F, and anti-ACTB antibodies. Representative results are displayed with graphs corresponding to the ratios of ATF6:ACTB or viral F:ACTB normalized to the control conditions. (E) Vero cells were transfected and infected as described in D. Progeny virus yields were examined by TCID50 at 72 hpi in Vero cells. Error bars, mean ± SD of 3 independent experiments. Two-way ANOVA test, #P > 0.05; *P < 0.05; ***P < 0.001, compared with the control group.
Discussion
Viruses have developed various complex strategies to regulate host cell responses to facilitate their replication and evade host defense mechanisms. aMPV is a causative agent for acute rhinotracheitis and swollen head syndrome.52 In the present work, a series of experiments was performed to identify what interactions exist between host cells and aMPV/C, with a particular focus on the effect of cellular autophagy on aMPV/C replication and the induction mechanisms of autophagy. Recently, an increasing number of studies have demonstrated that cellular autophagy plays a role in infections with Paramyxoviridae family members through different mechanisms in target cells.35,36 However, to date, no report has indicated whether cellular autophagy is involved in aMPV replication or which pathway of autophagy induction is activated in this context.
In the present study, we demonstrated that aMPV/C infection significantly increased the presence of single- or double-membrane vesicles in the perinuclear region of the cultured cells, as revealed by electron microscopic observation (Fig. 1A).20 Furthermore, the conversion of LC3 was analyzed by western blotting, and the results further confirmed that the formation of autophagic vesicles was related to cellular autophagy (Fig. 1B and C). Importantly, the conversion of LC3 was most evident at 72 hpi; therefore, we primarily focused on this time point of aMPV/C infection for further analysis of the mechanisms that induce autophagy in this context. Notably, LC3-II levels did not increase in Vero cells infected with heat-inactivated aMPV/C, which indicated that active replication of aMPV/C is required for the induction of cellular autophagy (Fig. 1E). In principle, the punctate accumulation of the exogenous protein GFP-LC3 represents a step in the cellular autophagy process. Subsequently, the confocal immunofluorescence analysis showed that GPF-LC3 displayed many positive puncta in the cytoplasm of aMPV/C-infected cells and that the viral N and F proteins were predominantly expressed in cytoplasm, representing the presence of aMPV/C virions, which colocalized with GFP-LC3 puncta (Fig. 1D).
To understand the role of cellular autophagy in aMPV/C replication, we examined how activation or inhibition of autophagy affected aMPV/C replication. First, our results showed that regulation of autophagy using the pharmacological compounds rapamycin or 3-methyladenine respectively, increased or inhibited viral F protein expression and also enhanced or reduced viral yields (Fig. 2). These data are concordant with studies of autophagy in the context of other viral species, such as NDV,34 encephalomyocarditis virus (EMCV),26 and porcine reproductive and respiratory virus (PRRSV).53,54 In addition, because the ATG7 and LC3 proteins play important roles in inducting autophagosome formation,26,41,55 we further analyzed how downregulating autophagy through silencing the ATG7 or LC3 proteins affected virus production. These results demonstrated that knockdown of ATG7 protein or LC3 protein significantly inhibited the expression of viral proteins as well as virus production (Fig. 3). Taken together, our findings illustrate that aMPV/C replication is influenced by changes in autophagy activity and not by the tested pharmacological compounds themselves.
During autophagy, autophagosomes fuse with lysosomes, leading to the degradation of the autophagic cargoes and cell substrates by lysosomal hydrolases.42 This series of events represents a complete autophagic process. Hepatitis C virus infection has been reported to enhance autophagic flux and induce complete autophagy.27,56,57 However, some viruses were believed to inhibit the maturation of autophagosomes in virus-infected cells. For example, hepatitis B virus, PRRSV, and coxsackievirus A16 can disrupt autophagosome trafficking to lysosomes, ultimately resulting in the accumulation of autophagosomes.25,53,58 In terms of lysosome-dependent signaling, the reduction of SQSTM1 protein expression during virus infection is actually considered as the ultimate outcome marking complete autophagy. Here, we demonstrated that aMPV/C infection induces autophagosomal maturation (Fig. 4). Our data suggest that SQSTM1 proteins are degraded during aMPV/C infection. Additionally, treatment with CQ, a specific inhibitor of autophagosome-lysosome fusion, restored SQSTM1 expression and inhibited virus production, which indirectly indicated that aMPV/C infection induces a complete autophagic process.
An increasing number of studies have reported that viruses can induce autophagy by activating diverse pathways. For instance, autophagy can be induced in host cells through the activation of the MAPK1/ERK2 (mitogen-activated protein kinase 1) by Epstein Barr virus,59 the modulation of MTOR (mechanistic target of rapamycin) by IBDV,39 the triggering of EIF2AK2/PKR (eukaryotic translation initiation factor 2 α kinase 2) expression by herpes simplex virus type 124 and the induction of ER stress by hepatitis C virus.27 In the present study, we found that HSPA5 protein expression significantly increased at 72 hpi in aMPV/C-infected Vero cells compared with the control cell groups (Fig. 5A). HSPA5 is a marker protein of ER stress,60 indicating that aMPV/C infection activates ER stress. In response to ER stress, the UPR pathways are induced to minimize ER malfunction. Therefore, we hypothesized that aMPV/C infection might induce autophagy through ER stress-associated UPR signaling.
Interestingly, we examined all 3 UPR pathways, namely, those modulated by EIF2AK3, ATF6 and ERN1, using western blotting or RT-PCR. The results showed that only the ATF6 pathway was activated and not the EIF2AK3 and ERN1 pathways (Fig. 5A). This pattern is different from the activation pathway produced by infection with NDV, a member of the family Paramyxoviridae.51 Both the ATF6 and EIF2AK3 pathways become activated in NDV-infected Vero cells (Fig. 5B) as observed for NDV infection in A549 cells.51 This further confirmed a unique event of aMPV/C-activated autophagy, but the precise mechanism underlying how aMPV/C induces autophagy needs to be studied in the future. In this study, our results also revealed that siRNA-mediated suppression of the ATF6 pathway reversed aMPV/C infection-induced accumulation of LC3-II and degradation of SQSTM1 (Fig. 6B). This result indicates that aMPV/C-induced autophagy might also be regulated by other signaling pathways independent of ER stress, and as such, the process was not fully inhibited in ATF6-knockdown cells. As shown in Fig. 6B and C, ATF6 gene knockdown significantly decreased viral yields in aMPV/C-infected Vero cells, suggesting that the ATF6 UPR pathway plays an important role in regulating virus production during aMPV/C infection. We also determined the effect of ATF6 overexpression on autophagy activation and aMPV/C yields and found that the level of autophagy activation and virus production were not significantly changed in ATF6-overexpressed cells (Fig. 6D to E). This might be associated with the fact that the level of autophagy activation parallels the amounts of ATF6 cleaved products (50 kD) but not its precursor (90 kD), because only the ATF6 cleaved products can translocate into the nucleus and activate transcription in response to ER stress.47,48 Furthermore, we found that the ATF6 precursor was not fully cleaved in aMPV/C-infected cells (Fig. 5A), and ATF6 overexpression only increased the levels of the ATF6 precursor but not its cleavage at the same dose of aMPV/C inoculation, therefore, ATF6 overexpression rarely affected the level of autophagy activation in aMPV/C-infected cells. Taken together, our study confirms the connection between aMPV/C infection and the ATF6 pathway in Vero cells. In other words, aMPV/C infection induces cellular autophagy by activating the ATF6 pathway in response to ER stress, which in turn benefits viral replication.
In summary, our results demonstrated for the first time that aMPV/C infection induces a complete autophagic process and that this event is partially regulated by ER stress. Our data further showed that the ATF6 UPR pathway is involved in autophagy induction, and silencing ATF6 protein inhibits aMPV/C replication. These results provide insights into the interactions that exist between aMPV/C and host cells and lay a foundation for further studies of the molecular mechanisms that underlie aMPV pathogenesis.
Materials and methods
Cell cultures and aMPV/C infection
Vero cells and DF-1 cells (immortal chicken embryo fibroblasts [CEF]) were originally purchased from the American Type Culture Collection (ATCC, CCL-81 and CRL-12203) and cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, 11995) supplemented with 100 mg/ml streptomycin, 100 units/ml penicillin and 5 to 10% fetal bovine serum (Gibco; Life Technologies, 10099–141) at 37°C in the presence of 5% CO2 in an incubator.
aMPV subgroup C (aMPV/C) strain JC is kept in our laboratory and was isolated from local Chinese meat-type chickens with respiratory syndrome as described previously.10 The viruses used to inoculate Vero cells were titrated by serial dilutions and used at 104.5 of the 50% tissue culture infectious dose (TCID50) per 0.1 mL. aMPV/C was heat inactivated at 90°C for 20 min in a water bath and then inoculated onto Vero cells to detect the infectivity of inactivated virus. Newcastle disease virus (NDV) strain LaSota was kindly provided by Dr. Zhenhua Zhang (Beijing Academy of Agriculture and Forestry, Beijing, China).
Antibodies and reagents
The following primary antibodies were used: rabbit anti-LC3 (L7543), rabbit anti-SQSTM1 (P0067), and mouse anti-ACTB (A5441). The following secondary antibodies were used: tetramethyl rhodamine isothiocyanate (TRITC)-conjugated goat anti-rabbit (T6778) or rabbit anti-mouse (T2402), and horseradish peroxidase (HRP)-conjugated goat anti-mouse (A9044), anti-rabbit (A0545) or rabbit anti-goat (A5402). The following reagents were used: rapamycin (Rap, R0395), 3-methyladenine (3-MA, M9281), thapsigargin (Tg, T9033) and chloroquine (CQ, C6628). All of the above antibodies and reagents were obtained from Sigma-Aldrich. Antibodies against EIF2AK3 (sc-13073), p-EIF2AK3 (sc-32577), EIF2S1 (sc-11386), p-EIF2S1 (sc-12412), ATF6 precursor (sc-22799), and cleaved ATF6 (sc-14253) were purchased from Santa Cruz Biotechnology. Mouse monoclonal antibodies against HSPA5 (3183s) and ATG7 (8558s) were obtained from Cell Signaling Technology. Mouse anti-F monoclonal antibodies and rabbit anti-N polyclonal antibodies were prepared in our laboratory.
Recombinant plasmid
The monkey LC3 gene and ATF6 gene from Vero cells was amplified by reverse transcription (RT)-PCR with gene-specific primers that were designed in accordance with the LC3 gene and ATF6 gene sequence in the GenBank database (accession no. NM_001193625 and XM_011952980, respectively, Table S1). The amplified cDNAs were then subcloned into pEGFP-C1 (Clontech, 6084–1) or pCMV-HA (Clontech, 631604) to generate the recombinant plasmid: pEGFP-LC3 and pCMV-HA-ATF6. These plasmids were sequenced to confirm that the amplified products had no errors introduced as a result of PCR amplification.
Virus infection and virus titeration
In accordance with the requirements of different experiments, the Vero cells or DF-1 cells were either infected with aMPV/C or NDV at a multiplicity of infection of 0.1 or 1.0 or mock-infected with phosphate-buffered saline (PBS; Beyotime, C0221A). Following a 1.5-h absorption time, the cells were then cultured in complete medium at 37°C for the indicated time points until the necessary samples had been harvested for further experiments. For the autophagy regulation experiments, Vero cells were treated with different concentrations of pharmacological compounds for 1 h or 4 h before viral infection.
The titer of aMPV/C produced was assayed on Vero cells monolayers. Cell supernatant was serially diluted and inoculated on Vero cells. Following 1.5-h incubation, fresh medium was added and incubated. Five d postinfection, cytopathic effect (CPE) was observed under a microscope and virus titer was determined as the 50% tissue culture infective dose (TCID50) per 0.1 mL.
ATF6 overexpression
Vero cells grown to 80% to 90% confluence on 6-well culture plates were transfected with pCMV-HA-ATF6 or pCMV-HA plasmid for 18 h, and then infected with aMPV/C for 72 h. The cells were harvested and analyzed by western blotting.
Knockdown of ATG7, LC3 and ATF6 by RNA interference (RNAi)
The following siRNAs were designed: siATG7 (sense, 5′-GCUGGUUUCCUUGCUUAAATT-3′; antisense, 5′-UUUAAGCAAGGAAACCAGCTT-3′), siLC3 (sense, 5′-CCUUCUUCCUGUUGGUGAATT-3′; antisense, 5′-UUCACCAACAGGAAGAAGGTT-3′), siATF6 (sense, 5′-GCUGUUCAAUACACAGAATT-3′; antisense, 5′-UUUCUGUGUAUUGAACAGCTT-3′) and scrambled-siRNAs (sense, 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense, 5′-ACGUGACACGUUCGGAGAATT-3′). These siRNAs were designed by the GenePharma Company (Suzhou, China) and used to silence the expression of ATG7, LC3 or ATF6 protein in Vero cells. The cells were transfected with different concentrations of siRNA using Lipofectamine RNAiMAX reagent according to the manufacturer's protocol and were then harvested for further analysis after 48 h.
RNA preparation and reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total cellular RNA from Vero cells was extracted with an RNeasy Mini Kit (Qiagen, 74104) according to the manufacturer's protocol. cDNA was reverse-transcribed with 2 μg of total RNA as the template, which was amplified by following the procedures described in the SuperScript III RT-PCR kit (Invitrogen, 18080–093) in MyCycler pro (Thermal Cycler, 563BR3037, California, USA).
The XBP1 (X-box binding protein 1) and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) genes were amplified by RT-PCR with the specific primers listed in Table S1, which were designed in accordance with gene sequences available in the GeneBank database (accession no. XM_010359220 and XM_010385947).
SDS-PAGE and western blotting
Whole cell lysates were prepared at the different time points after infection and transfection using RIPA lysis buffer (Beyotime, P0013B) and 1 mM phenylmethanesulfonyl fluoride (PMSF; Beyotime, ST506–2) in accordance with the manufacturer's protocol. The total protein concentration was measured using a bicinchoninic acid protein assay kit (Thermo Scientific, 23225). Equal amounts of total proteins were analyzed with SDS-PAGE and then transferred onto nitrocellulose membranes (Pall, 66485). After blocking with skim dry milk for 2 h at 37°C, the membranes were incubated with different primary antibodies for 2 h at 37°C, followed by incubation with horseradish peroxidase-conjugated secondary antibodies at 37°C for 2 h. The secondary antibodies were detected using a SuperSignal West Femto Substrate Trial Kit (Thermo Scientific, 34096) and then exposed in a chemiluminescence apparatus (Proteinsample, Santa Clara, CA, USA).
Confocal microscopy
Vero cells cultured until approximately 80% to 90% confluence in 24-well culture plates were infected with aMPV/C after transfecting with pEGFP-LC3 plasmids for 18 h. The cells were fixed with precooled 4% paraformaldehyde (Sigma-Aldrich, 16005) in PBS for 15 min and permeabilized using 0.1% Triton X-100 (Sigma-Aldrich, T8787) and in 2% BSA (Beyotime, ST023) in PBS for 30 min, and a mouse monoclonal antibody against viral F protein or a rabbit polyclonal antibody against viral N protein was incubated with the cells for 2 h at 37°C. After 3 washes with PBS-Tween-20 (PBST containing 0.05% Tween-20 [Sigma, P1379]), the cells were incubated with secondary TRITC-conjugated anti-mouse or anti-rabbit antibodies for 2 h at 37°C. Finally, the cells were washed with PBST and directly observed under a Nikon AIR confocal immunofluorescence microscope (Nikon Instruments, Inc., Melville, NY, USA).
Transmission electron microscopy
Subconfluent monolayers of Vero cells cultured in 6-well culture plates (Corning Inc.) were infected with aMPV/C for 72 h. The cells were then processed for transmission electron microscopy (TEM) observation as described previously.61 For immunoelectron microscopy (IEM), a preembedded silver enhancement immunogold method62 was used to determine localization of aMPV/C particles in virus-infected Vero cells. The processed frozen ultrathin sections were incubated with mouse anti-F monoclonal antibody (300 ×), and then incubated with goat anti-mouse IgG (100 ×) conjugated to colloidal gold particles 10 nm in diameter (Sigma, G7777). The gold labels were finally magnified by a silver enhancement kit (Sigma, SE100). Ultrathin sections were imaged using a Hitachi H-7500 transmission electron microscope (Hitachi Ltd, Tokyo, Japan).
Statistical analysis
The data are presented as the mean ± standard deviations (SD). One-way analysis of variance (ANOVA) and Dunnett and Tukey post hoc tests and 2-way ANOVA were used to analyze data. P < 0.05 was considered statistically significant.
Supplementary Material
Abbreviations
- ACTB
actin beta
- ATF6
activating transcription factor 6
- ATG7
autophagy-related 7
- BSA
bovine serum albumin
- CQ
chloroquine
- EIF2AK3/PERK
eukaryotic translation initiation factor 2 alpha kinase 3
- EIF2S1/EIF2A
eukaryotic translation initiation factor 2 subunit alpha
- ER
endoplasmic reticulum
- ERN1/IRE1
endoplasmic reticulum to nucleus signaling 1
- hpi
hours postinfection
- HSPA5/GRP78
heat shock protein family A (Hsp70) member 5
- MAP1LC3/LC3
microtubule associated protein 1 light chain 3
- Rap
rapamycin
- siRNA
small interfering RNA
- SQSTM1/p62
sequestosome 1
- Tg
thapsigargin
- UPR
unfolded protein response
- 3-MA
3-methyladenine
Disclosure of potential conflicts of interest
The authors declare that they have no conflicts of interest.
Funding
This study was supported by grants from the National Natural Science Foundation (31472212), China Agriculture Research System (CARS-42), the Beijing Municipal Science and Technology Project (Z141100002314013), and Innovative Capability Project of Beijing Academy of Agriculture and Forestry Sciences (KJCX20161503), People's Republic of China. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- [1].Aung YH, Liman M, Neumann U, Rautenschlein S. Reproducibility of swollen sinuses in broilers by experimental infection with avian metapneumovirus subtypes A and B of turkey origin and their comparative pathogenesis. Avian Pathol. 2008;37:65-74. doi: 10.1080/03079450701802222. PMID:18202952 [DOI] [PubMed] [Google Scholar]
- [2].Jones RC. Avian pneumovirus infection: Questions still unanswered. Avian Pathol. 1996;25:639-48. doi: 10.1080/03079459608419171. PMID:18645888 [DOI] [PubMed] [Google Scholar]
- [3].Easton AJ, Domachowske JB, Rosenberg HF. Animal pneumoviruses: Molecular genetics and pathogenesis. Clin Microbiol Rev. 2004;17:390-412. doi: 10.1128/CMR.17.2.390-412.2004. PMID:15084507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Buys SB, Du Preez JH. A preliminary report on the isolation of a virus causing sinusitis in turkeys in South African and attempts attenuate the virus. Turkeys. 1980;28:36-46 [Google Scholar]
- [5].Guionie O, Toquin D, Sellal E, Bouley S, Zwingelstein F, Allee C, Bougeard S, Lemiere S, Eterradossi N. Laboratory evaluation of a quantitative real-time reverse transcription PCR assay for the detection and identification of the four subgroups of avian metapneumovirus. J Virol Methods. 2007;139:150-8. doi: 10.1016/j.jviromet.2006.09.022. PMID:17126416 [DOI] [PubMed] [Google Scholar]
- [6].Bayon-Auboyer MH, Arnauld C, Toquin D, Eterradossi N. Nucleotide sequences of the F, L and G protein genes of two non-A/non-B avian pneumoviruses (APV) reveal a novel APV subgroup. J Gen Virol. 2000;81:2723-33. doi: 10.1099/0022-1317-81-11-2723. PMID:11038385 [DOI] [PubMed] [Google Scholar]
- [7].Cook JKA, Huggins MB, Orbell SJ, Senne DA. Preliminary antigenic characterization of an avian pneumovirus isolated from commercial turkeys in Colorado, USA. Avian Pathol. 1999;28:607-17. doi: 10.1080/03079459994407 [DOI] [PubMed] [Google Scholar]
- [8].Toquin D, Guionie O, Jestin V, Zwingelstein F, Allee C, Eterradossi N. European and American subgroup C isolates of avian metapneumovirus belong to different genetic lineages. Virus Genes. 2006;32:97-103. doi: 10.1007/s11262-005-5850-3. PMID:16525740 [DOI] [PubMed] [Google Scholar]
- [9].Lee E, Song MS, Shin JY, Lee YM, Kim CJ, Lee YS, Kim H, Choi YK. Genetic characterization of avian metapneumovirus subtype C isolated from pheasants in a live bird market. Virus Res. 2007;128:18-25. doi: 10.1016/j.virusres.2007.03.029. PMID:17485129 [DOI] [PubMed] [Google Scholar]
- [10].Wei L, Zhu S, Yan X, Wang J, Zhang C, Liu S, She R, Hu F, Quan R, Liu J. Avian metapneumovirus subgroup C infection in chickens, China. Emerg Infect Dis. 2013;19:1092-4. doi: 10.3201/eid1907.121126. PMID:23763901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Njenga MK, Lwamba HM, Seal BS. Metapneumoviruses in birds and humans. Virus Res. 2003;91:163-9. doi: 10.1016/S0168-1702(02)00256-3. PMID:12573494 [DOI] [PubMed] [Google Scholar]
- [12].van den Hoogen BG, Bestebroer TM, Osterhaus AD, Fouchier RA. Analysis of the genomic sequence of a human metapneumovirus. Virology. 2002;295:119-32. doi: 10.1006/viro.2001.1355. PMID:12033771 [DOI] [PubMed] [Google Scholar]
- [13].Yunus AS, Govindarajan D, Huang Z, Samal SK. Deduced amino acid sequence of the small hydrophobic protein of US avian pneumovirus has greater identity with that of human metapneumovirus than those of non-US avian pneumoviruses. Virus Res. 2003;93:91-7. doi: 10.1016/S0168-1702(03)00074-1. PMID:12727346 [DOI] [PubMed] [Google Scholar]
- [14].Xie Z, Klionsky DJ. Autophagosome formation: Core machinery and adaptations. Nat Cell Biol. 2007;9:1102-9. doi: 10.1038/ncb1007-1102. PMID:17909521 [DOI] [PubMed] [Google Scholar]
- [15].Klionsky DJ. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931-7. doi: 10.1038/nrm2245. PMID:17712358 [DOI] [PubMed] [Google Scholar]
- [16].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717-21. doi: 10.1126/science.290.5497.1717. PMID:11099404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304-12. doi: 10.1038/nrd2272. PMID:17396135 [DOI] [PubMed] [Google Scholar]
- [18].Yorimitsu T, Klionsky DJ. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542-52. doi: 10.1038/sj.cdd.4401765. PMID:16247502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Reggiori F. 1. Membrane origin for autophagy. Curr Top Dev Biol. 2006;74:1-30. doi: 10.1016/S0070-2153(06)74001-7. PMID:16860663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222. doi: 10.1080/15548627.2015.1100356. PMID:26799652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542-5. doi: 10.4161/auto.4600. PMID:17611390 [DOI] [PubMed] [Google Scholar]
- [22].Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201-14. doi: 10.1089/ars.2010.3482. PMID:20712405 [DOI] [PubMed] [Google Scholar]
- [23].Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23-35. doi: 10.1016/j.chom.2006.12.001. PMID:18005679 [DOI] [PubMed] [Google Scholar]
- [24].Lussignol M, Queval C, Bernet-Camard MF, Cotte-Laffitte J, Beau I, Codogno P, Esclatine A. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol. 2013;87:859-71. doi: 10.1128/JVI.01158-12. PMID:23115300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu B, Fang M, Hu Y, Huang B, Li N, Chang C, Huang R, Xu X, Yang Z, Chen Z, Liu W. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy. 2014;10:416-30. doi: 10.4161/auto.27286. PMID:24401568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang Y, Li Z, Ge X, Guo X, Yang H. Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy. 2011;7:613-28. doi: 10.4161/auto.7.6.15267. PMID:21460631 [DOI] [PubMed] [Google Scholar]
- [27].Wang J, Kang R, Huang H, Xi X, Wang B, Wang J, Zhao Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy. 2014;10:766-84. doi: 10.4161/auto.27954. PMID:24589849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mari M, Tooze SA, Reggiori F. The puzzling origin of the autophagosomal membrane. F1000 Biol Rep. 2011;3:25. doi: 10.3410/B3-25. PMID:22162728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hou L, Ge X, Xin L, Zhou L, Guo X, Yang H. Nonstructural proteins 2C and 3D are involved in autophagy as induced by the encephalomyocarditis virus. Virol J. 2014;11:156. doi: 10.1186/1743-422X-11-156. PMID:25178311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kania E, Pajak B, Orzechowski A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. Biomed Res Int. 2015;2015:352794. doi: 10.1155/2015/352794. PMID:25821797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al.. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220-31. doi: 10.1128/MCB.01453-06. PMID:17030611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299-304. doi: 10.1074/jbc.M607007200. PMID:16901900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann N Y Acad Sci. 2012;1271:20-32. doi: 10.1111/j.1749-6632.2012.06739.x. PMID:23050960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Meng C, Zhou Z, Jiang K, Yu S, Jia L, Wu Y, Liu Y, Meng S, Ding C. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch Virol. 2012;157:1011-8. doi: 10.1007/s00705-012-1270-6. PMID:22398914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Manuse MJ, Briggs CM, Parks GD. Replication-independent activation of human plasmacytoid dendritic cells by the paramyxovirus SV5 Requires TLR7 and autophagy pathways. Virology. 2010;405:383-9. doi: 10.1016/j.virol.2010.06.023. PMID:20605567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Joubert PE, Meiffren G, Gregoire IP, Pontini G, Richetta C, Flacher M, Azocar O, Vidalain PO, Vidal M, Lotteau V, et al.. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe. 2009;6:354-66. doi: 10.1016/j.chom.2009.09.006. PMID:19837375 [DOI] [PubMed] [Google Scholar]
- [37].Tiwari A, Patnayak DP, Chander Y, Goyal SM. Permissibility of different cell types for the growth of avian metapneumovirus. J Virol Methods. 2006;138:80-4. doi: 10.1016/j.jviromet.2006.07.020. PMID:16930732 [DOI] [PubMed] [Google Scholar]
- [38].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720-8. doi: 10.1093/emboj/19.21.5720. PMID:11060023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hu B, Zhang Y, Jia L, Wu H, Fan C, Sun Y, Ye C, Liao M, Zhou J. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy. 2015;11:503-15. doi: 10.1080/15548627.2015.1017184. PMID:25714412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963-6. doi: 10.1074/jbc.273.7.3963. PMID:9461583 [DOI] [PubMed] [Google Scholar]
- [41].Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al.. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425-34. doi: 10.1083/jcb.200412022. PMID:15866887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313-26. doi: 10.1016/j.cell.2010.01.028. PMID:20144757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, et al.. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025-40. doi: 10.1128/MCB.25.3.1025-1040.2005. PMID:15657430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Solomon VR, Lee H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625:220-33. doi: 10.1016/j.ejphar.2009.06.063. PMID:19836374 [DOI] [PubMed] [Google Scholar]
- [45].Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33-42. doi: 10.1247/csf.23.33. PMID:9639028 [DOI] [PubMed] [Google Scholar]
- [46].Nakano T, Watanabe H, Ozeki M, Asai M, Katoh H, Satoh H, Hayashi H. Endoplasmic reticulum Ca2+ depletion induces endothelial cell apoptosis independently of caspase-12. Cardiovasc Res. 2006;69:908-15. doi: 10.1016/j.cardiores.2005.11.023. PMID:16376871 [DOI] [PubMed] [Google Scholar]
- [47].Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787-99. doi: 10.1091/mbc.10.11.3787. PMID:10564271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99-111. doi: 10.1016/S1534-5807(02)00203-4. PMID:12110171 [DOI] [PubMed] [Google Scholar]
- [49].Zhang HM, Ye X, Su Y, Yuan J, Liu Z, Stein DA, Yang D. Coxsackievirus B3 infection activates the unfolded protein response and induces apoptosis through downregulation of p58IPK and activation of CHOP and SREBP1. J Virol. 2010;84:8446-59. doi: 10.1128/JVI.01416-09. PMID:20554776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yu CY, Hsu YW, Liao CL, Lin YL. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol. 2006;80:11868-80. doi: 10.1128/JVI.00879-06. PMID:16987981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cheng JH, Sun YJ, Zhang FQ, Zhang XR, Qiu XS, Yu LP, Wu YT, Ding C. Newcastle disease virus NP and P proteins induce autophagy via the endoplasmic reticulum stress-related unfolded protein response. Sci Rep. 2016;6:24721. doi: 10.1038/srep24721. PMID:27097866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].McDougall JS, Cook JK. Turkey rhinotracheitis: Preliminary investigations. Vet Rec. 1986;118:206-7. doi: 10.1136/vr.118.8.206. PMID:3716162 [DOI] [PubMed] [Google Scholar]
- [53].Sun MX, Huang L, Wang R, Yu YL, Li C, Li PP, Hu XC, Hao HP, Ishag HA, Mao X. Porcine reproductive and respiratory syndrome virus induces autophagy to promote virus replication. Autophagy. 2012;8:1434-47. doi: 10.4161/auto.21159. PMID:22739997 [DOI] [PubMed] [Google Scholar]
- [54].Liu Q, Qin Y, Zhou L, Kou Q, Guo X, Ge X, Yang H, Hu H. Autophagy sustains the replication of porcine reproductive and respiratory virus in host cells. Virology. 2012;429:136-47. doi: 10.1016/j.virol.2012.03.022. PMID:22564420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. PMID:15884975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest. 2011;121:37-56. doi: 10.1172/JCI41474. PMID:21135505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Huang H, Kang R, Wang J, Luo G, Yang W, Zhao Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy. 2013;9:175-95. doi: 10.4161/auto.22791. PMID:23169238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Shi Y, He X, Zhu G, Tu H, Liu Z, Li W, Han S, Yin J, Peng B, Liu W. Coxsackievirus A16 elicits incomplete autophagy involving the mTOR and ERK pathways. PloS One. 2015;10:e0122109. doi: 10.1371/journal.pone.0122109. PMID:25853521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hung CH, Chen LW, Wang WH, Chang PJ, Chiu YF, Hung CC, Lin YJ, Liou JY, Tsai WJ, Hung CL, et al.. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J Virol. 2014;88:12133-45. doi: 10.1128/JVI.02033-14. PMID:25122800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Benali-Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R, Ichas F, Rizzuto R, et al.. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921-33. doi: 10.1038/sj.onc.1208673. PMID:15897896 [DOI] [PubMed] [Google Scholar]
- [61].Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J Virol. 2007;81:12128-34. doi: 10.1128/JVI.01356-07. PMID:17855538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657-67. doi: 10.1083/jcb.152.4.657. PMID:11266458 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.