

ABSTRACT
Whereas the mechanisms involved in autophagosome formation have been extensively studied for the past 2 decades, those responsible for autophagosome-lysosome fusion have only recently begun to garner attention. In this study, we report that the multisubunit BORC complex, previously implicated in kinesin-dependent movement of lysosomes toward the cell periphery, is required for efficient autophagosome-lysosome fusion. Knockout (KO) of BORC subunits causes not only juxtanuclear clustering of lysosomes, but also increased levels of the autophagy protein LC3B-II and the receptor SQSTM1. Increases in LC3B-II occur without changes in basal MTORC1 activity and autophagy initiation. Instead, LC3B-II accumulation largely results from decreased lysosomal degradation. Further experiments show that BORC KO impairs both the encounter and fusion of autophagosomes with lysosomes. Reduced encounters result from an inability of lysosomes to move toward the peripheral cytoplasm, where many autophagosomes are formed. However, BORC KO also reduces the recruitment of the HOPS tethering complex to lysosomes and assembly of the STX17-VAMP8-SNAP29 trans-SNARE complex involved in autophagosome-lysosome fusion. Through these dual roles, BORC integrates the kinesin-dependent movement of lysosomes toward autophagosomes with HOPS-dependent autophagosome-lysosome fusion. These findings reveal a requirement for lysosome dispersal in autophagy that is independent of changes in MTORC1 signaling, and identify BORC as a novel regulator of autophagosome-lysosome fusion.
KEYWORDS: autophagosomes, autophagy, BORC, fusion, HOPS, kinesins, lysosomes, MTORC1, positioning, SNARE
Introduction
Autophagy is a catabolic process in which cytoplasmic components are engulfed into an autophagosome and delivered for degradation in the lysosome.1-3 This process is critical to many cellular functions, including the response to nutrient starvation, disposal of misfolded or aggregated proteins, clearance of damaged organelles, and degradation of intracellular pathogens, all of which promote cell survival. Abnormal autophagy, on the other hand, contributes to the pathogenesis of diseases such as cancer and neurodegeneration. The autophagic process involves many steps, including initiation, nucleation and elongation of the phagophore, engulfment of cytoplasmic materials into an autophagosome, autophagosome-lysosome fusion to form an autolysosome, and autophagic lysosome reformation (ALR). To date, most studies on the autophagic machinery have focused on the mechanisms of autophagosome formation,4 and only recently have the roles of the lysosome begun to be investigated in detail.5
As the final destination for the targeted materials, lysosomes are inextricably involved in the maintenance of autophagic flux. Indeed, lysosomes supply the hydrolytic enzymes and acidic luminal pH necessary for the degradation of the autophagic substrates. In addition, lysosomes serve as platforms for signaling molecules, such as the transcription factor TFEB (transcription factor EB)6 and MTOR (mechanistic target of rapamycin) complex 1 (MTORC1),7 which reversibly associate with the lysosomal membrane to regulate various steps of the autophagic process. MTORC1, in particular, is a direct regulator of the autophagic response to nutrient availability. When nutrients are abundant, MTORC1 is activated by recruitment to the lysosomal membrane, where it phosphorylates the autophagy-initiating ULK1 (unc-51 like autophagy activating kinase 1; yeast Atg1), leading to its inactivation and consequent inhibition of autophagy.8,9 Nutrient scarcity, on the other hand, causes release of inactive MTORC1 from the lysosomal membrane, enabling dephosphorylation and activation of ULK1, and thus triggering autophagy.8,9
Lysosomes also provide components involved in autophagosome-lysosome fusion. These include the membrane-anchored lysosomal R-SNARE VAMP8 (vesicle associated membrane protein 8), which forms a trans-SNARE complex with the autophagosomal Q-SNARE STX17 (syntaxin 17) and the ubiquitous Q-SNARE SNAP29 (synaptosome associated protein 29), for fusion of the lysosomal and autophagosomal membranes.10 In addition, lysosomes recruit the HOPS (homotypic fusion and vacuole protein sorting) complex, which tethers lysosomes to autophagosomes through direct or indirect interaction with the autophagosomal protein MAP1LC3/LC3 (microtubule associated protein 1 light chain 3; yeast Atg8),11,12 and subsequently coordinates assembly of the STX17-containing trans-SNARE complex.13-15 Another lysosome-associated protein, the RAB7 (RAB7, member RAS oncogene family)-effector EPG5 (ectopic P-granules autophagy protein 5 homolog) stabilizes this trans-SNARE complex and, in addition, imparts specificity to the fusion of autophagosomes with lysosomes.16
Recent studies have brought attention to another property of lysosomes that influences autophagy: their positioning within the cytoplasm.17 Lysosomes are dynamically distributed throughout the cytoplasm, moving bidirectionally between the center and the periphery of the cell by virtue of interactions with kinesin and dynein microtubule motors. Nutrient starvation induces concentration of lysosomes in the juxtanuclear area of the cell, a location that is thought to favor autophagy initiation and autophagosome-lysosome fusion.18-20 In line with this notion, siRNA-mediated silencing of the kinesin KIF2A (kinesin family member 2A) or the small GTPase ARL8B (ADP ribosylation factor like GTPase 8B) causes juxtanuclear clustering of lysosomes and enhancement of autophagy initiation.19 Conversely, overexpression of KIF1B (kinesin family member 1B), KIF2, or ARL8B disperses lysosomes to the cell periphery and inhibits autophagy, probably due to reduced autophagy initiation and autophagosome-lysosome fusion.19 These effects on autophagy are attributed largely to regulation of MTORC1 activity by lysosome positioning, such that juxtanuclear clustering inhibits MTORC1 whereas relocation to the periphery activates it.19 It remains to be determined, however, if factors other than changes in MTORC1 activity participate in the regulation of autophagy in connection to lysosome positioning.
We have recently described a lysosome-associated multiprotein complex named BLOC-1 related complex (BORC) that regulates lysosome positioning by promoting ARL8-dependent coupling to the kinesin-1 KIF5B (kinesin family member 5B) and kinesin-3 KIF1B proteins in non-neuronal cells (Fig. 1A).21,22 BORC comprises 8 subunits named BLOC1S1/BLOS1/BORCS1 (biogenesis of lysosomal organelles complex 1 subunit 1), BLOC1S2/BLOS2/BORCS2 (biogenesis of lysosomal organelles complex 1 subunit 2), SNAPIN/BORCS3 (SNAP associated protein), KXD1/BORCS4 (KxDL motif containing 1), BORCS5/myrlysin/LOH12CR1 (BLOC-1 related complex subunit 5), BORCS6/lyspersin/C17orf59 (BLOC-1 related complex subunit 6), BORCS7/diaskedin/C10orf32 (BLOC-1 related complex subunit 7), and BORCS8/MEF2BNB (BLOC-1 related complex subunit 8) (Fig. 1A). Knockout (KO) or knockdown (KD) of BORC subunits causes collapse of the lysosome population to the juxtanuclear area of the cell.21,22 Here we report that KO of any of several genes encoding BORC subunits increases the levels of lipidated LC3B (LC3B-II), a sign of altered autophagy. Surprisingly, this increase is not due to enhanced autophagy initiation, but to reduced lysosomal degradation of LC3B-II. Moreover, we find that BORC-subunit gene KO has no effect on basal MTORC1 activity, indicating that in this particular case, lysosome clustering inhibits autophagic flux independently of MTORC1. Further experiments show that BORC-subunit gene KO decreases the frequency of autophagosome-lysosome encounters, particularly in the peripheral cytoplasm. In addition, BORC gene KO impairs fusion of autophagosomes with lysosomes even when they are in close proximity of each other, as it happens in the juxtanuclear area. We show that this defect in autophagosome-lysosome fusion is likely due to a role of BORC in the ARL8-dependent recruitment of the HOPS complex to lysosomes. We conclude that BORC contributes to the maintenance of autophagic flux by promoting both encounter and fusion of lysosomes with autophagosomes. Through these dual roles, BORC coordinates peripheral deployment of lysosomes with autophagosome-lysosome fusion.
Figure 1.

Increased LC3B-II levels in BORC-KO cells. (A) Schematic representation of BORC-ARL8-KIF1 and BORC-ARL8-PLEKHM2/SKIP-KLC-KIF5 machineries for centrifugal movement of lysosomes in non-neuronal cells.21,22 (B) Confocal microscopy of WT, BORCS5-KO, BORCS5-rescue, BORCS6-KO, BORCS7-KO, and BORCS8-KO cells immunostained for endogenous LC3B and LAMP1. Nuclei were stained with DAPI. Scale bar: 10 μm. (C) Quantification of LC3B puncta from experiments as in B. Bars represent the mean ± SEM of puncta per cell from 20 cells in 3 independent experiments. **P < 0.001, ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Dunnett test. (D) Cell extracts of WT, BORCS5-KO, BORCS5-rescue, BORCS6-KO, BORCS7-KO, and BORCS8-KO cells were analyzed by immunoblotting with antibodies to the proteins indicated at right. The positions of molecular mass markers (in kDa) are indicated at left. (E) Quantification of LC3B-II levels (normalized to tubulin) from experiments as in E. Bars represent the mean ± SEM from 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Dunnett test.
Results
Increased levels of the autophagy marker LC3B-II in BORC-KO cells
The juxtanuclear clustering of lysosomes caused by KO of genes encoding BORC subunits21,22 provided an opportunity to examine the importance of lysosome dispersal for autophagy. Immunofluorescence microscopy of HeLa cells knocked out for the BORCS5, BORCS6, BORCS7 or BORCS8 genes encoding subunits of BORC (all collectively referred to as “BORC-KO cells”) showed that lysosome clustering was accompanied with increased staining for endogenous LC3B (Fig. 1B, C). In the process of autophagy, cytosolic LC3B-I is converted to autophagosome-bound LC3B-II by conjugation to phosphatidylethanolamine.23 In line with the immunofluorescence microscopy observations, immunoblot analysis revealed 2.4-4.4-fold increases in the steady-state levels of LC3B-II in the different BORC-KO cells (Fig. 1D, E). In contrast, the levels of the ATG12 (autophagy-related 12)–ATG5 (autophagy-related 5) conjugate, and ATG7 (autophagy-related 7) protein involved in LC3B-I lipidation,24 remained unchanged in the BORC-KO cells (Fig. 1D). Both the lysosome clustering and elevated LC3B-II phenotypes were reversed by stable transfection of a BORCS5-FOS (FLAG/One-STrEP) cDNA into the BORCS5-KO cells (Fig. 1B to E). These results indicated that BORC KO causes not only lysosome clustering but also altered autophagy.
Increased levels of the SQSTM1 autophagy receptor and reduced clearance of mutant HTT aggregates in BORC-KO cells
Damaged organelles or cytoplasmic protein aggregates undergo ubiquitination, leading to their recognition by the ubiquitin receptor SQSTM1/p62 (sequestosome 1).25 In turn, SQSTM1 binds to LC3B-II, guiding the ubiquitinated substrates to the phagophore for subsequent engulfment within the autophagosome.25 After fusion with lysosomes, both SQSTM1 and LC3B-II are degraded together with their cargos by lysosomal hydrolases.25,26 We observed that, in addition to elevated LC3B-II (Fig. 1B to E), BORC-KO cells exhibited 1.4- to 2.3-fold higher numbers of SQSTM1 cytoplasmic puncta (Fig. 2A, B) and 2.3–2.9 fold higher levels of SQSTM1 protein (Fig. 2C, D) relative to the parental HeLa cells. A well-characterized autophagic cargo of SQSTM1 is an aggregation-prone N-terminal fragment of HTT (huntingtin) with an expanded poly-glutamine tract (HTT103Q).27 We observed that BORCS5-KO increased the proportion of cells exhibiting HTT103Q-GFP aggregates from 13.7% to 21.4% (Fig. 2E, F). Stable transfection of the BORCS5-KO cells with BORCS5-FOS cDNA brought down the proportion of cells exhibiting HTT103Q-GFP aggregates to 13.3% (Fig. 2E, F). Taken together, these experiments demonstrated that BORC deficiency and the ensuing lysosome clustering were associated with increased accumulation of the autophagy protein LC3B-II and the receptor SQSTM1, and the autophagy substrate HTT103Q-GFP.
Figure 2.
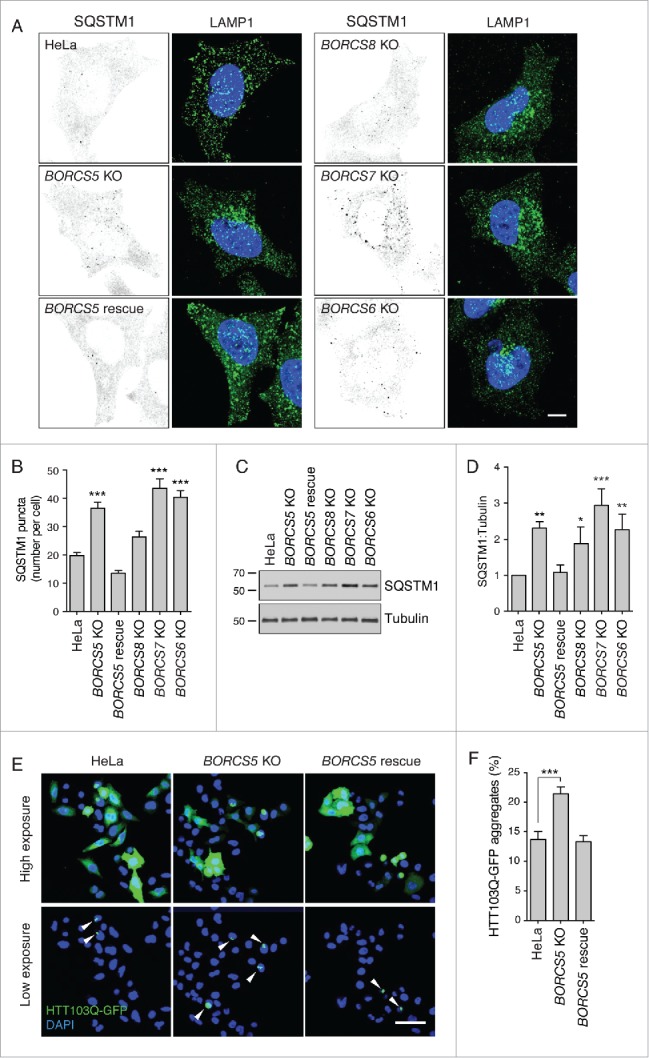
Increased SQSTM1 levels and decreased aggregate clearance in BORC-KO cells. (A) Confocal micrographs of WT, BORCS5-KO, BORCS5-rescue, BORCS6-KO, BORCS7-KO, and BORCS8-KO cells immunostained for SQSTM1 and LAMP1. Images of SQSTM1 staining are in negative grayscale for easier visualization. Nuclei were stained with DAPI. Scale bar: 10 μm. (B) Quantification of SQSTM1 puncta from experiment in A. Bars represent the mean ± SEM of SQSTM1 puncta per cell from 30 cells. ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Dunnett test. (C) Immunoblotting of extracts from WT, BORCS5-KO, BORCS5-rescue, BORCS6-KO, BORCS7-KO and BORCS8-KO cells with antibodies to SQSTM1 and tubulin (control). The positions of molecular mass markers (in kDa) are indicated at left. (D) Quantification of SQSTM1 normalized to tubulin from experiments as in (C). Bars represent the mean ± SEM from 3 independent experiments. *P < 0.05, **P < 0.001, ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Dunnett test. (E) Confocal images of WT, BORCS5-KO and BORCS5-rescue cells transfected with a plasmid encoding the aggregation-prone HTT103Q-EGFP for 48 h. Arrowheads indicate intracellular aggregates of HTT103Q-EGFP. Nuclei were stained with DAPI. Scale bar: 50 μm. (F) Percentage of cells with EGFP-positive aggregates. Over 600 GFP-positive cells from 3 independent experiments were analyzed. Bars represent the mean ± SEM of the percentage of cells with EGFP-positive aggregates. ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Dunnett test.
BORC KO abrogates changes in lysosome positioning and LC3B-II levels during starvation
To further investigate the role of BORC in autophagy, we examined the effects of incubating wild-type (WT), BORCS5-KO, and BORCS5-rescue HeLa cells in Dulbecco's modified Eagle's medium (DMEM; serum-free medium), a manipulation known to induce autophagy.28,29 We observed that short-term serum deprivation (15 to 30 min) caused lysosome clustering and increased LC3B staining (Fig. 3A) and LC3B-II levels (Fig. 3B, C). Upon longer incubations in serum-free medium (1 or 2 h), lysosomes returned to their normal distribution and LC3B-II levels decreased (Fig. 3A to C), indicating that these effects of serum deprivation are transient. In contrast, in BORCS5-KO cells, lysosomes were clustered and LC3B-II levels stayed high at all times independently of serum withdrawal (Fig. 3A to C). Low exposure of the immunoblots revealed that LC3B-II levels actually went up slightly over the already high levels in serum-deprived BORCS5-KO cells, consistent with an expected increase in autophagy initiation upon serum deprivation (Fig. 3B). Normal responses of LC3B-II to serum depletion could be restored by expression of BORCS5-FOS cDNA in the BORCS5-KO cells (Fig. 3A to C). Similar results were obtained by combined serum and amino-acid depletion, with the exception that lysosome dispersal did not recover after 1 h in WT cells (Fig. S1A,B). These results demonstrated that BORC is required for the changes in lysosome positioning and LC3B-II levels that occur upon autophagy induction by nutrient deprivation.
Figure 3.
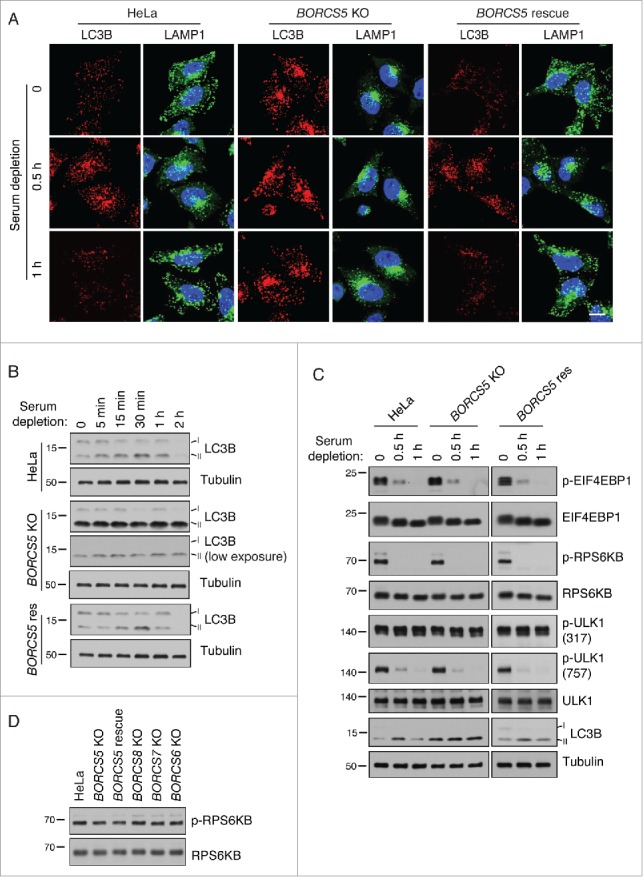
Changes in lysosome positioning induced by serum depletion are dependent on BORC. (A) Confocal microscopy of WT, BORCS5-KO and BORCS5-rescue cells placed in serum-free DMEM for the indicated times, and immunostained for endogenous LC3B and LAMP1. Nuclei were stained with DAPI. Scale bar: 15 μm. (B) WT, BORCS5-KO and BORCS5-rescue cells were placed in serum-free DMEM and collected at the indicated times. Cell lysates were subjected to immunoblotting with antibodies to LC3B and tubulin (control). (C) WT, BORCS5-KO and BORCS5-rescue cells were placed in serum-free DMEM for the indicated times, and cell lysates were analyzed by immunoblotting with antibodies to the indicated proteins. (D) WT, BORCS5-KO, BORCS5-rescue, BORCS6-KO, BORCS7-KO, and BORCS8-KO cells were lysed for immunoblotting with antibodies to RPS6KB and p-RPS6KB. In (B to D), the positions of molecular mass markers (in kDa) are indicated at left.
BORC KO does not affect basal MTORC1 activity and inactivation under starvation conditions
We next addressed whether the increased levels of LC3B-II in BORC-KO cells were due to changes in MTORC1 signaling. A key MTORC1 substrate in the regulation of the autophagic response is ULK1. Active MTORC1 inactivates ULK1 by phosphorylation on Ser757, thus inhibiting autophagy. In contrast, the AMP-activated protein kinase (AMPK) activates ULK1 by phosphorylation on Ser317 and Ser777, promoting autophagy initiation.9 Interestingly, we observed that WT, BORCS5-KO and BORCS5-rescue cell lines exhibited similar levels of Ser757- and Ser317-phosphorylated ULK1 under normal culture conditions (Fig. 3C). Moreover, serum starvation caused similar decreases in Ser757-phosphorylated ULK1 in all 3 cell lines (Fig. 3C). Likewise, the phosphorylation state of 2 other MTORC1 substrates, EIF4EBP1/4E-BP1 (eukaryotic translation initiation factor 4E binding protein 1) and RPS6KB/S6K (ribosomal protein S6 kinase B), under both basal and serum-depletion conditions was unaffected by the presence or absence of BORCS5 (Fig. 3C). Similar observations were made upon combined serum and amino acid depletion (Fig. S2). KO of the BORCS6, BORCS7 or BORCS8 subunits of BORC also had no effect on basal MTORC1 activity, as exemplified by the unchanged RPS6KB phosphorylation (Fig. 3D). Finally, immunofluorescence microscopy experiments showed that BORCS5 KO did not affect changes in MTORC1 association with lysosomes that occur during combined serum and amino acid depletion (Fig. S3). From these experiments, we concluded that juxtanuclear clustering of lysosomes and increased LC3B-II levels in BORC-deficient cells occurred without changes in basal MTORC1 activity and association with lysosomes.
BORC KO impairs autophagic flux
To determine if the increased levels of LC3B-II in BORCS5-KO cells resulted from increased synthesis (i.e., autophagy initiation) or decreased degradation (i.e., autophagic flux) of this protein, we examined the effect of the MTORC1 inhibitor and autophagy activator Torin2 (Fig. S4A), and the acidification and lysosomal degradation inhibitor bafilomycin A1 (Baf). We observed that treatment with Torin2 increased LC3B-II levels in both WT and BORCS5-KO cells, although the relative difference in LC3B-II levels between the 2 cell lines remained the same (Fig. 4A, B). In contrast, Baf treatment resulted in equally higher levels of LC3B-II in WT and BORCS5-KO cells (Fig. 4A, B). These experiments demonstrated that BORCS5 KO does not increase synthesis but decreases degradation of LC3B-II. The fact that incubation of BORCS5-KO cells with Baf still caused increased LC3B-II levels relative to untreated BORCS5-KO cells, however, indicated that the inhibition of autophagic flux by BORC KO was partial. These findings are consistent with the increased levels of SQSTM1 (Fig. 2A to D) and HTT103Q-GFP (Fig. 2E, F) in BORC-KO cells.
Figure 4.

Reduced autophagic flux in BORCS5-KO cells. (A) WT and BORCS5-KO cells were treated with no additions, 50 nM bafilomycin A1 (Baf), or 400 nM Torin2 for 2 h. LC3B-II and tubulin (control) levels were determined by immunoblotting. The positions of molecular mass markers (in kDa) are indicated at left. (B) Quantification of LC3B-II levels normalized to tubulin. Bars represent the mean ± SEM from 3 independent experiments. **P < 0.001, ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Tukey test. (C) WT, BORCS5-KO and BORCS5-rescue cells were transiently transfected with a plasmid encoding tandem GFP-mCherry-LC3B (tfLC3B). Lysosomes were labeled by internalization of Alexa Fluor 647-conjugated dextran (AF647-dextran) for 6 h, and chased overnight. mCherry, GFP and Alexa Fluor 647 (pseudocolored in blue) were visualized by confocal microscopy of live cells. Peripheral (box 1) and juxtanuclear (box 2) regions were magnified to show autophagosomes (yellow arrowheads), autolysosomes (magenta arrowheads) and lysosomes (blue arrowheads). Scale bar: 10 μm in low magnification, 1 μm in high magnification images. (D) The ratio of the number of autolysosomes (red-blue-positive puncta) to autophagosomes (red-green-positive puncta) was determined in the whole cell, the peripheral region and the juxtanuclear region. Bars represent the mean ± SEM of the ratio in 70 cells from 3 independent experiments. *P < 0.01, ***P < 0.0001, two-way ANOVA followed by multiple comparisons using the Tukey test.
WT and BORCS5-KO cells exhibited similar cathepsin L (CTSL) activity and lysosomal pH (Fig. S4B,C), suggesting that impaired turnover of LC3B-II was not due to altered degradative capacity of lysosomes in the KO cells. To determine if BORC depletion prevented delivery of LC3B-II from autophagosomes to lysosomes, we transfected WT, BORCS5-KO and BORCS5-rescue cells with a plasmid encoding a tandem GFP-mCherry-LC3B fusion protein (tf-LC3B), which serves as a probe for autophagic flux (Fig. 4C).30 At the neutral pH of autophagosomes both the GFP (green) and mCherry (red) proteins fluoresce, whereas in the acidic pH of autolysosomes the GFP fluorescence is quenched and only the red fluorescence is visible. In addition to expressing this probe, we allowed the cells to internalize Alexa Fluor 647-dextran (pseudocolored blue) for 16 h to mark lysosomes and autolysosomes. Cells were imaged by confocal microscopy, and green-red (autophagosomes) and red-blue (autolysosomes) puncta were quantified. We observed that in WT and BORCS5-rescue cells, red foci predominated, and the majority of these colocalized with blue but not green foci (Fig. 4C, D), indicating that most autophagosomes fused with lysosomes to form autolysosomes. In contrast, in BORCS5-KO cells most red foci colocalized with green but not blue foci (Fig. 4C, D), consistent with failure of autophagosome-lysosome fusion. The decreased number of autolysosomes in BORCS5-KO cells was particularly noticeable in the cell periphery (Fig. 4C, D). This observation was suggestive of an inability of lysosomes to reach peripherally-arising autophagosomes. Most of the accumulated mCherry-LC3B-labeled foci in BORCS5-KO cells were negative for the phagophore marker ATG16L1 (autophagy-related 16 like 1)31,32 (Fig. S5), confirming their identity as autophagosomes.
We also compared the colocalization of endogenous LC3B with LAMP1 in WT, BORCS5-KO and BORCS5-rescue cells, all of which were serum-deprived and treated with Baf to inhibit LC3B-II degradation. We observed a lower degree of LC3B-LAMP1 colocalization in BORCS5-KO relative to WT and BORCS5-rescue cells (Fig. 5A, B), also consistent with defective autophagosome-lysosome encounters. Finally, live-cell imaging of serum-deprived cells expressing mCherry-LC3B and LAMP1-GFP showed significantly more merging of autophagosomes and lysosomes in WT cells than in BORCS5-KO cells (Movie S1 and Fig. 5C-E). Similar results were obtained using a combination of GFP-LC3B and LAMP1-mCherry, in which the fluorescent tags were exchanged (Movie S2 and Fig. S6). Taken together, these results indicated that BORCS5 KO decreases encounter and fusion of autophagosomes with lysosomes, thus resulting in decreased LC3B turnover.
Figure 5.

Reduced autophagosome-lysosome encounters in BORCS5-KO cells. (A) Confocal microscopy of WT, BORCS5-KO and BORCS5-rescue cells placed in serum-free DMEM with 50 nM Baf for 30 min, and then immunostained with antibodies to LC3B and LAMP1. Nuclei were stained with DAPI. Scale bar: 10 μm. Images on the right column are magnifications of the boxed areas. Scale bar: 2.5 μm. (B) Pearson correlation coefficient of LC3B and LAMP1 colocalization from experiment in A. Bars represent the mean ± SEM of >80 cells from 3 independent experiments. ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Dunnett test. (C, D) WT (C) and BORCS5-KO (D) cells transiently expressing LAMP1-GFP and mCherry-LC3B were analyzed by time-lapse microscopy in serum-free DMEM. Images in (C and D) start at 25 min and 40 min after serum depletion, respectively. Scale bar: 10 μm. The left panels show magnified time-lapse images of the LC3B (red) and LAMP1 (green) vesicles indicated by arrowheads in the whole cell shown on the right. (E) Quantification of the number of autophagosome-lysosome merging events in 1 h from the experiments described in (C and D). Bars represent the mean ± SEM in 10 cells from 3 independent experiments. ***P < 0.0001, the unpaired Student t test.
Smaller LC3B-II increase in cells doubly mutated for the kinesins KIF5B and KIF1B
Our initial hypothesis was that decreased autophagosome-lysosome fusion and elevated LC3B levels in BORC-deficient cells were primarily due to defective lysosome positioning. Indeed, as shown above, the juxtanuclear clustering of lysosomes in the mutant cells decreased the frequency of encounters between lysosomes and autophagosomes, particularly those that formed in the cell periphery. However, 2 observations hinted at a more complex scenario: (i) autophagosomes accumulated not only in the periphery but also at the center of BORC-deficient cells, where lysosomes were abundant (Fig. 4C, D), and (ii) autophagosomes and lysosomes were sometimes observed to approach and move around each other, but not fuse, in BORC-deficient cells (Figs. 5D and S6).
To test more specifically for the importance of lysosome positioning in autophagic flux, we examined the effect of knocking out genes encoding 2 kinesins, KIF5B and KIF1B, which function downstream of BORC and ARL8 in centrifugal lysosome transport (Fig. 1A).21,22 We found that KIF5B KO had only a subtle effect on lysosome distribution, whereas KIF1B KO caused more appreciable clustering of lysosomes (Fig. 6A). Combination of KIF5B and KIF1B KO resulted in more dramatic juxtanuclear clustering of lysosomes, comparable to that caused by BORCS5 KO (Fig. 6A). These findings are consistent with the notion that both kinesins cooperate in lysosome movement to the periphery,22 with KIF1B having the most important role. Interestingly, the single kinesin-KO cells did not exhibit increases in LC3B staining (Fig. 6A, B) and LC3B-II levels (Fig. 6C, D), and the double kinesin-KO cells showed increases that were smaller than those in BORCS5-KO cells (Fig. 6A to D). Treatment with Baf increased the levels of LC3B-II 2.5 fold in WT cells and 1.6 fold in KIF5B KIF1B-KO cells, although a partial difference in LC3B-II levels between the 2 cell lines remained (Fig. 6E, D). These results are consistent with KIF5B KIF1B KO increasing both autophagy initiation and flux. Therefore, centrifugal transport of lysosomes is to some extent important for autophagic flux, likely by enabling encounter of lysosomes with peripheral autophagosomes. The larger increases of LC3B-II levels in BORC-deficient cells, however, suggested an additional role for this complex in autophagosome-lysosome fusion.
Figure 6.
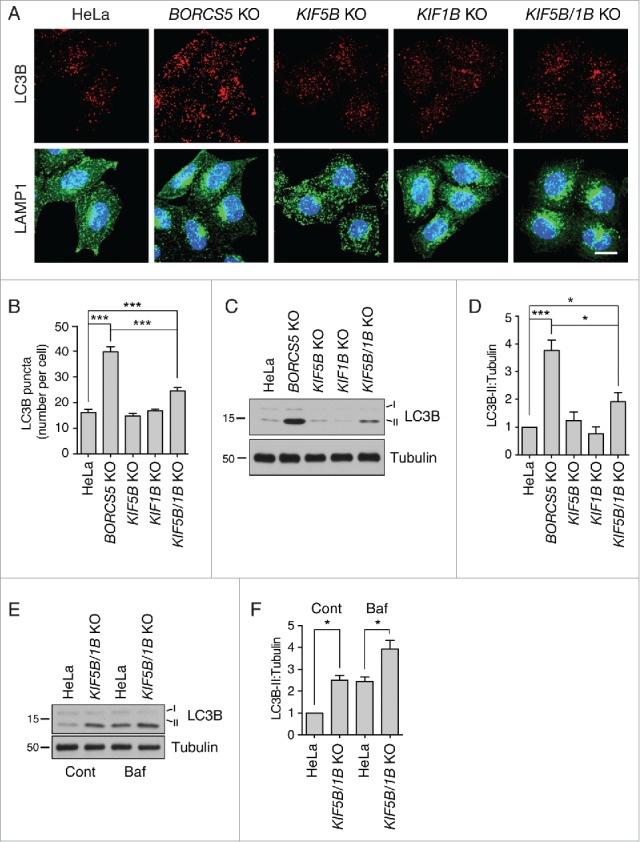
Slight increase in LC3B-II levels in KIF5B KIF1B-double-KO cells. (A) Confocal microscopy of WT, KIF5B-KO, KIF1B-KO and KIF5B KIF1B-double-KO cells immunostained for endogenous LC3B and LAMP1. Nuclei were stained with DAPI. Scale bar: 15 μm. (B) Quantification of LC3B puncta. Bars represent the mean ± SEM of LC3B puncta per cell in 25 cells from 3 independent experiments. ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Tukey test. (C) Cell extracts of WT, KIF5B-KO, KIF1B-KO and KIF5B KIF1B-double-KO cells were analyzed by immunoblotting with antibodies to LC3B and tubulin (control). The positions of molecular mass markers (in kDa) are indicated at left. (D) Quantification of LC3B-II normalized to tubulin levels. Bars represent the mean ± SEM LC3B-II/tubulin ratio from 3 independent experiments. *P < 0.01, ***P < 0.0001, one-way ANOVA, followed by multiple comparisons using the Tukey test. (E) WT and KIF5B KIF1B-double-KO cells were treated with no additions or 50 nM Baf for 2 h. LC3B-II and tubulin (control) levels were determined by immunoblotting. The positions of molecular mass markers (in kDa) are indicated at left. (F) Quantification of LC3B-II levels (normalized to tubulin) from experiments as in (E). Bars represent the mean ± SEM from 3 independent experiments. *P < 0.01, one-way ANOVA, followed by multiple comparisons using the Tukey test.
BORC promotes recruitment of HOPS to lysosomes and assembly of the STX17-containing SNARE complex
We considered the possibility that BORC could promote autophagosome-lysosome fusion by directly interacting with LC3. In this regard, we noticed that the KXD1 subunit of BORC contains 4 potential W/FxxL LC3-interacting region (LIR) motifs33 (Fig. 7A). Coimmunoprecipitation experiments showed that KXD1-GFP indeed interacted with endogenous LC3B in a manner dependent on the 4 LIR motifs (Fig. 7B). However, rescue of a KXD1-KO cell line with a KXD1 mutant having substitutions in all 4 LIR motifs restored normal levels of LC3B-II (Fig. 7C), indicating that these motifs are dispensable for the function of BORC in autophagy.
Figure 7.
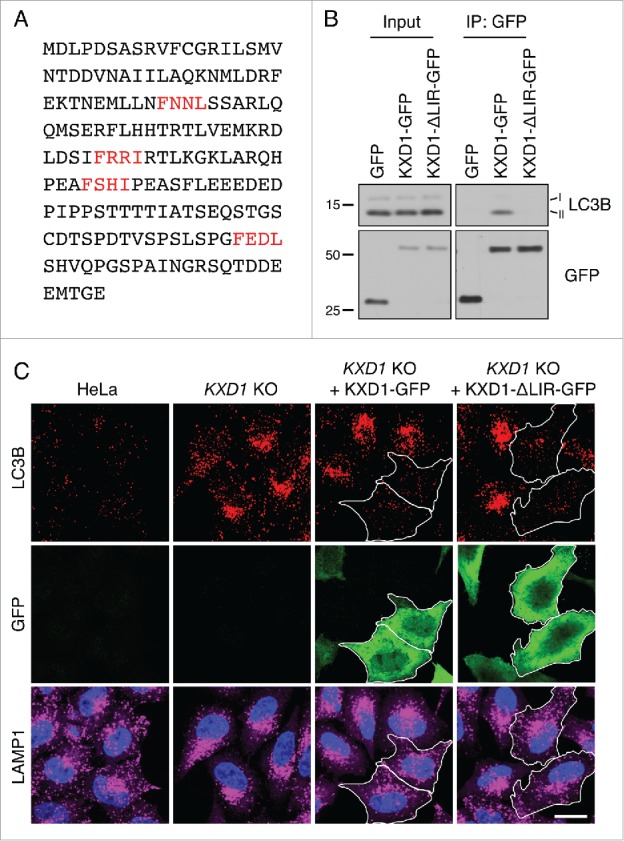
WT and ΔLIR-mutant KXD1 equally reduced LC3B accumulation in KXD1-KO cells. (A) Four potential LIR (LC3-interacting region) motifs in KXD1 are highlighted in red. LIR prediction was performed using an online database at http://repeat.biol.ucy.ac.cy/iLIR/. (B) Lysates of HeLa cells transfected with plasmids encoding GFP, GFP-tagged WT KXD1 or KXD1 with mutations in all 4 LIR motifs (ΔLIR) were immunoprecipitated with an antibody to GFP followed by immunoblotting with antibodies to GFP and LC3B. (C) WT, KXD1-KO cells, and KXD1-KO cells rescued with KXD1-GFP or KXD1(ΔLIR)-GFP were stained with antibody to LC3B. The outlines of the transfected cells are indicated. Nuclei were stained with DAPI. Cells were imaged by confocal microscopy. Scale bar: 20 μm.
In addition to regulating lysosome movement through engagement of the KIF5B34 and KIF1B motors35 (Fig. 1A), ARL8 recruits the HOPS tethering complex,36,37 which in turn promotes autophagosome-lysosome fusion through interaction with the Q-SNARE STX17.13-15,38 Because BORC functions to recruit both the ARL8A and ARL8B isoforms to lysosomes,21,22 we hypothesized that BORC-KO impaired association of HOPS with lysosomes, thus additionally contributing to the autophagosome-lysosome fusion defect in the KO cells. Immunofluorescence microscopy of WT HeLa cells showed that the mCherry-labeled VPS41 subunit of HOPS colocalized with ARL8B-GFP on LAMP1-containing lysosomes (Fig. 8A), as previously reported.36,37 In BORCS5-KO cells, however, we observed reduced lysosomal localization and increased cytosolic staining for both mCherry-VPS41 and ARL8B-GFP (Fig. 8A). Transfection with BORCS5-FOS rescued the association of both proteins with lysosomes (Fig. 8A). Similar results were obtained for FLAG-tagged VPS41 (Fig. S7A), and a mCherry-labeled form of another HOPS subunit, VPS39 (Fig. S7B).
Figure 8.
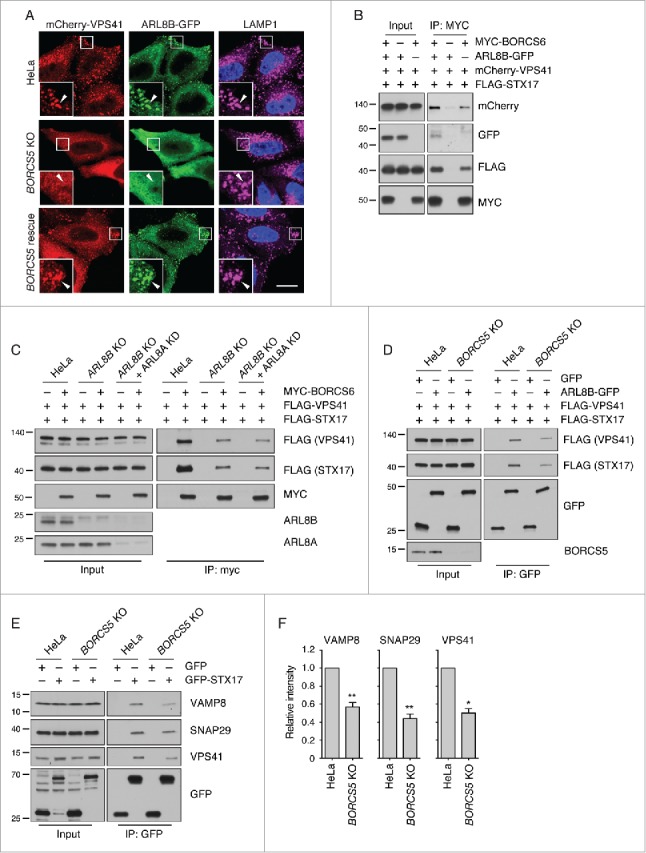
BORC promotes recruitment of HOPS to lysosomes and assembly of autophagic STX17 SNARE complex. (A) Confocal microscopy of WT, BORCS5-KO and BORCS5-rescue HeLa cells transiently transfected with plasmids encoding mCherry-VPS41 and ARL8B-GFP. Lysosomes were visualized by LAMP1 staining. Nuclei were stained with DAPI. Scale bar: 15 μm. (B) Lysates of HEK293T cells transfected with plasmids encoding mCherry-VPS41, FLAG-STX17 and either MYC-BORCS6, ARL8B-GFP or MYC-BORCS6+ARL8B-GFP were immunoprecipitated using antibody to the MYC epitope followed by immunoblotting with antibodies to the indicated proteins. (C) WT, ARL8B-KO and ARL8B-KO-ARL8A-KD HeLa cells were transfected with plasmids encoding FLAG-VPS41, FLAG-STX17 and MYC-BORCS6. Cells were lysed and immunoprecipitated with antibody to MYC and immunoblotted with antibodies to the indicated proteins. (D) WT and BORCS5-KO HeLa cells were transfected with plasmids encoding FLAG-VPS41, FLAG-STX17 and either GFP or ARL8B-GFP. Cells were lysed and immunoprecipitated with antibody to GFP and immunoblotted with antibodies to the indicated proteins. (E) WT and BORCS5-KO HeLa cells were transfected with plasmids encoding GFP or GFP-STX17. Cells were lysed and immunoprecipitated with antibody to GFP and immunoblotted for the indicated proteins. In (B to E), the positions of molecular mass markers (in kDa) are indicated at left. (F) Quantification of VAMP8, SNAP29 and VPS41 in immunoprecipitates (normalized to input) from experiments as in (E). Bars represent the mean ± SEM from 3 independent experiments. *P < 0.05, **P < 0.001, the unpaired Student t test.
To further investigate the involvement of BORC in HOPS recruitment and function, we examined the coimmunoprecipitation of the transiently expressed myc-tagged BORCS6 subunit of BORC with mCherry-VPS41 and FLAG-STX17 (Fig. 8B). The results showed that myc-BORCS6 indeed coprecipitated mCherry-VPS41 and FLAG-STX17, and that this coprecipitation was enhanced by expression of exogenous ARL8B-GFP (Fig. 8B). A similar, though quantitatively smaller, coprecipitation was observed using BORCS5-myc (Fig. S7C). In line with the intermediary role of ARL8B, KO of this gene and protein product decreased the amounts of FLAG-VPS41 and FLAG-STX17 that coprecipitated with myc-BORCS6 (Fig. 8C). KD of the ARL8A isoform in the ARL8B-KO cells did not completely abolish the coprecipitation of these proteins (Fig. 8C), suggesting that BORC might also interact with HOPS independently of ARL8. Moreover, ARL8B-GFP coprecipitated with FLAG-VPS41 and FLAG-STX17 to a larger extent in WT than in BORCS5-KO cells (Fig. 8D). Finally, BORCS5 KO decreased by ∼50% the coprecipitation of GFP-STX17 with its cognate SNAREs VAMP8 and SNAP29 (Fig. 8E, F), indicating that BORC is required for efficient assembly of these SNAREs. From these experiments, we concluded that BORC establishes a network of interactions with ARL8B, HOPS and STX17, promoting assembly of the STX17-VAMP8-SNAP29 trans-SNARE complex for efficient autophagosome-lysosome fusion (Fig. 9).
Figure 9.
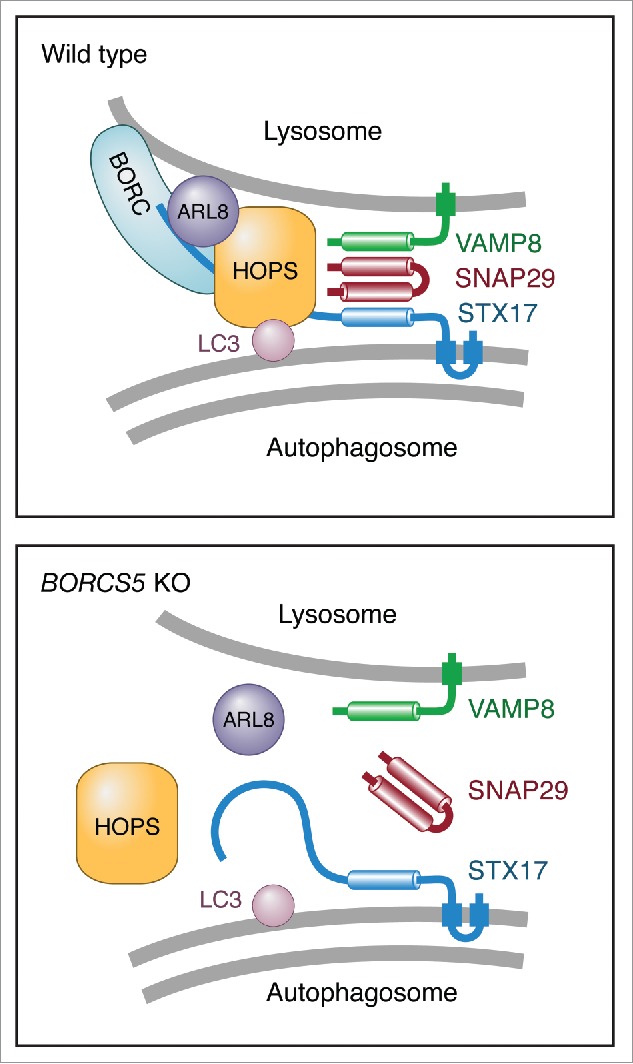
Schematic representation of the regulation of autophagosome-lysosome fusion by BORC. In WT cells, BORC recruits ARL8 and HOPS to the lysosomal membrane. HOPS in turn interacts with LC3B and STX17 on the autophagosomal membrane, promoting assembly of the STX17-VAMP8-SNAP29 trans-SNARE complex and autophagosome-lysosome fusion. In BORC-KO cells, ARL8B and HOPS remain in the cytosol, impairing STX17-VAMP8-SNAP29 trans-SNARE complex assembly and autophagosome-lysosome fusion.
Discussion
The results presented here demonstrate that BORC plays dual roles in autophagy by promoting both the kinesin-dependent encounter and HOPS-mediated fusion of lysosomes with autophagosomes. These roles of BORC are in line with its function in recruiting ARL8A and ARL8B to lysosomes, which in turn recruit both the kinesins and the HOPS complex responsible for the respective processes. Through these roles, BORC contributes to the maintenance of autophagic flux independently of changes in MTORC1 signaling.
BORC-dependent centrifugal transport of lysosomes is involved in autophagy independently of MTORC1
This study stemmed from our initial observation that juxtanuclear clustering of lysosomes caused by BORC-subunit KO was accompanied by increased levels of LC3B-II (Fig. 1). This effect was expected in light of previous work implicating ARL8B and the kinesin KIF2A in the regulation of autophagy by lysosome positioning.19 However, we were surprised to find that increases in LC3B-II in BORC-KO cells occurred without changes in MTORC1 activity under normal culture conditions (Fig. 3C, D). Moreover, BORC-KO did not affect MTORC1 inhibition upon serum depletion (Fig. 3C) or combined serum and amino acid depletion (Fig. S2). LC3B-II increases in BORC-KO cells were not due to enhanced autophagy initiation—an MTORC1-regulated process—but to a partial decrease in lysosomal degradation (Fig. 4A, B). Thus, juxtanuclear clustering of lysosomes does not necessarily lead to changes in MTORC1 activity. Nevertheless, we did observe delayed MTORC1 reactivation upon addition of serum to serum-starved BORC-KO cells (Fig. S8). In addition, double KO of KIF5B and KIF1B did cause an increase in autophagy initiation (Fig. 6E, F). Therefore, lysosome positioning can modulate MTORC1 activity and autophagy initiation under certain conditions, but the nature and extent of the changes appear to depend on the components of the lysosome-positioning machinery that are altered.
Bidirectional transport of both lysosomes and autophagosomes is critical for maintenance of autophagic flux
Having ruled out changes in basal MTORC1 activity as the cause for altered autophagy in BORC-KO cells, we hypothesized that the decreased LC3B-II degradation could be due to the inability of the centrally-clustered lysosomes to meet with autophagosomes arising in the cell periphery. Indeed, we observed a lower frequency of lysosome-autophagosome encounters (Fig. 5A to E), as well as reduced conversion of autophagosomes to autolysosomes, particularly in the peripheral cytoplasm (Figs. 4C, D and S6). Therefore, both centrifugal (this study) and centripetal19 movements of lysosomes are required for meetings with autophagosomes. The need for bidirectional movement also applies to autophagosomes, which undergo centrifugal transport mediated by the kinesin KIF5B and the adaptor protein FYCO1 (FYVE and coiled-coil domain containing 1),39,40 and centripetal transport driven by dynein and the adaptor proteins MAPK8IP1/JIP1 (mitogen-activated protein kinase 8 interacting protein 1),18,41 RILP (Rab interacting lysosomal protein) and OSBPL1A/ORP1L (oxysterol binding protein like 1A).42 Perturbation of either direction of autophagosome transport also impairs autophagic flux.18,40 Thus, maintenance of efficient autophagic flux requires that both lysosomes and autophagosomes are able to move throughout the cytoplasm.
BORC additionally controls autophagosome-lysosome fusion through ARL8-dependent and -independent interactions with HOPS and STX17
Several observations suggested that BORC does more than promote lysosome-autophagosome encounters. First, in BORC-KO cells many autophagosomes accumulated in the juxtanuclear area, in close proximity to the clustered lysosomes (Fig. 4C, D). Furthermore, lysosomes and autophagosomes in these cells were often seen to move around each other without consummating their fusion (Figs. 5D and S6). Finally, although juxtanuclear clustering of lysosomes in cells lacking both KIF5B and KIF1B also increased LC3B-II levels, it did so to a lesser extent than in BORC-KO cells (Fig. 6A to D). These observations pointed to an additional role of BORC in autophagosome-lysosome fusion, possibly by promoting the recruitment of the HOPS complex via ARL8.
ARL8 has previously been shown to mediate recruitment of HOPS to lysosomes through a direct interaction with the VPS41 subunit.36,37 In turn, HOPS interacts with LC3 either directly via the VPS39 subunit11 or indirectly through the adaptor protein PLEKHM1 (pleckstrin homology and RUN domain containing M1).12 In addition, HOPS interacts with the SNARE STX17 to promote assembly of the autophagy-specific trans-SNARE complex.13,14 Indeed, we found that BORC promotes recruitment of HOPS to lysosomes (Figs. 8A and S7A,B), and interacts with HOPS and STX17 (Fig. 8B to D). These interactions are largely dependent on ARL8, although they also have an ARL8-independent component. Furthermore, we found that BORC is required for efficient assembly of the autophagic STX17-VAMP8-SNAP29 trans-SNARE complex (Fig. 8E, F), thus demonstrating the functional significance of these interactions.
Like BORC and ARL8, RAB7 and its effector RILP (Rab-interacting lysosomal protein) have dual roles in late-endosome and lysosome centripetal transport and tethering to other endolysosomal organelles through interactions with dynein-dynactin and HOPS, respectively.43,44 The integration of late-endosome and lysosome transport and fusion with other organelles thus appears to be a common theme for BORC, ARL8 and RAB7, RILP function at both ends of the microtubule network.
Possible implications for BORC-dependent regulation of cell adhesion and migration
Previous work shows that BORC- and ARL8-dependent lysosome movement toward the cell periphery contributes to cell adhesion and cell migration.21,45 These effects were at least partly mediated by late-endosome-mediated dissociation of the signaling protein IQGAP1 (IQ motif containing GTPase activating protein 1) from focal adhesions, leading to focal adhesion turnover.45 Other studies suggested an additional mechanism for focal adhesion turnover and cell motility involving autophagy.46-48 This mechanism depends on interaction of LC3-II with an LIR motif in the focal adhesion protein paxillin48 and/or the autophagy receptor NBR1 (NBR1, autophagy cargo receptor).47 We observed that in BORC-KO cells some LC3B-positive autophagosomes accumulated at cell protrusions, suggesting that cell adhesion and migration might depend on BORC-dependent autophagic turnover of focal adhesion proteins.
Materials and methods
Cell culture and transfection
HeLa and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (Corning, 15-013-CV) supplemented with 10% fetal bovine serum (Corning, 35-011-CV), 100 IU/ml penicillin and 100 μg/ml streptomycin (Corning, 30-002-CI), and 2 mM L-glutamine (Corning, 25-005-CI) in a 37°C incubator (5% CO2, 95% air). For starvation, cells were briefly washed with Hank's balanced salt solution (HBSS; Corning, 20-023-CV) and incubated in HBSS (supplemented with 3 g/L sodium bicarbonate and 25 mM HEPES) or DMEM without serum for the periods indicated in the corresponding experiments. DNA and siRNA transient transfection was performed with Lipofectamine 2000 (Thermo Fisher Scientific, 11668-019).
Plasmids
Plasmids encoding HTT103Q-GFP (1385; from Michael Sherman), FLAG-STX17 (45911; from Noboru Mizushima,) and GFP-STX17 (45909; from Noboru Mizushima,) were obtained from Addgene. Plasmids encoding MYC-BORCS6 and LAMP1-GFP were previously described.21 A plasmid encoding GFP-mCherry-LC3B was generated by subcloning a mCherry-LC3B fragment into pEGFP-C1 (Clontech Laboratories, 6084). Plasmids encoding mCherry-VPS41 and mCherry-VPS39 were generated by subcloning the coding sequences from pSX-VPS41 and pSX-VPS3949 into pmCherry-C1 (Clontech Laboratories, 632524). A plasmid encoding KXD1-GFP was generated by subcloning a KXD1 fragment from MYC-KXD121 into pEGFP-N1 (Clontech Laboratories, 6085). The LIR-mutated KXD1 (KXD1(ΔLIR)-GFP) was generated by site-directed mutagenesis. The ARL8B-GFP plasmid was a gift from Dr. John Brumell (Hospital for Sick Children, Toronto, Canada). A FLAG-VPS41 plasmid was a gift from Dr. Chihiro Akazawa (Tokyo Medical and Dental University, Tokyo, Japan).
Antibodies and chemicals
We used primary antibodies to the following proteins: LC3B (Cell Signaling Technology, 3868), ATG5 (Cell Signaling Technology, 12994), ATG7 (Cell Signaling Technology, 8558), ATG12 (Cell Signaling Technology, 4180), TUBB/β-Tubulin (Cell Signaling Technology, 2146), p-EIF4EBP1/4E-BP1 (Cell Signaling Technology, 2855), EIF4EBP1/4E-BP1 (Cell Signaling Technology, 9452), RPS6KB/S6K (Cell Signaling Technology, 2708), p-RPS6KB/S6K (Cell Signaling Technology, 9234), p-ULK1 (Ser317; Cell Signaling Technology, 12753), p-ULK1 (Ser757; Cell Signaling Technology, 14202), ULK1 (Cell Signaling Technology, 8054), mCherry (Medical & Biological Laboratories, PM005), GFP (Thermo Fisher Scientific, A11122), FLAG epitope (Sigma-Aldrich, F1804), MYC epitope (Santa Cruz Biotechnology, sc-789), BORCS5 (Abgent, AP5806b), SNAP29 (Abcam, ab138500) and VAMP8 (Abcam, ab76021). HRP-conjugated secondary antibodies were from Perkin Elmer (NEF822001EA, NEF812001EA). Secondary antibodies conjugated to Alexa Fluor dyes were from Thermo Fisher Scientific. Baf (B1793) and Torin2 (SML1224) were from Sigma-Aldrich.
CRISPR/Cas9 KO
BORCS5-KO, BORCS7-KO and ARL8B-KO HeLa cells have been described previously.21,22 BORCS8-KO, BORCS6-KO, KXD1-KO, KIF5B-KO, KIF1B-KO and KIF5B KIF1B-KO HeLa cells were generated using CRISPR/Cas9.50 The targeting sequences for BORCS8 (TTTCCCGGTTCGCTCGGCCG/ CTTTAATTACCGGTCCCCCC) and KXD1 (GATACCACCACGGTTGGGTT/ GCTGGAACCTAACCCAACCG) were cloned separately into pSpCas9 (BB)-2A-GFP plasmid (Addgene, 48138; from Feng Zhang). HeLa cells were cotransfected with 2 plasmids containing the different targeting sequences for the same gene, and GFP-positive cells were selected on FACSAria II (BD Biosciences, San Jose, CA, USA) after 48 h. Single cell clones were isolated on 96-well plates. After 21 d, genomic DNA was extracted, the fragment containing mutant site was amplified by PCR on a S1000™ Thermal Cycler (Bio-Rad Laboratories, Hercules, USA), and the cleavage between 2 guide sequences was identified DNA sequencing. The targeting sequences for BORCS6 (GGAGGAGGAAGACAACGACG), KIF5B (CTCTCACGGCCCTCGCGACCACAAGCCCTCAG) and KIF1B (GTCGGGAGCCTCAGTGAAGG) were cloned into pSpCas9 (BB)-2A-GFP plasmid separately. HeLa cells were transfected with the plasmids containing the guide sequences, and GFP-positive cells were selected by flow cytometry after 48 h. Single cell clones were isolated on 96-well plates. KO cells were identified by DNA sequencing or immunoblotting. For KIF5B KIF1B double KO, the same protocol for KIF1B knockout was used but on a KIF5B KO cell line.
Immunoblotting
Cells transfected with the indicated plasmids were washed twice with ice-cold phosphate-buffered saline (PBS; Corning, 21-040-CV) and lysed in 1X LDS loading buffer (Thermo Fisher Scientific, B0007). The cell lysates were separated by SDS-PAGE and transferred to nitrocellulose membrane. The membrane was blocked with 5% nonfat milk (Bio-Rad Laboratories, 1706404XTU) before incubation with primary and secondary antibodies. Bound antibodies were visualized using SuperSignal™ Chemiluminescent Substrate (Thermo Fisher Scientific, 34080).
Immunoprecipitation
Transfected cells were washed with ice-cold PBS and lysed in lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 1% Triton X-100 [Sigma-Aldrich, T9284], 3% glycerol [Sigma-Aldrich, G6279]) with a protease inhibitor cocktail (Roche, 11697498001). Cell lysates were clarified by centrifugation at 13,000 g for 20 min and incubated with GFP-trap magnetic agarose beads (ChromoTek, gtma-20) or anti-MYC/c-myc agarose (Thermo Fisher Scientific, 20169) at 4°C for 4 h. After 3 washes with lysis buffer, the precipitates were eluted with 1X LDS loading buffer for GFP-trap precipitation or 3 × MYC peptide (Thermo Fisher Scientific, 20170) for anti-MYC precipitation. The precipitated samples were analyzed by immunoblotting with the indicated antibodies.
Immunofluorescence microscopy
Cells were seeded on 5 μg/ml FN (fibronectin; Sigma-Aldrich, F2006)-coated coverslips in 24-well plates at 40,000 cells per well in normal culture medium. After transfection or proper treatments, the cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, 15714) in PBS for 20 min, permeabilized with 0.2% Triton X-100 in PBS for 20 min, and blocked in 0.2% BSA (Sigma-Aldrich, A7030) in PBS for 20 min. For endogenous LC3B staining, cells were further fixed in methanol for 20 min at -20°C after the paraformaldehyde fixation. The cells were sequentially incubated with primary and secondary antibodies diluted with 0.2% BSA (Sigma-Aldrich, A7030) in PBS for 30 min at 37°C. Coverslips were mounted on glass slides using Fluoromount-G (Electron Microscopy Sciences, 17984-24). Cells were imaged using a Zeiss LSM780 confocal microscope (Carl Zeiss AG, Oberkochen, Germany). The final composite images were created using ImageJ (NIH).
Live-cell imaging
Cells were seeded on 5 μg/ml fibronectin-coated Lab-Tek Chambered Coverglass units (Thermo Fisher Scientific, 155383). Twenty-four h after transfection, cells were imaged using a spinning-disk confocal microscope (Intelligent Imaging Innovations, Denver, CO, USA). Labeling of lysosomes with internalized dextran was performed 4 h after transfection by loading the cells with Alexa Fluor 647-dextran (Thermo Fisher Scientific, D22914) for 16 h.
Statistics
One-way analysis of variance (ANOVA), followed by multiple comparisons using the Dunnett or the Tukey test, was used for statistical analysis of most data sets; the unpaired Student t test was used where indicated. All graphs show the mean ± SEM. The statistical significance is generally denoted as follows: ***P < 0.0001, **P < 0.001, *P < 0.01, except where indicated.
Supplementary Material
Abbreviations
- AMPK
AMP-activated protein kinase
- ARL8A
ADP ribosylation factor like GTPase 8A
- ARL8B
ADP ribosylation factor like GTPase 8B
- ATG
autophagy-related
- BLOC-1
biogenesis of lysosome-related organelles complex 1
- BORC
BLOC-1 related complex
- EIF4EBP1/4E-BP1
eukaryotic translation initiation factor 4E binding protein 1
- EPG5
ectopic P-granules autophagy protein 5 homolog
- FYCO1
FYVE and coiled-coil domain containing 1
- HOPS
homotypic fusion and vacuole protein sorting
- HTT
huntingtin
- IQGAP1
IQ motif containing GTPase activating protein 1
- KD
knockdown
- KIF
kinesin family
- KLC
kinesin light chain
- KO
knockout
- KXD1
KxDL motif containing 1
- LAMP1
lysosomal associated membrane protein 1
- LIR
LC3-interacting region
- MAPK8IP1/JIP1
mitogen-activated protein kinase 8 interacting protein 1
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3 beta
- NBR1
autophagy cargo receptor
- OSBPL1A
oxysterol binding protein like 1A
- PLEKHM1
pleckstrin homology and RUN domain containing M1
- PLEKHM2/SKIP
pleckstrin homology and RUN domain containing M2
- RAB7
member RAS oncogene family
- RILP
Rab interacting lysosomal protein
- RPS6KB
ribosomal protein S6 kinase B
- SNAP29
synaptosome associated protein 29
- SNAPIN
SNAP associated protein
- SNARE
SNAP (soluble NSF attachment protein) receptor
- SQSTM1
sequestosome 1
- STX17
syntaxin 17
- TFEB
transcription factor EB
- ULK1
unc-51 like autophagy activating kinase 1
- UVRAG
UV radiation resistance associated
- VAMP8
vesicle associated membrane protein 8
- VPS
vacuolar protein sorting
Disclosure of potential conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank X. Zhu for expert technical assistance, J. Brumell and C. Akazawa for kind gifts of reagents, and Amra Saric and Raffaella De Pace for critical review of the manuscript
Funding
This work was funded by the Intramural Program of NICHD, NIH (ZIA HD001607).
References
- [1].Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463-77. doi: 10.1016/S1534-5807(04)00099-1. PMID:15068787. [DOI] [PubMed] [Google Scholar]
- [2].Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664-73. doi: 10.1016/j.bbamcr.2008.07.014. PMID:18706940. [DOI] [PubMed] [Google Scholar]
- [3].Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol. 2011;76:397-402. doi: 10.1101/sqb.2011.76.011023. PMID:21813637. [DOI] [PubMed] [Google Scholar]
- [4].Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125-56. doi: 10.1146/annurev-biochem-052709-094552. PMID:21548784. [DOI] [PubMed] [Google Scholar]
- [5].Shen HM, Mizushima N. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci. 2014;39:61-71. doi: 10.1016/j.tibs.2013.12.001. PMID:24369758. [DOI] [PubMed] [Google Scholar]
- [6].Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. . A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095-108. doi: 10.1038/emboj.2012.32. PMID:22343943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290-303. doi: 10.1016/j.cell.2010.02.024. PMID:20381137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. . Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981-91. doi: 10.1091/mbc.E08-12-1248. PMID:19211835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132-41. doi: 10.1038/ncb2152. PMID:21258367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256-69. doi: 10.1016/j.cell.2012.11.001. PMID:23217709. [DOI] [PubMed] [Google Scholar]
- [11].Manil-Ségalen M, Lefebvre C, Jenzer C, Trichet M, Boulogne C, Satiat-Jeunemaitre B, Legouis R. The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev Cell. 2014;28:43-55. doi: 10.1016/j.devcel.2013.11.022. PMID:24374177. [DOI] [PubMed] [Google Scholar]
- [12].McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, et al. . PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015;57:39-54. doi: 10.1016/j.molcel.2014.11.006. PMID:25498145. [DOI] [PubMed] [Google Scholar]
- [13].Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, Mizushima N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014;25:1327-37. doi: 10.1091/mbc.E13-08-0447. PMID:24554770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Takáts S, Pircs K, Nagy P, Á Varga, Kárpáti M, Hegedűs K, Kramer H, Kovács AL, Sass M, Juhász G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell. 2014;25:1338-54. doi: 10.1091/mbc.E13-08-0449. PMID:24554766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhen Y, Li W. Impairment of autophagosome-lysosome fusion in the buff mutant mice with the VPS33A(D251E) mutation. Autophagy. 2015;11:1608-22. doi: 10.1080/15548627.2015.1072669. PMID:26259518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, et al. . The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell. 2016;63:781-95. doi: 10.1016/j.molcel.2016.08.021. PMID:27588602. [DOI] [PubMed] [Google Scholar]
- [17].Korolchuk VI, Rubinsztein DC. Regulation of autophagy by lysosomal positioning. Autophagy. 2011;7:927-8. doi: 10.4161/auto.7.8.15862. PMID:21521941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109-22. doi: 10.1247/csf.08005. PMID:18388399. [DOI] [PubMed] [Google Scholar]
- [19].Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al. . Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13:453-60. doi: 10.1038/ncb2204. PMID:21394080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jung J, Genau HM, Behrends C. Amino Acid-Dependent mTORC1 Regulation by the Lysosomal Membrane Protein SLC38A9. Mol Cell Biol. 2015; 35:2479-94. doi: 10.1128/MCB.00125-15. PMID:25963655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pu J, Schindler C, Jia R, Jarnik M, Backlund P, Bonifacino JS. BORC, a multiprotein complex that regulates lysosome positioning. Dev Cell. 2015;33:176-88.doi: 10.1016/j.devcel.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Guardia CM, Farías GG, Jia R, Pu J, Bonifacino J. BORC functions upstream of kinesins 1 and 3 to coordinate regional movement of lysosomes along different microtubule tracks. Cell Reports. 2016;17:1950-61. doi: 10.1016/j.celrep.2016.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805-12. doi: 10.1242/jcs.01131. PMID:15169837. [DOI] [PubMed] [Google Scholar]
- [24].Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298-302. doi: 10.1074/jbc.C700195200. PMID:17986448. [DOI] [PubMed] [Google Scholar]
- [25].Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603-14. doi: 10.1083/jcb.200507002. PMID:16286508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84-91. doi: 10.4161/auto.1.2.1697. PMID:16874052. [DOI] [PubMed] [Google Scholar]
- [27].Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A. 2000;97:1589-94. doi: 10.1073/pnas.97.4.1589. PMID:10677504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. . Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942-6. doi: 10.1038/nature09076. PMID:20526321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li X, Rydzewski N, Hider A, Zhang X, Yang J, Wang W, Gao Q, Cheng X, Xu H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol. 2016;18:404-17. doi: 10.1038/ncb3324. PMID:26950892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452-60. doi: 10.4161/auto.4451. PMID:17534139. [DOI] [PubMed] [Google Scholar]
- [31].Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679-88. doi: 10.1242/jcs.00381. PMID:12665549. [DOI] [PubMed] [Google Scholar]
- [32].Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497-510. doi: 10.1083/jcb.200712064. PMID:18443221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].ÅB Birgisdottir, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013;126:3237-47. doi: 10.1242/jcs.126128. PMID:23908376. [DOI] [PubMed] [Google Scholar]
- [34].Rosa-Ferreira C, Munro S. Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev Cell. 2011;21:1171-78. doi: 10.1016/j.devcel.2011.10.007. PMID:22172677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wu YE, Huo L, Maeder CI, Feng W, Shen K. The balance between capture and dissociation of presynaptic proteins controls the spatial distribution of synapses. Neuron. 2013;78:994-1011. doi: 10.1016/j.neuron.2013.04.035. PMID:23727120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Garg S, Sharma M, Ung C, Tuli A, Barral DC, Hava DL, Veerapen N, Besra GS, Hacohen N, Brenner MB. Lysosomal trafficking, antigen presentation, and microbial killing are controlled by the Arf-like GTPase Arl8b. Immunity. 2011;35:182-93. doi: 10.1016/j.immuni.2011.06.009. PMID:21802320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Khatter D, Raina VB, Dwivedi D, Sindhwani A, Bahl S, Sharma M. The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J Cell Sci. 2015;128:1746-61. doi: 10.1242/jcs.162651. PMID:25908847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wartosch L, Günesdogan U, Graham SC, Luzio JP. Recruitment of VPS33A to HOPS by VPS16 Is Required for Lysosome Fusion with Endosomes and Autophagosomes. Traffic. 2015; 16:727-42. doi: 10.1111/tra.12283. PMID:25783203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cardoso CM, Groth-Pedersen L, Hoyer-Hansen M, Kirkegaard T, Corcelle E, Andersen JS, Jaattela M, Nylandsted J. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS One. 2009;4:e4424. doi: 10.1371/journal.pone.0004424. PMID:19242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjorkoy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253-69. doi: 10.1083/jcb.200907015. PMID:20100911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fu MM, Nirschl JJ, Holzbaur EL. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell. 2014;29:577-590. doi: 10.1016/j.devcel.2014.04.015. PMID:24914561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wijdeven RH, Janssen H, Nahidiazar L, Janssen L, Jalink K, Berlin I, Neefjes J. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat Commun. 2016;7:11808. doi: 10.1038/ncomms11808. PMID:27283760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].van der Kant R, Fish A, Janssen L, Janssen H, Krom S, Ho N, Brummelkamp T, Carette J, Rocha N, Neefjes J. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J Cell Sci. 2013;126:3462-74. doi: 10.1242/jcs.129270. PMID:23729732. [DOI] [PubMed] [Google Scholar]
- [44].Lin X, Yang T, Wang S, Wang Z, Yun Y, Sun L, Zhou Y, Xu X, Akazawa C, Hong W, et al. . RILP interacts with HOPS complex via VPS41 subunit to regulate endocytic trafficking. Sci Rep. 2014;4:7282. doi: 10.1038/srep07282. PMID:25445562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schiefermeier N, Scheffler JM, de Araujo ME, Stasyk T, Yordanov T, Ebner HL, Offterdinger M, Munck S, Hess MW, Wickstrom SA, et al. . The late endosomal p14-MP1 (LAMTOR2/3) complex regulates focal adhesion dynamics during cell migration. J Cell Biol. 2014;205:525-40. doi: 10.1083/jcb.201310043. PMID:24841562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, Dinsdale D, Condorelli F, Brandner S, Campanella M, et al. . The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32:699-712. doi: 10.1038/onc.2012.111. PMID:22525272. [DOI] [PubMed] [Google Scholar]
- [47].Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, Ye J, Wittmann T, Debnath J. NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol. 2016;212:577-90. doi: 10.1083/jcb.201503075. PMID:26903539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M, Mui S, Macleod KF. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016;15:1660-72. doi: 10.1016/j.celrep.2016.04.065. PMID:27184837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Caplan S, Hartnell LM, Aguilar RC, Naslavsky N, Bonifacino JS. Human Vam6p promotes lysosome clustering and fusion in vivo. J Cell Biol. 2001;154:109-22. doi: 10.1083/jcb.200102142. PMID:11448994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. . Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819-23. doi: 10.1126/science.1231143. PMID:23287718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.