

Abstract
Cell-penetrating peptides (CPPs) have long held great promise for the manipulation of living cells for therapeutic and research purposes. They allow a wide array of biomolecules from large, oligomeric proteins to nucleic acids and small molecules to rapidly and efficiently traverse cytoplasmic membranes. With few exceptions, if a molecule can be associated with a CPP, it can be delivered into a cell. However, a growing realization in the field is that CPP-cargo fusions largely remain trapped in endosomes and are eventually targeted for degradation or recycling rather than released into the cytoplasm or trafficked to a desired subcellular destination. This ‘endosomal escape problem’ has confounded efforts to develop CPP-based delivery methods for drugs, enzymes, plasmids, etc. This conceptual overview provides a brief history of CPP research and discusses current issues in the field with a primary focus on the endosomal escape problem, of which several promising potential solutions have been developed. Are we on the verge of developing technologies to deliver therapeutics such as siRNA, CRISPR/Cas complexes and others that are currently failing because of an inability to get into cells, or are we just chasing after another promising but unworkable technology? We make the case for optimism.
Keywords: cell-penetrating peptides, protein transduction domains, endocytosis, endosomal escape, TAT
Introduction
Nearly thirty years ago, HIV researchers Frankel and Green independently stumbled upon a phenomenon that ultimately gave rise to a potentially transformative technology: the HIV protein transactivator of transcription (TAT) could readily pass through the plasma membrane of uninfected mammalian cells (1, 2). Further, and most importantly, TAT retained its normal functionality and subcellular localization post-entry as it readily entered the nucleus and promoted gene transcription. This surprising ability to traverse the cellular membrane was later mapped to a stretch of twelve basic amino acids and this penetrative property could be conferred onto other proteins to which that sequence was fused (3, 4). A new area of intense scientific research was thus born: the delivery of bioactive molecules into mammalian cells via direct penetration of the plasma membrane.
Since Frankel’s and Green’s discoveries, a large number of peptides that are rapidly internalized and enable transport of macromolecular cargos into mammalian cells in vitro and in vivo have been discovered or designed. These peptides came to be known as cell penetrating peptides (CPPs, also frequently referred to as protein transduction domains, or PTDs). The term CPP now refers to a broad grouping of non-homologous short peptides, the majority of which are hydrophilic and cationic in nature, though amphiphilic, anionic, and hydrophobic CPPs have been reported. A database of more than 1,600 CPPs is described by Agrawal et al. (5). Their unifying property is the ability to penetrate the plasma membrane for delivery of cargo into cells, from peptides and proteins both small and large to various nucleic acids, including DNA, RNA, siRNA, and peptide-nucleic acids (PNAs) (6 9).
Thirty years later, CPP-based technologies have largely failed. Frankel and Palo first reported that cellular entry of TAT was likely via an endosomal independent mechanism as introduction of the endosomal-blocking agent chloroquine enhanced TAT uptake into cells (1). However, much of the subsequent data supporting spontaneous membrane translocation were found to be the result of experimental artifacts and it was soon realized that majority of CPPs became trapped in endosomes following delivery into cells and, as a result, any associated cargo becomes trapped as well (10, 11). It is likely the case that, owing to endosomal entrapment, the vast majority of CPP-cargos are eventually targeted to either the lysosome for degradation or back to the plasma membrane for recycling and subsequent ejection from the cell. We have dubbed this the ‘endosomal escape problem’.
In spite of setbacks, CPPs still have significant therapeutic and investigative potential. Currently, more than 25 CPP clinical trials are underway, including a Phase III (12, 13). To successfully transition CPP-based technology into therapeutic delivery systems, several considerations need to be made. This review focuses on fundamental questions and the challenges faced by the field in this endeavor: How do CPPs get into the cell? Once inside, how is cargo released from the endosome into the cytosol? How do we ensure biological activity and correct subcellular localization of cargo following endosomal release? And finally, how can we ensure specificity in cellular targeting?
Breaking In: Mechanisms of cellular entry
Currently, the field recognizes endocytosis to be the primary mode of CPP cellular entry and subsequent endosomal escape the rate-limiting step for the effectiveness of CPPs to deliver cargo into living cells. However, there is still much controversy on how CPPs enter cells. This section of the review we will provide a brief history of 30 years of mechanistic insight on CPP cellular entry with a primary focus on endocytosis.
A brief glance at endocytic pathways
Endocytosis is a broad term encompassing a variety of pathways utilized by the cell to bring outside molecules in for a multitude of purposes – receptor & lipid recycling, inhibition of signal transduction, uptake of solutes/nutrients, and destruction of foreign/unwanted materials. The unifying property of endocytic pathways is, simply put, engagement of the plasma membrane for the formation of an intracellular membrane-bound organelle which will then transport the molecules to some destination within the cell. It is a tightly coordinated, energy-dependent process requiring extensive cellular adaptors and cytoskeletal rearrangements. As illustrated in Figure 1, following organelle formation, an endocytic vesicle undergoes a pH-dependent maturation process as it transitions from the early sorting endosome (pH ~6.5 – 6) into the late endosome or multivesicular body (MVB; pH ~5). The acidification process is dependent on ionic gradients, requiring calcium, sodium and potassium efflux as well as hydrogen and chloride influx. An endosome has three potential fates – it may be targeted to the lysosome from the MVB for destruction of its intraluminal contents or its contents may be recycled back to the plasma membrane either directly from the sorting endosome or after routing from the MVB to the trans golgi network. Alternatively, some endosomal contents such as nutrients are needed by the cell and thus must be released into the cytosol. This later process is mediated by the formation of intraluminal vesicles (ILVs) inside the endosome, essentially the formation of an exosome within the endosome. The ILV then undergoes a process known as ‘backfusion’ where it fuses with the endosomal membrane and its contents are released into the cytosol.
Figure 1.
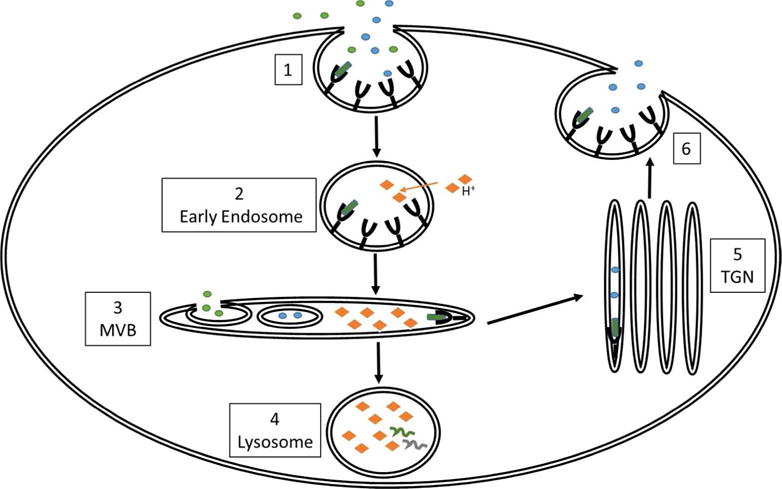
An overview of endocytosis. Endosome formation may be receptor (clathrin/caveolin) or non-receptor mediated (macropinocytosis). Following invagination (clatherin/cavaeolin) or extrvagination (macropinocytosis) of the plasma membrane, an early endosome is formed (1). As the endosome travels into the cell, it transitions to an early endosome and becomes increasingly acidified by pumping in cytosolic hydrogen ions into the vesicular lumen (2). As the early endosome matures, it begins to sort cargo by the formation of multiple smaller intracellular vesicles (ILVs) (3). Some of these vesicles may refuse with the endosomal membrane and deliver contents directly into the cytosol, called ‘backfusion’ while others will be destined for either the lysosome (4) or the trans-golgi network (TGN) (5). Cargo delivered to the lysosome will be degraded while cargo delivered to the TGN will be recycled back to the plasma membrane. Some receptors can bypass the TGN and be directly recycled back to the plasma membrane from the early endosome (6).
Endocytosis can further be broken down into phagocytosis and pinocytosis depending on whether entry is fluid-phase or not. We will limit our discussion to pinocytic pathways most commonly engaged by CPPs – macropinocytosis, clathrin-dependent and caveolin-dependent endocytosis, and lipid-raft endocytosis. A full description of these pathways is outside the scope of this review, however, a brief description of these pathways is provided below:
Macropinocytosis is defined as a receptor-independent and coat-independent process. First described by Warren Lewis in 1931, this is a form of cellular ‘pino’ (drinking) ‘cytosis’ (14). Macropinocytosis is classically thought to be a non-specific mechanism to bring solutes/nutrients into the cell via large endocytic vesicles. However, more recent studies point to a need for stimulation at the plasma membrane via the presence of extracellular growth factors (reviewed in (15)). Further, different cell types may utilize this process in different ways given their particular needs. For example, some cells use this process to obtain nutrients or to recycle portions of their membranes while immune cells utilize macropinocytosis for the regulation of antigen presentation.
Clathrin-dependent endocytosis is a receptor-driven process that results in the formation of a ‘coated’ vesicle. The trimeric coat protein, clathrin, for which this pathway is defined, was the first coat protein to be isolated from cellular membrane-bound vesicles in 1976 by Barbra Pearse (16). During endocytosis, three-footed triskelion subunits assemble via adaptor proteins at cholesterol-deficient regions of the plasma membrane, forming into a lattice work to create a highly-ordered ‘caged’ structure which is internalized. This pathway is considered ubiquitous across all cell types and is utilized in a variety of ways; transferrins, low-density lipoproteins, hormones and neurotransmitters (during reuptake) are a few examples of molecules taken up by this pathway.
Caveolin-mediated entry shares both similarities and distinctions from clathrin-mediated entry. Caveolins are also coat proteins that form tight associations with cholesterol present in the plasma membrane. Unlike the trimeric clathrins, following recruitment to the plasma membrane caveolins form into a distinct “U” shape, with both N-and C-termini pointing towards the cytoplasm. The resulting invaginations resemble cave-like structures called caveolae, for which they were originally named (17). Not surprisingly, caveolins are found localized to cholesterol-rich lipid rafts. Many growth factors utilize this pathway, as do some pathogens. Further, caveolin-dependent endocytosis is important in transendothelial transport. Unlike the more ubiquitous mechanism of clathrin-dependent endocytosis, caveolae formation is impacted by many cellular factors such as cell type as well as cell cycle progression. Further, some cells express caveolins at low levels or not at all.
Lipid-raft endocytosis is a non receptor mediated, concentration-dependent form of endocytosis occurring at cholesterol-enriched lipid rafts in the plasma membrane but does not rely on caveolin coat formation. In this form of endocytosis, glycosylphosphatidylinositol- anchored proteins (GPI-AP) group into distinct microdomains, invaginate and form into GPI-enriched intracellular vesicles. This pathway is primarily used as a constitutive means of bringing in extracellular fluids through lipid-raft mediated pathways based in membrane-molecule interactions and have been described for SV40-virions, vitamins, GPI-binding proteins, MHC-class I, IL-2, and IgE. Even though these pathways show significant variability for the requirement of local mediators and in cargo fate (recycling, degradation, intracellular release), they share commonalities in that they are receptor mediated and proceed via an absorptive fluid phase mechanism (reviewed in (18)).
Breaking in via endocytosis: The case and conundrum
Individual CPPs may engage one or more of the forms of pinocytosis defined above. With regards to TAT, different groups have reported cellular uptake by all of these endocytic mechanisms (19–25). Further, some CPPs such as the well-studied arginine-rich, 16-residue peptide corresponding to the third helix of the Drosophila melanogaster transcription factor Antennapedia homeodomain (Antp; penetratin) may enter via direct penetration (26). The debate is ongoing; CPP entry mechanism(s) is a heavily explored area with minimal consensus. Much of this likely owes to differences in laboratories, cell types, CPP constructs, and experimental conditions.
Frankel’s & Pabo demonstrated in their seminal paper that lysotrophic agents enhanced instead of blocked full-length TAT uptake into cells (1). This gave rise to the direct penetrance hypothesis was rapidly embraced by many in the field. Three years later, Mann & Frankel provided data that full-length TAT likely entered via an undefined absorptive-phase endocytic pathway. Namely, they found that uptake was significantly reduced when performed at 4°C (energy-independent) compared with 37°C (27). Interestingly, this was a cell-dependent effect as cervical cancer-derived cells (HeLa) showed a striking difference in energy-dependent uptake while temperature reduction had a notably smaller, though similar, impact on a T cell-derived cell line (27). This was this first illustration of a cell-type effect on TAT uptake and these findings hinted at complexities with which we are still coming to grips, including the importance of cell specific differences in CPP uptake.
The 1990s saw a flurry of reports of direct penetrance by CPPs. However, many of these observations were later found to be artifactual (10). In particular, Lundberg et al. demonstrated intracellular redistribution and dispersed cytoplasmic and nuclear distribution of CPPs resulted from cellular fixation whereas live cell imaging experiments indicated a punctate (likely endosomal) distribution (10). More recent work utilizing live cell imaging has shown evidence for the potential of non endocytic uptake of some CPP constructs, but these data are not without constraints on the concentration of the CPP and nature of the cargo (19, 28).
By the onset of the 21st century, over a decade from initial reports of TAT’s ability to traverse the plasma membrane, multiple conflicting reports were published in support of one endocytic pathway over another engaged by TAT as well as those of other newly discovered CPPs, as outlined in Table 1. In 2007, Duchardt et al. designed a series of experiments to clarify contradictory data on the use of endocytic pathways by CPPs that plagued the field. They studied concentration-dependent uptake mechanisms of three well-characterized cationic CPPs (penetratin, TAT, & R9) employing specific endosomal pathway inhibitors and tracers. It was demonstrated that all three of these CPPs engaged multiple forms of endocytosis (clathrin-dependent, caveolin-dependent, & macropinocytosis) and that concentration of the CPP played a role in which endocytic pathway was used (19). While these data are seemingly at odds with previous work, many proteins, receptors, and viruses make use of all of these forms of endocytosis. Further, while all of these pathways exhibit specificity, there exists a large degree of functional redundancy and cross-talk among them.
Table 1.
Experimental evidence for the use of different endocytic pathways by TAT in different cell lines.
| Endocytic Pathway | Cell Line | Endocytic Pathway Inhibitors / Tracers | Methodology |
|---|---|---|---|
| Macropinocytosis | Tex.Ioxp.EG (T – cells) | β-cyclodextrin; nystatin; EIPA; cytochalasin D [23] | Flow cytometry, live cell imaging [23] |
| Cos – 7 (fibroblasts) | EIPA; cytochalasin D / RFP-labeled caveolin [23] | Live cell imaging [23] | |
| CHO | Dyamin knock-down / transferrin [23] | Immunohistochemistry [23] | |
| K562 (lymphoblasts) | ATP – depletion; EIPA [37] | Flow cytometry [37] | |
| Clathrin-Dependent | HeLa | Chlorpromazine; potassium reduction / transferrin [38] Chlorpromazine (effective at high concentrations of TAT – 40 μM); EIPA; MβCD [19] | Flow cytometry [38] Flow cytometry, live cell imaging [19] |
| Jurkat T-cells | 4°C incubation; chlorpromazine; filipin; Eps15, dynamin, & intersectin dominant-negative mutants [25] | Radioactivity labeling, immunohistochemistry [25] | |
| CHO | Potassium reduction, nystatin / transferrin | Flow cytometry [38] | |
| Caveolin-Dependent | Hela | 4°C incubation / Cholera toxin, transferrin, CFP-labeled caveolin [20] Cytochalasin D, MβCD, 4°C incubation / transferrin, EEA-1, cholera toxin [24] | Live cell imaging [20] Live cell imaging, Immunohistochemisrty, reporter assays, flow cytometry [24] |
| Cos – 1 (fibroblasts) | Cytochalasin D, MβCD, 4°C incubation / caveloin-1 [24] | Flow cytometry, immunohistochemistry [24] | |
| Lipid Raft | Jurkat T – cells | Cytochalasin D, MβCD, 4°C incubation [24] | Flow cytometry, immunohistochemistry [24] |
| Undefined | PBM – MOs | ATP – depletion, 4°C incubation [38] | Flow cytometry [38] |
| HUVEC | ATP – depletion, 4°C incubation [38] | Flow cytometry [38] |
Inhibitors: Macropinocytosis – EIPA; Clathrin – cytochalasin D, chlorpromazine, potassium reduction; Caveolin – flipin, β-cyclodextrin, nystatin; Lipid Raft – β-cyclodextrin, nystatin, potassium reduction
Ten years later much remains unresolved concerning how CPPs enter a cell. Even though endocytosis gained the lime-light as the primary means of entry, direct penetrance still remains a distinct possibility and an active area of interest in the field. Determinants dictating CPP utilization of one endocytic pathway over another, or bypassing the endosome entirely, are thought be highly contextual and dependent on the nature of the CPP, concentration, receptor availability, media conditions and cell type. A better understanding of the factors influencing one form of uptake of another in different target cells and tissues will be paramount in optimizing targeting, uptake, and endosomal escape strategies to develop CPPs for effective cargo delivery in vivo and in vitro.
A receptor for CPPs?
Regardless if cellular entry is direct or endosomal, the CPP must come initially into contact with the plasma membrane to facilitate uptake. Thus, requirements for cellular entry are a primary question in the field. Currently, CPP-membrane interaction is thought to be governed by either non-specific electrostatic interactions or via ligand-receptor binding. Given that TAT, the prototypical CPP, is derived from a HIV protein, much of the work in this arena has been based on HIV research and has utilized either TAT or TAT-homologous arginine-rich CPPs. Early work on HIV entry mechanisms revealed that full-length TAT could bind, among other molecules, ubiquitously expressed cellular proteoglycans (namely heparan sulfates, HSPGs), integrins, and chemokine (C × C motif) receptor 4 (CXCR4), though in the latter case it serves as an antagonist (29–34). All of these molecules readily activate endocytic pathways in both clathrin-dependent and independent mechanisms. In particular, binding to HSPGs and integrins was mapped to the basic domain of TAT, which also confers its cell penetrating properties (29). In 1993, Vogel et al. reported that while the basic domain of TAT could readily bind integrin αvβ5, antibodies blocking this interaction had no effect on TAT uptake into cells (32). Since these observations, HSPGs have received prominent attention as potential TAT receptors (31, 35–39).
Interaction between TAT’s basic domain (its CPP sequence) and HS’s were originally elucidated by Rusnati et al. in 1998 (40). These observations were expanded upon in 2001 by Tyagi et al. who provided evidence for the need for cell surface proteoglycans for cellular internalization of TAT constructs, as cells defective in glycosaminoglycan (GAG) synthesis showed reduced TAT uptake (31). Mounting evidence for the roles of HSPGs in uptake and endocytosis of arginine-rich CPPs such as TAT have provided a new framework for the study of receptor-mediated entry of CPPs (31, 35–40). With respect to cationic CPPs, interactions with negatively charged GAGs comprising HSPGs are thought to concentrate these CPPs at the plasma membrane to facilitate endosomal uptake.
Identifying receptors for different CPPs will be paramount in untangling mechanisms of entry and may lead to the development of more efficient cargo-delivering CPPs. Expanding upon the HSPG-CPP interaction hypothesis, Letoha et al. in 2010 and Kawaguchi et al. in 2016 identified the ubiquitous HSPG syndecan 4 as a potential receptor for the classical arginine-rich CPPs (35, 37). Letoha et al. reported that syndecan 4-enhanced uptake of TAT, penetratin and the bioengineered octoarginine peptide R8 via an energy-dependent endocytic mechanism they attributed to macropinocytosis (37). This is not surprising as syndecan 4 is known to activate multiple downstream small GTPases involved in different endocytotic pathways including macropinocytosis, clathrin-dependent and caveolin-dependent endocytosis as well as lipid raft-dependent endocytosis (41, 42). A major limitation to this work is the use of a cell-based overexpression system in lymphoblasts, which normally do not express syndecans. There is some doubt as to whether a cell that does not normally express this molecule will recreate normal syndecan-mediated cellular uptake pathways following transfection.
Kawaguchi et al. later performed a screen for binding partners of R8 in HeLa cells (35). They identified 17 potential R8 binding patterns, 7 of which are proteoglycans and 2 of which are core components of the extracellular matrix (ECM). They confirmed that syndecan 4 facilitated cellular entry of R8 in a concentration-dependent manner, as siRNA-mediated knockdown of syndecan 4 impacted uptake at low concentrations (1 μM), but only had little effect at high concentration (10 μM) (35). In contrast to previous observations, R8 appeared to predominatly utilize a clathrin-dependent endocytic pathway instead of micropinocytosis though this may be a by-product of experimental conditions (22).
The other recently identified candidate receptor, CXCR4, is highly expressed on migratory cells such as immune and cancerous cells. CXCR4 was originally identified as a key receptor for HIV type X4 and works in concert with co-receptor CD4 and cell surface HSPGs to facilitate viral entry into target cells (33, 34). In 2012, Tanaka et al. provided evidence for the use of CXCR4 followed by subsequent macropinocytosis by the arginine-rich CPP R12 (43). In support of CXCR4 and syndecan 4 serving as receptors for arginine-rich CPPs, CXCR4 is normally complexed with syndecan 4 and that association promotes binding of its natural ligand, stromal cell-derived factor 1 (SDF-1) (44). At odds with this, however, Tanaka et al. found that CXCR4 facilitated uptake of R12 but not the other arginine-rich CPPs, TAT or R8, the later which is the proposed receptor of syndecan-4 (35, 43). The failure of TAT to bind CXCR4 likely owes to the fact that binding to CXCR4 has been mapped to TAT’s central ‘chemokine-like’ domain though direct analysis of its basic domain binding to CXCR4 has not been performed to our knowledge. Finally, CXCR4 is found in low abundance in normal, non-migratory, healthy cells and tissues so while CXCR4 may facilitate the uptake of at least one CPP, R12, as noted by the authors, it is likely not the sole receptor.
Identifying receptors for different CPPs will be paramount in untangling mechanisms of entry and may lead to the development of more efficient cargo delivering CPPs. However, if CPPs can readily utilize more ubiquitously expressed molecules such as HSPGs this may reduce therapeutic value without effective targeting strategies. Further, failure to control endocytic pathway engagement means a variety of fates may await any given molecule every time it is introduced.
Busting Out: Escape from the endosome
The field has increasingly recognized that the principal roadblock to development of therapeutics is cargo entrapment in the endocytic pathway. Almost all described CPP technologies are reliant on covalent crosslinking or nonspecific hydrophobic interactions (45). In our opinion, it is here that CPPs currently fail as a workable technology. If cellular entry is receptor-mediated, it could well be that high affinity of the CPP for its receptor is in part due to low off rates and hence trapping of its linked cargo in the endosomes may essentially be a kinetic problem. In this section we focus on some of the more promising approaches that have been developed to facilitate escape from the endosome.
Promising tricks for facilitating endosomal escape
The most promising ‘tricks’ to overcome entrapment include the use of endosomolytic agents, reversible covalent binding, and reversible high-affinity non-covalent binding. The Pellois group has developed an endosomolytic agent to promote release of cargo via endosomal leakage. Their dfTAT, a dimerized disulfide-linked TAT, can destabilize endosomes for the delivery of co-incubated cargo (46, 47). This methodology is particularly attractive as it allows for introduction of cargo, even multiple cargos, without direct interaction with TAT. Further, utilization of disulfide bonds should enhance stability for systemic delivery.
Several groups have developed novel reversible strategies that may have advantages over endosomolytic agents in toxicity, specificity and simplicity. Among the reversible strategies, the most common is the use of thiol coupling. The reducing environment of both the endosome and the cytoplasm should be an effective means to reduce disulfides and uncouple cargo from CPP. Further, as with the dfTAT model, the use of thiol coupling should provide stability and protect the cargo/CPP during systemic delivery. Another approach described by the Rossi lab utilizes an interesting photocleavable linkage to deliver cleaved peptides to the cytosol (48). While this may overcome entrapment, this strategy is seemingly impractical for many applications as it would be difficult to get light to many places within patients. The potential of this strategy as a research tool, however, may be high.
Models that utilize noncovalent CPP-cargo linkages can overcome problems associated with covalently bound cargo. Our group has recently described a novel CPP-adaptor method that exploits normal ionic gradients to free cargo following endosomal entry (49). We associated CPPs with their cargos via a reversible coupling between a CPP-containing calmodulin and cargo that contains a calmodulin binding site. Our initial findings, described in (49), showed that TAT-fused calmodulin (TAT-CaM) bound calmodulin binding site (CBS)-containing cargos with nanomolar affinity in the presence of calcium but not at all in its absence. In that study, three model cargos and three distinct cell lines were used, demonstrating general, efficient and rapid delivery of cargo to the cytoplasm at much lower dose (1 μM) than necessary using other means. Further, when calcium was removed from bound TAT-CaM-cargo complexes, very rapid dissociation ensued. As illustrated in Figure 2, the high calcium concentrations of the extracellular environment ensures high affinity CPP-cargo binding. Following entry into the cell, the extracellular calcium is rapidly lost as the endosome becomes increasingly acidified (50). We hypothesize that as calcium levels drop, the CPP adaptor releases its cargo from the early endosome prior to formation of the more acidic late endosome.
Figure 2.
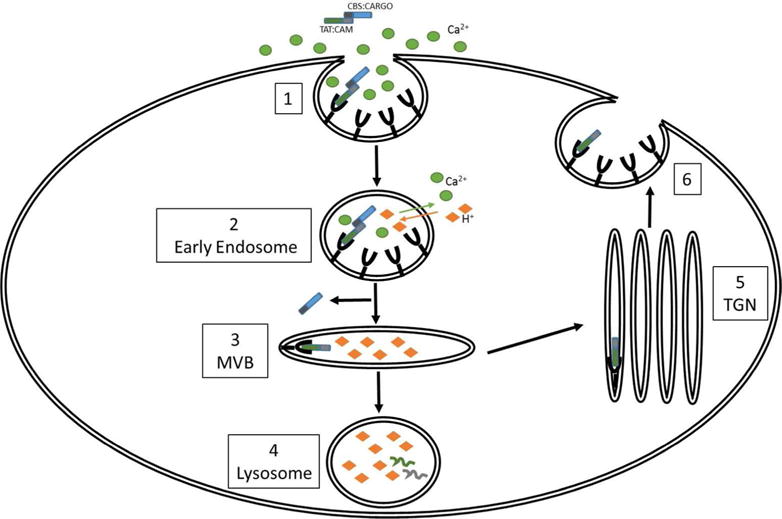
Proposed model for TAT:CAM-mediated intracellular delivery of CBS:CARGO. TAT fused to a calmodulin (TAT:CAM) will readily associate with cargo containing a calmodulin bind site (CBS:CARGO) in the extracellular environment owing to high levels of calcium (1). Following binding of TAT:CAM to a receptor, an endosome will form containing the TAT:CAM-CBS:CARGO as well as high levels of extracellular calcium. As the endosome matures, calcium will be pumped out of the intraluminal space while hydrogen ions are brought in (2). This shift in calcium concentrations within the endosome will cause the CBS:CARGO to disassociate from TAT:CAM, allowing for release of the cargo into the cytosol, presumably through IVLs, even if TAT:CAM remains tightly bound to its receptor (3). TAT:CAM itself may have multiple possible fates following formation of the late endosome. It may be sent to the lysosome for degradation if it retains its ability to bind its receptor in increasingly acidic conditions (4) or sent to the TGN (5) for recycling back to the plasma membrane (6).
Considerable attention has been given to the design of new, more efficient, delivery mechanisms. The use of non-covalent association of CPPs with their cargo is, in our opinion, a key strategy to enhance bioavailability of biologically active molecules. By noncovalently coupling the cargo from the CPP, problems with endosomal entrapment and the potential for a loss of biological activity are easily surmountable. Finally, these methodologies typically require reduced and more therapeutically feasible concentrations of protein for the desired effect.
Subcellular localization: targeting freed cargo to specific intracellular compartments
Once cargo has been delivered into the cell and successfully escaped the endosome, how does the cargo get targeted to a particular location within the cell? Much of our understating of subcellular localization of proteins is derived from basic knowledge of signal peptides that facilitate localization of proteins to intracellular compartments. Within the endomembrane system, such delivery is achieved through sorting of localization signals and subsequent vesicular-mediated targeted delivery. However, many subcellular compartments can directly import cytosolic proteins provided they contain a specific targeting sequence. Such examples include mitochondrial import, receptor mediated nuclear import-export, cotranslational entry into the ER, and import of cytosolic proteins, such as catalase, into peroxisomes post-budding off the ER membrane.
Protein-replacement therapy aims to deliver a fully functional protein to replace one that has been mutated, lost, or is underexpressed. Under ideal circumstances, a protein is successfully delivered into the cell, freed from the endosome, and behaves exactly like wild-type endogenous protein should. In the past decade, several groups have been successful in this endeavor. A group out of Hebrew University–Hadassah Medical School, led by Hala Lorberboum-Galski, has developed and successfully delivered functional proteins to the mitochondria in vitro to restore defects in the electron transport chain (51) and acetyl Co-A production (52) associated with life-threatening mitochondrial disease. Other groups have also demonstrated the ability to deliver functional copies of proteins into cells to alleviate dysfunctional proteins associated with mitochondrial and lysosomal diseases. For example, in 2012, Honda et al. were able to deliver functional subunits of the NADPH oxidase complex to the cytosol to restore its activity ex vivo in neutrophils from patients suffering from chronic granulomatous disease (CGD) (53), a disease characterized by improper ROS production resulting from defective NADPH complexes. While conceptually simple, these approaches are not without significant consideration to ensure the protein will behave as it should within the cell. The protein must be produced in highly purified, properly folded conformation, have its native subcellular localization signal accessible to subcellular import machinery (in the case of non-cytosolic proteins), show normal dynamics of stability and activity, be expressed at normal endogenous levels, engage with binding partners, and, in many cases, be able to undergo post-translational modification.
Delivery of therapeutic molecules into the cell often requires artificial means for intracellular localization of the molecule to the compartment of interest. The predominant approach for targeting CPP-bound cargo calls for the fusion of canonical signal sequences to cargo proteins, or even to the CPP tag itself (reviewed in (54)). Using this methodology, cargos have been delivered to the nucleus (55, 56), nucleolus (57), lysosome (58), peroxisome (55), the mitochondria (55, 59, 60) and the endoplasmic reticulum (55, 61), to name a few. Drawbacks to this approach include decreased uptake of tagged CPPs (61) and potential endosomal entrapment en route to the intended compartment. The underlying mechanism via which some of these CPPs-cargo complexes gain entry into specific subcellular compartments is less well understood. Within the endomembrane system, it is tempting to speculate that one might be able to exploit endosomal entry for cargo targeting though ensuring proper sorting within the late endosome may present challenges.
Targeting CPPs to specific cell types
The specific delivery of cargo to a particular target cell or tissue via CPP-mediated methods remains the Holy Grail of pharmacological work in this field (reviewed in (62)). The central problem is that of specificity: given that CPP uptake is thought to occur via a general mechanism with a ubiquitous cellular receptor such as HSPGs or membrane apparatus, it is extremely facile to deliver CPP-tagged cargo proteins systemically. The technical challenge, albeit not insurmountable, is to deliver them to a particular site, tissue, or organ system. Initial work in this area centered on localized injection of CPP-tagged cargo proteins. While these approaches yield the desired effect of delivery of CPP-cargo directly to tumor cells, they are only an effective for solid, localized tumors or easily isolated or discovered target regions.
Other strategies involve exploiting the biochemical nature of target cells/tissues. So-called activatable cell penetrating peptides (ACPPs) pair technical innovation with an improved working knowledge of affected cellular biochemistry and have been successful with some degree of target specificity by several groups (reviewed in (63)). These ACPPs are activatable in a stimulus-dependent manner and are caged or masked until they are in the vicinity of their target. This methodology employs an anionic inhibitor to shield cationic CPPs until they are near cells or tissues whose extracellular environment contains a product capable of cleaving the anionic inhibitor, thus freeing the CPP at a target site. This methodology was used successfully in 2004 to target tumor cells by Jiang et al., who used a proteolytic cleavage site targeted by metalloproteases secreted from tumor cells (64). These results were further confirmed in two reports in 2009 by Olsen et al. and Aguilera et al. of the Tsien lab (65, 66). However, due to endosomal entrapment (66) and significant off-target uptake by other tissues in vivo (67), this technology has developed only as a means to target tumors for imaging purposes, which in and of itself has significant value.
More recent studies have utilized pH-, photo- and hydrogen peroxide-sensitive CPP linkers to target desired tissues. In 2014, Weinstain et al. described a hydrogen peroxide-sensitive linker that was targeted to lung tissue following induction of LPS-mediated inflammation in a murine model (68). Given that infection results in high levels of reactive oxygen species, the constructs would ideally only be cleaved in regions undergoing an inflammatory response (perhaps cleverly harnessing the endosomal entrapment problem in a good way, withholding cargos from off-target cell cytoplasms and targeting them for destruction!). In 2016, Yang et al. successfully delivered siRNA in nanoparticles to tumors by exploiting lower pH conditions present in those tumors (69). However, that cleavage of the linker relied on both low pH conditions and exposure to near IR-light confers substantial accessibility limitations. While these studies, and others, yield clever and promising targeting mechanisms, they still lack the degree of specificity that may be needed for widespread therapeutic application as the potential for off target uptake by healthy cells remains a significant concern.
One means of overcoming off-target effects utilizing stimulus dependent methodology is to deliver an ACPP that can only be cleaved within targeted cells, provided they contain the product needed for cleavage. This so-called “Trojan Horse” strategy was first employed by Vocero-Abkani et al. in 1999 when TAT peptide was used to deliver proCaspase 3 to HIV-infected cells (70). Here, the proteolytic site was replaced with the proteolytic site for HIV-1 protease. Upon uptake of the cargo into HIV-infected cells, Caspase 3 is processed, killing the infected cells (70). And while this methodology may have significant therapeutic potential, it is certainly limited to specific diseases and lacks broader applications.
Perhaps most promisingly, recent biopanning strategies have identified a number of short peptides that confer cell type-specific delivery of cargo proteins via CPP-mediated transduction. Zahid et al. used this approach to identify a cardiac-specific peptide, termed Cardiac Targeting Peptide (CTP), that facilitated cargo uptake specifically to cardiomyocytes following systemic introduction (71). More recently, a cancer-specific CPP was designed by Lim et al. This CPP, called BR2, showed enhanced selectivity and nearly a 70% overall increase in cellular uptake in cancer cell lines in comparison with normal cells (72).
When moving from bench to bedside, enhancing specificity and decreasing off-target effects will be paramount. Considerable attention needs to be given the nature and goal of the treatment and optimization of the CPP construct to serve these purposes. What has become increasingly clear is that a one size-fits-all model will not be useful in this endeavour. As argued above, CPPs that bind ubiquitously expressed membrane proteins will likely have little promosie without coupling to a secondary targeting molecule, such as a nanoparticle. However, with significant advances in understanding CPP-cell interactions and the biochemical nature of afflicted cells of interest, designer therapeutic CPPs are clearly on the horizon.
Outlook: Cause for optimism
CPPs hold the immense promise of rapid, efficient, nontoxic delivery of biomolecules into living cells and thus represent great hope for development of enabling technologies for delivering therapeutics now stymied by poor cellular entry. They also may also confer other advantages such as fine control of dosing as compared to transfection or other disruptive delivery method. Technical problems with CPP delivery, while significant, may soon be solved with rather simple solutions that dissociate cargo from CPP via spontaneously cleavable or non-covalent linkages, opening the door to new generations of therapeutics. Prospects for even further enhanced utility by cell-specific targeting and increased ease of coupling, etc. may make them even more profoundly effective.
Highlights.
CPPs can readily enter a variety of cells utilizing multiple forms of endocytosis and, potentially, via direct penetrance
CPP-based technologies are one of the most promising means for delivery of a wide variety of cargo (proteins, peptides, DNA, siRNAs) into living cells
Endosomal entrapment of CPP-conjugated cargo is a limiting factor for the effectiveness of CPPs for protein delivery
Current approaches to achieve endosomal escape of CPP-associated cargo are destablization of endosomes, the use of cleavable linkers, and non-covalent attachments
Biological CPPs likely enter cells via ubiqutous mechanisms, however, designer CPPs are effective at targeting specific cells and tissues
Acknowledgments
Work in the McMurry Lab is funded by NIH grant R15 GM120691. Work in the Nowak lab is funded by R15 GM102826.
Footnotes
Conflicts of interest. The authors declare no conflicts of interest.
References
- 1.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55(6):1189–93. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 2.Green M, Lowenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus Tat trans-activator protein. Cell. 1988;55:1179–88. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 3.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, et al. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci (USA) 1994;91(2):664–8. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vives E, Charneau P, Van Rietschoten J, Rochat H, Bahraoui E. Effects of the Tat basic domain on human immunodeficiency virus type 1 transactivation, using chemically synthesized Tat protein and Tat peptides. J Virol. 1994;68(5):3343–53. doi: 10.1128/jvi.68.5.3343-3353.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agrawal P, Bhalla S, Usmani SS, Singh S, CHaudhary K, Raghava GPS, et al. CPPsite 2.0: a repository of experimentally validated cell-penetrating peptides. Nucl Acids Res. 2016;44:D1098–D103. doi: 10.1093/nar/gkv1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furuhata M, Kawakami H, RToma K, Hattori Y, Matitani Y. Intracellular delivery of proteins in complexes with oligoarginine-modified liposomes and the effect of oligoarginine length. Bioconjug Chem. 2006;17(4):935–42. doi: 10.1021/bc060034h. [DOI] [PubMed] [Google Scholar]
- 7.Henriques ST, Costa J, Castanho MA. Translocation of beta-galactosidase mediated by the cell-penetrating peptide of pep- 1 into lipid vesicles and human HeLa cells is drivven by membrane electrostatic potential. Biochemistry. 2005;44(30):10189–98. doi: 10.1021/bi0502644. [DOI] [PubMed] [Google Scholar]
- 8.Montrose K, Yang Y, Sun X, Wiles S, Krissansen GW. Xentry, a new class of cell-penetrating peptide uniquely equipped for delivery of drugs. Sci Rep. 2013;3:1661. doi: 10.1038/srep01661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Presente A, Dowdy SF. PTD/CPP peptide-mediated delivery of siRNAs. Curr Pharm Design. 2013;19:2943–7. doi: 10.2174/1381612811319160008. [DOI] [PubMed] [Google Scholar]
- 10.Lundberg M, Wikström S, Johansson M. Cell surface adherence and endocytosis of protein transduction domains. Mol Ther. 2003;8(1):143–50. doi: 10.1016/s1525-0016(03)00135-7. [DOI] [PubMed] [Google Scholar]
- 11.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, et al. Cell-penetrating peptides A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–90. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 12.Glogau R, Blitzer A, Brandt F, Kane M, Monheit GD, Waugh JM. Results of a randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of a botulinum toxin type A topical gel for the treatment of moderate-to-severe lateral canthal lines. J Drugs Dermatol. 2012;11(1):38–45. [PubMed] [Google Scholar]
- 13.Lonn P, Dowdy SF. Cationic PTD/CPP-mediated macromolecular delivery: charging into the cell. Expert Opin Drug Deliv. 2015;26:1–10. doi: 10.1517/17425247.2015.1046431. [DOI] [PubMed] [Google Scholar]
- 14.Lewis WH. Pinocytosis. B Johns Hopkins Hosp. 1931;49:17–27. [Google Scholar]
- 15.Lim JP, Gleeson PA. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol. 2011;89(8):836–43. doi: 10.1038/icb.2011.20. [DOI] [PubMed] [Google Scholar]
- 16.Pearse B. Clathrin: a unique protein associated with intracellular transfer of membrane by coated vesicles. P Natl Acad Sci USA. 1976;73(4):1255–9. doi: 10.1073/pnas.73.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada E. The fine structure of the gall bladder epithelium of the mouse. J Biophys Biochem Cy. 1955;1(5):445. doi: 10.1083/jcb.1.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayor S, Parton RG, Donaldson JG. Clathrin-independent pathways of endocytosis. CSH Perspect Biol. 2014;6(6):a016758. doi: 10.1101/cshperspect.a016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8(7):848–66. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 20.Ferrari A, Pellegrini V, Arcangeli C, Fittipaldi A, Giacca M, Beltram F. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol Ther. 2003;8(2):284–94. doi: 10.1016/s1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release. 2005;102(1):247–53. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 22.Nakase I, Niwa M, Takeuchi T, Sonomura K, Kawabata N, Koike Y, et al. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10(6):1011–22. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10(3):310–5. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 24.Fittipaldi A, Ferrari A, Zoppé M, Arcangeli C, Pellegrini V, Beltram F, et al. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem. 2003;278(36):34141–9. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 25.Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell. 2004;15(5):2347–60. doi: 10.1091/mbc.E03-12-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269(14):10444–50. [PubMed] [Google Scholar]
- 27.Mann DA, Frankel AD. Endocytosis and targeting of exogenous HIV-1 Tat protein. The EMBO J. 1991;10(7):1733. doi: 10.1002/j.1460-2075.1991.tb07697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiao C-Y, Delaroche D, Burlina F, Alves ID, Chassaing G, Sagan S. Translocation and endocytosis for cell-penetrating peptide internalization. J Biol Chem. 2009;284(49):33957–65. doi: 10.1074/jbc.M109.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS. 1997;11(12):1421–31. doi: 10.1097/00002030-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 30.Rusnati M, Coltrini D, Oreste P, Zoppetti G, Albini A, Noonan D, et al. Interaction of HIV-1 Tat protein with heparin. Role of the backbone structure, sulfation, and size. J Biol Chem. 1997;272(17):11313–20. doi: 10.1074/jbc.272.17.11313. [DOI] [PubMed] [Google Scholar]
- 31.Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. 2001;276(5):3254–61. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- 32.Vogel BE, Lee S-J, Hildebrand A, Craig W, Pierschbacher MD, Wong-Staal F, et al. A novel integrin specificity exemplified by binding of the alpha v beta 5 integrin to the basic domain of the HIV Tat protein and vitronectin. J Cell Biol. 1993;121(2):461–8. doi: 10.1083/jcb.121.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, et al. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. P Natl Acad Sci (USA) 2000;97(21):11466–71. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghezzi S, Noonan DM, Aluigi MG, Vallanti G, Cota M, Benelli R, et al. Inhibition of CXCR4-dependent HIV-1 infection by extracellular HIV-1 Tat. Biochem Biophys Res Comm. 2000;270(3):992–6. doi: 10.1006/bbrc.2000.2523. [DOI] [PubMed] [Google Scholar]
- 35.Kawaguchi Y, Takeuchi T, Kuwata K, Chiba J, Hatanaka Y, Nakase I, et al. Syndecan-4 Is a Receptor for Clathrin-Mediated Endocytosis of Arginine-Rich Cell-Penetrating Peptides. Bioconjugate Chem. 2016;27(4):1119–30. doi: 10.1021/acs.bioconjchem.6b00082. [DOI] [PubMed] [Google Scholar]
- 36.Kosuge M, Takeuchi T, Nakase I, Jones AT, Futaki S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjugate Chem. 2008;19(3):656–64. doi: 10.1021/bc700289w. [DOI] [PubMed] [Google Scholar]
- 37.Letoha T, Keller-Pintér A, Kusz E, Kolozsi C, Bozsó Z, Tóth G, et al. Cell-penetrating peptide exploited syndecans. BBA-Biomembranes. 2010;1798(12):2258–65. doi: 10.1016/j.bbamem.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 38.Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280(15):15300–6. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- 39.Console S, Marty C, García-Echeverría C, Schwendener R, Ballmer-Hofer K. Antennapedia and HIV TAT ‘protein transduction domains’ promote endocytosis of high Mr cargo upon binding to cell surface glycosaminoglycans. J Biol Chem. 2003 doi: 10.1074/jbc.M301726200. [DOI] [PubMed] [Google Scholar]
- 40.Rusnati M, Tulipano G, Urbinati C, Tanghetti E, Giuliani R, Giacca M, et al. The basic domain in HIV-1 Tat protein as a target for polysulfonated heparin-mimicking extracellular Tat antagonists. J Biol Chem. 1998;273(26):16027–37. doi: 10.1074/jbc.273.26.16027. [DOI] [PubMed] [Google Scholar]
- 41.Brooks R, Williamson RC, Bass MD. Syndecan-4 independently regulates multiple small GTPases to promote fibroblast migration during wound healing. Small GTPases. 2012;3(2):73–9. doi: 10.4161/sgtp.19301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christianson HC, Belting M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014;35:51–5. doi: 10.1016/j.matbio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka G, Nakase I, Fukuda Y, Masuda R, Oishi S, Shimura K, et al. CXCR4 stimulates macropinocytosis: implications for cellular uptake of arginine-rich cell-penetrating peptides and HIV. Chem Biol. 2012;19(11):1437–46. doi: 10.1016/j.chembiol.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 44.Hamon M, Mbemba E, Charnaux N, Slimani H, Brule S, Saffar L, et al. A syndecan-4/CXCR4 complex expressed on human primary lymphocytes and macrophages and HeLa cell line binds the CXC chemokine stromal cell–derived factor-1 (SDF-1) Glycobiology. 2004;14(4):311–23. doi: 10.1093/glycob/cwh038. [DOI] [PubMed] [Google Scholar]
- 45.Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18(7):385–93. doi: 10.1016/j.molmed.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 46.Erazo-Oliveras A, Najjar K, Dayani L, Wang TY, Johnson GA, Pellois JP. Protein delivery into live cells by incubation with an endosomolytic agent. Nat Methods. 2014;11(8):861–7. doi: 10.1038/nmeth.2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Najjar K, Erazo-Oliveras A, Pellois J. Delivery of proteins, peptides or cell-impermeable small molecules into live cells by incubation with the endosomolytic reagent dfTAT. J Vis Exp. 2015;103 doi: 10.3791/53175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baccile JA, Morrell MA, Falotico RM, Milliken BT, Drew DL, Rossi FM. Modular synthesis of photocleavable peptides using click chemistry. Tetrahedron Lett. 2012;53(15):1933–5. [Google Scholar]
- 49.Salerno JC, Ngwa VM, Nowak SJ, Chrestensen CA, Healey AN, McMurry JL. Novel cell penetrating peptides effect intracellular delivery and endosomal escape of desired protein cargos. J Cell Sci. 2016;129:893–7. doi: 10.1242/jcs.182113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerasimenko JV, Tepikin AV, Petersen OH, Gerasimenko OV. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr Biol. 1998;8(24):1335–8. doi: 10.1016/s0960-9822(07)00565-9. [DOI] [PubMed] [Google Scholar]
- 51.Rapoport M, Saada A, Elpeleg O, Lorberboum-Galski H. TAT-mediated delivery of LAD restores pyruvate dehydrogenase complex activity in the mitochondria of patients with LAD deficiency. Mol Ther. 2008;16(4):691–7. doi: 10.1038/mt.2008.4. [DOI] [PubMed] [Google Scholar]
- 52.Marcus D, Lichtenstein M, Saada A, Lorberboum-Galski H. Replacement of the C6ORF66 assembly factor (NDUFAF4) restores complex I activity in patient cells. Mol Med. 2013;19:124–34. doi: 10.2119/molmed.2012.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Honda F, Hane Y, Toma T, Yachie A, Kim E-S, Lee S-K, et al. Transducible form of p47 phox and p67 phox compensate for defective NADPH oxidase activity in neutrophils of patients with chronic granulomatous disease. Biochem Biophys Res Comm. 2012;417(1):162–8. doi: 10.1016/j.bbrc.2011.11.077. [DOI] [PubMed] [Google Scholar]
- 54.Cerrato CP, Künnapuu K, Langel Ü. Cell penetrating peptides with intracellular organelle targeting. Expert Opin Drug Del. 2017;14(2):245–55. doi: 10.1080/17425247.2016.1213237. [DOI] [PubMed] [Google Scholar]
- 55.Ngwa VM, Axford DS, Healey AN, Nowak SJ, Chrestensen CA, McMurry JL. A versatile cell penetrating peptide-adaptor system for efficient delivery of molecular cargos to subcellular destinations. PLoS One. 2017;12(5):e0178648. doi: 10.1371/journal.pone.0178648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang H-Y, Chen J-X, Sun Y-X, Deng J-Z, Li C, Zhang X-Z, et al. Construction of cell penetrating peptide vectors with N-terminal stearylated nuclear localization signal for targeted delivery of DNA into the cell nuclei. J Control Rel. 2011;155(1):26–33. doi: 10.1016/j.jconrel.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 57.Herce HD, Garcia AE, Cardoso MC. Fundamental molecular mechanism for the cellular uptake of guanidinium-rich molecules. JACS. 2014;136(50):17459–67. [Google Scholar]
- 58.Dekiwadia CD, Lawrie AC, Fecondo JV. Peptide-mediated cell penetration and targeted delivery of gold nanoparticles into lysosomes. J Pep Sci. 2012;18(8):527–34. doi: 10.1002/psc.2430. [DOI] [PubMed] [Google Scholar]
- 59.Cerrato CP, Pirisinu M, Vlachos EN, Langel Ü. Novel cell-penetrating peptide targeting mitochondria. FASEB J. 2015;29(11):4589–99. doi: 10.1096/fj.14-269225. [DOI] [PubMed] [Google Scholar]
- 60.Marcus D, Lichtenstein M, Cohen N, Hadad R, Erlich-Hadad T, Greif H, et al. Heterologous mitochondrial targeting sequences can deliver functional proteins into mitochondria. Int J Biochem Cell B. 2016;81:48–56. doi: 10.1016/j.biocel.2016.10.013. [DOI] [PubMed] [Google Scholar]
- 61.Zhang J, Sun A, Xu R, Tao X, Dong Y, Lv X, et al. Cell-penetrating and endoplasmic reticulum-locating TAT-IL-24-KDEL fusion protein induces tumor apoptosis. J Cell Phys. 2016;231(1):84–93. doi: 10.1002/jcp.25054. [DOI] [PubMed] [Google Scholar]
- 62.Vivès E, Schmidt J, Pèlegrin A. Cell-penetrating and cell-targeting peptides in drug delivery. BBA-Rev Cancer. 2008;1786(2):126–38. doi: 10.1016/j.bbcan.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 63.MacEwan SR, Chilkoti A. Harnessing the power of cell-penetrating peptides: activatable carriers for targeting systemic delivery of cancer therapeutics and imaging agents. Wires Nanomed Nanobi. 2013;5(1):31–48. doi: 10.1002/wnan.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell penetrating peptides. P Natl Acad Sci (USA) 2004;101(51):17867–72. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aguilera TA, Olson ES, Timmers MM, Jiang T, Tsien RY. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr Biol. 2009;1(5–6):371–81. doi: 10.1039/b904878b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olson ES, Aguilera TA, Jiang T, Ellies LG, Nguyen QT, Wong EH, et al. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr Biol. 2009;1(5–6):382–93. doi: 10.1039/b904890a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Duijnhoven SM, Robillard MS, Nicolay K, Grüll H. Tumor targeting of MMP 2/9 activatable cell-penetrating imaging probes is caused by tumor-independent activation. J Nucl Med. 2011;52(2):279–86. doi: 10.2967/jnumed.110.082503. [DOI] [PubMed] [Google Scholar]
- 68.Weinstain R, Savariar EN, Felsen CN, Tsien RY. In vivo targeting of hydrogen peroxide by activatable cell-penetrating peptides. JACS. 2014;136(3):874–7. doi: 10.1021/ja411547j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Y, Xie X, Yang Y, Li Z, Yu F, Gong W, et al. Polymer nanoparticles modified with photo-and pH-dual-responsive polypeptides for enhanced and targeted cancer therapy. Mol Pharm. 2016;13(5):1508–19. doi: 10.1021/acs.molpharmaceut.5b00977. [DOI] [PubMed] [Google Scholar]
- 70.Vocero-Akbani AM, Vander Heyden N, Lissy NA, Ratner L, Dowdy SF. Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat Med. 1999;5(1):29–33. doi: 10.1038/4710. [DOI] [PubMed] [Google Scholar]
- 71.Zahid M, Phillips BE, Albers SM, Giannoukakis N, Watkins SC, Robbins PD. Identification of a cardiac specific protein transduction domain by in vivo biopanning using a M13 phage peptide display library in mice. PLoS One. 2010;5(8):e12252. doi: 10.1371/journal.pone.0012252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lim KJ, Sung BH, Shin JR, Lee YW, Yang KS, Kim SC. A cancer specific cell-penetrating peptide, BR2, for the efficient delivery of an scFv into cancer cells. PLoS One. 2013;8(6):e66084. doi: 10.1371/journal.pone.0066084. [DOI] [PMC free article] [PubMed] [Google Scholar]