

Integrating comparative genomics and network science delineates the dynamics of retroelement activity in the genome
Abstract
Retroelements (REs) are mobile DNA sequences that multiply and spread throughout genomes by a copy-and-paste mechanism. These parasitic elements are active in diverse genomes, from yeast to humans, where they promote diversity, cause disease, and accelerate evolution. Because of their high copy number and sequence similarity, studying their activity and tracking their proliferation dynamics is a challenge. It is particularly difficult to pinpoint the few REs in a genome that are still active in the haystack of degenerate and suppressed elements. We develop a computational framework based on network theory that tracks the path of RE proliferation throughout evolution. We analyze SVA (SINE-VNTR-Alu), the youngest RE family in human genomes, to understand RE dynamics across hominids. Integrating comparative genomics and network tools enables us to track the course of SVA proliferation, identify yet unknown active communities, and detect tentative “master REs” that played key roles in SVA propagation, providing strong support for the fundamental “master gene” model of RE proliferation. The method is generic and thus can be applied to REs of any of the thousands of available genomes to identify active RE communities and master REs that were pivotal in the evolution of their host genomes.
INTRODUCTION
Retroelements (REs) are transposable elements that replicate and create new insertions throughout the genome. Using a “copy-and-paste” mechanism (1–3), these genomic parasites have eminently proliferated and spread throughout evolution and now constitute >40% of the DNA in the human genome (4–6). They generate genomic diversity in the human population (7–9), facilitate evolution (10), and cause disease by harmful de novo insertions in functional genomic regions (11). Thus, characterizing RE subfamilies and understanding their proliferation dynamics can illuminate the processes that forged the genome during evolution and persistently threaten its integrity (10, 12). Contemporarily, there are three families that contain active REs in the human genome (13, 14): LINE-1 (long interspersed nuclear element 1), Alu, and SVA (SINE-VNTR-Alu composite RE; Fig. 1A), which make up 17, 11, and 0.2% of the human genome, respectively (15). Only a small fraction of RE sequences within these families, belonging to a handful of subfamilies, can still proliferate (11, 13–15). Here, we focused on SVA, the youngest and least-studied active RE family in humans (16, 17). SVA emerged in the common ancestor of apes (hominoids) but successfully proliferated only in great apes or “hominids,” comprising human, chimpanzee, gorilla, and orangutan. The expansion dynamics of SVA throughout evolution has not yet been studied across hominids, and most importantly, the specific subset of active SVA subfamilies and elements that currently cause disease and drive their expansion across hominid genomes is still unknown.
Fig. 1. Network approach detects SVA communities with high precision.
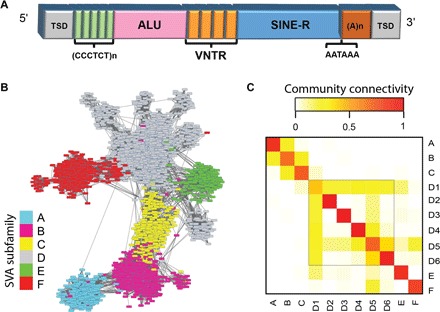
(A) Prototypical structure of an SVA element. In addition to the major SINE-R and ALU regions, repetitive VNTR and CCCTCT hexamer regions are presented. The SVA element ends with a characteristic polyadenylate tail and flanking target site duplications (TSDs), which form upon insertion of REs to the genome. The ~500-bp-long SINE-R region is used here to calculate SVA similarities, and the shorter ALU region is used for validation. (B) Similarity network of human SVA REs. The nodes represent REs, and the links are weighted by the strength of similarity between their SINE regions. Communities detected coincide with the commonly used SVA subfamily annotation assigned by RepeatMasker, which was used for node color-coding (Materials and Methods). (C) Intra- and intercommunity similarity. Calculated by the ratio of the number of present links to all possible links between communities (Materials and Methods). Red corresponds to strong linkage, which is the strongest along the diagonal, implying high-confidence separation between communities and, specifically, supporting our findings of the division of SVA_D to six communities.
Network science has been widely used to study the topology and community structure of real-world networks (18, 19). These analyses have provided new insights in sociology (20, 21), epidemiology (22), robustness (23), traffic (24), climate (25, 26), and neuroscience (27), among many other areas (28). Community structure analysis has been used for phylogenetic inference of biological sequences and, for protein sequences, has been shown to perform comparably to commonly used phylogenetic methods (29). Given that REs replicate in a manner where one element spawns the next, we advocate that networks are a natural scheme for representing RE relations and that network science embraces a large and yet unexploited potential for delineating RE activity and proliferation dynamics. To address these challenging issues, we developed a computational framework that combines complex network theory and computational genomics. We use hominid SVA for proof of concept, but the framework is modular and generic and hence can be applied to RE families of any genome.
The first step toward understanding RE activity is to identify the RE subfamilies. A commonly used method for subfamily identification, CoSeg (30), is based on multiple sequence alignments (MSAs) of a cohort of REs being analyzed. A recent method, based on MSAs as well, goes on to infer the ancestry relations within a family of REs using Bayesian statistics, from which it constructs an RE ancestry network (31). Here, we avoid using MSAs, which have some inherent limitations, such as being unable to properly align sets of sequences that have low global similarity (32), and use RE similarity networks that are constructed from RE pairwise similarity scores. The network approach, in combination with comparative genomics, enables us to study RE community structure, delineate evolutionary relations between communities, and, most importantly, identify active communities and REs that are responsible for RE proliferation in ancient and recent evolution.
RESULTS
SVA similarity networks identify SVA subfamily structure
To test the potential of network analysis to identify RE subfamilies or “communities” and describe their evolutionary dynamics, we constructed networks of SVA elements for four hominid reference genomes (human, chimpanzee, gorilla, and orangutan; see Materials and Methods). The networks were constructed from 2638 SVA elements in human, 2523 in chimp, 2333 in gorilla, and 1695 in orangutan (Materials and Methods and data set S1 of data file S1). To create similarity networks between SVAs, we produced similarity scores between each pair of elements. Using BLAST (basic local alignment search tool) (33), we aligned each pair of assembled SVA sequences within a genome and used the “bitscore” produced by BLAST as the similarity measure. The bitscore takes into account the amount of matches, mismatches, and insertions/deletions in the sequence alignment; therefore, it is a comprehensive measurement of the similarity of pairs of REs. The repetitiveness and variability in the VNTR (variable number tandem repeats) region hinder the ability to use it for accurate alignments, and VNTR alignment bitscores are biased by alignment length. Therefore, we focused on the SINE and Alu regions of SVA, which have nearly constant lengths of ~500 and ~360 base pairs (bp), respectively, creating separate networks for each (Materials and Methods). Comparing between the two types of networks will help validate our approach. Hence, links in the RE networks are weighted, where the nodes are the SVAs and the weights are BLAST alignment bitscores between pairs of REs. The weight values vary widely throughout the network and range from 300 to 900 and 200 to 500 for the SINE- and ALU-based networks, respectively. The strengths of matching links in the SINE and ALU networks are highly correlated (fig. S1). Notably, not all possible links were created, due to insignificant BLAST alignments between many pairs of REs in the network. In the human SINE network, for example, 510,045 links were created, which are ~15% of the 3,478,293 possible links.
The first step toward understanding RE proliferation dynamics is to identify RE subfamilies, which are sets of REs with highly similar genetic sequences. Within the networks, REs of the same subfamily are expected to be more densely connected than REs from different subfamilies in terms of number and strength of links. In network terminology, these can be viewed as communities, which are subsets of nodes that are more densely linked among themselves than they are to the rest of the network. The RE replication mechanism is such that a new RE is identical to its source element upon insertion. Over time, the new insertion accumulates mutations, as do all genomic sequences, which diverge it from its progenitor. Thus, young REs will be strongly linked to their sources and close “relatives,” and young communities will be more tightly linked than older ones (fig. S2). The community detection algorithm that we used, extremal optimization (EO) (34), is sensitive to the number of links, their strength, and topology. To better identify the communities and understand the network structure, we removed the weakest links of each node and retained only a fraction of the strongest links, denoted as Pr (Materials and Methods). For the RE networks, this method of disconnecting links is better than using a threshold, because it does not disconnect whole groups of more ancient REs from the network. After disconnecting the links, we applied EO and bootstrap algorithms for community detection (Materials and Methods). The bootstrap algorithm requires a predefined θ parameter, which denotes the fraction of runs that two nodes must be in the same community to be considered members of the same community. Increasing θ and decreasing Pr values increases the number of communities and, therefore, produces a finer resolution of community structure. Here, we used Pr = 0.25 and θ = 0.8, but consistent results were obtained for a broad range of different values (note S1). Moreover, we applied an additional community detection method (35), and the communities were highly consistent with the EO results for a variety of parameters (see note S2 and fig. S3). The bootstrapping of EO also identifies a small fraction of “unstable” nodes in the network (~1.8% in the human network; table S1), which lie on the border between two communities and hence resemble transitional states between communities (fig. S4).
To validate the communities identified, we compared our community assignment to subfamilies identified by RepeatMasker, the most widely used program for RE classification (36). We labeled each community based on the RepeatMasker SVA repeat annotation, which classifies SVA into six subfamilies (SVA_A to SVA_F), by using a majority rule for each community. Our network-based classification almost completely overlapped that of RepeatMasker, with 97.61% precision in human (Fig. 1B) and 98.99% in chimp (see Materials and Methods and note S3). This concordance allowed us to rely on the network’s further division of SVA_D, the largest SVA family in human, into six distinct subfamilies (SVA_D1 to SVA_D6; data set S2 of data file S1). These six subfamilies were also identified by a complementary community structure analysis using the Bornholdt method (note S4 and fig. S3). SVA_D1 is the largest subfamily (809 elements), and the others are much smaller (39 to 84 elements). The six SVA_D subfamilies are separated even more easily than SVA_B from SVA_C (table S2), which implies that the former (SVA_D) is more diverged than the latter (SVA_B and SVA_C). Intra- and intercommunity connectivity scores are used to determine the quality of community separation. We computed connectivity scores, and reassuringly, community intraconnectivity is found to be much larger than interconnectivity (see Materials and Methods and Fig. 1C). These scores can also be used to infer the evolutionary relations between communities, as discussed in the next section.
We built the human SVA similarity network based on the SINE regions. Our network approach is supported by the fact that there is a high agreement between the SINE-based communities and those identified by the ALU-based networks (fig. S5). Intersecting the two classifications enable us to further identify SVA subcommunities. SVA_F, for example, split into two subfamilies, separating the MAST2 transduction group (a set of SVAs that contains a segment of the MAST2 gene) (37, 38), SVA_F1, from the major SVA_F subfamily, which shows that our network approach can assist in identifying subsets of REs with distinct biological features.
Next, we wish to support the communities identified in the network by determining that the SVA communities represent groups of distinct sequences. To that end, we generated consensus sequences of the SINE regions for each community and constructed a neighbor-joining phylogenetic tree of them (fig. S6 and data set S5 of data file S1). The archetypical consensus sequences were distinct for each of the communities, including the six newly identified communities in SVA_D.
Comparison between hominid network structures delineates RE proliferation dynamics
The SVA family has been active in the lineage leading to humans for millions of years, since the common ancestor of hominids. Therefore, a genomic SVA network should contain ancient and young elements, with insertion times spanning from the hominid’s common ancestor to young species-specific ones. To compare SVA expansion in the different hominids and identify unique expansions in each genome, we construct SVA networks for each hominid separately and use comparative genomics to identify which communities are shared and which are species-specific (see Materials and Methods; Fig. 2, A to D; and data sets S2 and S3 of data file S1). The evolutionary relations between the hominids are such that orangutan branched off first from the common ancestor leading to the human lineage, followed by gorilla, and lastly chimp. As expected, the overall community structure and the number of shared or orthologous (existing in the same genomic location in multiple organisms due to insertion in a common ancestor) elements between hominids follow these phylogenetic relations. In addition to the shared ancient communities, the network analysis also revealed new species-specific communities of REs in each hominid, representing unique evolutionary expansions (Fig. 2, A to D). For nomenclature, we named these new communities in each hominid with prefixes denoting the A to F subfamily they belong to and enumerated the subfamilies by the communities’ age—higher numbers for younger communities (by activity times defined below). For species-specific communities in nonhuman primates, we added suffixes of acronyms resembling their genus and species (Pt for chimp, Gg for gorilla, and Pa for orangutan).
Fig. 2. SVA networks in hominids illustrate differential community structure and proliferation dynamics.
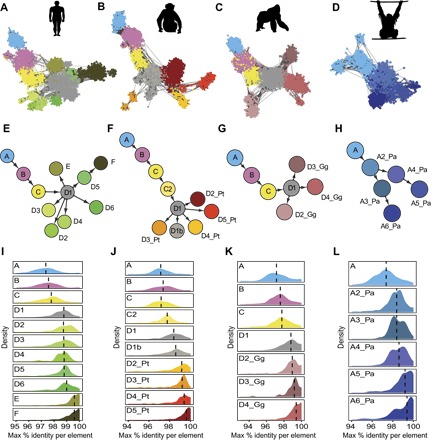
(A to D) SINE-based networks of SVA elements for each hominid. Common colors are assigned to communities A, B, C, and D1 corresponding to colors in (E) to (H). Unique colors are given to the remainder, which are primarily species-specific expansions. (E to H) Community-level directed networks describing SVA expansion in each hominid network presented in (A) to (D), respectively. (I to L) Density plots describing relative community activity periods for each community based on evolutionary distances between the REs within each community (Materials and Methods). The higher is the identity within a community, the younger is the community, indicating recent activity.
RE networks have a special property of “network expansion,” in which existing nodes in the network spawn new ones. This phenomenon can be viewed at two resolutions—either the community or element levels. Being initially interested in the macro level of SVA evolution, we constructed community-level networks for each hominid. To weigh the links in these networks, we devised an intercommunity similarity metric that represents the connectivity between each pair of communities (Materials and Methods). Then, applying Edmond’s algorithm (39) to each of these undirected networks, we calculated the optimal branching or “maximal spanning tree” describing the order of community spawning in each (Fig. 2, E to H). This algorithm requires defining a root community, which was set to the most ancient community in each network, systematically and unequivocally identified by using the comparative genomics data and intracommunity similarity rates described below (see Materials and Methods and Fig. 2, I to L).
The directed network structure revealed by Edmond’s algorithm illuminates different modes of community spawning throughout SVA evolution. In some cases, network expansion is linear, as seen for communities SVA_A to SVA_D. In other cases, there is an individual community that spawns multiple new communities, as exemplified by SVA_D1 in the human, chimp, and gorilla networks (Fig. 2, E to G). The general SVA network topology and these two modes of expansion infer different possible roles for communities in the network: (i) founder communities, which are the ancestral root community of an entire network (that is, SVA_A); (ii) transitional communities, which gave rise to a single community and have ceased to be active; (iii) spawning communities, which spawned multiple child communities; and (iv) terminal communities, which are current leaves in the directed network. These can be further classified as “dead-end” communities that were active in the past, or “active” communities that are still active. In the human network, D2, D3, and D4 are the dead-end communities, whereas E and F are the active communities (based on the method described in the next section).
Communities represent bursts of RE proliferation and highlight recent activity
There is an ongoing arms race between the genome and its resident REs. REs invade or emerge in a genome, and the latter must find the means for their restriction to maintain its integrity and proper function. In many cases, REs subsequently evolve to evade the host’s restriction, resulting in a new era of uncontrolled activity (40). This interplay causes REs to proliferate via intermittent bursts of activity. As is typical for active RE families, most SVAs in the genome are inactive due to truncation upon initial insertion or post-insertion degeneration, and only a limited set of source elements continues to be capable of mobilization. In essence, each community in the RE networks resembles a burst of activity of an individual or set of similar active source elements. The subset of REs that are still active are the ones that continue to create diversity and can cause disease in their host’s population. Therefore, we were most interested in developing a method that will infer the activity periods of each RE community and hence highlight the communities containing contemporarily active elements.
To this end, we devised a method based on intracommunity similarities that approximates the relative activity periods of each community. REs are identical to their progenitors or source elements upon insertion and accumulate random mutations that cause them to gradually diverge. Thus, the intracommunity sequence identity can be a proxy for activity time. Specifically, we calculated the percentage of sequence identity between each RE to its closest relative (by maximal bitscore) and used the median of these values per community to approximate the activity periods (Fig. 2, I to L). Although these are not exact estimations, they are sufficient to understand the relative activity periods of different communities. Reassuringly, this method accords with the known evolutionary relations of human SVA (fig. S6) (16) and an independent analysis obtained from comparative genomics data (Pearson’s r = 0.924 between the identity percent medians and the percentage of ancient “shared” elements per community). With regard to contemporary activity potential, the communities with the highest identity scores are the most likely to still be active today, especially if they have many pairs of REs with 100% identity. In agreement with previous knowledge, this method predicts that SVA_E and SVA_F are active in humans (Fig. 2O). We also identified a novel community within SVA_D, D6, which, with high probability, was active in recent human evolution (see additional support in the next section). For the other hominids, this approach reveals unique communities that are currently most probably active in each genome. To our knowledge, this is the first description of SVA subfamily activity capacities in nonhuman hominids (NHHs). Each NHH had two to four active communities: in chimpanzee, D4_Pt, D5_Pt, and possibly D2_Pt; in gorilla, all four SVA_D subfamilies (D1 to D4_Gg); and in orangutan, A5_Pa, A6_Pa, and possibly, albeit to a much lower extent, A2_Pa to A4_Pa as well. These species-specific communities contain the active elements driving diversity and disease in their respective hosts’ population.
A possible way to evaluate the extent of recent activity of a community is to search for sets of REs within a community that are 100% identical to each other and hence form fully connected cliques (Materials and Methods). By searching for cliques in the SVA network, we found that SVA_F has 6 large cliques, with a maximum clique size of 33; SVA_E has 5 cliques, with a maximum size of 5; and D1 also has a single small clique (fig. S7). These cliques most probably contain the most recently active REs in their respective communities.
RNA and DNA sequencing data support network-based community activity findings
To corroborate and further understand the activity capacities of the communities identified in the human genome, we analyzed comprehensive data sets of human RNA and DNA sequencing (Fig. 3). The subfamily expansion depicted by our genome-based networks reflects SVA proliferation specifically in the germ line or early embryonic development, which can result in heritable genomic insertions. RNA expression of an RE is a prerequisite for its ability to create new insertions. Therefore, we analyzed SVA RNA expression throughout early (preimplantation) human embryonic development (Materials and Methods), which is an ideal setting for creating new heritable insertions. A previous study (41) that analyzed this data showed that SVA expression strikingly increases at the eight-cell stage and further rises at the following morula stage. We wished to further examine the expression of each SVA community in the various developmental stages (Fig. 3, A and B). As expected, we find that SVA_E and SVA_F have elevated rates of RNA expression, but surprisingly, the greatest contributor to SVA expression is SVA_D1, identified by our community method as the founding community of SVA_D (Fig. 2E), despite being a relatively old and presumably inactive community. To understand this conundrum, we further divided SVA_D1 into subcommunities (using a higher θ = 0.9 in the EO algorithm; Materials and Methods) and identified four distinct communities in SVA_D1, with differential expression rates that are in general agreement with their estimated activity times (fig. S8). The youngest one, SVA_D1d, is responsible for 50% of SVA_D1’s expression and is more enriched in expression than SVA_E (but less than SVA_F) when normalized by its genomic element count. This subcommunity is also the largest, hence the most successful, within SVA_D1 (table S3).
Fig. 3. Network predictions of SVA activity are supported and refined by both RNA and DNA sequencing data.
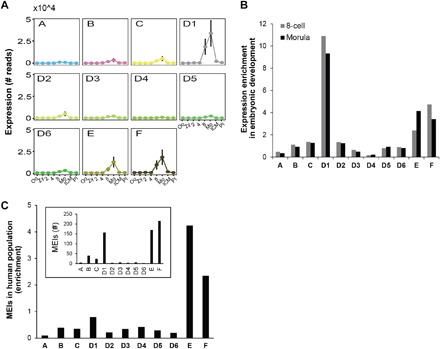
(A) SVA expression throughout early embryonic development derived from single-cell RNA sequencing (RNA-seq) data. Stages of development are as follows: oocyte (Oo), Zygote (Zy), two-cell (2), four-cell (4), eight-cell (8), morula (Mo), inner cell mass (ICM), and post-implantation (PI). SVA expression drastically rises at the eight-cell phase and peaks at the morula stage. (B) Enrichment of expression per human SVA community at peak expression stages. Enrichment was given by the number of RNA reads divided by the community size. (C) Enrichment of mobile element insertions (MEIs) generated by each human SVA community based on whole-genome sequencing (WGS) of 2422 individuals from the global human population. The inset contains the bulk number of MEIs per community, showing that D1, in addition to E and F, is responsible for a large fraction of recent SVA expansion.
Although RNA expression is necessary for RE mobilization, direct evidence for RE replication in the germ line can be best derived from MEIs in genomic sequencing data. Therefore, we analyzed WGS data of 2422 individuals from the 1000 Genomes Project (Materials and Methods) (9). Sudmant et al. (9) identified 818 SVA insertions in this genomic data that are not present in the human reference genome. These are annotated as “SVA,” and we were interested in further characterizing the source communities of these MEIs. As previously observed (14, 16), SVA_E and SVA_F were the most active communities, followed by SVA_D1, which was responsible for the greatest amount of MEIs among SVA_D subcommunities (Fig. 3C). Appealingly, further scrutinizing the four subcommunities within SVA_D1 discussed above shows that the same subcommunity, SVA_D1d, which we identified to be the youngest and the most expressed in embryonic development, is also the main contributor to MEIs in the human population (fig. S9). This cross-validation by multiple types of real biological data supports the ability of network analysis to identify active RE communities.
All-hominid network reveals key elements in SVA proliferation
The above network analysis reveals the expansion and activity of communities in each species. To compare and link between these communities, we applied the same methods used for community and ancestry detection to the SVA sequences of all four hominids merged together (Materials and Methods). By characterizing the communities in this all-hominid network and integrating the comparative genomics data (Materials and Methods), we can differentiate between communities that emerged in a common ancestor of two or more species (shared) and species-specific ones. The comparative genomics data also enable us to locate the root community of all SVA, which we use to infer and visualize the ancestry tree of SVA expansion throughout hominid evolution (Fig. 4). Our method infers tree structure primarily from sequence similarity. To see whether it corresponds to biological community properties that should gradually change with age, we compared several properties in parent and child community pairs in the tree (Materials and Methods). As hypothesized, we find that communities closer to the root are more ancient than their descendants by two criteria: (i) they are richer in shared elements and (ii) they have lower intracommunity similarity (Wilcoxon paired signed-rank test; P = 1.73 × 10−4 and 0.088, respectively).
Fig. 4. Integrated network from multiple hominids maps SVA expansion and uncovers probable source elements.
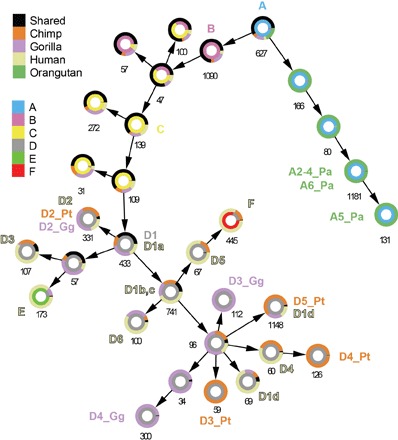
The Circos network demonstrates an approach to integrating RE data from multiple related organisms, describing the common and distinct evolutionary expansions of SVA in hominids. Each circle represents a community in the merged network (Materials and Methods), labeled with its respective size. The directional links between the communities represent the expansion of SVA throughout evolution, stemming from the ancestral SVA_A community, which emerged in the common ancestor of hominids. The inner ring is colored according to the subfamily annotation of the community (bottom legend), and the outer ring portrays the fraction of elements present in multiple species (shared) and the fraction of species-specific elements in each hominid (top legend). The shared elements in young communities are the most ancient in their communities and hence are the most probable active elements that founded them.
Next, we mapped the communities identified in the per-species network to the all-species one. As expected, the youngest communities identified in the per-hominid networks mapped to distal leaves in the tree. Intriguingly, these networks showed that communities such as human SVA_E and SVA_F, which were thought to be human-specific (16), contained elements from the chimp and gorilla genomes, too. This could occur because of two possible scenarios: either by the initial proliferation of an element that was present in a common ancestor or due to convergent evolution, where REs in multiple genomes independently underwent the same mutations to take the same form. Because we found multiple shared elements in both communities, the first explanation seems most likely. These shared elements most probably contain the founders of their entire respective communities.
Further analysis showed that human SVA_D2, an ancient and inactive family in human genomes, resides in the same all-hominid network community as communities that independently expanded in chimp and gorilla genomes (D2_Pt and D2_Gg) (Fig. 2, F and G). Moreover, the communities in these genomes expanded more successfully (33 and 100% more, respectively) than in human and are probably still active in the gorilla genome today (Fig. 2K). Another notable finding is that the four subcommunities of human D1 mapped to the all-hominid network in the expected order (Fig. 4). Once again, SVA_D1d is portrayed as a young community and is now found to represent a mostly human-specific expansion (table S3).
In addition to identifying tentative founders of young communities, we use the networks to identify possible “master SVAs” that played seminal roles in the initial proliferation of SVA in hominids. The linear evolution of SVA subfamilies A to D (Fig. 2, A to C) suggests that there could be a single “master RE” that was active over a long period in evolution, spawning subfamilies A to D subsequently, as it accumulated mutations over time. To identify such an element, we search for a full-length element in the human genome that has an ortholog (an SVA in the same genomic location) in the orangutan genome. We are specifically interested in elements that belong to SVA_D or an even younger subfamily in human and to any of the SVA_A-like subfamilies in orangutan. In the network, these would be orthologs that are present in different communities in the all-hominid SVA network (Fig. 5A). Rigorous computational and manual analyses reveal only two pairs of candidate elements that meet these criteria (note S5). Both elements reside within introns of genes (in reverse orientation): one in CABIN1 (calcineurin binding protein 1) and the other in NPLOC4 (nuclear protein localization protein 4 homolog). These elements respectively belong to SVA_A2_Pa and SVA_A in orangutan and to SVA_D1a in human. The locations of these SVAs are identical in the hominid genomes, as evinced by the similarity of their flanking genomic sequences and TSDs (that is, the short genomic sequence duplication that occurs during RE insertion; Fig. 1A and note S5). Because TSD lengths vary between insertion events, the identity of TSD length implies that these SVA elements are bona fide orthologs that resulted from a single insertion event in the hominid common ancestor (note S5). Put together with the unequivocal assignment of these orthologous SVA pairs to distinct and distant communities (Fig. 5B and figs. S9 to S11), one of these elements is the most probable master SVA behind the formation of subfamilies B, C, and D in the common ancestor of hominins (that is, hominids excluding orangutan).
Fig. 5. Networks, together with comparative genomics, pinpoint a tentative master SVA element in the CABIN1 gene.
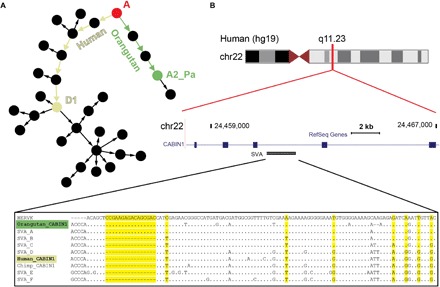
SVAs found in the exact same location in the human and orangutan genomes are most likely ancient SVAs that were inserted in the genome of the hominid’s common ancestor. One of the only examples for such an element is located in the CABIN1 gene. These ancient SVAs are expected to belong to the most ancient SVA subfamily, SVA_A. However, the “CABIN1 element” contains diagnostic mutations, unequivocally mapping it to distinct younger communities in the human and orangutan genomes. The most feasible explanation for this discrepancy is that this element was an active “master SVA” in the common ancestor of hominids that accumulated distinct mutations in the hominin and orangutan lineages after the latter’s speciation. This master element retained activity and passed on its new lineage-specific mutations to its offspring in each genome, fueling the formation of the communities found in the linear evolutionary path from SVA_A to the respective communities they currently map to in each genome. (A) Schematic of the all-hominid community network (as in Fig. 4), depicting the path of molecular evolution the SVA element in the CABIN1 gene underwent from SVA_A (red root node) to the SVA_D1a community in human (off-white node) and, in parallel, to the SVA_A2_Pa community in orangutan (green node). The accumulation of mutations necessary to transform from SVA_A to SVA_D in this ancient element suggests that it may be the master element that spawned SVA communities B to D. (B) Genomic location of the CABIN1 SVA element. The MSA compares the SINE regions of this element’s sequences from hominid genomes to the ancestral HERVK sequence, from which the SVA SINE region was derived, and to the SVA consensus sequences. The MSA, and especially the 16-bp deletion, supports the differential community classification of the orangutan element to SVA_A and of the other hominid genomes to SVA_D (yellow columns). The first 150 columns of the full alignment (fig. S9) are shown. Dots represent matches and hyphens represent deletions with respect to the HERVK sequence.
DISCUSSION
We hypothesized that networks, which naturally represent RE ancestry and similarities, are a powerful platform for studying RE activity. Here, we use hominid SVA to demonstrate that network analysis can reveal new RE community structure at a fine scale and delineate their expansion dynamics in the genome throughout evolution. Integrating networks with comparative genomics enables us to estimate activity potentials of RE communities, highlights specific elements that are active with high probability, and pinpoints elements that played key roles in the successful proliferation of an entire lineage of REs. These new RE communities, their activity time estimation, and the identification of the most probable master elements were supported with analyses of genomic sequencing data.
Subfamily classification algorithms regularly rely upon MSAs (30, 31). In many cases, clustering algorithms that detect sets of similar sequences are required as a preprocessing step for the MSA-based methods to work accurately (42). The first component in our network method is essentially a clustering algorithm that can uncover a spectrum of community structure resolutions, depending on the parameters used, enabling precise characterization of communities without necessitating MSAs, which have various constraints on the sets of sequences that can be aligned together (32). Using BLAST (and not a pattern-matching algorithm) for the distance metric enables us to take into account the specific processes of molecular evolution that shape genomic sequence over time.
Community detection in networks can be achieved in approximately linear run time (43, 44) and can therefore be applied to networks containing millions of nodes. The modularity of our approach, which enables us to substitute the community detection algorithm, also allows for cross-validation using multiple algorithms (as we show for SVA using the EO and Bornholdt methods). The pairwise similarity step required in our method before community detection is the computational bottleneck, but can also be parallelized or replaced by more efficient algorithms (45, 46), enabling us to optimize the efficiency of the entire pipeline. Thus, although we analyzed a data set of thousands of REs as a test set, the efficiency of network-based analysis makes it feasible to analyze even the largest RE families across more extensive sets of genomes, as we demonstrate in a preliminary analysis of the human Alu family, the RE family with the highest copy number in the human genome (note S4 and table S4).
The community-level directed networks reveal the evolutionary expansion of SVA in each hominid genome, allowing us to differentiate between successive (that is, linear) bursts of activity and the proliferation of multiple communities in parallel. The network topology reveals that different communities have played different roles in SVA expansion. Further, characterizing the intracommunity pairwise similarities enables us to estimate the period of activity and current activity potential of each community. Notably, the routine method for estimating the age of an RE is to calculate its divergence from its subfamily’s consensus sequence (4, 47). We find that considering only the maximal pairwise similarity per element can lead to new insights. For example, clique detection, based on pairwise similarity, enables us to identify the youngest and most probable active elements in communities, which are not necessarily the most similar to the community’s consensus, especially in heterogeneous communities (for example, SVA_D1). Supplementing the network analysis with RNA and DNA sequencing data validates and refines the understanding of RE community activity, supporting the predictive power of network analysis and demonstrating the advantage of combining network and biological analyses.
Integrating cross-species networks and comparative genomics data provides detailed understanding of shared and species-specific expansions in each genome. The Circos network (Fig. 4) enables us to effectively visualize the bird’s-eye view of the expansion of the entire RE family. Detailed inspection of specific elements in the network reveals the most probable key players in SVA evolution. The SVA elements residing in the CABIN1 and NPLOC4 genes, identified by crossing network and biological data, are the first discovered examples of a specific master RE that fueled the expansion of an entire RE family that is still active in humans. The existence of these specific elements in hominids and their absence from the Gibbon genome (fig. S13) (48) can explain the success of SVA, specifically in hominid genomes and not in more distant hominoids. One of these elements most probably drove the sequential expansion of SVA in hominins (human, chimp, and gorilla) from SVA_A until the formation of SVA_D, which proliferated to reach a critical mass that facilitated the parallel differential branching of SVA in each of these genomes.
It is most compelling to try to understand what is in the CABIN1 (or NPLOC4) element that enabled it to retain activity for such a long period of time, since the proliferation of SVA_A to that of SVA_D. CABIN1 and NPLOC4 are housekeeping genes (49) and are expressed throughout early embryonic development and in adult gonads (fig. S14), which supports the ability of these SVAs to drive SVA proliferation in the inherited genome. Genic REs that are in the same orientation as their host genes are removed by purifying selection at a faster rate than reverse-oriented elements, probably due to greater interference with gene function (4, 50). Thus, the fact that the CABIN1 and NPLOC4 elements are reverse-oriented may have contributed to their ability to stay under the radar of evolutionary forces. Furthermore, the CABIN1 element resides between two close-by exons, which impedes the genome’s ability to remove it by genomic deletion.
These elements are intriguing examples, but they are only representatives of an entire set of master elements that can be identified in the cross-hominid network. When seeking to identify elements that played similar roles in young communities, the shared elements in these communities are the immediate suspects, because these relatively ancient elements were the first to take the form of their respective communities and hence are the most probable founders. These primordial elements of contemporarily active communities may or may not still be active. The clique-based method we developed enables us to predict which elements have recently replicated. These active elements are of primary interest, because they are the elements that can still form disease-causing and evolution-promoting de novo insertions.
In conclusion, we present an approach that integrates network analysis and computational genomics to delineate RE dynamics and activity. The method could be used in the future to understand RE dynamics and identify active elements in any of the thousands of organisms whose genomes have been sequenced and to understand the evolution of viruses within hosts.
MATERIALS AND METHODS
SVA element retrieval and assembly
DNA sequences of SVA elements were downloaded from the RepeatMasker (36) table of the UCSC (University of California, Santa Cruz) Table Browser (51). The assemblies used were hg19 (human), panTro3 (chimp; Pan troglodytes), gorGor3 (gorilla; Gorilla gorilla), and ponAbe2 (orangutan; Pongo abelii). Poorly mapped elements, those assigned chromosome names of chr_random or chrUn, were discarded. RepeatMasker tends to split sequences of an individual SVA element in the genome into multiple fragments. Therefore, we first assembled SVA fragments into full-length elements with an empirically devised method: (i) SVA sequences within 500 bp apart were merged; (ii) sequences annotated by RepeatMasker as simple repeats (TCTCCCn or GGGAGAn) present within 100 bp of SVAs were merged with the SVA fragments because they are actually the SVAs’ 5′-hexamer repeats; (iii) for NHH genomes, VNTRs frequently had large gaps in the assembly (therefore, we merged SVA fragments with internal gaps, defined as fragments within 500 bp on each side of the gap); and (iv) SINE, VNTR, and ALU regions were identified within SVA sequences (as described below), and elements with SINE regions >600 bp were replaced with the original RepeatMasker-identified fragments, because they may resemble adjacent insertions that were mistakenly merged.
Identification of regions within SVA elements
SVA elements consist of three main regions: SINE, VNTR, and ALU (Fig. 1A). Most of our analyses focused on the SINE region, which is longer than the Alu region (~500 versus ~360 bp, respectively) and has constant length, unlike the VNTR. To identify these regions within genomic sequences, we first identified them in the six (SVA_A to SVA_F) consensus sequences found in Repbase Update (52). Next, for each hominid genome, genomic SVA sequences were aligned to each of the regions using BLAST (v2.2.23), and adjacent regions (≤50 bp) with similar region annotation and genomic orientation were merged. The longest region identified in this manner for each element was selected. For the SINE regions, subfamily annotation was assigned on the basis of the original annotation from the RepeatMasker table. This subfamily annotation was considered the gold standard and used to show the accuracy of the network-based annotation because RepeatMasker annotation is the most widely used and is the only comprehensive RE annotation that is publicly available.
RE network construction per hominid
Separate networks were constructed for SVAs of every hominid genome. Each node in the network represents a single genomic RE sequence. To generate weighted links, SVA sequences from each hominid genome were pairwise-aligned using BLAST (2.2.30+; “-strand plus -dust no -max_target_seqs 4000”). Links were drawn between pairs of sequences for which any alignment was produced. The best alignment for each pair was selected, and link weights were defined as alignment bitscores, which indicate the extent of sequence similarity. Only the SINE region was used in this analysis to avoid biases associated with VNTR lengths and alignment complication of this repetitive region. Additional networks were built from the Alu regions to validate and refine the SINE-based networks.
Filtering links from the network
The network community detection algorithm is sensitive to the number of links, their strength, and topology. To have a better resolution of the network community structure, we removed weak links from the network. Genomic sequences accumulate mutations over time; therefore, the older the nodes are, the weaker are their links. For this reason, we did not use a threshold to disconnect links, because it would disconnect whole groups of older REs from the network. Therefore, we disconnected the weakest links of each node and retained only the top fraction of strongest links, denoted as Pr. Decreasing Pr values removes more links and hence reveals more communities due to the better separation, whereas increasing Pr values causes communities to merge. In the Hominids networks, we used a Pr value of 0.25, which means that the strongest 25% of each node’s links were kept. Consistent results were obtained for a range of different Pr values (note S1).
Community detection in RE networks
Communities within networks can be defined as subsets of nodes that are more densely linked than the rest of the network (18, 19). We identified RE communities in the network using a Matlab implementation of the EO method (34), which takes into account the number of links in the network and their weights. EO was applied to both the initial and “filtered” link-disconnected networks with various values of Pr. Because EO contains random phases, there is some variation in the community detection in each run. Therefore, we ran the algorithm 100 times and used a bootstrap algorithm (53) for statistical validation of community assignment. The bootstrap algorithm requires a predefined θ parameter, which denotes the fraction of runs that two nodes must be in the same community to be considered bona fide members of the same community. Nodes that either were assigned to a community smaller than 20 nodes or were not assigned to a community were considered as unstable nodes. In the Hominids network, we used a default θ value of 0.8, but consistent results were obtained for a range of θ values. Increasing θ reveals more communities and more unstable nodes, whereas decreasing θ merges different communities (note S1). To further validate the SVA communities identified by EO, we applied the Bornholdt community detection method (35) to the SVA network with an array of parameters. This more efficient algorithm was used to show the upscaling of our network-based approach by applying it to a large network of human Alu (note S2).
Community connectivity scores
To characterize the relations and extent of separation between the communities, we used community connectivity scores to measure to what extent a community is connected within itself and separated from the others. The community separation measurement takes into account links within and between the communities. The measure is an n × n table, where n is the number of communities in the network and each cell notes the strength of the connectivity (0 ≤ Connectivity ≤ 1) between each pair of communities or within a certain community. The connectivity measurement is the ratio between the number of existing links between (or within) the communities and their possible number of links.
The formula for i ≠ j is
| (1) |
and the formula for i = j is
| (2) |
where Number_Of_Links(Commi, Commj) is the number of links between communities i and j. The number of links taken into account was different depending on the analysis the community separation was used for: All the links in the network were used for community connectivity scores (Fig. 1B), whereas for inferring community-level ancestry (Fig. 2, E to H), only a certain percentile of the strongest links were used, as described below in the section on Edmond’s algorithm.
Network visualization
The networks were plotted with Cytoscape (v3.4.0) (54) using “edge-weighted force directed layout” (55). For efficiency and improved perception, only the strongest links were presented, by setting the Pr parameter defined above to 0.03, which retains the strongest 3% of links per node. The unstable nodes and their links were not sketched.
Comparison of community annotation to known SVA subfamilies
To verify the communities detected by the network, we compared the network community assignment of the various nodes to the widely used RepeatMasker classification (36) provided in the UCSC Table Browser (51). Because SVA elements in this table tend to be fragmented into multiple sequences, we used the subfamily annotation of the SINE region, which is the one analyzed in the network. Our network-based classification is divided into more communities than the RepeatMasker classification. Therefore, we labeled each community based on the RepeatMasker SVA repeat annotation, which classifies SVA into six subfamilies (SVA_A to SVA_F), by using a majority rule for each community. Nodes assigned the same subfamily annotation as in RepeatMasker were considered correct. Thus, the true-positive (TP) and false-positive (FP) rates are the fraction of correctly and incorrectly annotated nodes, respectively (unstable nodes were disregarded in both).
Activity time approximation and clique finding
To approximate the relative activity time of each community, we analyzed the intracommunity sequence identity. We calculated the percentage of sequence identity between each RE to its closest relative (the one with maximal bitscore, which takes into account the alignment length) and used the median of these values per community to approximate the activity times (Fig. 2, I to L).
To find groups within the communities that have maximal identity levels, we generated a network in which its links are the percent identity between each pair of REs. We filtered out links with alignment lengths shorter than 450 or less than 100% identity.
Edmond’s optimum branching algorithm for community-level network analysis
A community-level network was constructed, in which nodes are communities and links between all pairs of communities are weighted by connectivity score matrices (see above). The networks analyzed for optimum branching were defined by Pr = 0.05, not the previously used Pr = 0.25, because evolutionary relations are most reliably inferred from the strongest links. To identify the ancestral relations between communities, we applied an automated optimum branching algorithm, Edmond’s algorithm (39), to the network. Edmond’s requires a predefined root, which was set to the most ancient community (SVA_A). This root was chosen on the basis of previous knowledge, although it complies with the percentage of divergence between the REs in each family and with the percentage of REs in each community shared by other organisms (that is, inserted in their common ancestor). Thus, these two measurements, for example, can be used to determine the root in other RE networks. This algorithm was applied to both the per-hominid (Fig. 2, E to H) and all-hominid networks (Fig. 4).
Construction of all-hominid SVA network
SINE regions of SVA sequences of all four hominids were pairwise-aligned using BLAST (2.2.30+; “-strand plus -dust no”). Communities were identified using the EO method (34), with Pr = 0.15 and θ = 0.92. Only communities with at least 20 elements were retained, which contained 8488 of the total 9184 elements in all hominids. The advantage of this approach is that it compares all sequences from all hominid genomes in an unbiased manner and hence enables linking between species-specific elements and communities from different genomes. The community-level network of Circos nodes (Fig. 4) was constructed by defining two tracks of information: the nodes’ presence/absence in different hominids and their A to F subfamily annotation. The former was derived from comparative genomics, which enables us to identify the presence/absence profiles of each genomic SVA element in the other three genomes. These shared elements between the species are termed orthologous, which implies that the element was inserted in these species’ common ancestor. Orthologs were identified using the UCSC Genome Browser’s executable “liftover” tool (June 2013; http://hgdownload.soe.ucsc.edu/admin/exe) (56) and whole-genome pairwise alignments. SINE regions of all SVAs were “lifted” (-minMatch = 0.5) for each genome to all other genomes. Only elements lifted to syntenic chromosomes, including the known Gorilla chr5:chr17 translocation (57), were accepted. In some cases, only part of the SINE region mapped to another genome and liftover does not provide target-genome coordinates. To obtain these for the partially lifted sequences, genomic sites at 50-bp intervals were selected per element and lifted independently. The genomic mapping assigned for these sites was then associated back to the full-length element. The second annotation, to subfamilies A to F, was given by the original RepeatMasker annotation. The plot was sketched in Cytoscape (v3.4.0) using the enhancedGraphics app (58).
Inferring age-related trends in the all-hominid community network
Two values were calculated per community: (i) the fraction of shared (orthologous) SVAs in each community and (ii) the median values of the maximum percent identity per element in the community (as calculated for per-hominid networks; Fig. 2, I to L). We hypothesized that the greater the distance of a community is from the SVA-founding root community, the lower its fraction of ancient elements and the higher its intracommunity sequence identity will be. Parent-child pairs in the network were compared using paired Wilcoxon’s signed-rank test in R.
RNA expression analysis of SVA in human preimplantation embryos
A previously published single-cell RNA-seq data set from human embryonic development (SRP011546) (59) was downloaded from the National Center for Biotechnology Information (NCBI) sequence read archive (www.ncbi.nlm.nih.gov/sra). Post-implantation RNA-seq data (SRR1295944, SRR1295945, and SRR1295946) were added from a later study (60). All reads were aligned to the human genome (hg19) using STAR (v2.4.2), with parameters that retain multimapped reads and hence enable accurate analysis of repetitive element expression (“--outFilterMultimapNmax 100--winAnchorMultimapNmax 100”). Expression per community was assessed on the basis of sequencing reads that mapped uniquely to REs of a single community. This is analogous to the common approach in RNA expression analysis to retain only uniquely mapped reads (that is, to a single genomic location) but is generalized to the community level as necessary for repetitive element analysis. This approach prevents erroneous intercommunity read mapping, which is common in young RE families, such as SVA. Cross-sample normalization was applied by DESeq normalization of gene expression based on the expression values of all RefSeq genes, which was quantified for each sample by featureCounts (v1.3.6-p1; default parameters) (61).
Community annotation of SVA MEIs in 1000 Genomes Project data
WGS reads were downloaded for 2504 human individuals from the phase 3 data of the 1000 Genomes Project (downloaded from fasp-g1k@fasp.1000genomes.ebi.ac.uk:vol1/ftp/phase3 via Aspera) (9). The analysis was restricted to the 2422 individuals screened for MEIs in the phase 3 structural variation analysis (9). Therein, 818 SVA MEIs were identified in the human population, but the insertions were annotated as SVA without further detail. We identify the specific communities that most probably spawned each MEI to understand the activity rates of each community. The 818 MEIs were previously identified primarily using discordant paired-end reads in the data, where one read (“read1”) maps to the genome at a specific locus and the other (“read2”) to an SVA, which is not present in the genome. These discordant reads imply that an MEI exists in proximity to the locus where read1 mapped to. We designed a pipeline to efficiently sift through 46 terabytes of raw data and focus on the reads supporting the SVA insertions, which could subsequently enable community annotation of MEIs. First, we used Kallisto (62) to identify all SVA reads (Kallisto v0.43.0; parameters: --fragment-length 100 --sd 1 --pseudobam). The index file used for SVA identification by Kallisto contained the six SVA consensus sequences from Repbase Update (52) and six consensus sequences we created from the SINE region of communities SVA_D1 to SVA_D6. Paired reads that either both or none matched SVA were discarded. For pairs with only one read (read1) mapping to SVA, read2 was mapped to the human genome (hg19) using STAR (v2.4.2a; parameters: --outFilterMultimapNmax 1 --outFilterMismatchNmax 999 --alignIntronMax 1) and was required to uniquely map within 750 bp of one of the 818 MEI sites. We validated that no SVAs were present in the reference genome within 500 bp of these 750-bp flanks; hence, any discordant read pairs mapping in this manner correspond to the SVA MEIs. Next, read1s were used for community annotation. We mapped them to the genome using STAR while allowing reads to map to multiple genomic locations (--winAnchorMultimapNmax 100 --outFilterMultimapNmax 100 --outFilterMismatchNmax 999 --alignIntronMax 1). Only reads entirely contained within a genomic SVA SINE region were retained. Next, similar to the commonly used method of selecting uniquely mapped reads, we retained only reads that mapped to a single SVA community in the genome. Because it was not uncommon for an MEI to have supporting reads uniquely mapping to multiple communities, the final community annotation per MEI was defined as that with maximum supporting reads.
Supplementary Material
Acknowledgments
We thank D. Pollock and A. Wacholder for help with running their software. Funding: S.H. acknowledges the MULTIPLEX (No. 317532) EU project, the Israel Science Foundation, the Italian-Israel and Japan-Israel MOST (Ministry of Science and Technology), ONR (Office of Naval Research), and DTRA (Defense Threat Reduction) for financial support. E.Y.L. was supported by the European Research Council (grant 311257) and the Israel Cancer Research Foundation. Author contributions: S.H. and E.Y.L. conceived the project. O.L. and B.A.K. performed the experiments. O.L. led the network analyses and B.A.K. led the bioinformatics analyses. All authors designed the experiments, analyzed the data, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/10/e1701256/DC1
note S1. Consistent results for different community detection parameters
note S2. Analysis of SVA using the Bornholdt community detection algorithm
note S3. Details of method for comparing different community structures
note S4. Analysis of human Alu communities
note S5. Identification of candidate master SVA elements
note S6. Identification and annotation of human Alu sequences
fig. S1. High correlation of SINE and ALU regions’ link strengths.
fig. S2. Network link strength histograms of the different SVA subfamilies in human.
fig. S3. Demonstration of the accuracy of Bornholdt’s scalable community detection method on SVA subfamilies.
fig. S4. Similarity network of human SVA REs with unstable nodes.
fig. S5. Concordance of human SVA communities based on similarity of SINE or Alu regions.
fig. S6. Neighbor-joining network comparing consensus sequences computed from each community.
fig. S7. Clique-based network.
fig. S8. SINE-based SVA RNA expression enrichment in early embryonic development per community (TH = 0.9).
fig. S9. SVA MEIs in 1000 genomes data per community (θ = 0.9).
fig. S10. Full alignment of SVA elements in the CABIN1 gene to SVA consensus sequences evinces the differential subfamily classification of human and orangutan SVA elements.
fig. S11. MSA of NPLOC4 SVA elements’ SINE regions to SVA consensus sequences.
fig. S12. MSA of CABIN1 and NPLOC4 SVA elements’ Alu regions to SVA consensus sequences.
fig. S13. Genomic loci of CABIN1 and NPLOC4 elements and conservation in other primates.
fig. S14. Expression of CABIN1 and NPLOC4 in human tissues.
table S1. Community list in different hominids for Pr = 0.25 and θ = 0.8.
table S2. Human community separation.
table S3. Community sizes for human SVA communities for Pr = 0.25 and θ = 0.9.
table S4. Computational resources.
table S5. Different human community detection comparisons based on the TP/FP ratio.
table S6. Comparison between SVA community structure as identified by Bornholdt and EO.
table S7. Human TP/FP ratio without assigning the unstable nodes.
table S8. Chimp TP/FP ratio without and with assigning the unstable nodes.
table S9. Comparison between known Alu subfamily structure and network’s community structure.
table S10. Comparison between known AluY subfamily structure and network’s community structure.
table S11. Human-orangutan orthologous SVA elements.
table S12. TSDs of the CABIN1 and NPLOC4 SVA elements in hominids.
table S13. Number of reads supporting the presence of CABIN1 and NPLOC4 SVA elements in each hominid genome.
data file S1. SVA_Supplementary_Data_v2.
REFERENCES AND NOTES
- 1.Luan D. D., Korman M. H., Jakubczak J. L., Eickbush T. H., Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell 72, 595–605 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Cost G. J., Feng Q., Jacquier A., Boeke J. D., Human L1 element target-primed reverse transcription in vitro. EMBO J. 21, 5899–5910 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dewannieux M., Esnault C., Heidmann T., LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 35, 41–48 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Smit A. F. A., Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev. 9, 657–663 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Cordaux R., Batzer M. A., The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Koning A. P. J., Gu W., Castoe T. A., Batzer M. A., Pollock D. D., Repetitive elements may comprise over two-thirds of the human genome. PLOS Genet. 7, e1002384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett E. A., Coleman L. E., Tsui C., Pittard W. S., Devine S. E., Natural genetic variation caused by transposable elements in humans. Genetics 168, 933–951 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prado-Martinez J., Sudmant P. H., Kidd J. M., Li H., Kelley J. L., Lorente-Galdos B., Veeramah K. R., Woener A. E., O’Connor T. D., Santpere G., Cagan A., Theunert C., Casals F., Laayouni H., Munch K., Hobolth A., Halager A. E., Malig M., Hernandez-Rodriguez J., Hernando-Herraez I., Prüfer K., Pybus M., Johnstone L., Lachmann M., Alkan C., Twigg D., Petit N., Baker C., Hormozdiari F., Fernandez-Callejo M., Dabad M., Wilson M. L., Stevison L., Camprubí C., Carvalho T., Ruiz-Herrera A., Vives L., Mele M., Abello T., Kondova I., Bontrop R. E., Pusey A., Lankester F., Kiyang J. A., Bergl R. A., Lonsdorf E., Myers S., Ventura M., Gagneux P., Comas D., Siegismund H., Blanc J., Agueda-Calpena L., Gut M., Fulton L., Tishkoff S. A., Mullikin J. C., Wilson R. K., Gut I. G., Gonder M. K., Ryder O. A., Hahn B. H., Navarro A., Akey J. M., Bertranpetit J., Reich D., Mailund T., Schierup M. H., Hvilsom C., Andrés A. M., Wall J. D., Bustamante C. D., Hammer M. F., Eichler E. E., Marques-Bonet T., Great ape genetic diversity and population history. Nature 499, 471–475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sudmant P. H., Rausch T., Gardner E. J., Handsaker R. E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M. H.-Y., Konkel M. K., Malhotra A., Stütz A. M., Shi X., Casale F. P., Chen J., Hormozdiari F., Dayama G., Chen K., Malig M., Chaisson M. J. P., Walter K., Meiers S., Kashin S., Garrison E., Auton A., Lam H. Y. K., Mu X. J., Alkan C., Antaki D., Bae T., Cerveira E., Chines P., Chong Z., Clarke L., Dal E., Ding L., Emery S., Fan X., Gujral M., Kahveci F., Kidd J. M., Kong Y., Lameijer E.-W., McCarthy S., Flicek P., Gibbs R. A., Marth G., Mason C. E., Menelaou A., Muzny D. M., Nelson B. J., Noor A., Parrish N. F., Pendleton M., Quitadamo A., Raeder B., Schadt E. E., Romanovitch M., Schlattl A., Sebra R., Shabalin A. A., Untergasser A., Walker J. A., Wang M., Yu F., Zhang C., Zhang J., Zheng-Bradley X., Zhou W., Zichner T., Sebat J., Batzer M. A., McCarroll S. A. The Genomes Project Consortium, Mills R. E., Gerstein M. B., Bashir A., Stegle O., Devine S. E., Lee C., Eichler E. E., Korbel J. O., An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chuong E. B., Elde N. C., Feschotte C., Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 18, 71–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solyom S., Kazazian H. H. Jr, Mobile elements in the human genome: Implications for disease. Genome Med. 4, 12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hancks D. C., Kazazian H. H. Jr, SVA retrotransposons: Evolution and genetic instability. Semin. Cancer Biol. 20, 234–245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns K. H., Boeke J. D., Human transposon tectonics. Cell 149, 740–752 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mills R. E., Bennett E. A., Iskow R. C., Devine S. E., Which transposable elements are active in the human genome? Trends Genet. 23, 183–191 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Hancks D. C., Kazazian H. H. Jr, Active human retrotransposons: Variation and disease. Curr. Opin. Genet. Dev. 22, 191–203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H., Xing J., Grover D., Hedges D. J., Han K., Walker J. A., Batzer M. A., SVA elements: A hominid-specific retroposon family. J. Mol. Biol. 354, 994–1007 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Ianc B., Ochis C., Persch R., Popescu O., Damert A., Hominoid composite non-LTR retrotransposons—Variety, assembly, evolution, and structural determinants of mobilization. Mol. Biol. Evol. 31, 2847–2864 (2014). [DOI] [PubMed] [Google Scholar]
- 18.M. E. J. Newman, Networks: An Introduction (Oxford Univ. Press, 2014), pp. 163–186.
- 19.R. Cohen, S. Havlin, Complex Networks: Structure, Robustness and Function (Cambridge Univ. Press, 2010). [Google Scholar]
- 20.Amaral L. A. N., Scala A., Barthélémy M., Stanley H. E., Classes of small-world networks. Proc. Natl. Acad. Sci. U.S.A. 97, 11149–11152 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watts D. J., Strogatz S. H., Collective dynamics of “small-world” networks. Nature 393, 440–442 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Pastor-Satorras R., Vespignani A., Epidemic spreading in scale-free networks. Phys. Rev. Lett. 86, 3200–3203 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Cohen R., Erez K., Ben-Avraham D., Havlin S., Resilience of the internet to random breakdowns. Phys. Rev. Lett. 85, 4626–4628 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Li D., Fu B., Wang Y., Lu G., Berezin Y., Stanley H. E., Havlin S., Percolation transition in dynamical traffic network with evolving critical bottlenecks. Proc. Natl. Acad. Sci. U.S.A. 112, 669–672 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamasaki K., Gozolchiani A., Havlin S., Climate networks around the globe are significantly affected by El Niño. Phys. Rev. Lett. 100, 228501 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Donges J. F., Zou Y., Marwan N., Kurths J., The backbone of the climate network. Europhys. Lett. 87, 48007 (2009). [Google Scholar]
- 27.Gallos L. K., Makse H. A., Sigman M., A small world of weak ties provides optimal global integration of self-similar modules in functional brain networks. Proc. Natl. Acad. Sci. U.S.A. 109, 2825–2830 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.A.-L. Barabási, Network Science (Cambridge Univ. Press, 2016). [Google Scholar]
- 29.Andrade R. F. S., Rocha-Neto I. C., Santos L. B. L., de Santana C. N., Diniz M. V. C., Lobão T. P., Goés-Neto A., Pinho S. T. R., El-Hani C. N., Detecting network communities: An application to phylogenetic analysis. PLOS Comput. Biol. 7, e1001131 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price A. L., Eskin E., Pevzner P. A., Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res. 14, 2245–2252 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wacholder A. C., Cox C., Meyer T. J., Ruggiero R. P., Vemulapalli V., Damert A., Carbone L., Pollock D. D., Inference of transposable element ancestry. PLOS Genet. 10, e1004482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D. Mount, Bioinformatics: Sequence and Genome Analysis (Cold Spring Harbor Laboratory Press, ed. 2, 2004). [Google Scholar]
- 33.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duch J., Arenas A., Community detection in complex networks using extremal optimization. Phys. Rev. E 72, 027104 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Reichardt J., Bornholdt S., Statistical mechanics of community detection. Phys. Rev. E 74, 016110 (2006). [DOI] [PubMed] [Google Scholar]
- 36.A. F. A. Smit, R. Hubley, P. Green, RepeatMasker Open-3.0 (1996–2007); www.repeatmasker.org
- 37.Damert A., Raiz J., Horn A. V., Löwer J., Wang H., Xing J., Batzer M. A., Löwer R., Schumann G. G., 5′-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Res. 19, 1992–2008 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hancks D. C., Ewing A. D., Chen J. E., Tokunaga K., Kazazian H. H., Exon-trapping mediated by the human retrotransposon SVA. Genome Res. 19, 1983–1991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edmonds J., Optimum branchings. J. Res. Natl. Bur. Stand. Sect. B 71B, 233–240 (1967). [Google Scholar]
- 40.Jacobs F. M. J., Greenberg D., Nguyen N., Haeussler M., Ewing A. D., Katzman S., Paten B., Salama S. R., Haussler D., An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 516, 242–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith Z. D., Chan M. M., Humm K. C., Karnik R., Mekhoubad S., Regev A., Eggan K., Meissner A., DNA methylation dynamics of the human preimplantation embryo. Nature 511, 611–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miele V., Penel S., Duret L., Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinf. 12, 116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clauset A., Newman M. E. J., Moore C., Finding community structure in very large networks. Phys. Rev. E 70, 066111 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Blondel V. D., Guillaume J.-L., Lambiotte R., Lefebvre E., Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008). [Google Scholar]
- 45.Edgar R. C., Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Blackshields G., Sievers F., Shi W., Wilm A., Higgins D. G., Sequence embedding for fast construction of guide trees for multiple sequence alignment. Algorithms Mol. Biol. 5, 21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.International Human Genome Sequencing Consortium Lander E. S., Linton L. M., Birren B., Nusbaum C., Zody M. C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J. P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange-Thomann N., Stojanovic N., Subramanian A. Dudley Wyman for Whitehead Institute for Biomedical Research Center for Genome Research Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J. C., Mungall A., Plumb R., Ross M., Shownkeen R. Sarah Sims for The Sanger Centre Waterston R. H., Wilson R. K., Hillier L. D. W., McPherson J. D., Marra M. A., Mardis E. R., Fulton L. A., Chinwalla A. T., Pepin K. H., Gish W. R., Chissoe S. L., Wendl M. C., Delehaunty K. D., Miner T. L., Delehaunty A., Kramer J. B., Cook L. L., Fulton R. S., Johnson D. L., Minx P. J. Sandra W. Clifton for Washington University Genome Sequencing Center Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J.-F., Olsen A., Lucas S., Elkin C., Uberbacher E. Marvin Frazier for US DOE Joint Genome Institute Gibbs R. A., Muzny D. M., Scherer S. E., Bouck J. B., Sodergren E. J., Worley K. C., Rives C. M., Gorrell J. H., Metzker M. L., Naylor S. L., Kucherlapati R. S., Nelson D. L. George M. Weinstock for Baylor College of Medicine Human Genome Sequencing Center Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y. Todd Taylor for RIKEN Genomic Sciences Center Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C. Patrick Wincker for Genoscope and CNRS UMR-8030 Rosenthal A., Platzer M., Nyakatura G., Taudien S. Andreas Rump for Department of Genome Analysis Institute of Molecular Biotechnology Smith D. R., Doucette-Stamm L., Rubenfield M., Weinstock K., Mei Lee H. JoAnn Dubois for GTC Sequencing Center Yang H., Yu J., Wang J., Huang G. Jun Gu for Beijing Genomics Institute/Human Genome Center Hood L., Rowen L., Madan A. Shizen Qin for Multimegabase Sequencing Center; The Institute for Systems Biology Davis R. W., Federspiel N. A., Abola A. P. Michael J. Proctor for Stanford Genome Technology Center Roe B. A., Chen F. Huaqin Pan for University of Oklahoma’s Advanced Center for Genome Technology Ramser J., Lehrach H. Richard Reinhardt for Max Planck Institute for Molecular Genetics McCombie W. R., de la Bastide M. Neilay Dedhia for Cold Spring Harbor Laboratory Lita Annenberg Hazen Genome Center:, Helmut Blöcker, Klaus Hornischer Gabriele Nordsiek for GBF—German Research Centre for Biotechnology Agarwala R., Aravind L., Bailey J. A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D. G., Burge C. B., Cerutti L., Chen H.-C., Church D., Clamp M., Copley R. R., Doerks T., Eddy S. R., Eichler E. E., Furey T. S., Galagan J., Gilbert J. G. R., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Steven Johnson L., Jones T. A., Kasif S., Kaspryzk A., Kennedy S., James Kent W., Kitts P., Koonin E. V., Korf I., Kulp D., Lancet D., Lowe T. M., McLysaght A., Mikkelsen T., Moran J. V., Mulder N., Pollara V. J., Ponting C. P., Schuler G., Schultz J., Slater G., Smit A. F. A., Stupka E., Szustakowki J., Thierry-Mieg D., Thierry-Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y. I., Wolfe K. H., Yang S.-P. Ru-Fang Yeh for Genome Analysis Group (listed in alphabetical order, also includes individuals listed under other headings) Collins F., Guyer M. S., Peterson J., Felsenfeld A. Kris A. Wetterstrand for Scientific management: National Human Genome Research Institute US National Institutes of Health Myers R. M., Schmutz J., Dickson M., Grimwood J. David R. Cox for Stanford Human Genome Center Olson M. V., Kaul R. Christopher Raymond for University of Washington Genome Center Shimizu N., Kawasaki K. Shinsei Minoshima for Department of Molecular Biology Keio University School of Medicine Evans G. A., Athanasiou M. Roger Schultz for University of Texas Southwestern Medical Center at Dallas Aristides Patrinos for Office of Science US Department of Energy Michael J. Morgan for The Wellcome Trust , Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).11237011 [Google Scholar]
- 48.Carbone L., Harris R. A., Gnerre S., Veeramah K. R., Lorente-Galdos B., Huddleston J., Meyer T. J., Herrero J., Roos C., Aken B., Anaclerio F., Archidiacono N., Baker C., Barrell D., Batzer M. A., Beal K., Blancher A., Bohrson C. L., Brameier M., Campbell M. S., Capozzi O., Casola C., Chiatante G., Cree A., Damert A., de Jong P. J., Dumas L., Fernandez-Callejo M., Flicek P., Fuchs N. V., Gut I., Gut M., Hahn M. W., Hernandez-Rodriguez J., Hillier L. W., Hubley R., Ianc B., Izsvák Z., Jablonski N. G., Johnstone L. M., Karimpour-Fard A., Konkel M. K., Kostka D., Lazar N. H., Lee S. L., Lewis L. R., Liu Y., Locke D. P., Mallick S., Mendez F. L., Muffato M., Nazareth L. V., Nevonen K. A., O’Bleness M., Ochis C., Odom D. T., Pollard K. S., Quilez J., Reich D., Rocchi M., Schumann G. G., Searle S., Sikela J. M., Skollar G., Smit A., Sonmez K., ten Hallers B., Terhune E., Thomas G. W. C., Ullmer B., Ventura M., Walker J. A., Wall J. D., Walter L., Ward M. C., Wheelan S. J., Whelan C. W., White S., Wilhelm L. J., Woerner A. E., Yandell M., Zhu B., Hammer M. F., Marques-Bonet T., Eichler E. E., Fulton L., Fronick C., Muzny D. M., Warren W. C., Worley K. C., Rogers J., Wilson R. K., Gibbs R. A., Gibbon genome and the fast karyotype evolution of small apes. Nature 513, 195–201 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eisenberg E., Levanon E. Y., Human housekeeping genes, revisited. Trends Genet. 29, 569–574 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Nellåker C., Keane T. M., Yalcin B., Wong K., Agam A., Belgard T. G., Flint J., Adams D. J., Frankel W. N., Ponting C. P., The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 13, R45 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karolchik D., Hinrichs A. S., Furey T. S., Roskin K. M., Sugnet C. W., Haussler D., Kent W. J., The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bao W., Kojima K. K., Kohany O., Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gfeller D., Chappelier J.-C., De Los Rios P., Finding instabilities in the community structure of complex networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 72, 56135 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.J. Heer, S. K. Card, J. A. Landay, Prefuse: A toolkit for interactive information visualization, CHI ’05 Proceedings of the SIGCHI Conference on Human Factors in Computing Systems, Portland, OR, 2 to 7 April 2005. [Google Scholar]
- 56.Karolchik D., Baertsch R., Diekhans M., Furey T. S., Hinrichs A., Lu Y. T., Roskin K. M., Schwartz M., Sugnet C. W., Thomas D. J., Weber R. J., Haussler D., Kent W. J., The UCSC Genome Browser Database. Nucleic Acids Res. 31, 51–54 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yunis J. J., Prakash O., The origin of man: A chromosomal pictorial legacy. Science 215, 1525–1530 (1982). [DOI] [PubMed] [Google Scholar]
- 58.Morris J. H., Kuchinsky A., Ferrin T. E., Pico A. R., enhancedGraphics: A Cytoscape app for enhanced node graphics. F1000Research 3, 147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan L., Yang M., Guo H., Yang L., Wu J., Li R., Liu P., Lian Y., Zheng X., Yan J., Huang J., Li M., Wu X., Wen L., Lao K., Li R., Qiao J., Tang F., Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 20, 1131–1139 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Guo H., Zhu P., Yan L., Li R., Hu B., Lian Y., Yan J., Ren X., Lin S., Li J., Jin X., Shi X., Liu P., Wang X., Wang W., Wei Y., Li X., Guo F., Wu X., Fan X., Yong J., Wen L., Xie S. X., Tang F., Qiao J., The DNA methylation landscape of human early embryos. Nature 511, 606–610 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Bray N. L., Pimentel H., Melsted P., Pachter L., Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Danon L., Díaz-Guilera A., Duch J., Arenas A., Comparing community structure identification. J. Stat. Mech. 2005, P09008 (2005). [Google Scholar]
- 64.Han K., Konkel M. K., Xing J., Wang H., Lee J., Meyer T. J., Huang C. T., Sandifer E., Hebert K., Barnes E. W., Hubley R., Miller W., Smit A. F. A., Ullmer B., Batzer M. A., Mobile DNA in Old World monkeys: A glimpse through the rhesus macaque genome. Science 316, 238–240 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Tyner C., Barber G. P., Casper J., Clawson H., Diekhans M., Eisenhart C., Fischer C. M., Gibson D., Gonzalez J. N., Guruvadoo L., Haeussler M., Heitner S., Hinrichs A. S., Karolchik D., Lee B. T., Lee C. M., Nejad P., Raney B. J., Rosenbloom K. R., Speir M. L., Villarreal C., Vivian J., Zweig A. S., Haussler D., Kuhn R. M., Kent W. J., The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 45, D626–D634 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ichiyanagi K., Nakajima R., Kajikawa M., Okada N., Novel retrotransposon analysis reveals multiple mobility pathways dictated by hosts. Genome Res. 17, 33–41 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee E., Iskow R., Yang L., Gokcumen O., Haseley P., Luquette L. J. III, Lohr J. G., Harris C. C., Ding L., Wilson R. K., Wheeler D. A., Gibbs R. A., Kucherlapati R., Lee C., Kharchenko P. V., Park P. J. Cancer Genome Atlas Research Network , Landscape of somatic retrotransposition in human cancers. Science 337, 967–971 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Helman E., Lawrence M. S., Stewart C., Sougnez C., Getz G., Meyerson M., Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 24, 1053–1063 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burton F. H., Loeb D. D., Edgell M. H., Hutchison C. A. III, L1 gene conversion or same-site transposition. Mol. Biol. Evol. 8, 609–619 (1991). [DOI] [PubMed] [Google Scholar]
- 70.Hardies S. C., Martin S. L., Voliva C. F., Hutchison C. A. III, Edgell M. H., An analysis of replacement and synonymous changes in the rodent L1 repeat family. Mol. Biol. Evol. 3, 109–125 (1986). [DOI] [PubMed] [Google Scholar]
- 71.Myers J. S., Vincent B. J., Udall H., Watkins W. S., Morrish T. A., Kilroy G. E., Swergold G. D., Henke J., Henke L., Moran J. V., Jorde L. B., Batzer M. A., A comprehensive analysis of recently integrated human Ta L1 elements. Am. J. Hum. Genet. 71, 312–326 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/10/e1701256/DC1
note S1. Consistent results for different community detection parameters
note S2. Analysis of SVA using the Bornholdt community detection algorithm
note S3. Details of method for comparing different community structures
note S4. Analysis of human Alu communities
note S5. Identification of candidate master SVA elements
note S6. Identification and annotation of human Alu sequences
fig. S1. High correlation of SINE and ALU regions’ link strengths.
fig. S2. Network link strength histograms of the different SVA subfamilies in human.
fig. S3. Demonstration of the accuracy of Bornholdt’s scalable community detection method on SVA subfamilies.
fig. S4. Similarity network of human SVA REs with unstable nodes.
fig. S5. Concordance of human SVA communities based on similarity of SINE or Alu regions.
fig. S6. Neighbor-joining network comparing consensus sequences computed from each community.
fig. S7. Clique-based network.
fig. S8. SINE-based SVA RNA expression enrichment in early embryonic development per community (TH = 0.9).
fig. S9. SVA MEIs in 1000 genomes data per community (θ = 0.9).
fig. S10. Full alignment of SVA elements in the CABIN1 gene to SVA consensus sequences evinces the differential subfamily classification of human and orangutan SVA elements.
fig. S11. MSA of NPLOC4 SVA elements’ SINE regions to SVA consensus sequences.
fig. S12. MSA of CABIN1 and NPLOC4 SVA elements’ Alu regions to SVA consensus sequences.
fig. S13. Genomic loci of CABIN1 and NPLOC4 elements and conservation in other primates.
fig. S14. Expression of CABIN1 and NPLOC4 in human tissues.
table S1. Community list in different hominids for Pr = 0.25 and θ = 0.8.
table S2. Human community separation.
table S3. Community sizes for human SVA communities for Pr = 0.25 and θ = 0.9.
table S4. Computational resources.
table S5. Different human community detection comparisons based on the TP/FP ratio.
table S6. Comparison between SVA community structure as identified by Bornholdt and EO.
table S7. Human TP/FP ratio without assigning the unstable nodes.
table S8. Chimp TP/FP ratio without and with assigning the unstable nodes.
table S9. Comparison between known Alu subfamily structure and network’s community structure.
table S10. Comparison between known AluY subfamily structure and network’s community structure.
table S11. Human-orangutan orthologous SVA elements.
table S12. TSDs of the CABIN1 and NPLOC4 SVA elements in hominids.
table S13. Number of reads supporting the presence of CABIN1 and NPLOC4 SVA elements in each hominid genome.
data file S1. SVA_Supplementary_Data_v2.