

The classical strong metal–support interaction between TiO2 and IB metals was demonstrated.
Abstract
Supported metal catalysts play a central role in the modern chemical industry but often exhibit poor on-stream stability. The strong metal–support interaction (SMSI) offers a route to control the structural properties of supported metals and, hence, their reactivity and stability. Conventional wisdom holds that supported Au cannot manifest a classical SMSI, which is characterized by reversible metal encapsulation by the support upon high-temperature redox treatments. We demonstrate a classical SMSI for Au/TiO2, evidenced by suppression of CO adsorption, electron transfer from TiO2 to Au nanoparticles, and gold encapsulation by a TiOx overlayer following high-temperature reduction (reversed by subsequent oxidation), akin to that observed for titania-supported platinum group metals. In the SMSI state, Au/TiO2 exhibits markedly improved stability toward CO oxidation. The SMSI extends to Au supported over other reducible oxides (Fe3O4 and CeO2) and other group IB metals (Cu and Ag) over titania. This discovery highlights the general nature of the classical SMSI and unlocks the development of thermochemically stable IB metal catalysts.
INTRODUCTION
Supported metal catalysts, comprising metal particles finely dispersed on high–surface area materials, are the most widespread form of heterogeneous catalysts (1–3). They play a central role in the modern chemical industry, and, hence, are of vital importance to the global economy (4). The principal role of the support was initially ascribed to stabilization of the active component and/or increase in the metal dispersion and surface area (5, 6). However, it was soon realized that physicochemical interactions between the metal and the support can also influence catalytic performance through so-called carrier effects (1, 5). These metal-support interactions have a long history of controlling catalyst structure and reactivity (7).
A specific form of these carrier effects came to prominence in the late 1970s, where an unusual interaction was observed between platinum group metals (PGMs) and TiO2, which markedly suppressed small-molecule (such as CO and H2) adsorption following high-temperature reduction (HTR) treatments. This discovery by Tauster et al. (8) was termed the strong metal–support interaction (SMSI; now referred to as classical SMSI). Subsequently, SMSI effects have been the subject of intensive study (9–12). Although suppressed H2 and CO adsorption was initially ascribed to electronic perturbation of the PGM atoms by interaction with Ti cations (8), a consensus has now been reached that this SMSI effect originates from metal encapsulation by a titania overlayer (11, 13). Induction of the SMSI markedly modifies catalytic performance in diverse reactions (11, 14–16) and has been exploited to improve catalyst stability (17–19), identify reaction mechanisms (20, 21), and enhance activity (14, 22). Consequently, research into catalyst systems that can manifest SMSI effects has undergone rapid expansion (15). However, to date, classical SMSI has never been reported for Au/TiO2 catalysts, and there is only one such example where SMSI has been inferred for any group IB metal based on x-ray photoelectron spectroscopy (XPS) and ion-scattering spectroscopy (ISS) measurements of Ag/TiO2 (23).
Oxide-supported gold catalysts have attracted global attention in recent decades, with Au/TiO2 probably the most extensively investigated (6, 24–26). Supported Au catalysts have shown promise for several industrial processes because of their unique catalytic performance, including a new commercial process for vinyl chloride monomer production (27); however, practical applications have been hindered by poor on-stream stability (28, 29). The genesis of an SMSI between Au and titania would offer a route to stabilize Au nanoparticles (NPs) (18, 19), but surface science studies of model catalysts have found no evidence for this phenomenon (30–32). Although Goodman (33) noted electron transfer between Au and TiO2 upon reduction, this was accompanied by increased CO adsorption, contrary to classical SMSI. Au NPs are not believed to undergo SMSI with titania (34, 35), variously attributed to gold’s relatively low work function and surface energy compared with other noble metals (31, 36) or (supposed) inability to dissociate molecular hydrogen and thus initiate titania reduction and subsequent oxide migration to the metal surface. However, recent theoretical and experimental studies demonstrate that gold can dissociate H2 and, hence, potentially promote the reduction of oxide supports (37–39); there is no scientific basis for the notion that Au/TiO2 should not exhibit an SMSI.
Progress on practical catalyst systems has uncovered an O2-induced SMSI for gold supported on ZnO nanorods (40) and hydroxyapatite (18, 19), the reverse of conditions required for classical SMSI. However, decreased CO adsorption was reported following the reduction of Au/CeO2 (41), hinting at the existence of classical SMSI effects in gold catalysis. Here, we report the unequivocal discovery of a classical SMSI in Au/TiO2, evidenced by encapsulation of gold particles by partially reduced titania and accompanied by suppressed CO adsorption and enhanced catalytic stability for CO oxidation. The SMSI extends to Au supported over other reducible oxide supports and other group IB metals (Cu and Ag) over titania. For Au/TiO2, titania reduction is identified as critical for inducing the SMSI state. This finding highlights the generality of the classical SMSI phenomena and associated difficulties that arise when seeking parallels between surface science and practical catalyst studies (due to pressure and material gaps), and unlocks new opportunities to design thermally stable, supported group IB metal catalysts.
RESULTS
The Au/TiO2 sample of RR2Ti was supplied by Haruta Gold Inc. and denoted as RR2Ti-fresh. It was subsequently reduced under 10 volume % H2/He flow at temperatures between 200° and 500°C for 1 hour, and each was denoted as RR2Ti-HX (X, the reduction temperature). The RR2Ti-H500 sample was also reoxidized under 10 volume % O2/He flow at 400°C for 1 hour and denoted as RR2Ti-(H500+O400).
The main characteristics of classical SMSI following HTR are (i) the significant suppression of small-molecule adsorption (such as CO and H2), (ii) electron transfer from the support to metal resulting in a negatively charged metal species, (iii) mass transport from the support to encapsulate metal NPs with a thin overlayer of the support materials, and (iv) a reversal of the preceding phenomena upon reoxidation. Each of these characteristics is evidenced below.
Fourier transform infrared study of CO adsorption
The mean diameters of Au NPs within the RR2Ti-HX (X = 200 to 500) family determined by high-resolution transmission electron microscopy (HRTEM) were 4.8 to 5.2 nm (fig. S1), which were only slightly larger than the mean diameter of the RR2Ti-fresh sample (4.1 nm). These values were consistent with the x-ray powder diffraction (XRD) (fig. S2A) result, where the diffractogram of RR2Ti-H500 is similar to that of RR2Ti-fresh, with no reflections due to any gold phase visible and a common ratio of anatase (101)/rutile (110) reflections. Together, these show that neither Au NP size nor TiO2 phase composition was influenced by HTR treatments. This is in line with the previous study on a real catalyst (34) but is in contrast to surface studies on model catalysts (42, 43), suggesting that the interaction between gold and TiO2 during ambient pressure reduction over inhomogeneous catalysts differs from that under ultrahigh vacuum over single crystals. These observations also show that the RR2Ti samples provide an excellent testing ground for exploring Au NP and support interactions because possible particle size and titania phase differences between samples before and after HTR are minimal.
The suppression of chemisorption of small molecules on metals and electron transfer between metals and supports following HTR are the primary characteristics of SMSI. In situ diffuse reflectance infrared (IR) Fourier transform spectroscopy (DRIFTS) was therefore used to explore both effects because this technique is sensitive to both adsorption and electronic properties of metal surfaces with a suitable probe molecule (18, 19). Figure 1 shows the DRIFTS of CO adsorption on RR2Ti catalysts as a function of various pretreatments. Three bands at 2175, 2120, and 2103 cm−1 are observed for the RR2Ti-fresh sample. The first two are attributed to gaseous CO, and the latter is attributed to CO adsorbed on metallic Au (CO-Au0) (44). The CO-Au0 band intensity decreased monotonically with increasing reduction temperature and disappeared after a 500°C reduction, evidencing a loss in CO adsorption sites over gold precisely as anticipated for classical SMSI, where H2 and CO adsorption are markedly suppressed (8, 45). Although this decreased CO adsorption has been occasionally reported for Au NPs (41), complete loss of CO adsorption capacity has never been observed to our knowledge. Because the Au NP dimensions for all Au/TiO2 samples are similar (figs. S1 and S2A), the loss of CO adsorption cannot arise from a change in the gold dispersion. This conclusion is supported by the observation that CO adsorption was recovered for RR2Ti-(H500+O400) (which had a similar Au NP size distribution; fig. S1F) obtained from in situ O2 treatment of RR2Ti-H500 at 400°C for 1 hour under 10 volume % O2/He. Note that the recovered CO adsorption is blue-shifted relative to that of RR2Ti-fresh, suggesting that surface Au species have a slight positive charge after oxidation. This indicates that the Au-TiO2 interaction after a redox cycle slightly differs from that in the fresh RR2Ti material; however, we show later that this interaction can be fully reversed under repeated redox cycling. It should also be noted that the RR2Ti-H400 material exhibited a new band around 2070 cm−1, ascribed to CO adsorbed on a negatively charged Au surface (CO-Aux−) (44). The existence of such a negative Au surface species evidences electron transfer from TiO2 to Au following a ≥400°C reduction, as observed for the classical SMSI in TiO2-supported PGMs. Additional insight into the electronic properties of the RR2Ti-fresh, RR2Ti-H500, and RR2Ti-(H500+O400) samples was obtained by XPS; this was especially important for the RR2Ti-H500 material for which no CO was adsorbed.
Fig. 1. In situ DRIFT spectra of CO adsorption over RR2Ti-fresh, RR2Ti-HX (X = 200 to 500), and RR2Ti-(H500+O400) samples.
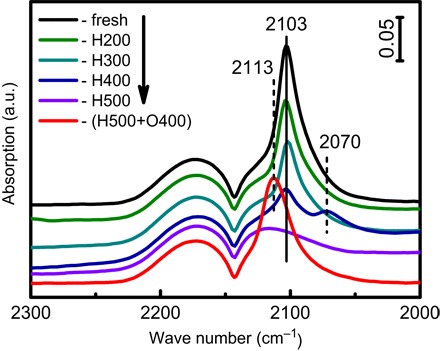
a.u., arbitrary units.
XPS and EPR studies
Figure 2A shows that the binding energy of the Au 4f7/2 spin-orbit split component for RR2Ti-fresh was 83.5 eV, typical of metallic gold (19). The Au 4f7/2 shifted to 83.1 eV following a 500°C reduction, indicating the formation of electron-rich Au species and consistent with the RR2Ti-H400 DRIFT spectrum (Fig. 1). After a 400°C oxidation, the Au 4f7/2 shift was reversed, returning to 83.5 eV and demonstrating a return of the negatively charged Au species to the metallic state. Thus, XPS evidences reversible electron transfer between TiO2 and Au NPs under redox treatments. Although the latter observation appears at odds with CO DRIFTS, which indicates that surface Au species have a slight positive charge after oxidation, these IR measurements are more surface-sensitive than XPS (for Au 4f7/2 photoelectrons, the inelastic mean free path is ~1.3 nm), and hence, partially charged Auδ+ atoms likely only reside in the terminating layer of the NPs in direct contact with titania. No reduced Ti3+ species were observed in the corresponding Ti 2p XP spectra of these samples (fig. S3), in contrast to the case of TiO2-supported PGMs (30, 36), suggesting either a smaller magnitude of charge transfer from titania or smaller number of reduced TiOx species in our gold materials. Gold encapsulation by titania in the SMSI state following HTR should also decrease the Au/Ti surface ratio, and a 33% decrease was observed between the RR2Ti-fresh and RR2Ti-H500 samples. This decrease was reversed upon subsequent reoxidation at 400°C, with the Au/Ti surface ratio of RR2Ti-(H500+O400) returning to that of the fresh sample. This magnitude decrease is close to ~38% attenuation calculated for the complete encapsulation of Au NPs by a single monolayer of 0.7-nm-thick titania (46), assuming an inelastic mean free path of 1.45 nm for 1402-eV kinetic energy Au 4f7/2 electrons. The return of the Au/Ti surface ratio to its original value following reoxidation supports the idea that the reversible attenuation of gold XP signal resulted from a classical SMSI (metal encapsulation by a thin oxide overlayer following HTR, reversed upon subsequent oxidation). Evidence for Ti3+ species was thus explored by more sensitive electron paramagnetic resonance (EPR) measurements (47–49). Figure 2B shows that signals with g = 1.975, attributed to bulk Ti3+ defects (48, 50), were present in all samples. The RR2Ti-H500 sample exhibited a marked increase in this signal intensity by a factor of 6 to 7, coincident with the appearance of a new signal with g = 1.930, attributed to surface Ti3+ (51), highlighting a significant increase in overall Ti3+ species following HTR. Subsequent oxidation suppressed the g = 1.975 signal and eliminated the g = 1.930 signal, restoring the RR2Ti-fresh spectrum. Thus, EPR provides compelling evidence for TiO2 reduction after HTR and, hence, H2 dissociation and concomitant charge transfer from TiOx to Au, as expected for a classical SMSI.
Fig. 2. Electronic properties of RR2Ti samples.
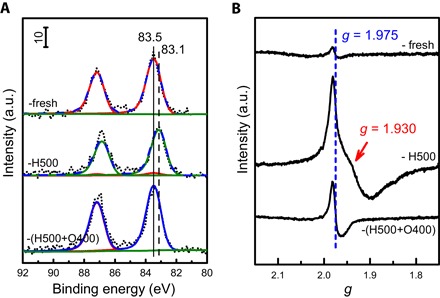
(A) Au 4f XP spectra of RR2Ti-fresh, RR2Ti-H500, and RR2Ti-(H500+O400) samples. (B) EPR spectra of the RR2Ti-fresh, RR2Ti-H500, and RR2Ti-(H500+O400) samples obtained at 100 K.
HRTEM and EELS studies
The above results demonstrate the suppression of CO adsorption and accompanying electron transfer from TiO2 to Au after reduction at elevated temperatures (especially 500°C). This loss of CO adsorption capacity does not originate from a dispersion change, is reversible (most likely arising from the encapsulation of Au NPs by a thin overlayer of TiO2 support), and has never been reported previously (32, 33, 36), except under long-term electron irradiation in an environmental TEM (52). Potential gold encapsulation was therefore explored through HRTEM, as shown in Fig. 3. As expected, Au NPs in the RR2Ti-fresh samples were naked, but were covered with a thin layer of low-contrast material following subsequent reduction, where the overlayer coverage/thickness was proportional to the reduction temperature. For samples reduced at 200° and 300°C, naked and covered Au NPs coexist (Fig. 3, B and C, and corresponding insets), whereas for RR2Ti-H400 and RR2Ti-H500 samples, only encapsulated Au NPs were observed. Hence, in accordance with DRIFTS, we propose that all Au NPs in RR2Ti-H500 were completely encapsulated, and for RR2Ti-H400, either a small proportion of Au was not fully encapsulated or the encapsulating titania overlayer was not crystalline and remained gas-permeable (53). According to a classical SMSI, this overlayer would comprise TiOx species (with x < 2) (10, 54). The nature of the overlayer in RR2Ti-H500 was therefore probed by electron energy loss spectroscopy (EELS). Figure 3G and fig. S4 reveal clear Ti L-edge signals for all measured Au NPs, proving their coverage by a Ti-containing coating. Spectral fitting of Fig. 3G (shown in fig. S5) revealed the presence of two chemically distinct Ti species: those on the support (region III) characteristic of Ti4+ and those on Au NPs (region II) characteristic of Ti3+ species, consistent with recent reports (21, 53). In the case of the reoxidized RR2Ti-(H500+O400) sample (where CO chemisorption was restored), the thin overlayer has retreated, although not completely (Fig. 3F and fig. S6), in line with previous reports of classical SMSI for PGM catalysts (55–58).
Fig. 3. HRTEM images and EELS spectra.
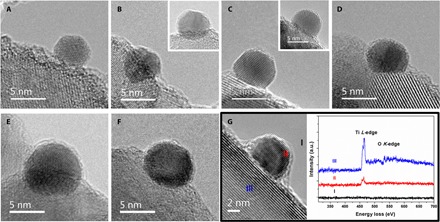
(A to F) HRTEM images of (A) RR2Ti-fresh, (B) RR2Ti-H200, (C) RR2Ti-H300, (D) RR2Ti-H400, (E) RR2Ti-H500, and (F) RR2Ti-(H500+O400). (G) EELS spectra of the RR2Ti-H500 sample. Spectra were background-subtracted.
CO oxidation catalysis
One of the key features of classical SMSI behavior is that the aforementioned HTR phenomena are reversible upon O2 treatment, as is observed for RR2Ti-(H500+O400). For example, the restoration of CO adsorption for RR2Ti-(H500+O400) (Fig. 1) and HRTEM (Fig. 3F) evidences the reversibility of CO adsorption and gold encapsulation. XPS (Fig. 2A) and CO DRIFTS (band position) suggest that electron transfer between titania and gold is also reversible. The reversible nature of the classical SMSI effect following HTR and subsequent reoxidation is expected to strongly affect catalyst activity (18, 19, 40). The catalytic performance of the present Au/TiO2 samples was therefore assessed toward CO oxidation, an industrially important reaction for pollution abatement, over two cycles of redox treatments under a model CO/O2 feedstream and compared with analogous in situ CO DRIFTS measurements (Fig. 4). Suppressed CO chemisorption following HTR was mirrored by a marked decrease in CO conversion, with catalyst reoxidation restoring both CO adsorption capacity and oxidation activity, providing strong evidence for a reversible, classical SMSI in the commercial RR2Ti system (18, 40, 56).
Fig. 4. Reversibility of RR2Ti properties under redox treatment.
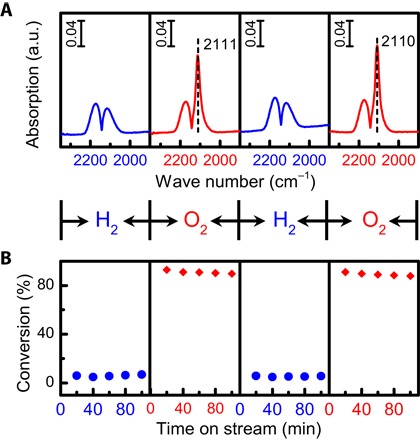
(A) In situ DRIFT spectra of CO adsorption over the RR2Ti-fresh sample following alternating pretreatment with 10 volume % H2/He at 500°C (for the first and third cycles) or 10 volume % O2/He at 400°C (for the second and fourth cycles) for 1 hour. (B) CO conversion at 80°C for the RR2Ti-fresh sample as a function of pretreatments described in (A). Reaction gas composition: 1 volume % CO + 1 volume % O2 balanced with He; gas flow rate, 33.3 ml/min; space velocity (SV), 64,452 ml gcat−1 hour−1.
The above unambiguously demonstrates that classical SMSI occurs in the commercial RR2Ti catalyst. To ascertain whether classical SMSI extended to related materials, we synthesized three Au/TiO2 catalysts using a deposition-precipitation method (59) and three popular forms of titania—P25 (Degussa), anatase, and rutile—as supports, as described in the Supplementary Materials. XRD (fig. S2, B to D) again confirmed the absence of significant Au NP size or titania phase differences within each catalyst family. In all cases, gold encapsulation (fig. S7) and the reversible suppression and recovery of CO chemisorption (fig. S8) following respective reductive and oxidation pretreatments indicate a common classical SMSI between Au NPs and these three titania supports, akin to that of RR2Ti. CO DRIFTS also highlights anatase as the most susceptible to encapsulation, followed by P25, with rutile as the least susceptible (showing only slightly suppressed CO adsorption). Similar observations were also obtained with 2- to 3-nm colloidal Au NPs on P25 (figs. S9 and S10), evidencing the generality of the SMSI following HTR of Au/TiO2 catalysts. Because the classical SMSI was caused by a reduction treatment, we hypothesized that this phenomenon may extend to other reducing gases, such as CO, at high temperature, and HRTEM and DRIFTS (fig. S11) confirmed that this high-temperature CO pretreatments of the RR2Ti catalyst induced reversible CO adsorption and Au NP encapsulation analogous to a classical SMSI effect.
The classical SMSI in PGM systems is not confined to titania and is reported over a range of reducible oxides (45). In light of this consideration, synthetic Au/Fe3O4 (prepared as described in the Supplementary Materials) and commercial Au/CeO2 (RR2Ce-2, supplied by Haruta Gold Inc.) were also subjected to similar H2 (HTR) and O2 redox protocols and characterized by XRD, HRTEM (figs. S12 and S13, respectively), and DRIFTS of chemisorbed CO (fig. S14). In both cases, neither Au NP size nor support phase exhibited significant changes following reduction or reoxidation, except for the appearance of a certain amount of Fe and α-Fe2O3, respectively, for the Au/Fe3O4 material. Reversible CO adsorption and gold encapsulation were again observed in both supports (figs. S14 and S15), similar to those seen in titania as detailed above, again evidencing a classical SMSI. Note that the threshold temperature for Au NP encapsulation of 500°C in the Au/CeO2 system is far lower than that for Pt/CeO2 and Rh/CeO2 (≥700°C) (58, 60), suggesting that, in some cases, classical SMSI is more facile in supported Au catalysts than in supported PGM catalysts.
The above results demonstrated that classical SMSI can occur in reducible oxide-supported Au catalysts. Grünert et al. (23) have previously inferred classical SMSI from surface-sensitive XPS and ISS spectroscopies for Ag/TiO2 catalysts for acrolein hydrogenation, although they did not visualize metal encapsulation, hinting that classical SMSI may extend to other IB metals. We therefore synthesized and investigated TiO2-supported Ag and Cu analogs in our Au/TiO2 systems. Figures S16 to S18 confirm that reversible CO adsorption and encapsulation of Ag NPs occurred following redox treatments, which were not attributable to particle size changes and hence classical SMSI between Ag NPs and TiO2. However, classical SMSI could not be induced for Cu/TiO2 by the same 500°C H2 reduction, where increased CO adsorption was observed (fig. S19): Corresponding EPR spectra (fig. S20) identified significant surface Ti3+ (g = 1.95) species for Ag/TiO2 but not for Cu/TiO2. Together, these results suggest that Cu is less effective in promoting TiO2 reduction, which is regarded as a prerequisite to induce an SMSI. The impact of IB metals on the reducibility of titania was further explored by temperature-programmed reduction (TPR). Figure S21 highlights only a weak HTR (804°C) for the parent TiO2 (P25), indicating minimal titania reduction. In contrast, RR2Ti-fresh exhibited a low-temperature (515°C) reduction, evidencing gold-promoted reduction of TiO2. For Cu/TiO2-fresh, this reduction shifted >700°C, indicating that Cu is a less efficient promoter of TiO2 reduction than gold but nonetheless still facilitates support reduction, and indicating that higher-temperature reduction protocols might induce an SMSI state for Cu/TiO2. To test this hypothesis, we reduced Cu/TiO2-fresh at 800°C and found complete suppression of CO adsorption (fig. S22); this could be partially reversed by subsequent 600°C oxidation. Considering that Cu can be totally oxidized to Cu2+ by calcination at 600°C and CO cannot be absorbed on Cu2+ (61), we in situ reduced the oxidized Cu/TiO2 at 200°C and discovered a significant CO adsorption. These results unambiguously demonstrate the occurrence of a high-temperature (800°C) SMSI for Cu/TiO2 and reaffirm that the reduction of surface Ti4+ is critical to induce the SMSI state (although other factors such as the surface energy/work function of metals may also play a role).
IB group metals, such as Au and Ag, are much less thermodynamically stable than PGMs when dispersed on supports and readily sinter at high temperature (28, 62). The general discovery of a classical SMSI between IB metals (Au, Ag, and Cu) and reducible oxides may facilitate the design and development of high-stability IB metal catalysts (18, 19). It is generally accepted that the low stability of supported Au catalysts has been a major challenge hindering their practical application (63), with previous studies indicating that an SMSI between Au and support could significantly improve their reaction stability (18, 19). We therefore explored whether the induction of a classical SMSI in Au/TiO2 could enhance catalyst stability toward CO oxidation, a key reaction in emission control systems, using a simulated mixture of 1.6 volume % CO, 1 volume % O2, 0.01 volume % propene, 0.0051 volume % toluene, and 10 volume % water balanced with He at 300°C; this relatively high temperature is expected to promote undesired sintering of Au NPs. For the RR2Ti-fresh catalyst, as expected, CO conversion decreased gradually during the 100-hour test, as shown in Fig. 5. High-angle annular dark field (HAADF)–scanning TEM (STEM) analysis of the fresh and used samples revealed the Au NPs sintered slightly during reaction (fig. S23). Control experiments show that deactivation of the RR2Ti-fresh catalyst was irreversible (fig. S24) and, hence, likely due to this sintering rather than the accumulation of surface carbonate (64, 65). In contrast, the RR2Ti-H400 sample, in which an SMSI operated, exhibited excellent stability, with negligible loss of CO oxidation activity throughout the test. To confirm that the excellent stability of the RR2Ti-H400 sample arose from the SMSI between Au and titania, the RR2Ti-H500 and RR2Ti-(H500+O400) samples were also explored for CO oxidation. Figure S25 shows that RR2Ti-H500 exhibited high stability (albeit a slightly lower activity) similar to the other HTR-treated sample, whereas the RR2Ti-(H500+O400) sample still underwent slow deactivation (albeit less than observed for the fresh sample). The latter findings are as expected because oxidation will reverse the SMSI, and hence, these treatments will be less effective in inhibiting Au NP sintering. Note that the 300°C reaction conditions did not reverse the SMSI in either RR2Ti-H400 or RR2Ti-H500 catalyst despite the net (slightly) oxidizing conditions, presumably reflecting the low O2 concentration (<1 volume %) and strong competitive adsorption of CO. The observation that the RR2Ti-H500 catalyst exhibits excellent activity for CO oxidation is, at first, surprising given its poor room temperature CO adsorption (Fig. 1). However, the work of several groups, including our own, have shown that complete encapsulation of Pt (66, 67) or Ir (68) by a reducible metal oxide (ceria), resulting in materials with very low CO titratable areas, produces a more active catalyst than the bare precious metal itself; metal-promoted oxygen vacancy formation in the oxide overlayer is postulated to markedly enhance reactivity.
Fig. 5. The CO conversion curves as a function of reaction time on the RR2Ti-fresh and RR2Ti-H400 samples tested at 300°C.
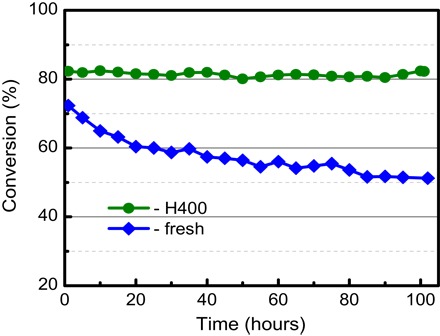
Reaction gas composition: 1.6 volume % CO, 1 volume % O2, 0.01 volume % propene, 0.0051 volume % toluene, and 10 volume % water balanced with He. For RR2Ti-H400: 40.5 mg of sample diluted with 120 mg of SiO2 powder; gas flow rate, 33.3 ml/min; SV, 49,000 ml gcat−1 min−1. For RR2Ti-fresh: 15.9 mg of sample diluted with 60 mg of SiO2 powder; gas flow rate, 33.3 ml/min; SV, 127,000 ml gcat−1 min−1.
In summary, we have shown the existence of a classical SMSI between Au and TiO2 that is relatively insensitive to the oxide phase (anatase or rutile) or reducing gas (H2 or CO) used to induce the SMSI. This classical SMSI extends to other reducible oxides, such as Fe3O4 and CeO2, and other titania-supported group IB group metals, such as Ag and even Cu. Genesis of the SMSI state in Au/TiO2 significantly enhances long-term catalyst stability toward CO oxidation. These discoveries may lead to a deeper understanding of the SMSI mechanism and its generalization to other transition metals, and unlock new design strategies for the development and operation of thermochemically stable supported group IB metal catalysts through precise engineering of the interaction between metal NPs and reducible oxide supports.
MATERIALS AND METHODS
Raw materials
Sodium hydroxide (NaOH, 98%), anatase (TiO2, 99.8%), and rutile (TiO2, 99.8%) were purchased from Aladdin Company. Poly(vinyl alcohol) [PVA; molecular weight (Mw), 10,000; 80% hydrolyzed] was purchased from Aldrich. P25 (TiO2) was purchased from Evonik Degussa. Hydrogen terachloroaurate (IV) hydrate (HAuCl4·4H2O), ferroferric oxide (Fe3O4; 99.95%), sodium borohydride (NaBH4; 98%), silver nitrate (AgNO3; 99.8%), and copper(II) nitrate hemi(pentahydrate) [Cu(NO3)2·5H2O; 98%] were purchased from Alfa Aesar. The gold samples of RR2Ti and RR2Ce-2 were supplied by Haruta Gold Inc.
Preparation of gold samples
Au samples were prepared by deposition-precipitation method (59) or colloidal deposition method (69). For the deposition-precipitation method, in a typical procedure, the pH value of 40.0-ml HAuCl4 aqueous solution (1.25 mgAu/ml) was adjusted to ~9 with NaOH solution (0.1 M) under vigorous stirring. One gram of support was added to the above HAuCl4 aqueous solution, and pH was kept at ~9 by adding NaOH solution. After vigorous stirring for 1 hour, the product was heated to 65°C and stirred for another 1 hour. The precipitates were filtered, washed with deionized water for several times to eliminate Cl− ions, dried at 60°C overnight, and denoted as Au/S-fresh (where S represents the support; S = P25, anatase, rutile, and Fe3O4).
For the colloidal deposition method, in a typical procedure, 0.2 ml of 0.25 M HAuCl4 solution and 1.33 ml of 0.5 weight % PVA (Aldrich; Mw, 10,000; 80% hydrolyzed) solution (weight ratio of Au/PVA, 1.5:1) were added to 100 ml of water under vigorous stirring. After 10 min, 2.54 ml of the 0.1 M NaBH4 solution was rapidly injected into the solution to obtain a dark orange–brown solution, indicating the formation of gold colloid. One gram of P25 was then added immediately. After vigorous stirring for 6 hours, the solids were collected by filtration after washing and dried at 60°C overnight, followed by heating in 10 volume % O2/He flow at 300°C for 1 hour with the purpose of removing the PVA. The obtained sample was denoted as C-Au/P25-fresh.
Preparation of Ag/TiO2 and Cu/TiO2 samples
Silver and copper samples were prepared by a facile adsorption method: 0.5 g of TiO2 was added to 120 ml of fresh AgNO3 aqueous solution (0.42 mgAg/ml) or 100 ml of fresh Cu(NO3)2·5H2O aqueous solution (0.39 mgCu/ml) at 30°C and stirred overnight. Then, the precipitates were filtered and washed with deionized water for several times. The samples were dried at 60°C overnight, followed by calcination in air flow at 500°C for 1 hour, and denoted as Ag/TiO2-fresh or Cu/TiO2-fresh.
Pretreatment of the fresh samples
These fresh samples were further reduced under 10 volume % H2/He or 10 volume % CO/He flow for 1 hour at different temperatures and denoted as sample-HX or sample-COX. For comparison, some reduced samples were further oxidized under 10 volume % O2/He flow for 1 hour and denoted as sample-(HX+OY) (Y, the oxidation temperature) or sample-(COX+OY). For example, RR2Ti-(H500+O400) means the RR2Ti-fresh sample was first reduced in 10 volume % H2/He flow at 500°C for 1 hour and then oxidized in 10 volume % O2/He flow at 400°C for 1 hour.
Characterization
HRTEM and HAADF-STEM analyses were performed with a JEOL JEM-2100F microscope operated at 200 kV. The EELS analysis was conducted on an FEI Tecnai G2 F20 microscope equipped with a Gatan Imaging Filter system operated at 200 kV. The chemical compositions of the covering layer of Au NPs were characterized by directly putting electron beam at the Au NPs in a STEM mode.
XRD patterns were collected at a PW3040/60 X’Pert PRO (PANalytical) diffractometer equipped with a Cu Kα radiation source (λ = 0.15432 nm), operating at 40 kV and 40 mA. A continuous mode was used to collect data in the 2θ range of 20° to 90°.
XPS was performed on a Kratos Axis HSi spectrometer with a monochromated Al Kα x-ray source operated at 90 W and a magnetic charge neutralizer. Samples were resistively heated in bespoke Ta crucible held within the analysis chamber. Reduction was performed under 1 × 10−7 torr H2 at 500°C for 5 hours, and reoxidation was performed under 1 × 10−7 torr O2 at 400°C for 5 hours, where the sample was cooled to room temperature and the analysis chamber was evacuated to 1 × 10−9 torr before spectral acquisition. Spectral processing was performed using CasaXPS version 2.3.16, where energy referencing to adventitious carbon at 284.6 eV as well as surface compositions and peak fitting were derived using appropriate instrumental response factors and common line shapes for each element.
EPR measurements were performed in a quasi in situ model using a bespoke U-shape reaction tube connected to a quartz EPR tube equipped with a greaseless high-vacuum stopcock. Samples were loaded into the U-shape reaction tube and exposed to various pretreatments. After pretreatment, samples were transferred to the EPR tube without air exposure. The EPR tube was subsequently connected to a vacuum system and evacuated at room temperature to pressures <10−4 torr. The EPR tube was then sealed and transferred for measurements at 100 K.
In situ DRIFTS spectra were acquired with a Bruker Equinox 70 spectrometer equipped with a mercury cadmium telluride detector and operated at a resolution of 4 cm−1. Twenty milligrams of the sample was loaded into the ZnSe window, which can work at a high temperature. Before CO adsorption, the sample was in situ pretreated in a flow (33.3 ml/min) of He at 120°C (for the fresh samples) or 10 volume % CO/He (for the CO atmosphere reduction sample of RR2Ti) or 10 volume % H2/He at different temperatures for 1 hour and then cooled to room temperature. The gas flow was switched to pure He to collect background spectrum. Subsequently, a mixture gas of 3 volume % CO/He (33.3 ml/min) was introduced into the reaction cell, and the spectra were collected with time until there was no change in it. For the reoxidation samples, an in situ heating progress under 10 volume % O2/He (33.3 ml/min) at a certain temperature for 1 hour was also performed before background spectrum was collected. For the reversibility spectra, before every cycle of CO adsorption, the sample was pretreated at 500°C under 10 volume % H2/He (for the first and third cycles) or at 400°C under 10 volume % O2/He atmosphere (for the second and fourth cycles) for 1 hour. After the sample was cooled to room temperature, the gas flow was switched to pure He and the background spectrum was collected.
Catalytic performance test
CO oxidation under model and simulated exhaust emission control conditions was conducted in a fixed-bed quartz reactor with a length of 200 mm and an inner diameter of 8 mm. To test the reversibility of CO oxidation under model conditions, 31 mg of RR2Ti-fresh diluted with 100 mg of SiO2 powder was first purged with He for 30 min and then pretreated at 500°C under 10 volume % H2/He (for the first and third cycles of CO conversion test) or 10 volume % O2/He atmosphere (for the second and fourth cycles of CO conversion test) for 1 hour. After cooling the sample to 80°C and purging with He for 30 min, a feed gas (1 volume % CO + 1 volume % O2 balanced with He) was passed over the catalyst bed with a flow rate of 33.3 ml/min, resulting in an SV of 64,452 ml gcat−1 hour−1. For the simulated exhaust conditions, CO oxidation was first performed under an atmosphere with water, the feed gas was 1.0 volume % CO + 1 volume % O2 + 10 volume % water balanced with He, using 10.8 mg of the catalyst diluted with 40 mg of SiO2. A full-simulated CO emission control test feed gas of 1.6 volume % CO + 1 volume % O2 + 0.01 volume % propene + 0.0051 volume % toluene + 10 volume % water balanced with He was subsequently used with a specified amount of the catalyst (diluted threefold in SiO2). The inlet and outlet gas compositions were analyzed by an on-line gas chromatograph (Hewlett Packard 6890) equipped with a TDX-01 column.
Supplementary Material
Acknowledgments
We thank Q. Fu at the Dalian Institute of Chemical Physics (DICP) for his useful discussion. We also thank H. Han at the DICP for his help in analyzing the EPR measurements. Funding: This work was supported by National Key R&D Program of China (2016YFA0202801 and 2016YFA0202804), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB17020100), the National Natural Science Foundation of China (21776270, 21476232, and 21303184), and the Department of Science and Technology of Liaoning Province (2015020086-101). B.Z. acknowledges the financial support provided by the National Natural Science Foundation of China (91545119), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (CAS) (2015152), and the Strategic Priority Research Program of the CAS (XDA09030103). Author contributions: H.T. performed the catalyst preparation, characterizations, and catalytic tests. Y.S. conducted the HRTEM examination. B.Z. and D.S. conducted the EELS examination and analysis. A.F.L., M.A.I., and K.W. conducted the XPS examination and analysis. L.L. conducted the in situ DRIFTS characterization. Y.R., J.H., M.H., X.L., and C.J. helped with the catalyst characterizations and the data analysis. B.Q., J.W., and T.Z. conceived the idea, designed the study, analyzed the data, and cowrote the paper (assisted by A.F.L.). All the authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors at wangjh@dicp.ac.cn.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/10/e1700231/DC1
fig. S1. TEM images of various Au samples and the corresponding size distributions of Au NPs.
fig. S2. XRD patterns.
fig. S3. Ti 2p XP spectra of RR2Ti-fresh, RR2Ti-H500, and RR2Ti-(H500+O400) samples.
fig. S4. EELS spectra of RR2Ti-H500 sample.
fig. S5. Fitted EELS spectra as shown in Fig. 3G with a range of 450 to 475 eV.
fig. S6. HRTEM images of the RR2Ti-(H500+O400) sample.
fig. S7. HRTEM images.
fig. S8. In situ DRIFT spectra of CO adsorption.
fig. S9. In situ DRIFT spectra of CO adsorption on C-Au/P25-H500 and C-Au/P25-(H500+O400).
fig. S10. HRTEM images.
fig. S11. SMSI behavior of RR2Ti catalyst under CO treatment.
fig. S12. XRD patterns.
fig. S13. TEM images.
fig. S14. In situ DRIFT spectra of CO adsorption.
fig. S15. HRTEM images.
fig. S16. XRD patterns of Ag/TiO2 series samples with pretreatment under various conditions.
fig. S17. HRTEM images.
fig. S18. In situ DRIFT spectra of CO adsorption on Ag/TiO2-H500 and Ag/TiO2-(H500+O500).
fig. S19. In situ DRIFT spectra of CO adsorption on Cu/TiO2-fresh and Cu/TiO2-H500.
fig. S20. EPR spectra of the Ag/TiO2 series samples and Cu/TiO2 series samples obtained at 100 K.
fig. S21. Mass spectrometry signals of H2 for H2-TPR profiles of the samples of TiO2, RR2Ti-fresh, and Cu/TiO2-fresh.
fig. S22. In situ DRIFT spectra of CO adsorption on Cu/TiO2-H800, Cu/TiO2-(H800+O600), and Cu/TiO2-(H800+O600+H200).
fig. S23. HAADF-STEM images of RR2Ti-fresh before and after CO oxidation at 300°C for 100 hours and the corresponding Au size distributions.
fig. S24. CO conversion curves as a function of reaction time on the RR2Ti-fresh tested at 300°C.
fig. S25. CO conversion curves as a function of reaction time on RR2Ti-H500 and RR2Ti-(H500+O400) tested at 300°C.
REFERENCES AND NOTES
- 1.Boudart M., Catalysis by supported metals. Adv. Catal. 20, 153–166 (1969). [Google Scholar]
- 2.Cinneide A. D. O., Clarke J. K. A., Catalysis on supported metals. Catal. Rev. 7, 213–232 (1972). [Google Scholar]
- 3.Bell A. T., The impact of nanoscience on heterogeneous catalysis. Science 299, 1688–1691 (2003). [DOI] [PubMed] [Google Scholar]
- 4.G. Ertl, H. Knözinger, F. Schüth, J. Weitkamp, Eds., Handbook of Heterogeneous Catalysis (Wiley-VCH Verlag GmbH & Co. KGaA, ed. 2, 2008), vol. 8, p. 4270. [Google Scholar]
- 5.Solymosi F., Importance of the electric properties of supports in the carrier effect. Catal. Rev. 1, 233–255 (1968). [Google Scholar]
- 6.Haruta M., Nanoparticles can open a new world of heterogeneous catalysis. J. Nanopart. Res. 5, 3–4 (2003). [Google Scholar]
- 7.Liu J., Advanced electron microscopy of metal-support interactions in supported metal catalysts. ChemCatChem 3, 934–948 (2011). [Google Scholar]
- 8.Tauster S. J., Fung S. C., Garten R. L., Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 100, 170–175 (1978). [Google Scholar]
- 9.Tauster S. J., Fung S. C., Baker R. T. K., Horsley J. A., Strong interactions in supported-metal catalysts. Science 211, 1121–1125 (1981). [DOI] [PubMed] [Google Scholar]
- 10.Sadeghi H. R., Henrich V. E., SMSI in Rh/TiO2 model catalysts: Evidence for oxide migration. J. Catal. 87, 279–282 (1984). [Google Scholar]
- 11.Tauster S. J., Strong metal-support interactions. Acc. Chem. Res. 20, 389–394 (1987). [Google Scholar]
- 12.Belzunegui J. P., Sanz J., Rojo J. M., Contribution of physical blocking and electronic effect to establishment of strong metal–support interaction in rhodium/titanium dioxide catalysts. J. Am. Chem. Soc. 114, 6749–6754 (1992). [Google Scholar]
- 13.Roberts S., Gorte R. J., A study of the migration and stability of titania on a model Rh catalyst. J. Catal. 124, 553–556 (1990). [Google Scholar]
- 14.Vannice M. A., Garten R. L., Metal-support effects on the activity and selectivity of Ni catalysts in COH2 synthesis reactions. J. Catal. 56, 236–248 (1979). [Google Scholar]
- 15.Haller G. L., Resasco D. E., Metal-support interaction: Group VIII metals and reducible oxides. Adv. Catal. 36, 173–235 (1989). [Google Scholar]
- 16.Fan L., Fujimoto K., Promotive SMSI effect for hydrogenation of carbon dioxide to methanol on a Pd/CeO2 catalyst. J. Catal. 150, 217–220 (1994). [Google Scholar]
- 17.Lee J., Burt S. P., Carrero C. A., Alba-Rubio A. C., Ro I., O’Neill B. J., Kim H. J., Jackson D. H. K., Kuech T. F., Hermans I., Dumesic J. A., Huber G. W., Stabilizing cobalt catalysts for aqueous-phase reactions by strong metal–support interaction. J. Catal. 330, 19–27 (2015). [Google Scholar]
- 18.Tang H., Wei J., Liu F., Qiao B., Pan X., Li L., Liu J., Wang J., Zhang T., Strong metal–support interactions between gold nanoparticles and nonoxides. J. Am. Chem. Soc. 138, 56–59 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Tang H., Liu F., Wei J., Qiao B., Zhao K., Su Y., Jin C., Li L., Liu J. J., Wang J., Zhang T., Ultrastable hydroxyapatite/titanium-dioxide-supported gold nanocatalyst with strong metal–support interaction for carbon monoxide oxidation. Angew. Chem. Int. Ed. 55, 10606–10611 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Bonanni S., Aït-Mansour K., Harbich W., Brune H., Effect of the TiO2 reduction state on the catalytic CO oxidation on deposited size-selected Pt clusters. J. Am. Chem. Soc. 134, 3445–3450 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Matsubu J. C., Zhang S., DeRita L., Marinkovic N. S., Chen J. G., Graham G. W., Pan X., Christopher P., Adsorbate-mediated strong metal–support interactions in oxide-supported Rh catalysts. Nat. Chem. 9, 120–127 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Sonström P., Arndt D., Wang X., Zielasek V., Bäumer M., Ligand capping of colloidally synthesized nanoparticles—A way to tune metal-support interactions in heterogeneous gas-phase catalysis. Angew. Chem. Int. Ed. 50, 3888–3891 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Grünert W., Brückner A., Hofmeister H., Claus P., Structural properties of Ag/TiO2 catalysts for acrolein hydrogenation. J. Phys. Chem. B. 108, 5709–5717 (2004). [Google Scholar]
- 24.Stephen A., Hashmi K., Homogeneous catalysis by gold. Gold Bull. 37, 51–65 (2004). [Google Scholar]
- 25.Chen M. S., Goodman D. W., The structure of catalytically active gold on titania. Science 306, 252–255 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Hutchings G., A golden future. Nat. Chem. 1, 584 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Johnston P., Carthey N., Hutchings G. J., Discovery, development, and commercialization of gold catalysts for acetylene hydrochlorination. J. Am. Chem. Soc. 137, 14548–14557 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Wolf A., Schüth F., A systematic study of the synthesis conditions for the preparation of highly active gold catalysts. Appl. Catal. A 226, 1–13 (2002). [Google Scholar]
- 29.Zanella R., Louis C., Influence of the conditions of thermal treatments and of storage on the size of the gold particles in Au/TiO2 samples. Catal. Today 107–108, 768–777 (2005). [Google Scholar]
- 30.Pesty F., Steinrück H.-P., Madey T. E., Thermal stability of Pt films on TiO2(110): Evidence for encapsulation. Surf. Sci. 339, 83–95 (1995). [Google Scholar]
- 31.Gao Y., Liang Y., Chambers S. A., Thermal stability and the role of oxygen vacancy defects in strong metal support interaction—Pt on Nb-doped TiO2(100). Surf. Sci. 365, 638–648 (1996). [Google Scholar]
- 32.Zhang L., Persaud R., Madey T. E., Ultrathin metal films on a metal oxide surface: Growth of Au on TiO2 (110). Phys. Rev. B 56, 10549–10557 (1997). [Google Scholar]
- 33.Goodman D. W., “Catalytically active Au on Titania:” Yet another example of a strong metal support interaction (SMSI)? Catal. Lett. 99, 1–4 (2005). [Google Scholar]
- 34.Shastri A. G., Datye A. K., Schwank J., Gold-titania interactions: Temperature dependence of surface area and crystallinity of TiO2 and gold dispersion. J. Catal. 87, 265–275 (1984). [Google Scholar]
- 35.Meyer R., Lemire C., Shaikhutdinov S. K., Freund H.-J., Surface chemistry of catalysis by gold. Gold Bull. 37, 72–124 (2004). [Google Scholar]
- 36.Fu Q., Wagner T., Olliges S., Carstanjen H.-D., Metal–oxide interfacial reactions: Encapsulation of Pd on TiO2 (110). J. Phys. Chem. B. 109, 944–951 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Dyrbeck H., Hammer N., Rønning M., Blekkan E. A., Catalytic oxidation of hydrogen over Au/TiO2 catalysts. Top. Catal. 45, 21–24 (2007). [Google Scholar]
- 38.Panayotov D. A., Burrows S. P., Yates J. T. Jr, Morris J. R., Mechanistic studies of hydrogen dissociation and spillover on Au/TiO2: IR spectroscopy of coadsorbed CO and H-donated electrons. J. Phys. Chem. C 115, 22400–22408 (2011). [Google Scholar]
- 39.Sun K., Kohyama M., Tanaka S., Takeda S., A study on the mechanism for H2 dissociation on Au/TiO2 catalysts. J. Phys. Chem. C 118, 1611–1617 (2014). [Google Scholar]
- 40.Liu X., Liu M.-H., Luo Y.-C., Mou C.-Y., Lin S. D., Cheng H., Chen J.-M., Lee J.-F., Lin T.-S., Strong metal–support interactions between gold nanoparticles and ZnO nanorods in CO oxidation. J. Am. Chem. Soc. 134, 10251–10258 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Cíes J. M., del Río E., López-Haro M., Delgado J. J., Blanco G., Collins S., Calvino J. J., Bernal S., Fully reversible metal deactivation effects in gold/ceria–zirconia catalysts: Role of the redox state of the support. Angew. Chem. Int. Ed. 49, 9744–9748 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Mitchell C. E. J., Howard A., Carney M., Egdell R. G., Direct observation of behaviour of Au nanoclusters on TiO2(110) at elevated temperatures. Surf. Sci. 490, 196–210 (2001). [Google Scholar]
- 43.Kitchin J. R., Barteau M. A., Chen J. G., A comparison of gold and molybdenum nanoparticles on TiO2(110) 1×2 reconstructed single crystal surfaces. Surf. Sci. 526, 323–331 (2003). [Google Scholar]
- 44.Mihaylov M., Knözinger H., Hadjiivanov K., Gates B. C., Characterization of the oxidation states of supported gold species by IR spectroscopy of adsorbed CO. Chem. Ing. Tech. 79, 795–806 (2007). [Google Scholar]
- 45.Tauster S. J., Fung S. C., Strong metal-support interactions: Occurrence among the binary oxides of groups IIA–VB. J. Catal. 55, 29–35 (1978). [Google Scholar]
- 46.Sasaki T., Ebina Y., Kitami Y., Watanabe M., Oikawa T., Two-dimensional diffraction of molecular nanosheet crystallites of titanium oxide. J. Phys. Chem. B. 105, 6116–6121 (2001). [Google Scholar]
- 47.Howe R. F., Gratzel M., EPR observation of trapped electrons in colloidal titanium dioxide. J. Phys. Chem. 89, 4495–4499 (1985). [Google Scholar]
- 48.Okumura M., Coronado J. M., Soria J., Haruta M., Conesa J. C., EPR study of CO and O2 interaction with supported Au catalysts. J. Catal. 203, 168–174 (2001). [Google Scholar]
- 49.Coronado J. M., Maira A. J., Conesa J. C., Yeung K. L., Augugliaro V., Soria J., EPR study of the surface characteristics of nanostructured TiO2 under UV irradiation. Langmuir 17, 5368–5374 (2001). [Google Scholar]
- 50.Conesa J. C., Malet P., Munuera G., Sanz J., Soria J., Magnetic resonance studies of hydrogen-reduced rhodium/titanium dioxide catalysts. J. Phys. Chem. 88, 2986–2992 (1984). [Google Scholar]
- 51.Li G., Dimitrijevic N. M., Chen L., Nichols J. M., Rajh T., Gray K. A., The important role of tetrahedral Ti4+ sites in the phase transformation and photocatalytic activity of TiO2 nanocomposites. J. Am. Chem. Soc. 130, 5402–5403 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Kuwauchi Y., Yoshida H., Akita T., Haruta M., Takeda S., Intrinsic catalytic structure of gold nanoparticles supported on TiO2. Angew. Chem. Int. Ed. 51, 7729–7733 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Zhang S., Plessow P. N., Willis J. J., Dai S., Xu M., Graham G. W., Cargnello M., Abild-Pedersen F., Pan X., Dynamical observation and detailed description of catalysts under strong metal–support interaction. Nano Lett. 16, 4528–4534 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Takatani S., Chung Y.-W., Strong metal–support interaction in NiTiO2: Auger and vibrational spectroscopy evidence for the segregation of TiOx (x ≈ −1) on Ni and its effects on CO chemisorption. J. Catal. 90, 75–83 (1984). [Google Scholar]
- 55.Logan A. D., Braunschweig E. J., Datye A. K., Smith D. J., Direct observation of the surfaces of small metal crystallites: Rhodium supported on titania. Langmuir 4, 827–830 (1988). [Google Scholar]
- 56.Braunschweig E. J., Logan A. D., Datye A. K., Smith D. J., Reversibility of strong metal–support on RhTiO2. J. Catal. 118, 227–237 (1989). [Google Scholar]
- 57.Bernal S., Botana F. J., Calvino J. J., López C., Pérez-Omil J. A., Rodríguez-Izquierdo J. M., High-resolution electron microscopy investigation of metal-support interactions in Rh/TiO2. J. Chem. Soc. Faraday Trans. 92, 2799–2809 (1996). [Google Scholar]
- 58.Bernal S., Blanco G., Calvino J. J., López-Cartes C., Pérez-Omil J. A., Gatica J. M., Stephan O., Colliex C., Electron microscopy (HREM, EELS) study of the reoxidation conditions for recovery of NM/CeO2 (NM: Rh, Pt) catalysts from decoration or alloying phenomena. Catal. Lett. 76, 131–137 (2001). [Google Scholar]
- 59.Zhao K., Qiao B., Wang J., Zhang Y., Zhang T., A highly active and sintering-resistant Au/FeOx–hydroxyapatite catalyst for CO oxidation. Chem. Commun. 47, 1779–1781 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Bernal S., Calvino J. J., Cauqui M. A., Gatica J. M., López Cartes C., Pérez Omil J. A., Pintado J. M., Some contributions of electron microscopy to the characterisation of the strong metal–support interaction effect. Catal. Today 77, 385–406 (2003). [Google Scholar]
- 61.Qiao B., Wang A., Lin J., Li L., Su D., Zhang T., Highly effective CuO/Fe(OH)x catalysts for selective oxidation of CO in H2-rich stream. Appl. Catal. B 105, 103–110 (2011). [Google Scholar]
- 62.Amin S. A., Pazouki M., Hosseinnia A., Synthesis of TiO2-Ag nanocomposite with sol-gel method and investigation of its antibacterial activity against E. coli. Powder Technol. 196, 241–245 (2009). [Google Scholar]
- 63.Corti C. W., Holliday R. J., Thompson D. T., Progress towards the commercial application of gold catalysts. Top. Catal. 44, 331–343 (2007). [Google Scholar]
- 64.Qiao B., Wang A., Yang X., Allard L. F., Jiang Z., Cui Y., Liu J., Li J., Zhang T., Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Qiao B., Wang A., Li L., Lin Q., Wei H., Liu J., Zhang T., Ferric oxide-supported Pt subnano clusters for preferential oxidation of CO in H2-rich gas at room temperature. ACS Catal. 4, 2113–2117 (2014). [Google Scholar]
- 66.Hardacre C., Ormerod R. M., Lambert R. M., Platinum-promoted catalysis by ceria: A study of carbon monoxide oxidation over Pt(111)/CeO2. J. Phys. Chem. 98, 10901–10905 (1994). [Google Scholar]
- 67.Hardacre C., Rayment T., Lambert R. M., Platinum/ceria CO oxidation catalysts derived from Pt/Ce crystalline alloy precursors. J. Catal. 158, 102–108 (1996). [Google Scholar]
- 68.Huang Y., Wang A., Li L., Wang X., Su D., Zhang T., “Ir-in-ceria”: A highly selective catalyst for preferential CO oxidation. J. Catal. 255, 144–152 (2008). [Google Scholar]
- 69.Liu Y., Jia C.-J., Yamasaki J., Terasaki O., Schüth F., Highly active iron oxide supported gold catalysts for CO oxidation: How small must the gold nanoparticles be? Angew. Chem. Int. Ed. 49, 5771–5775 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/10/e1700231/DC1
fig. S1. TEM images of various Au samples and the corresponding size distributions of Au NPs.
fig. S2. XRD patterns.
fig. S3. Ti 2p XP spectra of RR2Ti-fresh, RR2Ti-H500, and RR2Ti-(H500+O400) samples.
fig. S4. EELS spectra of RR2Ti-H500 sample.
fig. S5. Fitted EELS spectra as shown in Fig. 3G with a range of 450 to 475 eV.
fig. S6. HRTEM images of the RR2Ti-(H500+O400) sample.
fig. S7. HRTEM images.
fig. S8. In situ DRIFT spectra of CO adsorption.
fig. S9. In situ DRIFT spectra of CO adsorption on C-Au/P25-H500 and C-Au/P25-(H500+O400).
fig. S10. HRTEM images.
fig. S11. SMSI behavior of RR2Ti catalyst under CO treatment.
fig. S12. XRD patterns.
fig. S13. TEM images.
fig. S14. In situ DRIFT spectra of CO adsorption.
fig. S15. HRTEM images.
fig. S16. XRD patterns of Ag/TiO2 series samples with pretreatment under various conditions.
fig. S17. HRTEM images.
fig. S18. In situ DRIFT spectra of CO adsorption on Ag/TiO2-H500 and Ag/TiO2-(H500+O500).
fig. S19. In situ DRIFT spectra of CO adsorption on Cu/TiO2-fresh and Cu/TiO2-H500.
fig. S20. EPR spectra of the Ag/TiO2 series samples and Cu/TiO2 series samples obtained at 100 K.
fig. S21. Mass spectrometry signals of H2 for H2-TPR profiles of the samples of TiO2, RR2Ti-fresh, and Cu/TiO2-fresh.
fig. S22. In situ DRIFT spectra of CO adsorption on Cu/TiO2-H800, Cu/TiO2-(H800+O600), and Cu/TiO2-(H800+O600+H200).
fig. S23. HAADF-STEM images of RR2Ti-fresh before and after CO oxidation at 300°C for 100 hours and the corresponding Au size distributions.
fig. S24. CO conversion curves as a function of reaction time on the RR2Ti-fresh tested at 300°C.
fig. S25. CO conversion curves as a function of reaction time on RR2Ti-H500 and RR2Ti-(H500+O400) tested at 300°C.