

Abstract
Long QT syndrome (LQT) is a pro-arrhythmogenic condition with life threatening complications. Fifteen genes have been associated with congenital LQT however, the genetic causes remain unknown in more than 20% of cases.
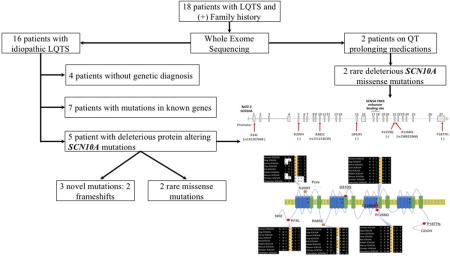
Eighteen patients with history of palpitations, presyncope, syncope and prolonged QT were referred to the Yale Cardiovascular Genetics Program. All subjects underwent whole exome sequencing (WES) followed by confirmatory Sanger sequencing. Mutation burden analysis was carried out using WES data from sixteen subjects with no identifiable cause of LQT.
Deleterious and novel SCN10A mutations were identified in three of the sixteen patients (19%) with idiopathic LQT. These included two frameshifts and one missense variants (p.G810fs, p.R1259Q, and p.P1877fs). Further analysis identified two damaging SCN10A mutations with allele frequencies of ~0.2% (p.R14L, p.R1268Q) in two independent cases. None of the SCN10A mutation carriers had mutations in known arrhythmia genes. Damaging SCN10A mutations (p.R209H, p.R485C) were also identified in the two subjects on QT prolonging medications.
Our findings implicate SCN10A in LQT. The presence of frameshift mutations suggests loss of function as the underlying disease mechanism. The common association with AF suggests a unique mechanism of disease for this LQT gene.
Graphical Abstract
1. Introduction
Long QT syndrome (LQT) is a pro-arrhythmogenic condition that increases the risk of a unique life threatening polymorphic ventricular tachycardia known as “torsades de pointes”, and sudden cardiac death (SCD). Congenital LQTs are inherited disorders caused by mutations in cardiac conduction channels or associated proteins, and are estimated to affect 0.005% to 0.05% of the general population (1). LQT is also accounted for by QT prolonging drugs, electrolyte abnormalities (2), ischemic heart disease (3) or structural heart disease; a condition that is often referred to as acquired LQT. With the advent of genome sequencing it is evident that genetic variants with small effects also account for some, if not all, subclinical acquired LQTs that manifest themselves in the presence of additional precipitating factors (4, 5). It has also been estimated that 10 to 36% of patients with LQT genotypes are silent mutation carriers (6, 7).
To this date fifteen different types of congenital LQT have been characterized and account for about 80% of inherited long QT cases. These correspond to mutations in genes encoding potassium channels (8–13), calcium channels (14), calcium signaling proteins (15–17), anchoring proteins (18, 19), transport proteins (20, 21), and voltage gated sodium channels (22, 23). Most of these genes have been established as casual genes for LQT based on linkage or segregation analysis, or in the case of KCNE2, CAV3, SNTA1 and CALM2 genes by association studies. The genetic cause of congenital LQT remains unknown in at least 20% of cases (24).
The SCN10A gene encodes the alpha subunit of a voltage-gated sodium channel (Nav1.8), which is expressed in the peripheral nervous system but also has low expression in atrial and ventricular cardiomyocytes and neurons of the heart (25–28). Genome wide association studies implicated the SCN10A gene in cardiac conduction (29–31). In addition, mutations in the SCN10A gene have been linked to cardiac arrhythmias such as Brugada syndrome (32–36), atrial fibrillation (37–40) and sudden cardiac death (41).
SCN10A influences cardiac conduction via three proposed mechanisms based on a multitude of in-vitro and in-vivo studies (42). Given the expression of Nav1.8 in cardiac myocytes, it can have direct effects on cellular physiology (25, 26, 30). Alternatively, indirect effects could be mediated via modifying the expression of SCN5A gene, located immediately downstream of its 3′ end. The presence of an enhancer binding domain of SCN5A within the SCN10A gene, encompassing exons 17 and 18, supports this concept (43–45). Lastly, SCN10A, which has robust expression in the cholinergic vagal neurons and dorsal root ganglia, has been associated with modulation of cardio-vagal input from the peripheral nervous system (27, 46, 47).
No prior studies attributed QT prolongation to mutations in SCN10A. In this study, we report a series of patients who were referred to our cardiovascular genetics clinic for genetic screening for prolonged QT associated with either syncope, pre-syncope, SCD, or AF and were found by whole exome sequencing (WES) to have damaging mutations in the SCN10A gene.
2. Materials and Methods
Study Subjects
Patients were referred to the Yale Cardiovascular Genetics team for genetic screening of SCD and/or cardiac arrhythmias. The protocols were approved by the institutional review board at Yale University School of Medicine. Written consent to participate in the study and to undergo genetic sequencing was obtained from all patients. Detailed clinical information, including laboratory data and clinical imaging were collected. Potential non-genetic causes for prolonged QT were excluded. Out of the eighteen patients referred for prolonged QT, palpitation and syncope/presyncope, two were on QT prolonging medications and hence were excluded from the initial mutation burden analysis. Five out of the sixteen patients had already undergone targeted panel genotyping for LQT in previous years with negative results.
Genomic DNA was extracted from peripheral blood leukocytes and sent for exome sequencing. Family history was obtained from the index cases, and pedigrees were constructed based on self-reported phenotypes. Family members were not available for segregation analysis. Thus, we proceeded with a mutation burden analysis.
Whole Exome Sequencing and Targeted Sequence Capture
Genomic DNA was captured on exomes at the W.M. Keck Facility at Yale University using Roche NimbleGen 2.1M Human Exome Array, as described earlier (48). In brief, DNA libraries were prepared and sequenced on the Illumina Genome Analyzer, followed by image analysis and base calling. Sequences were aligned against human reference genome (UCSC Genome Browser hg19) and processed using MAQ program SAMtools. SAMtools was also used for the single-nucleotide variant detection and filtering against the reference genome as described earlier. Filters were applied against published databases. A computer script was designed for variants annotation based on the novelty, conservation, tissue expression and their effect on protein function. They were considered nonconservative if the substituted amino acid was conserved in all species. Polyphen, SIFT and CADD scoring were used for in silico prediction of pathogenicity of the mutations (49). Genetic intolerance score was calculated using RVIS (Residual Variation Intolerance Score) (50).
We first screened for all variants in known LQT-associated genes with allele frequencies < 1% in the ExAC database. Based on the prevalence of congenital long QT syndrome (1:2,000 to 1:20,000) (1) and percentage of unknown genes (20%) we used a stringent filtering for discovery of novel variants. All variants with allele frequencies greater than 1:100,000 in the ExAC database, or variants considered benign either by by PolyPhen or SIFT prediction software were filtered out. Once the disease associated variant(s) were identified we screened the database for presence of variants with allele frequencies of less 1% in EXAC database in the same gene(s). In addition, we screened for variants in genes encoding cardiac voltage gated calcium, sodium, or potassium channels, with allele frequencies less than 1%.
The 1,000 Genomes Project and an exome database of 2,000 healthy white subjects were also used as reference. Confirmatory Sanger sequencing was carried out for all variants of interest.
Literature and Database Review
We queried existing genetic databases (EXAC) for mutation burden analysis in the SCN10A gene. EXAC contained 121,412 alleles in 60,706 subjects with available SCN10A sequences. We filtered the database for rare damaging mutations that have allele frequency less than 0.001%. The number of mutation carriers was calculated using the Hardy-Weinberg equilibrium model. Assuming low prevalence of prolonged QT in the general population, we used the EXAC database as a control and examined the association between rare deleterious SCN10A mutations and long QT. The odds ratio, standard error and 95% confidence interval were calculated (51) and the p-value was calculated using a two-tailed Chi-Squared test with Yates’ correction. We also performed a literature review on all reported SCN10A mutations. All the reported cases, disease associations and corresponding ECG parameters were summarized.
3. Results
SCN10A Mutations Among Patients with QT Prolongation Referred for Genetic Testing
Of the 18 patients referred for QT prolongation, 16 were not on any QT-prolonging medications and did not have any other identifiable cause for prolonged QT. Mutations in known arrhythmia genes were identified in 7 out of the 16 patients with idiopathic LQT. Five of those patients had pathogenic variants in known LQT genes; AKAP9 (p.Q3520H), ANK2 (p.V3632I), ANK2 (p.E1449G), CAV3 (p.T78M), and KCNQ1 (p.L266P). One had mutations in RBM20 (p.D996Y) and TTN (c.32562-insAGA) that are associated with arrhythmias and dilated cardiomyopathy (52, 53). Another patient had a mutation in CTNNA3 (p.H727R), a gene associated with arrhythmogenic right ventricular cardiomyopathy (54).
The mutation burden analysis of WES using all variants with allele frequencies <1:100,000 revealed 3 independent novel heterozygous deleterious mutations in SCN10A gene in 3 subjects without mutations in known LQT genes with a P-value<0.0001 and an odds ratio of 90.9 (95% confidence interval of 31.4 to 263.2; supplemental table 1). No other mutations were found in the same gene in more than 2 subjects.
Two patients had completely novel frameshift mutations (p.G810fs and p.P1877fs). The subject with p.G810fs variant was a 53 years old patient with palpitations, pre-syncope, paroxysmal AF, and a biphasic T-wave on ECG. His corrected QT (QTc) was prolonged at 466 msec. The proband with the p.P1877fs variant presented at 33 years of age due to palpitations and a QTc of 519 msec. He had shortening of his QTc during an exercise stress test, and did not have AF. In this subject, genetic variants in AKAP-9 (p.R3704Q) and ANK-2 (p.D955G) were also identified. The variant in AKAP-9 was predicted to be benign, whereas the ANK-2 variant falls in the non-canonical transcript of the protein. A third completely novel variant was identified in a patient with pre-syncope and palpitations. The mutation, which results in p.R1259Q substitution was predicted to be damaging by PolyPhen and SIFT (table 1). None of the SCN10A mutations carriers had neuropathy. Of the five patients that had previously undergone panel genotyping, only one had an SCN10A mutation, while the disease gene for the other four remains unidentifiable at this point.
Table 1.
Population characteristics of SCN10A mutation carriers. Clinical data, ECG parameters along with the genetic variants in the studied patients with idiopathic QT prolongation.
| Protein Variant |
Incidence in EXAC |
Age1 | Sex | Fam hx2 |
Afib3 | Symptoms4 | ECG characteristics5 | PolyP8 | SIFT9 | CADD10 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||||||
| HR | PR | QRS | QT | QTc | TdP6 | T-wave7 | |||||||||||||
| R14L | 0.001928 | 48 | F | Yes | No | Syncope | 81 | 168 | 102 | 444 | 516 | No | Normal | D | D | 27 | |||
| G810fs | Novel | 53 | M | Yes | Yes | Pre-syncope | 60 | 200 | 98 | 460 | 466 | No | Biphasic | D | D | – | |||
| R1259Q | Novel | 71 | F | Yes | Yes | Pre-syncope, palpitations | 73 | N/A | 106 | 488 | 535 | No | Normal | D | D | 35 | |||
| R1268Q | 0.001765 | 48 | M | Yes | Yes | Pre-syncope, palpitations | 59 | 78 | 104 | 444 | 437 | No | Normal | B | D | 28 | |||
| P1877fs | Novel | 33 | M | No | No | Palpitations | 84 | 158 | 102 | 460 | 519 | No | Notched | D | D | – | |||
Age at the time of diagnosis is reported in years.
Family history of syncope, long QT syndrome, or sudden cardiac death.
Presence of atrial fibrillation (Afib).
Symptoms experienced by the patient without stress. Stress tests were performed in all patients except R1268Q. QTc lengthening was observed only with R14L with the stress test.
All ECG intervals are reported in msecs.
TdP refers to torsades de pointes.
T-wave morphology, checked for T-wave alternans, and notched T-waves.
PolyP: Polyphen in silico prediction software, D= damaging, B= benign.
SIFT in silico prediction software, D= damaging
CADD score, a score of more than 15 is considered pathogenic.
Given that nonsynonymous SCN10A mutations with allele frequencies as high as 1% have been associated with Brugada syndrome and AF, we screened the remaining subjects for SCN10A mutations using a less stringent frequency of <1%. The analysis revealed 2 additional missense mutations. One mutation leading to p.R14L amino acid substitution in SCN10A was identified in a 48 years old woman who presented with syncope and LQT and had no identifiable mutation in known LQT genes. The p.R14L variant, which based on PolyPhen and SIFT is predicted to be damaging (table 1) has an allele frequency of 0.19% in EXAC database and has been previously associated with AF and Brugada but not long QT (table 2). The second missense mutation in SCN10A resulted in p.R1268Q substitution and was identified in a subject with familial AF, long QT, pre-syncope and palpitations of no identifiable cause, who also had no identifiable mutation in known LQT genes. The p.R1268Q variant has an allele frequency of 0.18% in EXAC database (table 1), but its disease association was not known. The variant was predicted to be damaging by SIFT only.
Table 2. Summary of reported SCN10A mutations with known clinical significance.
All reported cases are summarized along with electrocardiographic characteristics when available.
| ECG characteristics7
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation (dbSNP)1 |
Ref2 | n3 | G4 | Age5 | Phenotype | Family Hx6 | HR | PR | QRS | QTc | SIFT | Polyphen | Conservation | MAF8 |
| R14L (rs141207048) | 1 | 1 | M | 20 | Persistent AF | – | – | – | – | – | Damaging | Probably-damaging | Conserved | 1.93E-03 |
| 3 | 1 | – | 31 | AVNRT, paroxysmal AF | – | 112 | 206 | 98 | 434 | Damaging | Probably Damaging | Conserved | 1.93E-03 | |
| 4 | 1 | F | 50 | BrS, dizziness/palpitations | Yes | 73 | 234 | 112 | 443 | Damaging | Probably Damaging | Conserved | 2.00E-03 | |
| 4 | 1 | M | 19 | BrS | No | 66 | 185 | 113 | 416 | Damaging | Probably Damaging | Conserved | 2.00E-03 | |
| 4 | 1 | M | 65 | BrS, SCD, syncope | Yes | 54 | 194 | 94 | 378 | Damaging | Probably Damaging | Conserved | 2.00E-03 | |
| 4 | 1 | M | 49 | BrS, dizziness/palpitations | N/A | 88 | 130 | 110 | 411 | Damaging | Probably Damaging | Conserved | 2.00E-03 | |
| V94G (rs202143516) | 3 | 1 | – | 28 | Paroxysmal AF | PPM in dad and sister | 57 | 144 | 102 | 414 | Damaging | Probably Damaging | Conserved | 8.11E-04 |
| 3 | 1 | – | 31 | Paroxysmal AF, non-hemorrhagic stroke | – | 64 | 132 | 84 | 396 | Damaging | Probably Damaging | Conserved | 8.11E-04 | |
| T137M (rs148663098) | 4 | 1 | M | 37 | BrS | No | 59 | 205 | 72 | 348 | Tolerated | Benign | Conserved | 7.70E-05 |
| Y158D (rs202192818) | 1, 8 | 1 | F | 48 | Paroxysmal AF | – | – | – | – | NP | Damaging | Probably Damaging | Conserved | 2.64E-04 |
| 1, 8 | 1 | M | 52 | Persistent AF | – | – | – | – | NP | Damaging | Probably Damaging | Conserved | 2.64E-04 | |
| 3 | 1 | – | 32 | Persistent AF | – | 103 | 186 | 86 | 418 | Damaging | Probably Damaging | Conserved | 2.64E-04 | |
| W189R (−) | 7 | 1 | F | 63 | – | no | 77 | 180 | 60 | 453 | Damaging | Damaging | Conserved | 0.00E+00 |
| A200V (−) | 5 | – | – | – | BrS association | – | – | – | – | – | damaging | Probably Damaging | conserved | 1.08E-04 |
| I206M (rs74717885) | 4 | 1 | M | 39 | BrS, syncope | No | 78 | 168 | 80 | 388 | Damaging | Benign | Not conserved | 1.16E-02 |
| 4 | 1 | M | 58 | BrS, syncope | No | 84 | 210 | 100 | 408 | Damaging | Benign | Not conserved | 1.16E-02 | |
| S242T (rs140288103) | 4 | 1 | M | 20 | BrS, SCD, syncope | No | 67 | 170 | 105 | 396 | Damaging | Probably Damaging | Conserved | 2.31E-04 |
| E285K (rs146151670) | 1 | 1 | F | 48 | Paroxysmal AF | – | – | – | – | – | Tolerated | Benign | Not conserved | 3.71E-04 |
| F386C (rs78555408) | 4 | 1 | M | 65 | BrS, SCD | No | 55 | 200 | 116 | 464 | Damaging | Probably Damaging | Conserved | 2.00E-03 |
| L388P (rs199734710) | 4 | 1 | M | 56 | BrS, dizziness/palpitations | N/A | 86 | 180 | 112 | 418 | Damaging | Probably Damaging | Conserved | 2.31E-04 |
| 417delK (−) | 1 | 1 | M | 29 | Permanent AF, SVR | – | – | – | – | – | – | – | – | – |
| H506N (−) | 1 | 1 | M | 60 | Paroxysmal AF | – | – | – | – | – | Tolerated | Benign | Conserved | 8.40E-06 |
| c.599+1 (rs75991777) | 3 | 1 | – | 35 | Paroxysmal AF, aflutter, inducible VT and ICD, normal coronary angiography | SCD and AF | 63 | 162 | 116 | 421 | – | – | – | – |
| V620I (rs151303346) | 4 | 1 | F | 46 | BrS, SCD | No | 53 | 280 | 84 | 406 | Damaging | Probably Damaging | Conserved | 3.84E-04 |
| I671V (−) | 5 | – | – | – | BrS association | – | – | – | – | – | N/A | Benign | conserved | 0.00E+00 |
| F719S (−) | 1 | 1 | M | 25 | Persistent AF | – | – | – | – | – | Damaging | Possibly-damaging | Conserved | 4.13E-05 |
| G810W (rs145712124) | 1 | 1 | M | 51 | Paroxysmal AF | – | – | – | – | – | Damaging | probably-damaging | Conserved | 1.89E-04 |
| R814H (rs139861061) | 1, 8 | 1 | F | 48 | Paroxysmal AF | – | – | – | – | NP | Damaging | Possibly-damaging | Not conserved | 3.13E-04 |
| 1, 8 | 1 | M | 52 | Persistent AF | – | – | – | – | NP | Damaging | Possibly-damaging | Not conserved | 3.13E-04 | |
| 3 | 1 | – | 32 | persist AF | – | 103 | 186 | 86 | 418 | Damaging | Possibly Damaging | Not Conserved | 3.13E-04 | |
| E825D (rs146028829) | 3 | 1 | – | 18 | Paroxysmal AF, AVNRT | – | 84 | 172 | 72 | 423 | Tolerated | Benign | Not Conserved | 7.66E-04 |
| R844H (−) | 7 | 1 | M | 35 | SCD | SCD | 60 | 144 | 72 | 336 | Damaging | Damaging | Conserved | 1.65E-05 |
| 7 | 1 | M | 56 | – | No | 77 | 160 | 80 | 430 | Damaging | Damaging | Conserved | 1.65E-05 | |
| F938YFSX12 (−) | 4 | 1 | F | 42 | BrS, syncope, dizziness/palpitations | No | 68 | 160 | 120 | 426 | – | – | – | – |
| I999L (−) | 3 | 1 | – | 35 | Paroxysmal AF, IRBBB | Mother with SVPCs | 61 | 160 | 100 | 412 | Tolerated | Benign | Conserved | 0.00E+00 |
| P1045T (rs73062575) | 6 | 1 | F | 25 | AVNRT, concealed BrS, chest pain, migraines | – | – | – | – | – | Tolerated | Benign | Not conserved | 5.00E-01 |
| A1073V (rs6795970) | 2 | 743 | – | – | PR interval duration in all patients, and heart rate response in Afib patients. | – | – | – | – | – | benign | Benign | Not conserved | 3.41E-01 |
| 5 | – | – | – | BrS association | – | – | – | – | – | benign | Benign | Not conserved | 3.41E-01 | |
| V1078A (−) | 6 | 1 | M | 44 | BrS, AVNRT, PVC, VT, syncope | – | – | – | – | – | Tolerated | Benign | Not conserved | 0.00E+00 |
| D1080N (−) | 4 | 1 | M | 28 | BrS | No | 71 | 240 | 158 | 409 | Tolerated | Probably Damaging | Conserved | 7.70E-05 |
| F1115L (−) | 1 | 1 | M | 58 | Persistent AF | – | – | – | – | – | Damaging | Probably Damaging | Conserved | 0.00E+00 |
| W1135R (−) | 1 | 1 | F | 34 | Persistent AF | – | – | – | – | – | Damaging | Probably Damaging | Conserved | 0.00E+00 |
| I1225T (rs139638446) | 4 | 1 | M | 44 | BrS, dizziness/palpitations | N/A | 80 | 180 | 110 | 404 | Damaging | Probably Damaging | Conserved | 4.61E-04 |
| 4 | 1 | M | 44 | BrS, syncope | No | 3 | 200 | 116 | 525 | Damaging | Probably Damaging | Conserved | 4.61E-04 | |
| R1268Q (rs138832868) | 1 | 1 | M | 16 | Paroxysmal AF | – | – | – | – | – | Damaging | Benign | Conserved | 1.77E-03 |
| 1 | 1 | M | 46 | Paroxysmal AF | – | – | – | – | – | Damaging | Benign | Conserved | 1.77E-03 | |
| 3 | 1 | – | 30 | Paroxysmal AF, IRBBB | – | 59 | 152 | 94 | 394 | Damaging | Probably Damaging | Conserved | 1.77E-03 | |
| 3 | 1 | – | 23 | AF, Aflutter | – | 96 | – | – | – | Damaging | Probably Damaging | Conserved | 1.77E-03 | |
| 4 | 1 | F | 49 | BrS, syncope, dizziness/palpitations | Yes | 79 | 170–200 | 102 | 436 | Damaging | Probably Damaging | Conserved | 2.08E-03 | |
| 6 | 1 | F | 28 | AVNRT, suspected BrS | – | – | – | – | – | Damaging | Benign | Conserved | 1.00E-01 | |
| V1287I (rs145032037) | 1 | 1 | M | 31 | Persistent AF | – | – | – | – | – | Damaging | Probably Damaging | Conserved | 5.03E-04 |
| 1 | 1 | M | 52 | Persistent AF | – | – | – | – | – | Damaging | Probably Damaging | Conserved | 5.03E-04 | |
| G1299A (−) | 5 | – | – | – | BrS association | – | – | – | – | – | damaging | Probably Damaging | conserved | 0.00E+00 |
| N1328K (−) | 7 | 1 | M | 68 | Syncope | no | 76 | 140 | 160 | 450 | Damaging | Damaging | Conserved | 0.00E+00 |
| S1337T (rs11711062) | 4 | 1 | M | 46 | BrS, syncope, dizziness/palpitations | No | 69 | 148 | 105 | 448 | Damaging | Benign | Not conserved | 4.54E-03 |
| R1380Q (rs149155352) | 1 | 1 | M | 60 | Paroxysmal AF | – | – | – | – | – | Damaging | Probably Damaging | Conserved | 9.08E-05 |
| 7 | 1 | M | 50 | VF | Seizure, syncope, SCD | 59 | 200 | 60 | 388 | Damaging | Damaging | Conserved | 9.08E-05 | |
| G1406D (−) | 4 | 1 | M | 36 | BrS, dizziness/palpitations | No | 73 | 150 | 110 | 442 | Damaging | Probably Damaging | Conserved | 8.26E-06 |
| C1523Y (rs142217269) | 1 | 1 | M | 47 | Persistent AF | – | – | – | – | – | Damaging | Probably Damaging | Conserved | 1.11E-03 |
| 3 | 1 | – | 30 | pAF | AF | 54 | 154 | 96 | 398 | Damaging | Probably Damaging | Conserved | 1.11E-03 | |
| V1548F (−) | 1 | 1 | M | 28 | Paroxysmal AF, SVR | – | – | – | – | – | Damaging | Benign | Not conserved | 0.00E+00 |
| R1588Q (−) | 3 | 1 | – | 28 | Paroxysmal AF, AVNRT | – | 55 | 156 | 96 | 419 | Damaging | Probably Damaging | Conserved | 1.65E-05 |
| G1662S (rs151090729) | 1 | 1 | M | 46 | Persistent AF | – | – | – | – | – | Damaging | Probably Damaging | Conserved | 1.57E-03 |
| 4 | 1 | M | 44 | BrS, syncope | No | 79 | 190 | 120 | 436 | Damaging | Probably Damaging | Conserved | 1.31E-03 | |
| 4 | 1 | M | 19 | BrS, SCD, syncope, dizziness/palpitations | Yes | 44 | 200 | 120 | 352 | Damaging | Probably Damaging | Conserved | 1.31E-03 | |
| 4 | 1 | M | 27 | BrS, SCD | No | 120 | 220 | 120 | 387 | Damaging | Probably Damaging | Conserved | 1.31E-03 | |
| V1697I (rs77804526) | 4 | 1 | M | 39 | BrS, syncope | No | 78 | 168 | 80 | 388 | Tolerated | Benign | Conserved | 1.01E-02 |
| 4 | 1 | M | 31 | BrS | Yes | 75 | 200 | 120 | 403 | Tolerated | Benign | Conserved | 1.01E-02 | |
| 4 | 1 | M | 58 | BrS, syncope | No | 84 | 210 | 100 | 408 | Tolerated | Benign | Conserved | 1.01E-02 | |
| 6 | 1 | M | 60 | AVNRT, suspected BrS | – | – | – | – | – | Tolerated | Benign | Conserved | 4.10E-01 | |
| 6 | 1 | F | 41 | AVNRT, concealed BrS, chest pain | – | – | – | – | – | Tolerated | Benign | Conserved | 4.10E-01 | |
| N1715T (−) | 4 | 1 | M | 65 | BrS, induced VT/VF, dizziness/palpitations | No | 71 | 220 | 72 | 463 | Damaging | Probably Damaging | Conserved | 8.24E-06 |
| R1863Q (rs191869263) | 7 | 1 | M | 51 | Syncope | no | 57 | 190 | 70 | 351 | Damaging | Damaging | Conserved | 1.65E-05 |
| R1869C (rs141648641) | 1 | 1 | M | 25 | Persistent AF | – | – | – | – | – | Damaging | Probably-damaging | Conserved | 8.07E-04 |
| 4 | 1 | M | 43 | BrS, SCD, dizziness/palpitations | No | 64 | 192 | 84 | 385 | Damaging | Probably Damaging | Conserved | 9.23E-04 | |
| 4 | 1 | F | 47 | BrS, dizziness/palpitations | No | 56 | 180 | 88 | 408 | Damaging | Probably Damaging | Conserved | 9.23E-04 | |
| A1886V (rs142653846) | 1, 8 | 1 | M | 17 | Paroxysmal AF, SVR | – | – | – | – | NP | Tolerated | Benign | Not conserved | 1.18E-03 |
Mutation; designated amino-acid changes and corresponding dbSNP annotation when available. Of note, none of these mutations with QT prolongation fell in the enhancer region.
References; 1: (Savio-Galimberti, et al., 2014), 2: (Delaney, et al., 2014), 3: (Jabbari, et al., 2015), 4: (Hu, et al., 2014), 5: (Behr, et al., 2015), 6: (Hasdemir, et al., 2015), 7: (Fukuyama, et al., 2016), 8: (Bezzina, et al., 2013).
n; number of patients.
G; gender, M: male and F: female.
Age; age in years
Family Hx; family history.
ECG characteristics; HR: heart rate in beats per minute, PR: PR interval in milliseconds, QRS: QRS interval in milliseconds, QTc: corrected QT interval in milliseconds, borderline elevated QTc interval is highlighted in grey and high QTc interval is highlighted in yellow.
MAF; minor allele frequency based on the EXAC database.
Conservation analysis showed that all of the substituted amino-acids are evolutionarily highly conserved (figure 1b). There was no clustering of the mutations observed, hence a genotype phenotype correlation could not be established (figure 1b). For instance, the p.R1268Q variant is located in the cytoplasmic portion of the protein, while the p.R1259Q variant is in the transmembrane portion between the voltage sensing domain and the channel pore domain. Interestingly, none of the above mutations were near the enhancer-binding domain of the SCN10A gene (figure 1a). There was strong family history of arrhythmias and LQT among SCN10A mutation carriers (figure 2). Careful review of the pedigree suggests pleotropic effect of these mutations, resulting in trait that range from atrial fibrillation to supraventricular tachycardia, long QT and syncope with an inheritance that is consistent with an autosomal dominant pattern. First degree relatives were not available for genetic testing.
Figure 1.
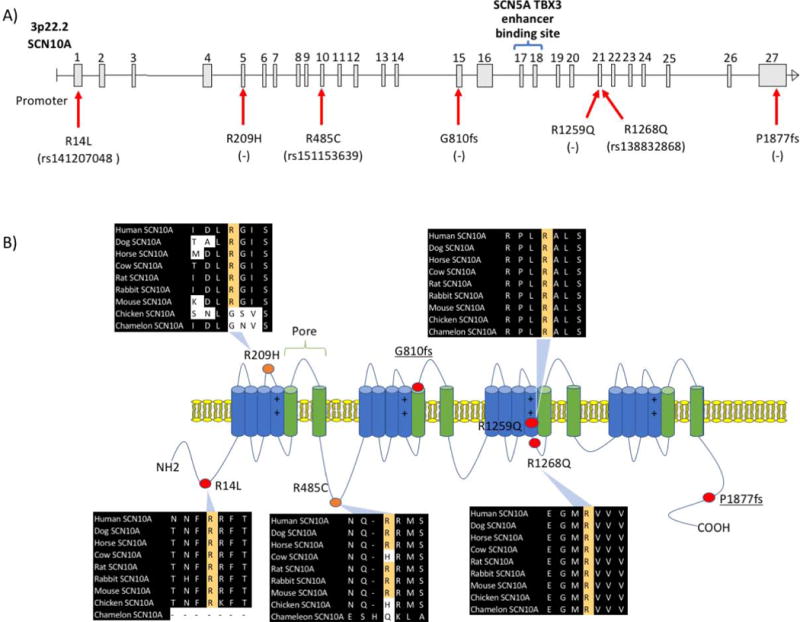
Nav1.8 voltage gated sodium channel encoding SCN10A gene domains, long QT associated mutations and conservation analysis. (A) SCN10A gene is comprised of 27 exons located on the short arm of chromosome 3. The enhancer-binding domain encompasses exons 17, 18 and the intronic region in between. The mutations are labelled along the gene with the corresponding dbSNP (rs) number when available. (B) Several of the mutations fall within the transmembrane alpha helical domains (G810fs, R1259Q, and R1268Q), the R14L is in the N-terminal domain, and the P1877fs is in the C-terminal domain. The two mutations in the patients who were on QT prolonging medications are labelled in orange, the R209H in proximity to the voltage sensing domain and R485C within the channel modulation domain. All the mutation occurred at evolutionarily conserved site.
Figure 2.
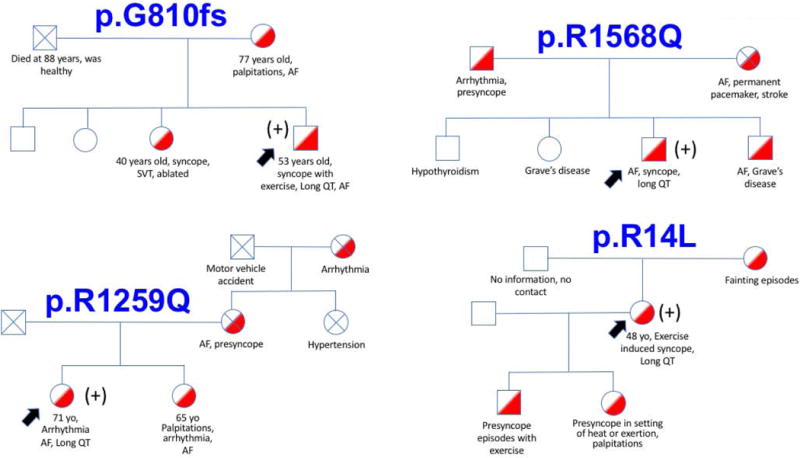
Family pedigrees of index cases with SCN10A mutations. There was strong family history among SCN10A mutation carriers concerning for arrhythmias and underlying long QT syndrome (labelled in red). The black arrows show index cases. The (+) sign indicates that the subject is heterozygous for an SCN10A mutation.
SCN10A Mutations Among Patients on QT Prolonging Medications
Two patients, who were taking QT prolonging medications also underwent exome sequencing. The first patient was a 59 years old man with a complex medical history including hypertrophic cardiomyopathy (HCM), on metoclopramide who had QT prolongation, and was found to have a genetic variant in the SCN10A gene leading to p.R485C substitution. Interestingly, he later developed persistent AF. He had also a non-conservative MYBPC3 mutation as the underlying cause of his HCM. The SCN10A p.R485C is a rare variant with an allele frequency of 7.0×10−4 in the EXAC database, which is also predicted to be damaging. The second patient was a 57 years old woman, who presented with persistent AF and had been placed on Sotalol. Her QTc on this drug ranged from 459 to 615 msecs. Given the strong family history of arrhythmia and AF she underwent WES, which revealed a missense mutation in SCN10A gene leading to p.R209H substitution. There were no other mutations identified in known arrhythmia genes. The p.R209H is a rare variant with an allele frequency of 3.3×10−5 in the EXAC database and is predicted to be damaging. Both substituted amino-acids were highly conserved (figure 1a) and none was located near the enhancer-binding domain of the SCN10A gene (figure 1b). Moreover, the CADD scores for p.R485C and p.R209H were 34 and 33, respectively which suggest a very high likelihood of pathogenicity.
Review of Rare Damaging SCN10A Mutations in EXAC and Mutation Burden Analysis
We queried the EXAC database for rare damaging mutations in the SCN10A gene. Out of the 60,706 total subjects, there were 303 damaging mutations at an allele frequency of less <1:100,000 (figure 3).
Figure 3.
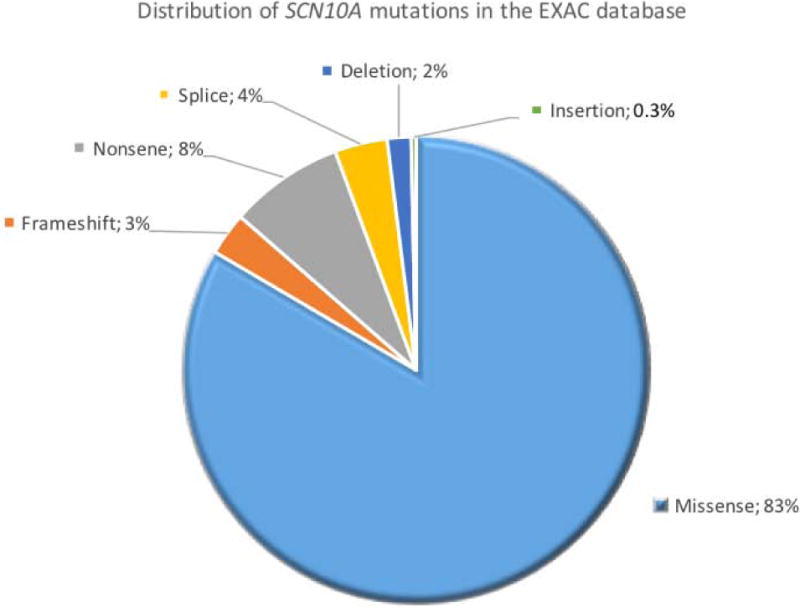
Distribution of SCN10A mutations exomes of 60,706 subjects in EXAC database. Overall there were 303 rare (minor allele frequency< 0.001%) and damaging mutations. The majority of the mutations were miss-sense (n= 253, 83%), followed by 24 non-sense (8%), 11 splice site (4%), and 9 frameshift (3%) mutations, 5 were deletion (2%) and 1 was an insertion mutation (0.3%).
Mutation burden analysis (supplementary table 1) using the EXAC database as control showed an odds ratio of 90.9 for having prolonged QT if the patient harbors a rare and deleterious SCN10A mutation (95% confidence interval of 31.4 to 263.2, P-value < 0.0001).
In a second analysis using an allele frequency of <1:100 two more variants in the SCN10A gene were identified in our patient cohort. Additionally, out of the 60,706 total subjects in the EXAC database, there were 574 damaging mutations at an allele frequency <1:100. These included missense (n=501), non-sense (n=32), splice (n=17), frameshift (n=15), deletion (n=8), and insertion (n=1). Mutation burden analysis at this allele frequency showed an odds ratio of 4.9 for having prolonged QT if the patient harbors a deleterious SCN10A mutation (95% confidence interval of 1.7 to 14.2, P-value = 0.0047; supplemental table 1). Interestingly, SCN10A’s RVIS score of −1.32 places this gene among the 4.74% most intolerant genes. Taken together, our analyses provide strong statistical evidence for association between SCN10A mutations and long QT trait.
Systematic Literature Review of All Reported SCN10A Mutations and Available ECG Characteristics
In our review of the published literature we found 46 different SCN10A reported mutations. The available clinical and ECG data are summarized in table 2. Interestingly, heterozygous SCN10A mutations causing p.F385C, p.I1225T, p.N1328K, p.N1715T amino acid substitutions were detected in four independent patients with QT prolongation. All four variants had been reported in independent publications, hence they lacked the power to establish association between SCN10A mutations and QT prolongation.
The subject with the deleterious p.F385C SCN10A variant is reported as a 65-year-old man with a history of sudden cardiac death, Brugada syndrome, and a QTc of 464 msec (table 2).
One of the two subjects with symptomatic Brugada syndrome and p.I1225T variants had been reported to have a prolonged QTc of 525 msec (table 2).
The third deleterious SCN10A mutation had been reported in a 68-year-old man with syncope and a prolonged QTc of 450 msec. He had a novel missense mutation in SCN10A that substituted a highly conserved amino acid (p.N1328K).
The fourth reported deleterious SCN10A mutation substitutes a highly conserved amino acid (p.N1715T) with a rare allele frequency of 8.1×10−4 in the EXAC database. This mutation had been identified in a 65-year-old man with presyncope and palpitations, inducible ventricular tachycardia, and ventricular fibrillation, diagnosis of Brugada syndrome and a QTc of 463 msec.
In addition, there has been report of 6 different SCN10A mutations in 6 subjects with borderline prolonged QTc, resulting in p.R14L, p.W189R, p.R844H, p.S1337T, p.G1406D, and p.G1662S amino acid substitutions (table 2). All of these mutations substituted highly conserved amino acids and were predicted to be damaging. Of note, there have been 6 patients with the p.R14L variant, 4 patients with the p.G1662S variant and 2 patients with the p.R844H variant reported and only one from each group has had borderline prolonged QTc (table 2).
4. Discussion
Voltage gated sodium channels (Nav) conduct inward sodium currents that are regulated by the transmembrane potential. These channels are composed of the pore forming or α-subunit, and a β regulatory subunit. The α-subunit possesses a voltage sensing domain, a pore domain with a filter selective for sodium ions, an activation gate, and an inactivation gate (55). There are nine different Navs identified in humans with different tissue distribution and functionality based on the α-subunit (55). The α-subunits of Nav1.1 to Nav1.9 are encoded by nine different genes.
Gain of function mutations in SCN5A have been previously implicated in congenital LQT type 3 (56). The SCN5A gene encodes Nav1.5, which is the most abundant voltage gated sodium channel in cardiac tissue. Nav1.5 is responsible for the depolarization phase of the cardiac action potential, characterized by rapid inward sodium current that is quickly inactivated by the inactivation gate (57). Congenital LQT type 3 mutations cause a gain of function in Nav1.5 due to failure of the inactivation gate to terminate the influx of sodium at the appropriate time. Hence, sodium entry slowly continues during the subsequent phases of the action potential and results in prolonged repolarization (58). This is in contrast to Brugada associated mutations in SCN5A that cause loss of function via premature closure of Nav1.5 channel without effect on the QT interval (59). An overlap syndrome of long QT and Brugada has also been described as some SCN5A genotypes can cause features of both syndromes (60).
In parallel, SCN10A mutations have been strongly associated with Brugada syndrome and other cardiac conduction abnormalities in humans. In fact, up to 10% of Brugada cases are attributed to SCN10A mutations (34).
A recent fine-mapping study implicated the SCN5A-SCN10A locus in QT prolongation (61). In-vitro studies have shown that the Nav1.8 contributes to late cardiac sodium current and displays marked differences in gating compared to Nav1.5 (26). While the enhancer hypothesis offered a mechanism whereby polymorphisms in SCN10A could contribute to SCN5A expression levels (45), the majority of the arrhythmia associated SCN10A mutations fall far from the enhancer-binding domain. However, a direct effect through the sympathetic nervous system is another plausible mechanism. Although Nav1.8 has low expression in cardiomyocytes, its expression levels are high in intra-cardiac ganglia and neurons (27, 62). Loss of Nav1.8 current had low effect on cardiomyocyte conduction (27, 62). However, Nav1.8 blockade significantly reduces the sodium current and firing frequency of intra-cardiac neurons (27). Similarly, Nav1.8 channel blockade in cardiac ganglionated plexi has been shown to suppress cardiac conduction and atrial fibrillation inducibility (46). Accordingly, left cardiac sympathetic neuron denervation (LCSD) has been successfully used to treat LQT syndrome (63, 64).
In our study, we observed a remarkably high prevalence of deleterious SCN10A mutations in patients with prolonged QT, an association that hasn’t been made before. All the observed mutations affected the amino-acid sequence of Nav1.8, either through deleterious amino-acid substitutions at conserved sites or frameshift mutations. None of the observed mutations resides near the enhancer-binding domain, which means that they are less likely to disturb cardiac conduction via indirect effects on SCN5A expression levels. Although, one cannot exclude an allosteric effect of these mutations on the enhancer-binding domain. More likely these mutations affect Nav1.8 function in regulation of cardiac sodium current and repolarization. The amino acid substitutions involve the voltage sensing domain (p.R209, p.R1259Q and p.R1268Q), channel modulation domain (p.R485C), and N-terminal domain (p.R14L).
While the three deleterious and entirely novel variants (p.G810fs, p.R1259Q, and p.P1877fs) in SCN10A provided a strong signal of association with LQT, the subsequent mutations were present at higher allele frequencies. For instance, the p.R1268Q variant was present at an allele frequency of 0.18% that is high compared to the prevalence of LQT. However, the same mutation had been associated with Brugada syndrome (34, 36), which is also a rare disease (65). Similarly, the p.R14L variant with an allele frequency of 0.19% had been previously associated with Brugada syndrome (34, 38, 39). We speculate that these are disease-contributing variants that are not sufficient to independently cause disease, which explains their pleotropic effects and incomplete penetrance. The two variants (p.R209H, p.R485C) identified in the two patients who were on QT prolonging medications are also most likely not sufficient to cause LQT and require the contribution of environmental factors such as QT prolonging drugs. Although mutations in known LQT genes account for about 80% of LQT cases, our study was enriched for patients who had previously undergone targeted genotyping panels (5 out of 16 patients) with negative results for LQT gene mutations. This has resulted in the lower yield for mutations in known LQT genes in our WES and likely higher SCN10A mutation rate compared to the general population.
Of interest is the prior association between SCN10A mutations and unexplained nocturnal sudden cardiac death (41). Although the SCD had been attributed to a possible underlying Brugada syndrome, the current findings suggest that LQT induced torsades could be another mechanism of SCD in patients harboring SCN10A mutations. Overall, these findings are of great significance to the study of cardiac electrophysiology and prevention of fatal arrhythmias in at-risk subjects.
5. Conclusion and Future Prospects
To this date there is no definitive therapy available for LQT, therefore, screening and early detection of prolonged QT remains a central approach for risk stratification and primary prevention against fatal arrhythmias in affected subjects and their extended families. Hence, it is imperative to identify all genetic culprits in abnormal cardiac repolarization and long QT. Strikingly, the genetic cause in congenital LQT remains elusive in about 20% of inherited cases, while many of the acquired LQTs are also accounted for by predisposing genetic mutations.
Our study implicates SCN10A mutations as an underlying cause of LQT, adding to the list of genes that should be screened for in patients with prolonged QT. The presence of frameshift mutations, which result in premature stop codon and potentially non-sense mediated decay, suggest that the disease mechanism is due to loss of function of the encoded protein. In addition, the common association with AF suggests that SCN10A may represent a unique subtype of LQT genes.
Our findings advance the identification of genetic causes of LQT and improve the capability of screening in individuals at risk. Further investigations into genotype-phenotype correlation may improve our ability to predict the development of isolated atrial fibrillation, Brugada syndrome, LQT or combination thereof in SCN10A mutation carriers. In addition, future investigations are necessary to examine the co-expression and interaction of SCN10A with SCN5A in regulation of sodium current in cardiomyocytes.
Supplementary Material
Acknowledgments
Financial support and sponsorship:
This manuscript was supported by grants from the National Institutes of Health (NIH) (RHL135767A to Arya Mani).
Footnotes
The authors have no conflicts of interest to report.
References
- 1.Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kay GN, Plumb VJ, Arciniegas JG, et al. Torsade de pointes: the long-short initiating sequence and other clinical features: observations in 32 patients. J Am Coll Cardiol. 1983;2:806–817. doi: 10.1016/s0735-1097(83)80226-5. [DOI] [PubMed] [Google Scholar]
- 3.Kenigsberg DN, Khanal S, Kowalski M, et al. Prolongation of the QTc interval is seen uniformly during early transmural ischemia. J Am Coll Cardiol. 2007;49:1299–1305. doi: 10.1016/j.jacc.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 4.Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 5.Yang P, Kanki H, Drolet B, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 6.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 7.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 8.Russell MW, Dick M, 2nd, Collins FS, et al. KVLQT1 mutations in three families with familial or sporadic long QT syndrome. Hum Mol Genet. 1996;5:1319–1324. doi: 10.1093/hmg/5.9.1319. [DOI] [PubMed] [Google Scholar]
- 9.Benson DW, MacRae CA, Vesely MR, et al. Missense mutation in the pore region of HERG causes familial long QT syndrome. Circulation. 1996;93:1791–1795. doi: 10.1161/01.cir.93.10.1791. [DOI] [PubMed] [Google Scholar]
- 10.Duggal P, Vesely MR, Wattanasirichaigoon D, et al. Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation. 1998;97:142–146. doi: 10.1161/01.cir.97.2.142. [DOI] [PubMed] [Google Scholar]
- 11.Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 12.Tristani-Firouzi M, Jensen JL, Donaldson MR, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) J Clin Invest. 2002;110:381–388. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Yang Y, Liang B, et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010;86:872–880. doi: 10.1016/j.ajhg.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Crotti L, Johnson CN, Graf E, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makita N, Yagihara N, Crotti L, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet. 2014;7:466–474. doi: 10.1161/CIRCGENETICS.113.000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pipilas DC, Johnson CN, Webster G, et al. Novel calmodulin mutations associated with congenital long QT syndrome affect calcium current in human cardiomyocytes. Heart Rhythm. 2016;13:2012–2019. doi: 10.1016/j.hrthm.2016.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Marquardt ML, Tester DJ, et al. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104:20990–20995. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 21.Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 23.Medeiros-Domingo A, Kaku T, Tester DJ, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–142. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 25.Chambers JC, Zhao J, Terracciano CM, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152. doi: 10.1038/ng.516. [DOI] [PubMed] [Google Scholar]
- 26.Yang T, Atack TC, Stroud DM, et al. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111:322–332. doi: 10.1161/CIRCRESAHA.112.265173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verkerk AO, Remme CA, Schumacher CA, et al. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res. 2012;111:333–343. doi: 10.1161/CIRCRESAHA.112.274035. [DOI] [PubMed] [Google Scholar]
- 28.Facer P, Punjabi PP, Abrari A, et al. Localisation of SCN10A gene product Na(v)1.8 and novel pain-related ion channels in human heart. Int Heart J. 2011;52:146–152. doi: 10.1536/ihj.52.146. [DOI] [PubMed] [Google Scholar]
- 29.Holm H, Gudbjartsson DF, Arnar DO, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 30.Sotoodehnia N, Isaacs A, de Bakker PI, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denny JC, Ritchie MD, Crawford DC, et al. Identification of genomic predictors of atrioventricular conduction: using electronic medical records as a tool for genome science. Circulation. 2010;122:2016–2021. doi: 10.1161/CIRCULATIONAHA.110.948828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bezzina CR, Barc J, Mizusawa Y, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. doi: 10.1038/ng.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukuyama M, Ohno S, Makiyama T, et al. Novel SCN10A variants associated with Brugada syndrome. Europace. 2016;18:905–911. doi: 10.1093/europace/euv078. [DOI] [PubMed] [Google Scholar]
- 34.Hu D, Barajas-Martinez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. 2014;64:66–79. doi: 10.1016/j.jacc.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behr ER, Savio-Galimberti E, Barc J, et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res. 2015;106:520–529. doi: 10.1093/cvr/cvv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasdemir C, Payzin S, Kocabas U, et al. High prevalence of concealed Brugada syndrome in patients with atrioventricular nodal reentrant tachycardia. Heart Rhythm. 2015;12:1584–1594. doi: 10.1016/j.hrthm.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Ritchie MD, Denny JC, Zuvich RL, et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127:1377–1385. doi: 10.1161/CIRCULATIONAHA.112.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savio-Galimberti E, Weeke P, Muhammad R, et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc Res. 2014;104:355–363. doi: 10.1093/cvr/cvu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jabbari J, Olesen MS, Yuan L, et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ Cardiovasc Genet. 2015;8:64–73. doi: 10.1161/HCG.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delaney JT, Muhammad R, Shi Y, et al. Common SCN10A variants modulate PR interval and heart rate response during atrial fibrillation. Europace. 2014;16:485–490. doi: 10.1093/europace/eut278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L, Zhou F, Huang L, et al. Association of common and rare variants of SCN10A gene with sudden unexplained nocturnal death syndrome in Chinese Han population. Int J Legal Med. 2016 doi: 10.1007/s00414-016-1397-1. [DOI] [PubMed] [Google Scholar]
- 42.Park DS, Fishman GI. Nav-igating through a complex landscape: SCN10A and cardiac conduction. J Clin Invest. 2014;124:1460–1462. doi: 10.1172/JCI75240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van den Boogaard M, Barnett P, Christoffels VM. From GWAS to function: genetic variation in sodium channel gene enhancer influences electrical patterning. Trends Cardiovasc Med. 2014;24:99–104. doi: 10.1016/j.tcm.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 44.van den Boogaard M, Smemo S, Burnicka-Turek O, et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest. 2014;124:1844–1852. doi: 10.1172/JCI73140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van den Boogaard M, Wong LY, Tessadori F, et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012;122:2519–2530. doi: 10.1172/JCI62613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qi B, Wei Y, Chen S, et al. Nav1.8 channels in ganglionated plexi modulate atrial fibrillation inducibility. Cardiovasc Res. 2014;102:480–486. doi: 10.1093/cvr/cvu005. [DOI] [PubMed] [Google Scholar]
- 47.Blasius AL, Dubin AE, Petrus MJ, et al. Hypermorphic mutation of the voltage-gated sodium channel encoding gene Scn10a causes a dramatic stimulus-dependent neurobehavioral phenotype. Proc Natl Acad Sci U S A. 2011;108:19413–19418. doi: 10.1073/pnas.1117020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keramati AR, Fathzadeh M, Go GW, et al. A form of the metabolic syndrome associated with mutations in DYRK1B. N Engl J Med. 2014;370:1909–1919. doi: 10.1056/NEJMoa1301824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petrovski S, Wang Q, Heinzen EL, et al. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DG A. Practical statistics for medical research. London: Chapman and Hall; 1991. [Google Scholar]
- 52.Li D, Morales A, Gonzalez-Quintana J, et al. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci. 2010;3:90–97. doi: 10.1111/j.1752-8062.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Hengel J, Calore M, Bauce B, et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:201–210. doi: 10.1093/eurheartj/ehs373. [DOI] [PubMed] [Google Scholar]
- 55.de Lera Ruiz M, Kraus RL. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J Med Chem. 2015;58:7093–7118. doi: 10.1021/jm501981g. [DOI] [PubMed] [Google Scholar]
- 56.Rivolta I, Abriel H, Tateyama M, et al. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J Biol Chem. 2001;276:30623–30630. doi: 10.1074/jbc.M104471200. [DOI] [PubMed] [Google Scholar]
- 57.Antzelevitch C, Nesterenko V, Shryock JC, et al. The role of late I Na in development of cardiac arrhythmias. Handb Exp Pharmacol. 2014;221:137–168. doi: 10.1007/978-3-642-41588-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khan IA, Nair CK. Brugada and long QT-3 syndromes: two phenotypes of the sodium channel disease. Ann Noninvasive Electrocardiol. 2004;9:280–289. doi: 10.1111/j.1542-474X.2004.93533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naccarelli GV, Antzelevitch C. The Brugada syndrome: clinical, genetic, cellular, and molecular abnormalities. Am J Med. 2001;110:573–581. doi: 10.1016/s0002-9343(01)00625-8. [DOI] [PubMed] [Google Scholar]
- 60.Veldkamp MW, Viswanathan PC, Bezzina C, et al. Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ Res. 2000;86:E91–97. doi: 10.1161/01.res.86.9.e91. [DOI] [PubMed] [Google Scholar]
- 61.Avery CL, Wassel CL, Richard MA, et al. Fine-mapping of QT regions in global populations refines previously identified QT loci and identifies signals unique to african and hispanic descent populations. Heart Rhythm. 2016 doi: 10.1016/j.hrthm.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stroud DM, Yang T, Bersell K, et al. Contrasting Nav1.8 Activity in Scn10a−/− Ventricular Myocytes and the Intact Heart. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.115.002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109:1826–1833. doi: 10.1161/01.CIR.0000125523.14403.1E. [DOI] [PubMed] [Google Scholar]
- 64.Schwartz PJ, Locati EH, Moss AJ, et al. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome. A worldwide report. Circulation. 1991;84:503–511. doi: 10.1161/01.cir.84.2.503. [DOI] [PubMed] [Google Scholar]
- 65.Kamakura S. Epidemiology of Brugada syndrome in Japan and rest of the world. Journal of Arrhythmia. 2013;29:52–55. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.