

Dear Editor, X-linked reticulate pigmentary disorder (XLPDR, MIM 301220) is a rare syndrome first recognized by Partington1. The cardinal manifestations of the disorder are diffuse reticulate hyperpigmentation, hypohidrosis, and unique facial features, as well as recurrent pneumonias and sterile inflammation in various organs1–9. The syndrome is exceedingly rare and only 20 patients have been reported worldwide. Recently, we identified that the disorder is associated with a recurrent intronic mutation in POLA1, the gene encoding the catalytic subunit of DNA Polymerase-α9. Here, we report a new case of XLPDR and include the first detailed description of the evolution of its dermatologic features, as well as confirmation of the same intronic POLA1 mutation.
The patient is the fourth child of four and has an unaffected older sister (Fig. 1a). His older brother has an atrial septal defect, but no other abnormalities. Another brother was born with Potter syndrome and died at 2 hours of age. The patient’s mother has restricted hyperpigmentation along Blaschko’s lines (Fig. 1b), which is also observed in his grandmother and aunt, but no other males in the family appear clinically affected.
Figure 1. General clinical characteristics of the new XLPDR proband.
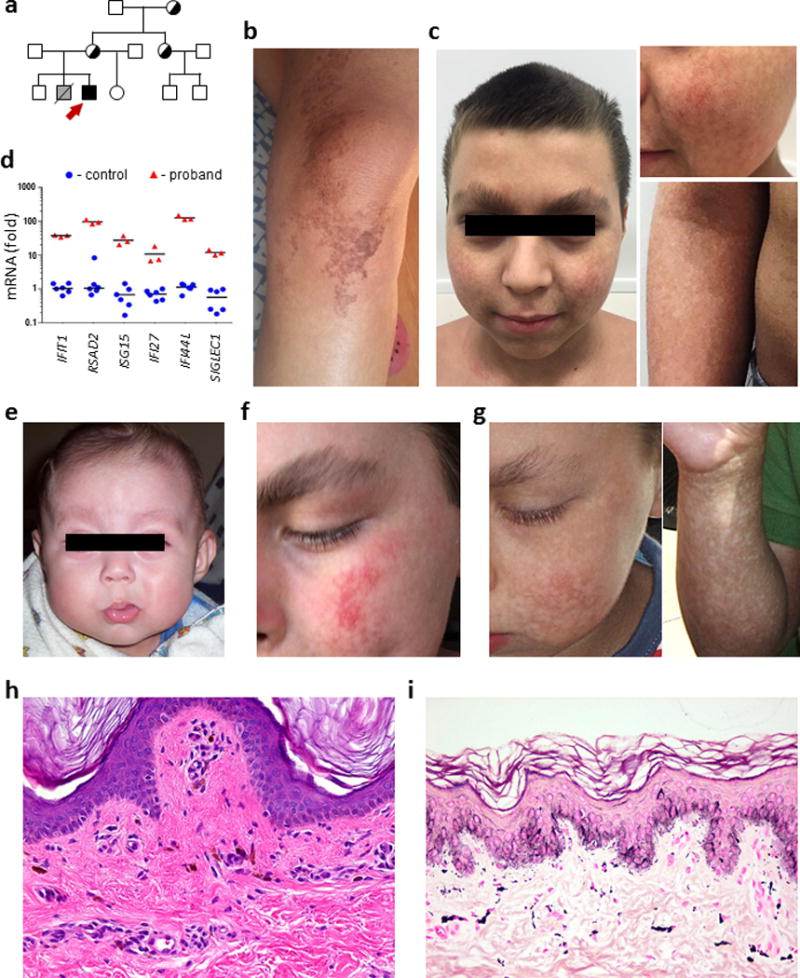
a) Family pedigree of the XLPDR proband (red arrow). Carriers are noted by half-shaded symbols.
b) Patchy hyperpigmentation on the knee of the proband’s mother.
c) Facial and skin features of the proband at age 12, including upswept hair, flared eyebrows, and skin hyperpigmentation.
d) Transcript levels for interferon-stimulated genes were determined by qRT-PCR in whole blood-derived RNA from XLPDR proband (n=1), and unaffected ‘travel’ controls (n=2). All samples were analyzed in triplicate according to a previously published protocol9. Transcript abundance is displayed as the fold rate compared to the same reference control sample. Black bar depicts the mean value. For all samples unpaired Student’s t-test p<0.01.
e) Lack of skin pigmentary changes before age of 1 year.
f) Facial telangiectasias at 4 years of age.
g) Skin hyperpigmentation at 8 years of age.
h) Skin histopathology at 7 years of age (haematoxylin and eosin, original magnification ×100).
i) Skin histopathology at 12 years of age (haematoxylin and eosin, original magnification ×100).
During his first year of life, the patient developed recurrent pneumonia, urethral strictures with occasional bleeding after urination, and bloody diarrhea (which eventually resolved while on hydrolyzed formula). However, he had no recurrent or unusual non-pulmonary infections and did not have growth delay. At the age of 7, he developed bronchiectasis in the left lower lobe (requiring pulmozyme treatment) and was noted to have decreased visual acuity and photophobia due to bilateral erosive keratitis. At 13 years of age, the typical facial features of XLPDR (upswept hair and flared eyebrows) along with reticulate hyperpigmentation were noted (Fig. 1c). Laboratory studies (Suppl Table 1) were otherwise within normal ranges.
The diagnosis of XLPDR was confirmed at age 13 by genetic and immunological tests. The patient was found to be hemizygous for a POLA1 mutation (NC_000023.10:g.24744696A>G) previously reported9. This non-coding mutation in intron 13 of the POLA1 gene is known to be associated with activated type I interferon responses9, and indeed the patient displayed dramatic elevation of interferon-stimulated genes (Fig. 1d).
While it has been recognized that the pigmentary changes in XLPDR are not present at birth, the evolution of the skin phenotype has not been reported. Retrospective analysis of the new proband indicates no overt changes in skin pigmentation in the first year of life (Fig. 1e). The first dermatologic manifestations were noted at 14 months of age, when he developed facial telangiectasias (Fig. 1f). At the same age, the patient developed hypopigmented spots on the face, extremities, and ultimately the trunk (Fig. 1g). These skin changes were not associated with pruritus or pain, but the skin was persistently dry. Frank hypohidrosis was noted in infancy, selectively affecting trunk and extremities. In contrast, the head and neck presented profuse sweating since this age. In view of these changes, a skin biopsy was obtained from his left thigh at age 7, which revealed epidermal hypomelanosis with orthokeratosis, focal acanthosis, focal lymphocytosis, and mild hypopigmentation (Fig. 1h). The dermis layer contained few melanophages and minimal superficial lymphocytic perivascular infiltrate. Direct immunofluorescence revealed granular deposits of C3 (+) at the dermo-epidermal junction, but no deposition of IgG, IgA, IgM, or fibrin. Hyperpigmentation was first noted by the parents at 8 years of age. At the age of 12, a new skin biopsy revealed orthokeratosis with areas of hyperpigmentation and hypopigmentation of the basal layer of the epidermis. The dermis had a mild perivascular inflammatory infiltrates composed by lymphocytes and some melanophages (Fig. 1i).
Our findings in this case further confirm that XLPDR is associated with constitutive activation of type I interferon responses9. Moreover, these data demonstrate lack of genetic heterogeneity in XLPDR, meaning that the exact same mutation has been seen in all cases thus far. We speculate that the unique features of the syndrome may be directly related to the specific effects induced by this intronic variant, such as possible variability in missplicing among tissues. Unlike Aicardi-Goutieres syndrome (AGS), the best known interferonopathy, XLPDR does not involve the brain and is associated with a unique set of manifestations including the characteristic facial features and hypohidrosis, which remain unexplained. Among type I interferonopathies, skin findings are seen in activating mutations in STING, which lead to pulmonary and skin vasculitis, predominantly affecting digits10. Additionally, ADAR mutations, which have been linked to AGS, can also result in dyschromatosis symmetrica hereditaria (MIM 127400), characterized by the development in early childhood of symmetric hyper and hypopigmented macules on the face and extremities, much like our patient described here10. The recognition that hyperpigmentation develops during childhood is a point that will be important in the early identification of XLPDR. Moreover, several XLPDR cases, including this one, were misdiagnosed as having cystic fibrosis, and such prominent pulmonary phenotype is unique among type I interferonopathies. Assessed in aggregate, XLPDR is highly likely when a patient has a combination of diffuse hyperpigmentation, typical facial features, and recurrent lung infections, as in this case (Table 1). Future clinical reports will be required in order to expand our understanding of the clinical evolution of the dermatologic manifestations of XLPDR.
Table 1.
Clinical features in the proband compared to previously reported frequency of these features in XLPDR.
| Manifestations | Reported Proband | Occurrence in previously reported cases9 |
|---|---|---|
| Diffuse Hyperpigmentation | + | 100% |
| Facies | + | 100% |
| Lung infections/Bronchiectasis | + | 92% |
| Hypohidrosis | + | 64% |
| Enterocolitis and diarrhea | + | 64% |
| Failure to thrive | − | 57% |
| Corneal inflammation/scarring | + | 50% |
| Recurrent non-pulmonary infections | − | 38% |
| Urethral strictures | + | 35% |
| Digital Clubbing | + | 21% |
| Toe abnormalities | − | 21% |
| Mycobacterial infection | − | 14% |
Supplementary Material
Acknowledgments
We thank our patient and his family for agreeing to participate in this study.
Funding sources: This work was supported by the NIH through the following grants: R56 AI113274 to E.B. and A.R.Z., and R01 DK073639 to E.B., and by Iniciativa Científica Milenio through grant P09/016-F to A.B.
Footnotes
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Partington MW, Marriott PJ, Prentice RS, Cavaglia A, Simpson NE. Familial cutaneous amyloidosis with systemic manifestations in males. Am J Med Genet. 1981;10:65–75. doi: 10.1002/ajmg.1320100109. [DOI] [PubMed] [Google Scholar]
- 2.Ades LC, Rogers M, Sillence DO. An X-linked reticulate pigmentary disorder with systemic manifestations: report of a second family. Pediatr Dermatol. 1993;10:344–351. doi: 10.1111/j.1525-1470.1993.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 3.Anderson RC, Zinn AR, Kim J, Carder KR. X-linked reticulate pigmentary disorder with systemic manifestations: report of a third family and literature review. Pediatr Dermatol. 2005;22:122–126. doi: 10.1111/j.1525-1470.2005.22206.x. [DOI] [PubMed] [Google Scholar]
- 4.Duman N, Ersoy-Evans S, Gokoz O. Reticulate Pigmentation with Systemic Manifestations in a Child. Pediatr Dermatol. 2015;32:871–872. doi: 10.1111/pde.12646. [DOI] [PubMed] [Google Scholar]
- 5.Fraile G, Norman F, Reguero ME, Defargues V, Redondo C. Cryptogenic multifocal ulcerous stenosing enteritis (CMUSE) in a man with a diagnosis of X-linked reticulate pigmentary disorder (PDR) Scand J Gastroenterol. 2008;43:506–510. doi: 10.1080/03014460701723912. [DOI] [PubMed] [Google Scholar]
- 6.Gedeon AK, Mulley JC, Kozman H, Donnelly A, Partington MW. Localisation of the gene for X-linked reticulate pigmentary disorder with systemic manifestations (PDR), previously known as X-linked cutaneous amyloidosis. Am J Med Genet. 1994;52:75–78. doi: 10.1002/ajmg.1320520115. [DOI] [PubMed] [Google Scholar]
- 7.Megarbane H, Boehm N, Chouery E, Bernard R, Salem N, Halaby E, Levy N, Megarbane A. X-linked reticulate pigmentary layer. Report of a new patient and demonstration of a skewed X-inactivation. Genet Couns. 2005;16:85–89. [PubMed] [Google Scholar]
- 8.Pezzani L, Brena M, Callea M, Colombi M, Tadini G. X-linked reticulate pigmentary disorder with systemic manifestations: a new family and review of the literature. Am J Med Genet A. 2013;161:1414–1420. doi: 10.1002/ajmg.a.35882. [DOI] [PubMed] [Google Scholar]
- 9.Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, Pokatayev V, Dozmorov I, Khan S, Miyata N, et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nature immunology. 2016;17:495–504. doi: 10.1038/ni.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyts I, Casanova JL. A human inborn error connects the α’s. Nature immunology. 2016;17:472–474. doi: 10.1038/ni.3420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.