

Abstract
Marine mussels use catechol-rich interfacial mussel foot proteins (mfps) as primers that attach to mineral surfaces via hydrogen, metal coordination, electrostatic, ionic, or hydrophobic bonds, creating a secondary surface that promotes bonding to the bulk mfps. Inspired by this biological adhesive primer, it is shown that a ≈1 nm thick catecholic single-molecule priming layer increases the adhesion strength of crosslinked polymethacrylate resin on mineral surfaces by up to an order of magnitude when compared with conventional primers such as noncatecholic silane- and phosphate-based grafts. Molecular dynamics simulations confirm that catechol groups anchor to a variety of mineral surfaces and shed light on the binding mode of each molecule. Here, a ≈50% toughness enhancement is achieved in a stiff load-bearing polymer network, demonstrating the utility of mussel-inspired bonding for processing a wide range of polymeric interfaces, including structural, load-bearing materials.
Keywords: adhesion, dynamic bonding, mussels, primer, surfaces
Adhesion and coating technologies often employ the processing strategy of priming a surface prior to the application of a bulk resin to enhance bonding performance. The most common primers for mineral fillers in load-bearing polymer composites, such as those used in dental restorative applications, rely on silane-based chemistries. However, silane grafting involves uses of toxic chemicals[1,2] and difficult processing[3,4] and therefore there is a great demand for alternative strategies for surface priming.
Inspiration for our approach is found in marine mussels (Figure 1a) which use primers to improve holdfast bonding to rock surfaces. Of the more than 10 known holdfast proteins, including mussel foot proteins (mfp) 1–6 and collagens,[5–7] mfp-3 and -5 are found at the interface between the byssal plaque (the disk-shaped adhesive pad) and the rock surface (Figure 1b) where they act as surface primers.[5,8,9] These interfacial mfps have unusually high abundance (28–34 mol%) of aromatic residues including tyrosine (Y), tryptophan (W), and a posttranslationally modified form of tyrosine: 3,4-dihydroxyphenyl-L-alanine (DOPA, Y′). Of these, DOPA is now accepted as one of the key functional groups for wet-adhesion due to its strong, bidentate binding to oxide mineral surfaces.[5] Inspired by the natural DOPA chemistry, numerous laboratories have exploited catechol functionalization in the translation of this bioadhesion to synthetic systems[10–12] and for various applications[1,13–15] such as functional materials; for example, catechol-functionalized polymers were used as binders for silicon lithium battery anodes,[15] and catecholic zwitterionic surfactants were employed as surface modifiers on nanogrooved dielectric substrates for organic field-effect transistors.[1] Although previous studies using catechol-decorated polymers have demonstrated improvements in adhesion,[5,12,16] the performance of such manmade polymers,[16] as well as isolated native mussel and engineered proteins[17] is still far lower than that of whole natural mussel plaques, in terms of the total energy required to dislodge a plaque from a surface.[18] These differences may arise from the lack of consideration given in previous work to the heterogeneous nature of the mussel plaque structures[19] and their impact on load transfer within the material. For example, although catechol residues are highly enriched (20–28 mol%) in interfacial mfps, they are much less abundant in bulk mfps (2–5 mol%). Nonetheless, the majority of efforts in mussel-inspired wet-adhesion have focused on applying catechol functionalities, which promote nanoscale interfacial adhesion, into the bulk phase of synthetic polymers.[20–22] By contrast, very few mussel-mimetic adhesion studies[23,24] have attempted to create surface primers to improve bond performance, although strong catecholic bonding to metal surfaces has been demonstrated[25] and utilized.[26] In general, bonding to hydrated mineral surfaces remains extremely challenging. By simply shifting the paradigm of translation, we here apply the discovery of catechol-rich biological priming to synthetic catecholic priming at the same nanoscopic length scales, and achieve a significant (i.e., an order of magnitude), enhancement in the adhesive strength of a conventional polymethacrylate (PMA) resin bonded to mineral surfaces.
Figure 1.
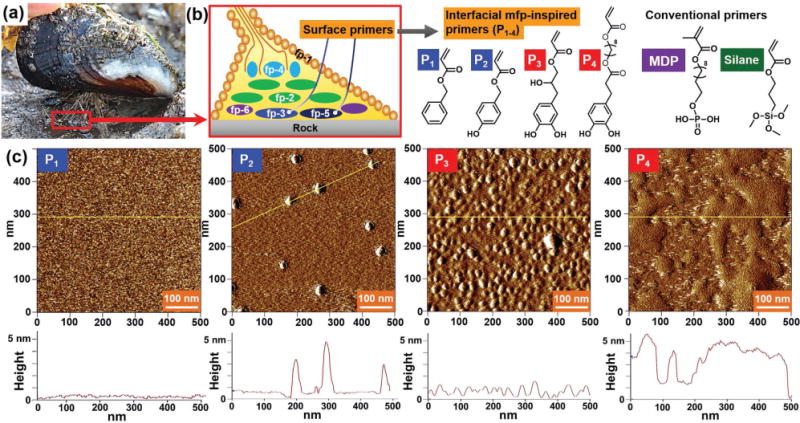
Surface priming by mussel-inspired primers. a) A mussel attached to a rock surface using its byssus (Goleta Pier, California). b) Schematics showing the location of interfacial mfps within a plaque, and the chemical structures of the mussel-inspired primers and conventional primers. c) AFM images and height profiles of the mussel-inspired primers adsorbed to mica.
Our strategy of using a priming layer to enhance bonding performance of conventional PMA resins[23,27] contrasts with the few prior approaches that used polydopamine or DOPA-containing polymethacrylate. Our proposed biomimetic coating operations use simple dip-coating of surfaces into dopamine-containing aqueous solutions at pH values of ≈8–8.5; the surfaces are then soaked for several hours to allow the auto-oxidative cross-linking reaction to come to completion.[13] Coating with polydopamine[23] or catechol-polymethacrylate[27] does improve adhesion performance due to improved wettability or cohesion with iron-catechol coordination within the priming polymer layer, respectively, but neither approach has been shown to achieve direct chemical bridging[8,28] between the substrate and PMA resin. Indeed, such coating efforts differ from the mussel’s native strategy, and perhaps not surprisingly, the mussel’s strong wet adhesion and high toughness have not yet been replicated with synthetic materials using this approach.
In this study, instead of using the brittle polymer coating (i.e., polydopamine), we have used self-assembled monolayers to provide dynamic interactions (i.e., via hydrogen bonds) at the molecular scale interface, as nature does. In particular, we endeavored to test whether mimicking the priming strategy of mussels with a very simplified molecular analog could enhance the performance of a conventional PMA resin. First, we prepared acrylate primers (small bifunctional molecules containing aromatic and acrylic groups at opposite ends) to mimic catecholic interfacial interactions to mineral surfaces and crosslinking interactions to the bulk PMA resin. The primers were designed to anchor to the substrate, providing chemical bridging between the two dissimilar materials. Rather than synthesizing perfectly matching homologs with exactly the same linkers, we prepared primers using commercially available, cheap, and naturally abundant compounds (e.g., eugenol and dihydrocaffeic acid derivatives) to create a more practical and versatile synthetic system. Four different acrylic surface primers functionalized with benzene (P1), phenol (P2), and catechol (P3 and P4) were synthesized as analogs of aromatic residues in mfps (Figure 1b). P4 differs from P3 by its longer alkyl spacer (with an additional eight hydrocarbons) between the catechol and acrylate groups.
To assess surface binding and morphology, each primer solution (at 1 wt% in methanol or DMSO) was applied onto a mica or glass surface. After a 30 s incubation, the excess of each primer was rinsed away, and the height profile of each surface investigated by atomic force microscopy (AFM). Whereas noncatecholic primers showed no adsorption or aggregate formation, it was confirmed that the catecholic primers P3 and P4 densely primed the mica (Figure 1c). However, due to the roughness of glass surfaces (root mean square, RMS R ≈ 5 nm), which overwhelmed molecular roughness of bound primers (RMS R ≈ 1 nm), clear distinctions between the glass surfaces were not discernible (Figure S5, Supporting Information). The quantitative adsorption of the primers to SiO2 surfaces evaluated by quartz crystal microbalance with dissipation (QCM-D) confirmed the adsorption of monolayers of the catecholic primers on mica and glass surfaces (data are shown in Figure 2a and Figure S6, Supporting Information). The experimental methods are fully described in the Supporting Information.
Figure 2.
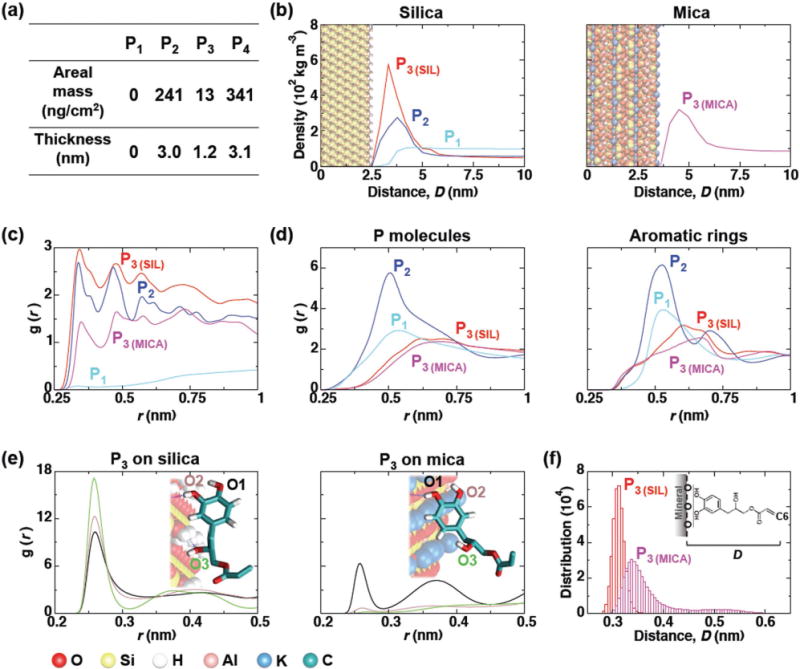
Computational modeling of primer adsorption onto silica and mica surfaces. a) Calculated mass and thickness of adsorbed layers of P1–4 based on QCM-D results (see Supporting Information for more details). b) Density profiles of primers along the axis normal to the mineral surfaces. All three primers were simulated on the silica surfaces, and only P3 was simulated on mica to confirm the universal mineral adsorption. Curves represent the average densities as a function of distance D from the oxygen atoms on the mineral surface to each primer molecule. The position of the mineral (silica and mica) surfaces in the graphs are shown as CPK model (as illustrated in the bottom left of the figure). c) The time-averaged radial distribution functions between the aromatic rings for each primer type and the mineral surface oxygen atoms are shown. The radial distribution function is given by g(r) = 4πr2ρ dr, which represents a normalized histogram constructed from the calculated radial distances r between all molecular pairs of a given number density ρ. d) Radial distribution functions between the center of mass of primer molecules and aromatic groups (represented as carbon atoms) over time. e) Radial distribution function between the oxygen atoms from mineral surface and hydroxyl groups of P3. A representative snapshot of the preferred adsorption configuration is shown as an inset in each case each hydroxyl group labeled. f) Histogram of the average minimum distance between the plane formed by the O atoms of the mineral surface and the C6 carbon atom. All analyses were performed using the last 400 ns time interval of each simulation.
Molecular dynamics (MD) simulations were carried out to characterize the molecular adsorption of the primers on model crystalline silica and mica surfaces. Such modeling can elucidate how small changes in molecular structure and surface characteristics can affect adsorption. The computational methods are fully described in the Supporting Information. For this work, P1, P2, and P3 were selected to assess the influence of their aromatic groups on mineral surface adsorption as their similar structure and contour length allow more convenient comparisons in MD simulations. The simulations were initiated with a dense layer of the primers, ≈2.5 molecules nm−2, placed above the mineral surfaces, and were run for 500 ns. The results of the MD simulations agreed with those of the AFM and QCM-D studies: the catecholic moieties in P3 were densely packed at both the mica and silica surfaces, whereas P1 did not adsorb (Figure 2a,b). In addition, the MD simulations allow a more detailed assessment of the differences between the phenol and catechol groups of the P2 and P3 molecules, respectively. We found that both P2 and P3 were recruited to oxide surfaces via hydrogen bond formation; however, although the P2 hydroxyl groups showed a well-defined interaction with silica surface, the molecules possessed a higher diffusion coefficient in the plane normal to the mineral surface (Table 2, Supporting Information). This behavior indicates multiple binding and unbinding events that, in the case of P2, resulted in a significantly less dense binding layer than was achieved with P3, in agreement with experimental observations. This finding also agrees with previous studies[5] that showed that two or more vicinal hydroxyl groups are crucial for the effective adsorption of DOPA molecules in a bidentate fashion. P2 molecules also show the strongest interaction between aromatic rings of neighboring molecules, leading to the formation of aggregates (Figure 2c), in agreement with AFM and QCM-D studies. Notably, all three of the hydroxyl groups of P3 contributed significantly to hydrogen bonding to the silica surface, whereas only the catecholic hydroxyl groups of P3 interacted effectively with mica (Figure 2d). As a result, we observed a larger contact area for each P3 molecule to silica and consequently, a smaller thickness of the P3 molecular layer as compared to that observed on mica (Figure 2e).
To explore the effects of this mussel-inspired priming layer of molecular thickness (1–5 nm) on adhesion enhancement, we investigated the performance of a crosslinked PMA resin, a common crosslinked copolymer resin used in dental restoration and medical bone cement applications[29] (Figure S7, S8, Supporting Information). The resin was prepared by a photoinitiated radical polymerization of four comonomers: triethylene glycol dimethacrylate, bisphenol A glycerolate dimethacrylate, 2-dimethylaminoethyl methacrylate, camphoquinone. Each primer was applied onto various mineral substrates prior to the application of the PMA resin. Two conventional priming layers were used as comparisons for the new mussel-inspired priming system: a phosphate-based primer (10-methacryloyloxydecyl dihydrogen phosphate, herein called “MDP”) and a silane-based primer (3-trimethoxysilylpropyl acrylate, herein called “Silane”).
First, we investigated the effect of the mussel-inspired priming on the lap shear strength of the PMA resin bonded to mica, glass, or a silicon wafer. Although our initial focus was on the dynamic interactions between silicate mineral surfaces and PMA resins, we also conducted a lap shear test using tooth enamel surfaces (consisting of >90% hydroxyapatite, the chief mineral component of human bone) to explore potential clinical applications, for example as a tooth bonding primer that could eliminate harmful acid etching that causes permanent damage to native dental tissues as well as other complications such as cavities and hypersensitivities.[30,31] Primer adsorption was performed as described, then the PMA resin was cured on the primed substrates (Figure 3a, see Supporting Information for details). The measured maximum lap shear force, Fmax, was converted to shear strength, σshear, max = Fmax/πR2, where R is the radius of the glued surface. Adhesive failure between polymer and substrate was confirmed with optical microscopy. On silicate materials (e.g., mica and silica), the lap shear strength of cured PMA using P1 (σshear, max = 0.4 ± 0.1 and 2.0 ± 0.3 MPa, n = 10; the mean of maximum shear strength on mica and glass, respectively, ±as standard deviation, and the n is number of experiments), P2 (σshear, max = 0.4 ± 0.1 and 2.0 ± 0.3 MPa, n = 10) and MDP primers (σshear, max = 0.5 ± 0.1 and 1.9 ± 0.3 MPa, n = 10) was similar to that of PMA cured with no primer (σshear, max = 0.4 ± 0.1 and 1.9 ± 0.4 MPa, n = 10) at all. Indeed, all four values agreed within experimental uncertainty. By contrast, the use of the catecholic primers P3 (σshear, max = 0.8 ± 0.2 and 4.2 ± 0.7 MPa, n = 10), P4 (σshear, max = 0.9 ± 0.1 and 4.5 ± 0.5 MPa, n = 10) doubled the lap shear strength compared with the no-primer case. In fact, the measured strengths of the catechol-primed samples were equal, within experimental uncertainty, to that of the silane-primed surface (σshear, max = 0.9 ± 0.4 and 3.8 ± 1.1 MPa, n = 10), which is used as the industry standard despite of its low surface coverage, high energy consumption and toxic chemical usage[1,2] (Figure 3d,e; Figure S9, Supporting Information). On tooth enamel (Figure 3f), the lap shear strength measured using surfaces primed with P3 (σshear, max = 3.0 ± 0.3 MPa, n = 10), and P4 (σshear, max = 3.0 ± 0.3 MPa, n = 10) was almost double that of the no-primer case (σshear, max = 1.8 ± 0.1 MPa, n = 10), and equal, within experimental uncertainty, to that of MDP (σshear, max = 3.1 ± 0.2 MPa, n = 10), which is used as the dental industry standard despite the unfavorable pH dependency of phosphate-calcium binding.[31,32] By contrast, enamel surfaces primed with P1, P2, and silane showed no increase in the lap shear strength compared with the no primer control. Most importantly, the catecholic primer shows a universal increase in lap shear strength on both silicate and human mineral surfaces, in contrast to current treatments which are optimized to a specific surface chemistry. We also varied conditions from pH 3–10 after deposition of the molecules, and found that the lap shear strength was unchanged, since the unbound excess of all catechol primers was removed with the rinsing step. All sample preparation and testing were carried out at room temperature.
Figure 3.
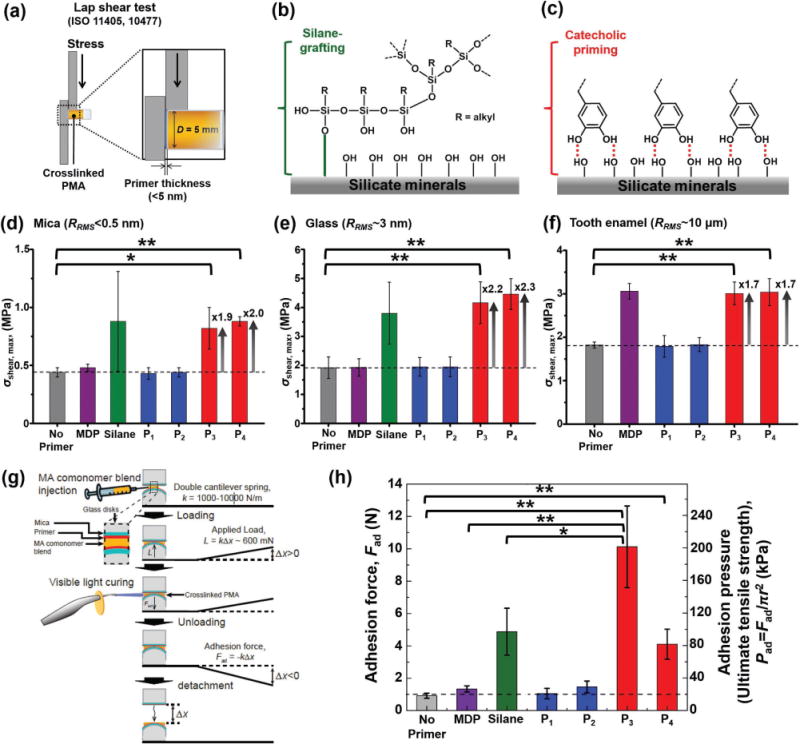
Effects of surface priming on adhesion of a crosslinked PMA resin. a) Schematic of the experimental procedure used in the lap shear test. b,c) Schematic drawing of silane-grafted (b) and catechol-primed (c) silicate mineral surfaces. Lap shear strengths of cured PMA on: d) mica, e) glass, and f) tooth enamel (IRB No. S-D20 160003) surfaces treated with different primers (MDP, silane, P1, P2, P3, and P4). In each case, the RMS surface roughness RRMS (measured in AFM) is shown. g) Schematic showing the experimental procedure used in the SFA measurements. h) Adhesion force (left axis) and pressure (right axis) between two identical primer-treated mica surfaces. P-values were calculated using the Student’s t-test to assess statistical certainty of the comparisons (*p < 0.01; **p < 0.001).
Additionally, we premixed the catechol primers with bulk PMA resin to test if bonding performance can be further improved, despite reports that catechols can act as radical scavengers.[33,34] As anticipated, the curing of the PMA resin was not completed in the given period of time, <1–5 min, which is not acceptable for practical dental applications.
To further investigate the effects of primers on pure adhesion, we used a surface forces apparatus (SFA) (see Figure 3g and Supporting Information for details) as a complement to the lap shear tests, which also involve cohesive failure and friction. To eliminate surface roughness that can contribute to mechanical interlocking, each primer was applied onto an atomically smooth mica surface before application of the PMA resin. The measured adhesion force, Fad, was converted to adhesion pressure (Pad = Fad/πr2), where r is the radius of the surface area at the adhesive contact. It was confirmed that the adhesive failure occurred almost always between the resin and mica, and never in the bulk phase of the cured PMA resin. Occasionally, with surfaces primed with P3, the adhesion between the mica and PMA resin exceeded the adhesion between the mica and epoxy resin, which was used to fix the mica substrate to the glass cylinder in the SFA, and hence the adhesive failure occurred between the mica and epoxy resin (the values obtained from mica-epoxy resin failure were excluded). As shown in Figure 3h, compared with plain mica (with no primer), surfaces primed with P1 and P2 showed no increase in adhesion pressure (Pad), whereas P3 (Pad = 201.5 ± 50.2 kPa, n = 5) and P4 (Pad = 81.7 ± 18.5 kPa, n = 5) exhibited a ≈10-fold and approximately fourfold increase in adhesion, respectively. Moreover, when the adhesion pressure of P3 was compared to that of the conventional MDP or silane primers, we observed an approximately eightfold or approximately twofold increase, respectively. In the case of silane (the most popular surface coupling reagent), we attribute this enhancement to the higher surface physicochemical binding density of the catecholic bidentate hydrogen bonding[35] as compared with silane-based coupling agents that typically provide only 10–20% binding efficiency during covalent bonding[4,36,37] (shown schematically in Figure 3b,c). Moreover, the measured adhesion pressure of P3 (≈200 kPa) is comparable with the estimated interfacial stress achieved just before adhesive failure of actual mussel plaques (Pad ≈ 270 kPa).[18]
PMA resins are widely used and in particular in medical and dental restorative materials and adhesives.[38,39] For dental applications, the PMA resins are typically formed with glass fillers that are treated by silane grafting to increase the rigidity and hardness of PMA resin composites. Next, we tested the use of the catecholic primers for PMA resin composites to eliminate the use of toxic chemicals for silane-based primer treatment and moreover offer the promise of dynamic[40] and self-repairing[29] bonds in such composites. Catecholic primer P3, which exhibited higher adsorption and adhesion performance compared P4, was chosen for further scrutiny. To rule out contact cytotoxicity a P3-primed glass surface was prepared, and nontoxicity was confirmed in a cell viability assay based on ISO 10993 (Figure S11, Supporting Information). The contact cytotoxicity results show high biocompatibility of catechol-primed glass surfaces, with similar results as those achieved using pristine and conventional silane-treated glass surfaces, suggesting high potential for biomedical and dental applications.
The mechanical properties of PMA resin composites containing P3 primed glass fillers were tested in compressive loading and compared to those of composites containing either conventional silane-primed fillers or without fillers (Figure 4a), respectively. To rule out the possibility that changes in the filler distribution under various treatments[41] could affect the observed mechanical properties, the topology and thermal properties of the various composites were investigated. Scanning electron microscopy (SEM) showed the glass fillers to be densely packed in all the composites (Figure 4a), and no obvious differences in packing were observed between the silane- and P3-treated samples. Unfortunately, the nonuniformity of the crushed glass fillers and the highly irregular surfaces of the fracture debris after compressive failure did not allow us to distinguish whether failure occurred cohesively within the bulk of the resin, or at the glass–polymer interface (Figure S15, Supporting Information). However, we observed similar glass transition temperatures (Tg ≈ 156 °C, Figure S16, Supporting Information) for all three composites using differential scanning calorimetry (DSC) and similar thermal decomposition patterns for the glass-filled composites through thermogravimetric analysis (TGA) (Figure S17, Supporting Information). These results suggest that the mechanical and thermal properties of the PMA composites are not strongly affected by fillers, but rather dominated by bulk polymer network.
Figure 4.
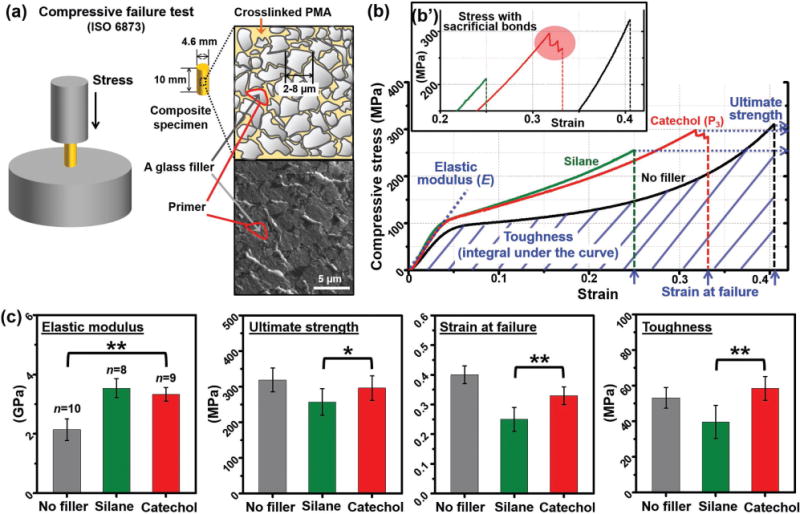
Effects of surface priming on the mechanical properties of rigid glass-filled PMA resin composites. a) Schematic showing the experimental procedure used in the compressive failure test, and schematic drawing and SEM image of a primer-treated glass-filled composite. b) A representative stress–strain curve of the mineral–PMA composite. b′) Zoom-in of the stress–strain curve (x-axis: 0.2–0.42, y-axis: 225–320 MPa) emphasizing the jagged stress decrease during failure of the P3-treated sampled (red circle). c) Elastic modulus, ultimate strength, strain at failure, and toughness of the composites. P-values were calculated using the Student’s t-test to assess statistical certainty of the comparisons (*p < 0.05; **p < 0.001).
In the mechanical testing in addition to the lap shear and SFA pulling adhesion tests of PMA resin on mineral surfaces, we chose to study the PMA composites containing glass fillers in compressive failure mode since in general such rigid composites are used in medical and dental load-bearing systems to resist compressive strain. Figure 4b shows a representative stress–strain curve for each of the three composite samples. As expected, the silane-treated glass-filled PMA composite exhibited ≈50% increase in elastic modulus as compared to the no-filler PMA sample; this enhancement is the rationale for adding fillers into PMA composites. However, in such rigid synthetic polymer composites, this increase in modulus always results in a reduction in extensibility (as determined by the strain at failure) and the resultant brittleness often leads to a reduction in ultimate strength, as shown here in the silane-treated glass-filled PMA composites (≈20% decrease in ultimate strength and ≈40% decrease in strain at failure). By contrast, the catechol-treated glass-filled PMA composites exhibited the same enhancement in elastic modulus but with a negligible reduction in the ultimate strength and only a modest ≈15% decrease in the strain at failure. This leads to a significant ≈50% enhancement in toughness compared to that of the brittle silane-treated PMA composite sample (Figure 4c). Thus, with the more ductile catechol-based priming system, we can achieve the requisite increase in resin stiffness while better maintaining the strain at failure of the PMA composite. We also conducted cyclic loading and oscillatory shear rheology tests at temperatures above Tg in an attempt to further investigate the properties of the catecholic dynamic bonding (see Supporting Information for more details). However, the observed differences between the silane- and catechol- filler-containing PMA composite samples were negligible (Figure S13, S14, Supporting Information); we attribute this to the high modulus of the samples, which leads to very small sample strains, at or below the spatial resolution of our instruments.
Interestingly, the stress–strain curve for the P3-coated glass-filled samples exhibited a noticeable pattern of jagged stress decreases after reaching its ultimate strength value (Figure 4b′) in eight out of nine samples whereas the silane-treated and no-filler samples typically exhibited abrupt and immediate fracture (Figure S12, Supporting Information). Such jagged features in the stress response have been observed in other biomaterials (i.e. bone) and in soft polymer networks[42] and have been attributed to the nanoscale rupture of sacrificial bonds which dissipate energy and provide hidden length and thus ductility.[1,43,44] We hypothesize that this pattern of saltatory stress decrease in the P3-treated samples might be correlated to the failure of the dynamic bonds formed between the catechol and mineral surfaces.[26,40,45]
In biological organisms, energy dissipation has been demonstrated to occur by sacrificial bonds and hidden lengths even in rigid tissues such as bone.[43] However, in man-made systems these toughening phenomena have been shown only in relatively soft materials to date. For example, although it has been recently reported that sample toughness increases can be achieved with the addition of catechol-mediated dynamic bonds[46] and double networks,[47] these studies have been limited to hydrogels and elastomers, where the elastic modulus E is less than 300 kPa.[48] In this study, we present significant toughness enhancement of a highly rigid synthetic polymer resin composite by a biologically inspired adhesive primer, providing strong and dynamic binding. The elastic modulus, E > 3.5 GPa, of the bioinspired composites we present here is substantially (up to a million fold) stiffer than those tough elastomers found in literature.[46,47] This study thus demonstrates the utility of mussel-inspired dynamic bonding for processing a wide range of practical polymeric interfaces, including structural, load-bearing materials.
In summary, the catecholic surface priming mechanism of mussels was successfully translated to a synthetic system. The bioinspired primer forms a ≈1 nm thick self-assembled molecular layer within 30 s. The binding mechanism onto different mineral surfaces was revealed by MD simulations, in combination with AFM and QCM-D. Building on this fundamental understanding of molecular adsorption and adhesion of newly designed catecholic primers, we enhanced the adhesion performance of a PMA resin by up to an order of magnitude on mica, glass, silica, and tooth enamel. The strong and dynamic catecholic binding also led to significant toughness enhancement (≈50%) of a highly rigid (E ≈ 3.5 GPa) polymer resin composite, providing particular promise for use as a structural material, particularly for biomedical applications. Finally, this study suggests the enormous potential of the next generation of bioinspired surface primers to replace the toxic, time and energy consuming silane-based coupling agents currently in use.
Experimental Section
All experimental procedures are reported in the Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by the Office of Naval ResearchN000141310867 (S.S., J.H.W., B.K.A.), National Institute of HealthR01 DE018468 (D.W.L., J.H.W., J.N.I.), and a generous gift from the Valois Family (J.H.W.). Several experiments made use of the MRL Shared Experimental Facilities supported by the MRSEC Program of the National Science Foundation (NSF) under Award No. DMR 1121053; a member of the NSF-funded Materials Research Facilities Network. Authors wish to thank Osaka Organic Chemical Industry LTD for providing the triethylsilane-protected eugenol acrylate. S.S. was supported by Pusan National University Research Grant, 2017. D.W.L., J.S.A., S.W.J., and E.S., B.S.K. were supported by the National Research Foundation (NRF) 2016R1C1B2014294, the Marine Biotechnology Program D11013214H480000110 and NRF2010-0028684, respectively, funded by the South Korean Government. E.F. was supported by the MRSEC Program of the NSF under Award No. DMR-1121053. M.T.V. acknowledges support of the NSF through Award No. DMR-1410985. K.C. and R.D.L. acknowledge support from FACEPE, CAPES, and CNPq. J.E.S. acknowledges support from the Center for Scientific Computing at the California Nanosystems Institute (NSF Grant CNS-0960316), and the NSF through Award No. MCB-1158577. This work also used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF Grant ACI-1053575 and the computational capabilities of the Texas Advanced Computing Center at the University of Texas at Austin (Grant MCA05S027). Partial computational resources were also provided by LNCC, the Brazilian Scientific Computing Center. Authors thank Alain Dequit from the Institut de Chemie de Clermont-Ferrand for providing the mica coordinates for molecular dynamics analyses and Wei Wei for her assistance in conducting and analyzing the QCM-D experiments.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Dr. Sungbaek Seo, Marine Science Institute, University of California, Santa Barbara, CA 93106, USA Materials Research Laboratory, Materials Research Science and Engineering Center, University of California, Santa Barbara, CA 93106, USA; Biomaterials Science, Pusan National University, Miryang 627-706, South Korea.
Dr. Dong Woog Lee, Energy and Chemical Engineering, Ulsan National Institute of Science and Technology, Ulsan 689-798, South Korea Chemical Engineering, University of California, Santa Barbara, CA 93106, USA.
Dr. Jin Soo Ahn, Marine Science Institute, University of California, Santa Barbara, CA 93106, USA Dental Research Institute and Biomaterials Science, Dentistry, Seoul National University, Seoul 110-749, South Korea.
Dr. Keila Cunha, Marine Science Institute, University of California, Santa Barbara, CA 93106, USA Fundamental Chemistry, Federal University of Pernambuco, Recife, PE 50740-670, Brazil; Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106, USA.
Dr. Emmanouela Filippidi, Materials Research Laboratory, Materials Research Science and Engineering Center, University of California, Santa Barbara, CA 93106, USA Mechanical Engineering, University of California, Santa Barbara, CA 93106, USA.
Dr. Sung Won Ju, Dental Research Institute and Biomaterials Science, Dentistry, Seoul National University, Seoul 110-749, South Korea
Eeseul Shin, Chemistry, Ulsan National Institute of Science and Technology, Ulsan 689-798, South Korea.
Dr. Byeong-Su Kim, Chemistry, Ulsan National Institute of Science and Technology, Ulsan 689-798, South Korea
Dr. Zachary A. Levine, Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106, USA
Dr. Roberto D. Lins, Fundamental Chemistry, Federal University of Pernambuco, Recife, PE 50740-670, Brazil Aggeu Magalhaes Institute, Oswaldo Cruz Foundation, Recife, PE 50670-465, Brazil.
Dr. Jacob N. Israelachvili, Materials Research Laboratory, Materials Research Science and Engineering Center, University of California, Santa Barbara, CA 93106, USA Chemical Engineering, University of California, Santa Barbara, CA 93106, USA.
Dr. J. Herbert Waite, Marine Science Institute, University of California, Santa Barbara, CA 93106, USA Materials Research Laboratory, Materials Research Science and Engineering Center, University of California, Santa Barbara, CA 93106, USA.
Dr. Megan T. Valentine, Materials Research Laboratory, Materials Research Science and Engineering Center, University of California, Santa Barbara, CA 93106, USA Mechanical Engineering, University of California, Santa Barbara, CA 93106, USA.
Dr. Joan Emma Shea, Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106, USA
Dr. B. Kollbe Ahn, Marine Science Institute, University of California, Santa Barbara, CA 93106, USA Materials Research Laboratory, Materials Research Science and Engineering Center, University of California, Santa Barbara, CA 93106, USA.
References
- 1.Das S, Lee BH, Linstadt RTH, Cunha K, Li Y, Kaufman Y, Levine ZA, Lipshutz BH, Lins RD, Shea JE, Heeger AJ, Ahn BK. Nano Lett. 2016;16:6709. doi: 10.1021/acs.nanolett.6b03860. [DOI] [PubMed] [Google Scholar]
- 2.Dupraz AMP, Meer SATvd, De Wijn JR, Goedemoed JH. J Mater Sci: Mater Med. 1996;7:731. [Google Scholar]
- 3.Lee JH, Um CM, Lee I. Dent Mater. 2006;22:515. doi: 10.1016/j.dental.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Gooding JJ, Ciampi S. Chem Soc Rev. 2011;40:2704. doi: 10.1039/c0cs00139b. [DOI] [PubMed] [Google Scholar]
- 5.Lee BP, Messersmith PB, Israelachvili JN, Waite JH. Annu Rev Mater Res. 2011;41:99. doi: 10.1146/annurev-matsci-062910-100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silverman HG, Roberto FF. Mar Biotechnol. 2007;9:661. doi: 10.1007/s10126-007-9053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waite JH, Andersen NH, Jewhurst S, Sun C. J Adhes. 2005;81:297. [Google Scholar]
- 8.Ahn BK, Das S, Linstadt R, Kaufman Y, Martinez-Rodriguez NR, Mirshafian R, Kesselman E, Talmon Y, Lipshutz BH, Israelachvili JN, Waite JH. Nat Commun. 2015;6:8663. doi: 10.1038/ncomms9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang DS, Zeng H, Masic A, Harrington MJ, Israelachvili JN, Waite JH. J Biol Chem. 2010;285:25850. doi: 10.1074/jbc.M110.133157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H, Lee BP, Messersmith PB. Nature. 2007;448:338. doi: 10.1038/nature05968. [DOI] [PubMed] [Google Scholar]
- 11.Ye Q, Zhou F, Liu W. Chem Soc Rev. 2011;40:4244. doi: 10.1039/c1cs15026j. [DOI] [PubMed] [Google Scholar]
- 12.Faure E, Falentin-Daudré C, Jérôme C, Lyskawa J, Fournier D, Woisel P, Detrembleur C. Prog Polym Sci. 2013;38:236. [Google Scholar]
- 13.Lee H, Dellatore SM, Miller WM, Messersmith PB. Science. 2007;318:426. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yah WO, Xu H, Soejima H, Ma W, Lvov Y, Takahara A. J Am Chem Soc. 2012;134:12134. doi: 10.1021/ja303340f. [DOI] [PubMed] [Google Scholar]
- 15.Ryou MH, Kim J, Lee I, Kim S, Jeong YK, Hong S, Ryu JH, Kim TS, Park JK, Lee H, Choi JW. Adv Mater. 2013;25:1571. doi: 10.1002/adma.201203981. [DOI] [PubMed] [Google Scholar]
- 16.Seo S, Das S, Zalicki PJ, Mirshafian R, Eisenbach CD, Israelachvili JN, Waite JH, Ahn BK. J Am Chem Soc. 2015;137:9214. doi: 10.1021/jacs.5b03827. [DOI] [PubMed] [Google Scholar]
- 17.Zhong C, Gurry T, Cheng AA, Downey J, Deng Z, Stultz CM, Lu TK. Nat Nanotechnol. 2014;9:858. doi: 10.1038/nnano.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desmond KW, Zacchia NA, Waite JH, Valentine MT. Soft Matter. 2015;11:6832. doi: 10.1039/c5sm01072a. [DOI] [PubMed] [Google Scholar]
- 19.Filippidi E, DeMartini DG, Malo de Molina P, Danner EW, Kim J, Helgeson ME, Waite JH, Valentine MT. J R Soc, Interface. 2015;12 doi: 10.1098/rsif.2015.0827. 20150827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matos-Pérez CR, White JD, Wilker JJ. J Am Chem Soc. 2012;134:9498. doi: 10.1021/ja303369p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehdizadeh M, Weng H, Gyawali D, Tang L, Yang J. Biomaterials. 2012;33:7972. doi: 10.1016/j.biomaterials.2012.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Meng H, Liu Y, Narkar A, Lee BP. ACS Appl Mater Interfaces. 2016;8:11980. doi: 10.1021/acsami.6b01364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi M, Sun H, Zhang H, Deng X, Cai Q, Yang X. Mater Sci Eng C. 2016;58:742. doi: 10.1016/j.msec.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 24.Hamming L, Fan X, Messersmith P, Brinson L. Compos Sci Technol. 2008;68:2042. doi: 10.1016/j.compscitech.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Scherer NF, Messersmith PB. Proc Natl Acad Sci USA. 2006;103:12999. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stepuk A, Halter JG, Schaetz A, Grass RN, Stark WJ. Chem Commun. 2012;48:6238. doi: 10.1039/c2cc31996a. [DOI] [PubMed] [Google Scholar]
- 27.Lee SB, González-Cabezas C, Kim KM, Kim KN, Kuroda K. Biomacromolecules. 2015;16:2265. doi: 10.1021/acs.biomac.5b00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin Q, Gourdon D, Sun C, Holten-Andersen N, Anderson TH, Waite JH, Israelachvili JN. Proc Natl Acad Sci USA. 2007;104:3782. doi: 10.1073/pnas.0607852104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn BK, Lee DW, Israelachvili JN, Waite JH. Nat Mater. 2014;13:867. doi: 10.1038/nmat4037. [DOI] [PubMed] [Google Scholar]
- 30.Baumann T, Carvalho TS, Lussi A. Sci Rep. 2015;5:15194. doi: 10.1038/srep15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Landuyt KL, Snauwaert J, De Munck J, Peumans M, Yoshida Y, Poitevin A, Coutinho E, Suzuki K, Lambrechts P, Van Meerbeek B. Biomaterials. 2007;28:3757. doi: 10.1016/j.biomaterials.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 32.Emmett M. Kidney Int. 2004;66:S25. doi: 10.1111/j.1523-1755.2004.09005.x. [DOI] [PubMed] [Google Scholar]
- 33.Thavasi V, Leong LP, Bettens RPA. J Phys Chem A. 2006;110:4918. doi: 10.1021/jp057315r. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Q, Nurumbetov G, Simula A, Zhu C, Li M, Wilson P, Kempe K, Yang B, Tao L, Haddleton DM. Polym Chem. 2016;7:7002. [Google Scholar]
- 35.Yu J, Kan Y, Rapp M, Danner E, Wei W, Das S, Miller DR, Chen Y, Waite JH, Israelachvili JN. Proc Natl Acad Sci USA. 2013;110:15680. doi: 10.1073/pnas.1315015110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huheey RL, Keiter JE, Keiter EA. Inorganic Chemistry, Principles of Structure and Reactivity. Prentice Hall; Upper Saddle River, NJ, USA: 1993. [Google Scholar]
- 37.Naik VV, Crobu M, Venkataraman NV, Spencer ND. J Phys Chem Lett. 2013;4:2745. [Google Scholar]
- 38.Kuehn KD, Ege W, Gopp U. Orthop Clin North Am. 2005;36:17. doi: 10.1016/j.ocl.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Roeters JJM, Shortall ACC, Opdam NJM. Br Dent J. 2005;199:73. doi: 10.1038/sj.bdj.4812520. [DOI] [PubMed] [Google Scholar]
- 40.Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC, Waite JH. Proc Natl Acad Sci USA. 2011;108:2651. doi: 10.1073/pnas.1015862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pu Z, Mark JE, Jethmalani JM, Ford WT. Chem Mater. 1997;9:2442. [Google Scholar]
- 42.Vaca C, Shlomovitz R, Yang Y, Valentine MT, Levine AJ. Soft Matter. 2015;11:4899. doi: 10.1039/c5sm00262a. [DOI] [PubMed] [Google Scholar]
- 43.Fantner GE, Hassenkam T, Kindt JH, Weaver JC, Birkedal H, Pechenik L, Cutroni JA, Cidade GAG, Stucky GD, Morse DE, Hansma PK. Nat Mater. 2005;4:612. doi: 10.1038/nmat1428. [DOI] [PubMed] [Google Scholar]
- 44.Lieou CKC, Elbanna AE, Carlson JM. Phys Rev E. 2013;88:12703. doi: 10.1103/PhysRevE.88.012703. [DOI] [PubMed] [Google Scholar]
- 45.Menyo MS, Hawker CJ, Waite JH. Soft Matter. 2013;9:10314. doi: 10.1039/C3SM51824H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grindy SC, Learsch R, Mozhdehi D, Cheng J, Barrett DG, Guan Z, Messersmith PB, Holten-Andersen N. Nat Mater. 2015;14:1210. doi: 10.1038/nmat4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ducrot E, Chen Y, Bulters M, Sijbesma RP, Creton C. Science. 2014;344:186. doi: 10.1126/science.1248494. [DOI] [PubMed] [Google Scholar]
- 48.Skelton S, Bostwick M, O’Connor K, Konst S, Casey S, Lee BP. Soft Matter. 2013;9:3825. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.