

Abstract
Congenital dyserythropoietic anemia type I (CDAI) is an autosomal recessive inherited haematological disorder associated with moderate-to-severe anemia characterized by ineffective erythropoiesis with distinct morphological abnormalities in erythroid precursors. We present two case of congenital dyserythropoietic anemia type I in two Sicilian patients heterozygous for β0 39 globin gene cod 39 C > T with marked bone marrow abnormalities, responding to treatment with alpha interferon. The diagnosis was established using routine haematological and biochemical test, light and electron microscopy; molecular analysis of the CDAN1 gene associated to the CDAI disease was performed. The response to the treatment was monitored using the hemoglobin levels, the red cell count, the reticulocyte count and the transfusional requirement. This report points out the usefulness of the treatment with interferon alpha in two Sicilian beta thalassemia carriers, in which the therapy was well tolerated without producing any side effects; in these patients the transfusion requirements after the initiation of interferon therapy decreased.
Keywords: Congenital dyserythropoietic anemia type I, Interferon α, Spongy heterochromatin
The congenital dyserythropoietic anemia type I (CDAI) is a rare inherited haematological disorder characterized by moderate-to-severe anemia, ineffective erythropoiesis with distinct morphological features that include internuclear chromatin bridges, spongy heterochromatin, and invagination of cytoplasm into nuclear area seen in erythroid precursors [1]. Approximately 90% of patients with a bone marrow examination that suggests CDAI have mutations in the CDAN1 gene [2]. Lavabre-Bertrand et al. [3] first reported the efficacy of therapy with α-interferon (IFN-α) in increasing hemoglobin levels.
We reported the response to IFN-α treatment into two Sicilian patients affected with morphologically confirmed CDAI and beta thalassemia carriers (heterozygous for β0 39 globin mutation). The suspicion of CDAI in these patients came with the observation of smear bone marrow at light microscopy with the finding of megaloblastic erythroid hyperplasia, binucleated red cell precursors and inter-chromatin bridges (Fig. 1a).
Fig. 1.
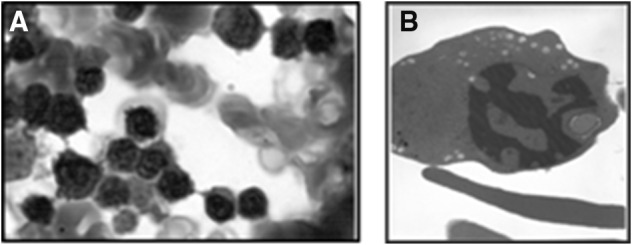
a Light microscopy of bone marrow: erythroblasts obtained by aspiration, indicating internuclear chromatin bridges. b Electron microscopy of bone marrow: CDA1 erythroblast with characteristic “Swiss cheese” (spongy) heterochromatin
The diagnosis of CDAI was then made based on the following findings: congenital microcytic anemia because they were beta thalassemia carriers, jaundice and splenomegaly. In the patient 1 the percentage of reticulocytes was 2.38 and the reticulocyte production index was 1.02 while, in the patient 2 they were 3.38 and 0.83 respectively. Hereditary spherocytosis (HS), myelodysplasia, hereditary hemochromatosis and unclassified congenital haemolytic anaemia and folate deficiency were ruled out. Hb fractions determined by high-performance liquid chromatography (HPLC) not reveal the presence of pathologic peak. The molecular analysis of beta-globin genes showed a heterozygous status for the nonsense codon39 C > T mutation. Molecular study for alpha-thalassaemia genes did not show deletions; the ααα anti3.7 and ααα anti4.2 arrangements was not found. The electronic microscopy (EM) confirmed the presence of erythroblasts with invagination of the nuclear membrane and the characteristic pattern of spongy “swiss cheese” heterochromatin (Fig. 1b). Mutations in the CDAN1 gene, which encodes Codanin-1, are responsible for the majority of CDAI. However, no likely pathogenic CDAN1 mutation has been detected in approximately 20% of cases, suggesting the presence of at least one other locus [4]. Moreover, mutations in CDAN1 gene were not identified in our patients.
Patient 1, a 15-year-old woman, with moderate congenital microcytic anemia (Hb 8.4 ± 0.24 g/dL) started IFN α2b therapy in March 2014 (4.5 × 106 subcutaneous IU weekly). Patient 2, a 50-year-old woman, was admitted to hospital for evaluation of persistent severe microcytic hypochromic anemia since birth. She underwent sporadic transfusions from the age of 8 to the age of 43 years. Treatment with IFN α2b began in March 2014 with 9 × 106 subcutaneous IU weekly. Pre-treatment data showed Hb of 6.62 g/dL and an average transfusional interval of 30 days (18 blood bags infused/year).
The favorable response to IFN-α2 in the above mentioned patients demonstrates that interferon remains an effective treatment of anemia in CDAI for a long period and free of relevant side effects. Patient 1 showed an increasing on haemoglobin level (1 g/dL) since the beginning of treatment (pre-treatment Hb level 8.5 versus 9.44 g/dL post-treatment). The treatment was well tolerated; the only symptom was flu-like syndrome on the first day of drug administration. There were no changes in the indices of hemolysis. Patient 2 was treated for several years as a thalassemia patient and received monthly blood transfusions (18 blood bags infused/year) with pre-treatment Hb level of 6.62 g/dL. During the year of treatment with IFNα2b the patient was transfused with pre-transfusional Hb of 7.17 g/dL and with an average transfusional interval of 49 days. These data showed a reduction of transfusion requirement. The treatment was well tolerated, apart a 2-weeks drug interruption due to transaminases increase (AST × 5, ALT × 2), resolved with IFN-α discontinuation. Optimization of the dose and duration of treatment may be necessary to minimize the side effects associated with prolonged treatment (Fig. 2a, b).
Fig. 2.

a Comparison between pre-treatment Hb levels vs follow-up Hb levels. b Haemoglobin levels in response to IFN α2b treatment
The favorable clinical response observed confirms the previously published data on the efficacy of IFN-α. Similarly to the others rare anemias, we suggest a multidisciplinary approach for treating patients with CDAI in cooperation with specialized centers in order to recognize disease-specific complications and to avoid useless diagnostic procedures. A collection of all cases in a national and international registry should be pursued to define evidence-based recommendations for the management of these patients.
Compliance with Ethical Standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
V. Agrigento and R. Barone have contributed equally to this work.
References
- 1.Yarali N, Fişgin T, Duru F, Atilla P, Müftüoğlu SF, Kaymaz SF. Successful management of congenital dyserythropoietic anemia type I with interferon alpha in a child. Pediatr Hematol Oncol. 2005;22(4):265–270. doi: 10.1080/08880010590935149. [DOI] [PubMed] [Google Scholar]
- 2.Dgany O, Avidan N, Delaunay J, Krasnov T, Shalmon L, Shalev H, Eidelitz-Markus T, Kapelushnik J, Cattan D, Pariente A, Tulliez M, Crétien A, Schischmanoff PO, Iolascon A, Fibach E, Koren A, Rössler J, Le Merrer M, Yaniv I, Zaizov R, Ben-Asher E, Olender T, Lancet D, Beckmann JS, Tamary H. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet. 2002;71(6):1467–1474. doi: 10.1086/344781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavabre-Bertrand T, Blanc P, Navarro R, Saghroun M, Vannereau H, Braun M, Wagner A, Taïb J, Lavabre-Bertrand C, Navarro M. Alpha-Interferon therapy for congenital dyserythropoiesis type I. Br J Haematol. 1995;89(4):929–932. doi: 10.1111/j.1365-2141.1995.tb08442.x. [DOI] [PubMed] [Google Scholar]
- 4.Babbs C, Roberts NA, Sanchez-Pulido L, McGowan SJ, Ahmed MR, Brown JM, Sabry MA, WGS500 Consortium. Bentley DR, Mc Vean GA, Donnelly P, Gileadi O, Ponting CP, Higgs DR, Buckle VJ. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica. 2013;98(9):1383–1387. doi: 10.3324/haematol.2013.089490. [DOI] [PMC free article] [PubMed] [Google Scholar]