

Abstract
Background and aims
Next generation sequencing approaches have tremendously improved the diagnosis of rare genetic diseases. It may however be faced with difficult clinical interpretation of variants. Inherited enzymatic diseases provide an invaluable possibility to evaluate the function of the defective enzyme in human cell biology. This is the case for respiratory complex III, which has 11 structural subunits and requires several assembly factors. An important role of complex III in liver function is suggested by its frequent impairment in human cases of genetic complex III defects.
Methods
We report the case of a child with complex III defect and acute liver dysfunction with lactic acidosis, hypoglycemia, and hyperammonemia. Mitochondrial activities were assessed in liver and fibroblasts using spectrophotometric assays. Genetic analysis was done by exome followed by Sanger sequencing. Functional complementation of defective fibroblasts was performed using lentiviral transduction followed by enzymatic analyses and expression assays.
Results
Homozygous, truncating, mutations in LYRM7 and MTO1, two genes encoding essential mitochondrial proteins were found. Functional complementation of the complex III defect in fibroblasts demonstrated the causal role of LYRM7 mutations. Comparison of the patient’s clinical history to previously reported patients with complex III defect due to nuclear DNA mutations, some actually followed by us, showed striking similarities allowing us to propose common pathophysiology.
Conclusions
Profound complex III defect in liver does not induce actual liver failure but impedes liver adaptation to prolonged fasting leading to severe lactic acidosis, hypoglycemia, and hyperammonemia, potentially leading to irreversible brain damage.
Graphical abstract
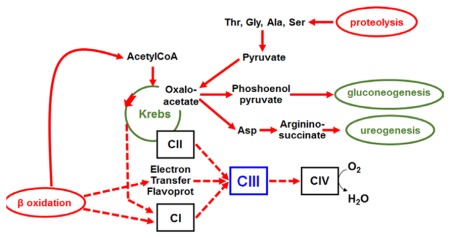
During prolonged fasting, β oxidation and proteolysis are activated to produce both energy and gluconeogenic substrates. High fluxes of electrons are generated (
 ) that all converge towards the respiratory complex III (CIII) explaining the sensitivity to fast of patients with hepatic complex III defect
) that all converge towards the respiratory complex III (CIII) explaining the sensitivity to fast of patients with hepatic complex III defect
Introduction
Classical mitochondrial diseases result from defects of the mitochondrial oxidative phosphorylation pathway (OXPHOS). They present with diverse clinical features [1]. Liver failure however is frequently reported in severe pediatric cases [2]. Its mitochondrial origin is often diagnosed by the presence of defective OXPHOS activities in liver. However, because of the possibility of improper sample preservation or nature, the diagnosis cannot solely rely on these defects but requires identification of the genetic cause of the disease. That identification has recently been tremendously facilitated by next generation sequencing. Genetic causes of mitochondrial liver failure may be classified with respect to the type of OXPHOS defect. The most frequent are combined defects involving OXPHOS complexes with mitochondrial DNA (mtDNA)-encoded subunits, caused often by mtDNA depletion (profound decrease of the mtDNA amount) [3–6] or defects of the mtDNA translation [7–9]. In this group liver functions are globally altered associating synthesis failure with coagulopathy, cytolysis, and cholestasis often progressing towards cirrhosis. These diseases are often multisystem disorders, associating neurological deficits to the liver failure. Much rarer cases of liver failure have been associated with an isolated OXPHOS defect, which is most often respiratory complex III (ubiquinol cytochrome c oxidoreductase, EC 1.10.2.2). In these cases the liver alterations seemed to differ according to the causal gene. Global liver failure, resembling that observed with multiple OXPHOS defects, has been described in severe, early onset, diseases due to mutations of BCS1L gene encoding a complex III assembly factor [10, 11], acute episodes of hypoglycemia and hyperammonemia have been associated with mutations of UQCRB, CYC1 and UQCRC2 genes encoding complex III structural subunits [12–14], and normal liver functions have been reported in diseases due to mutations of genes encoding either complex III assembly factors (TTC19 [15], UQCC2 [16], UQCC3 [17] and LYRM7 [18, 19]) or structural subunits (MT-CYB [20], UQCRQ [21]). The role of complex III in liver function thus appears disputable. We here report a patient with liver complex III defect. Although exome sequencing revealed two genes, MTO1 and LYRM7, both carrying homozygous truncating mutations and both encoding essential mitochondrial proteins, functional complementation by lentiviral vectors demonstrated that the LYRM7 mutations were causing the complex III defect. Comparison of the patient’s clinical history to previously reported patients with complex III defect, some actually followed by us, led us to propose common pathophysiology for several complex III defects whereby profound complex III defect in liver does not induce actual liver failure but impedes liver adaptation to prolonged fasting. Lack of appropriate metabolic treatment of the fasting-induced lactic acidosis, hypoglycemia, and hyperammonemia may result in irreversible brain damage.
Material, patient and methods
Patient
The patient was the third child of first cousins parents. She first presented at 6 months of age with ketoacidosis, hyperglycemia and glycosuria. Intravenous injection of insulin induced hypoglycemia without correcting ketonuria. The child was transferred to La Rabta hospital in Tunis. At her arrival she was hypotonic, polypneic and presented with mild hepatomegaly. Biological tests revealed compensated metabolic acidosis (pH 7.42, bicarbonate 15.7 mM, pCO2 24 mmHg), hyperlactatemia (5.2 mM), and hyperlactatorrachia (8 mM) with mild hyperammonemia (64 μM, N<50), but normal liver enzymes and coagulation factors. Very high lactate, 3 OH butyrate and 2-ketoglutarate were found on urinary organic acids profiling. Electrocardiogram, echocardiography, kidney functions, and electroencephalogram were normal.
The further disease course was marked by episodes of metabolic crises associating metabolic acidosis with hyperlactatemia, ketonuria and hypoglycemia; ammonia was not measured. A severe episode, lasting three days and associated with coma, occurred at one year of age. It was followed by irreversible neurological damage with loss of contact, profound axial hypotonia, peripheral hypertonia, horizontal nystagmus, and deglutition troubles. Hepatic and skin biopsies were performed at that period. The child died shortly afterwards during a recurrent metabolic crisis at 20 months of age.
Samples
All analyses were performed after written informed consent from the parents of the child according to the local institution rules. A needle liver biopsy was immediately frozen in liquid nitrogen, stored at −80°C until enzymatic assays. Primary fibroblasts were derived from a forearm skin biopsy as described in [22].
DNA samples were obtained from liver and fibroblasts using standard procedure with SDS and proteinase K digestion followed by phenol extraction and isopropanol precipitation. DNA samples from blood were obtained using QiaAmp DNA minikit (Qiagen, Courtaboeuf, France). RNA samples from fibroblasts were obtained using the miRNeasy kit (Qiagen, Courtaboeuf, France).
Mitochondrial fractions were prepared from 2 T175 fibroblasts flasks according to [23].
Mitochondrial activities
Spectrophotometric assays for the respiratory complexes I, II, III and IV, as well as citrate synthase activities were performed according to [24].
Western blot analysis
Proteins from mitochondrial pellets were separated by blue native polyacrylamide gel electrophoresis [25] or by SDS-PAGE on a linear 4 to 20% gradient polyacrylamide gel (Biorad, Hertfordshire, UK). Antibodies against UQCRC2 and UQCRFS1 were kindly given by Dr Catherine Godinot, France [26], antibodies against MT-ND1 and against MT-CO2 were produced in our group [27]; the other antibodies were commercially available: anti-SDHA, anti-ATP5B and anti-LYRM7 were from Abcam (France), anti-MTO1 from Proteintech (USA) and anti-ACTB from Sigma-Aldrich (USA). They were visualized using peroxidaseconjugated secondary antibodies (Sigma-Aldrich, USA) and Pierce™ ECL Western Blotting Substrate (Life Technologies, USA). Western blot signals were recorded on a Fusion ultrasensitive camera platform (Vilber Lourmat, Germany) and quantified using the Fusion attached software. Results were normalized to the control run on the same gel and to complex II, either the whole complex II after blue native gel electrophoresis or SDHA subunit after SDS gel electrophoresis.
Exome sequencing
Exome sequencing and variant filtering was essentially performed as described previously [28]. In brief, coding DNA fragments were enriched with the SureSelect Human All Exon 50Mb V4 Kit (Agilent, Santa Clara, CA, USA) and sequencing was performed on a HiSeq2000 system (Illumina, San Diego, CA, USA). Reads were aligned to the human genome assembly hg19 (UCSC Genome Browser) with Burrows-Wheeler Aligner (BWA, version 0.7. 5) and detection of genetic variation was performed using SAMtools (version 0.1.19), PINDEL (version 0.2.5a7), and ExomeDepth (version 1.0.0). 96.9 % of the target was covered at least 20-fold.
Lentiviral gene transfer
LYRM7 cDNA was purchased from DNASU Plasmid Repository (Clone ID HsCD00514704) and cloned into the pLenti6.3/V5-TOPO vector system (Invitrogen, Life technologies) for subsequent lentiviral-mediated expression in skin fibroblast cell lines using the ViraPower HiPerform Lentiviral TOPO Expression Kit (Invitrogen, Life technologies) [29]. The corresponding MTO1 clone was already available [30]. We also constructed a third lentivirus for expression of the MTO1 cDNA sequence but with Geneticin resistance [31]. Stably transduced fibroblasts were selected using Blasticidin (Invitrogen) and/or Geneticin (Sigma-Aldrich).
RNA analyses
Reverse transcription of 2 micrograms total RNA was performed with the Transcriptor First strand cDNA synthesis kit (Roche Diagnostics, USA). The cDNA steady-state was quantified using LightCycler 480 SybrGreen master mix (Roche Diagnostics, USA). Serial dilutions of linearized pGEM-T plasmids with the amplification products as insert, allowed absolute quantification of the cDNA in copy numbers per μL. Results were expressed as copy numbers per 1000 ACTB copies.
Statistical methods
Statistics were performed with SigmaPlot 12.5 software. Non parametric tests were used unless the data were shown to have a normal distribution allowing the use of parametric tests. The numbers given indicate independent measurements. Mean comparisons were performed with Mann and Whitney test or unpaired Student t test. The Holm-Sidak test for multiple comparisons was used to address the relative influence of lentivirus infection, genotype and their interaction.
Results
Complex III defect was present in the patient’s liver and fibroblasts
Spectrophotometric assays of the respiratory chain activities in the patient’s liver biopsy disclosed a profound defect in complex III (Table 1). Other mitochondrial activities were either normal or elevated, in particular rotenone sensitive complex I activity, which is highly sensitive to improper preservation. Significant decrease of complex III activity was also observed in cultured skin fibroblasts from the patient but with complex III residual activity higher than in liver, without significant impact on the combined complex II+III activity. Complex IV activity appeared mildly decreased in fibroblasts, the defect only being significant when complex IV activity was normalized to citrate synthase activity (p=0.031). Analysis of the OXPHOS composition of fibroblasts by blue native polyacrylamide gel electrophoresis showed that the amount of complex III was decreased while that of complexes I, II, IV and V was close to controls (Figure 1A). SDS-PAGE of complex III subunits showed an almost normal amount of UQCRC2 that contrasted with a severe decrease of UQCRFS1 (Figure 1B). Unassembled subunits are efficiently degraded by the mitochondrial quality control [32]. UQCRC2 is one of the first subunits assembled to the complex whereas UQCRFS1 is the last subunit added [33]. The observed pattern could therefore suggest either an alteration of UQCRFS1 itself or a complex III assembly defect.
Table 1.
OXPHOS activities in liver and fibroblasts
| Assay | Patient’s liver | Control livers [n=28] | Patient’s fibroblasts [n=4] | Control fibroblasts [n=70] |
|---|---|---|---|---|
|
| ||||
| CI | 54 | 29±10 (19–44) | NA | NA |
|
| ||||
| CII | 105 | 137±48 (77–198) | 27±3 (24–30) | 28±10 (15–40) |
|
| ||||
| CIII | 0 | 63±21 (41–89) | 14±11 (2–21) | 54±21 (30–79) |
|
| ||||
| CIV | 88 | 75±27 (47–117) | 55±20 (37–77) | 82±21 (57–106) |
|
| ||||
| II+III | 16 | 59±22 (38–81) | 25±3 (22–27) | 30±8 (20–41) |
|
| ||||
| CS | 93 | 68±29 (29–107) | 68±8 (60–75) | 78±25 (51–106) |
|
| ||||
| III/CS | 0 | 1.13±0.81 (0.45–2.10) | 0.20±0.15 (0.03–0.29) | 0.78±0.33 (0.32–1.42) |
| IV/CS | 0.95 | 1.26±0.55 (0.58–1.17) | 0.76±0.24 (0.62–0.99) | 1.11±0.29 (0.81–1.47) |
| III/II | 0 | 0.52±0.32 (0.22–0.91) | 0.51±0.37 (0.09–0.74) | 2.06±0.97 (0.85–3.98) |
| III/IV | 0 | 0.93±0.53 (0.48–1.60) | 0.20±0.17 (0.06–0.37) | 0.72±0.30 (0.40–1.09) |
Apart from the results in the Patient’s liver that were obtained only once, values are given as median ±standard deviation and as range between 10th and 90th centile between brackets; the number of different control values is given between square brackets in the column title; numbers in bold significantly differed from controls using mean comparison with Mann and Whitney test; CI=complex I or rotenone sensitive NADH ubiquinone oxidoreductase activity; CII= complex II or succinate ubiquinone oxidoreductase activity; CIII= complex III or antimycine sensitive ubiquinol cytochrome C oxidoreductase activity; CIV= complex IV or cytochrome C oxidase activity; II+III= combined II+III or succinate cytochrome C oxidoreductase activity; CS= citrate synthase, a Krebs cycle enzyme used as an indicator of the mitochondrial content; apart from the ratios to citrate synthase, results are expressed as nanomoles per minute and milligram protein of either the liver post-nuclear supernatant or the whole fibroblast homogenate.
Figure 1. Decreased amount of fully assembled respiratory complex III and altered ratio of complex III subunits in the patient’s fibroblasts.
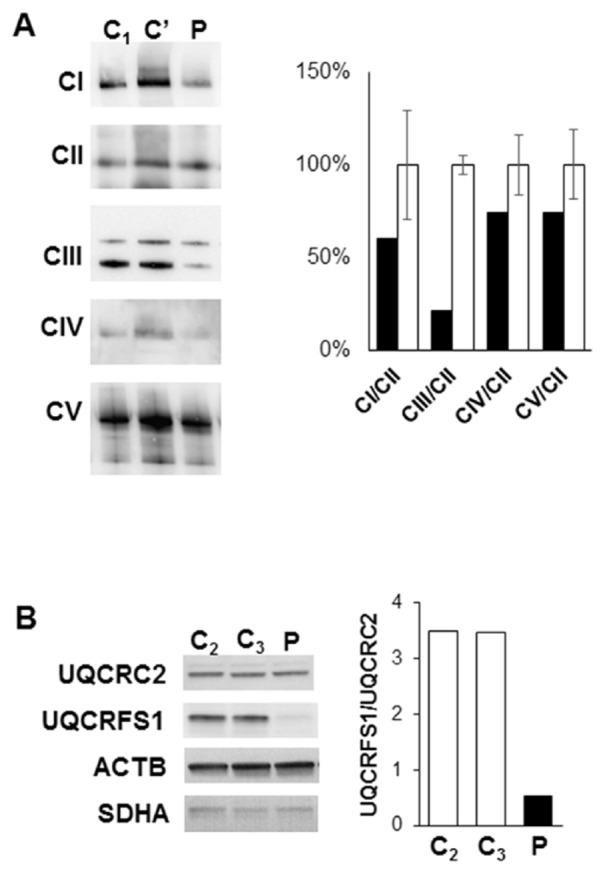
C1,2,3= control fibroblasts, C’= fibroblasts from a patient with complex III defect associated with a MT-CYB variant, P=fibroblasts from the patient.
(A) Blue native PAGE of crude mitochondrial pellets, CI = respiratory complex I, CII = complex II, CIII = complex III, CIV = complex IV, CV = complex V were revealed with antibodies against MT-ND1, SDHA, UQCRC2, MT-CO2 and ATPB respectively; the two bands of complex III correspond to monomers and dimers, both included in quantification; Quantification of Patient values (black bars), expressed as % of the mean value of controls of the same gel (white bars, error bars showing the maximum and minimum values).
(B) SDS PAGE of whole cells homogenates, showing UQCRC2 and UQCRFS1, two complex III subunits respectively involved in early and late complex assembly; ACTB and SDHA are loading controls; quantification of the UQCRFS1/UQCRC2 ratio with white bars= controls and black bar=Patient.
Search for the genetic alteration causing complex III defect disclosed two candidate genes
Direct sequencing of the coding sequences of UQCRFS1 and other genes implicated in human complex III defects (BCS1L, MT-CYB, UQCRB, UQCRQ and CYC1) disclosed only normal sequence or known polymorphisms. We thus turned to whole exome sequencing. Given that the observed pattern was in line with autosomal recessive inheritance and the clinical phenotype was very rare, we focused on genes carrying homozygous or possible compound heterozygous rare variants (Minor Allele Frequency <0.1 % in 5,000 in house controls and the Washington Exome Variant Server). This search identified 22 genes. Two of these genes, LYRM7 and MTO1, encode mitochondrial proteins [34] and variants in both have been linked to OXPHOS defects. Moreover, in both genes, mutations were homozygous predicted truncating variants. In LYRM7 (NM_181705.3) it was a c.[52delA];[52delA], p.[Arg18Aspfs*12];[Arg18Aspfs*12] frameshift variant and in MTO1 (NM_012123.3) it was a c.[1996C>T];[1996C>T], p.[Arg666*];[Arg666*] stop variant (Figure 2). The modification induced by the mutation and the phylogenetic conservation of the proteins are shown in Table 2. LYRM7 encodes for a small mitochondrial protein (104 aminoacids, molecular weight 11.9 kDa) belonging to the LYR family. It has recently been identified as a complex III late assembly factor [35] and found mutated in a severe infantile case of complex III defect with early onset severe encephalopathy and lactic acidosis [18]. MTO1 encodes for a mitochondrial protein (732 aminoacids, molecular weight after mitochondrial processing 74 kDa [36]), which has been shown to play a role in mitochondrial tRNA post transcriptional modifications [37]. It has first been thought to be involved in human pathology as a potential modifying factor for mtDNA rRNA mutations [38] but was later associated with cases of hypertrophic cardiomyopathy with combined OXPHOS defect and lactic acidosis [36].
Figure 2. Both LYRM7 and MTO1 homozygous deleterious mutation segregated with the disease.

Electrophoregram of the gene sequence in the blood DNA of the different family members; MTO1 and LYRM7 mutations are homozygous in the patient, heterozygous in her two parents and one of her healthy sibling and absent in her second healthy sibling.
Table 2.
Expected consequences of the LYRM7 and MTO1 genes mutations on the proteins

|
Only the region affected by the mutation are shown (N′-terminus for LYRM7 and C′-terminus for MTO1). LYRM7 frameshift mutation induces a change of 10 of the aminoacids from position 18 to 28 followed by a stop codon at amino acid position 29; it therefore drastically modifies the protein that normally comprises 104 aminoacids. MTO1 mutation changes Arg666 into a stop, it thus truncates the protein of its 27 last amino acids. Both LYRM7 and MTO1 proteins are, as a whole, very well conserved among mammals. The C′-terminal domain of MTO1 does not appear conserved. The patient’s deletion however removes the last part of MTO1 GidA-associated domain, which has been proposed to bind tRNA [43]
We verified the presence of the LYRM7 and MTO1 mutations by Sanger sequencing and analyzed their segregation within the family (Figure 2). For both genes, the mutation was found in a homozygous state in the patient, heterozygous in both her parents as well as in one of her healthy siblings, and absent in her second healthy sibling. Familial analysis thus could not help to exclude one of the two genes.
Complementation assays demonstrated that LYRM7 mutations were causing the complex III defect
To assess the causal role of the mutation we constructed lentiviral vectors carrying the wild type cDNA sequence of LYRM7 (LYRM7-lentivirus) or MTO1 (MTO1-lentivirus) associated with blasticidin resistance. In order to test the combined effect of the two mutations, we also constructed a third lentivirus carrying the wild type MTO1 cDNA sequence but with neomycin resistance in order to be independently selected from the virus carrying LYRM7 cDNA. Control transfection were performed with lentiviruses only carrying blasticidin or neomycin resistance.
Transduction was performed in fibroblasts from the patient and a control. Its efficacy was shown in all transduced cells by the significant expression of the messenger RNA coding either for resistance to blasticidin or neomycin or for LYRM7 or MTO1 (Figure 3A). Of note both LYRM7 and MTO1 messenger RNA were not significantly decreased in the non-transduced patient’s cells as compared to non-transduced control cells suggesting absence of nonsense-mediated decay of the mutant messenger RNA.
Figure 3. Only LYRM7 expression restored complex III activity.

Transduced lentivirus are indicated in the upper panel; A= Transcription assays by quantitative RT PCR, RNA data from two biological replicates were analyzed in triplicates, normalized to ACTB expression and expressed as fold of expression to non-transduced control cells; B= Assays of mitochondrial activities, data from two biological replicates analyzed twice
C= Control cells; P=Patient’s cells; non-transduced cells = white bars for the control, black bars for the patient; cells transduced with LYRM7-lentivirus = bars with horizontal lines, with MTO1-lentivirus = bars with vertical lines, with both LYRM7- and MTO1-lentiviruses = bars with diagonal lines *=p<0.05, **=p<0.01, ***=p<0.001 as compared to non-transduced control cells unless otherwise specified with brackets.
Mitochondrial activities were assayed after more than ten generations under antibiotic selection (Figure 3B). Significant interaction between genotype and lentiviral infection was only shown for complex III activity (two way ANOVA p=0.006). Infection with the LYRM7-lentivirus, whether alone or in combination with the MTO1-lentivirus, significantly increased complex III activity in patient’s but not in control cells. Complex II or citrate synthase activities did not show any significant variation among genotypes or after lentiviral infection, whatever viruses used. The combined transduction with both MTO1- and LYRM7-lentivirus had significant impact on complex IV activity in both patient and control cell lines. The initially mild complex IV defect of the patient’s cells was further decreased while control cells presented with significant decrease of their complex IV activity. The toxicity of the repeated lentivirus infection or of long-term antibiotics treatment was ruled out by the normal mitochondrial activities of cells transduced with empty lentiviruses encoding either blasticidin or neomycin resistance, alone or in combination (Supplemental Figure 1). The relatively high passage number reached by the cells when combined transduction was applied could underlie the observation. In any case these difficulties prevented to elucidate the potential relative influence on the biochemical phenotype of MTO1 and LYRM7 mutations.
Functional complementation was further confirmed by western blot analysis showing restoration of UQCRFS1 protein steady-state by LYRM7-lentivirus infection, either alone or in combination with MTO1-lentivirus, but not by MTO1-lentivirus alone (Figure 4).
Figure 4. LYRM7 expression restored UQCRFS1 expression.

A= Representative western blot analysis of purified mitochondria from control (C) and Patient (P) fibroblasts transduced with either LYRM7- or MTO1-lentivirus, alone or in association, as indicated below the western blot; specific bands are indicated with an arrow on the left side; B=quantification of the protein steady-state normalized to SDHA and to the non-transduced control mitochondria of the same gel; data from five independent experiments for LYRM7, MTO1, and UQCRFS1 and from two for UQCRC2; C= Control; P=Patient; mitochondria from non-transduced cells = white bars for control cells and black bars for patient’s cells; mitochondria from cells transduced with LYRM7-lentivirus = bars with horizontal lines, with MTO1-lentivirus = bars with vertical lines, with both LYRM7- and MTO1-lentiviruses = bars with diagonal lines; *=p<0.05, **=p<0.01, ***=p<0.001 as compared to non-transduced control cells.
Discussion
This study exemplifies some of the difficulties encountered with next generation sequencing. The unbiased and efficient whole exome-next generation sequencing is bound to find numerous homozygous genetic variants in consanguineous families, such as the one herein reported. Filtering is mandatory to reduce the number of candidate genes. Based on bi-allelic variants plus the nature of the mutation and a known mitochondrial function of the encoded protein in our case, it reduced the number of candidate genes to only two, both already associated with OXPHOS defects. Both mutations are predicted to result in protein truncation. However, the LYRM7 mutation leaves only 17 amino acids unchanged, while the MTO1 mutation leaves a 665 amino acids shortened by 27 amino acids. Western blot analysis showed that LYRM7 protein could not be detected in the patient’s samples in contrast to control. This was not the case for MTO1 protein. The patient’s phenotype resembled that of other complex III deficiencies while her normal heart function contrasted with the cardiomyopathy usually reported in patients with MTO1 mutations. The patient’s complex III defect was directly explained by the role of LYRM7 in complex III assembly while her normal complex I activity contrasted with the complex I defect, isolated or combined with complex IV, which is associated with MTO1 mutations. Finally functional complementation was decisive in linking the complex III defect with LYRM7 mutations while showing no impact of transduction with MTO1-lentivirus. Despite the clear causative link between LYRM7 mutation and complex III deficiency, a potential harmful effect of the MTO1 variant on the patient clinical phenotype could not be excluded.
The severe metabolic crises without actual liver failure observed with LYRM7 defect exemplify OXPHOS contribution to liver role during prolonged fasting when both β oxidation and gluconeogenesis are concomitantly activated in liver (Figure 5). Their electrons then converge towards complex III either via complex II (oxidizing succinate into fumarate) or complex I (oxidizing the NADH coming from the Krebs cycle or β oxidation). A defective complex III, unable to sustain the increased electron flux, will provoke the slowing down of the Krebs cycle at its four oxido-reduction steps, succinate dehydrogenase and the three NAD-dependent dehydrogenases. Because all their intermediary metabolites pass through the Krebs cycle, gluconeogenic substrates are produced to a lesser extent explaining the occurrence of hypoglycemia. Hyperammonemia on the other hand is explained by the decrease of oxaloacetate, which reduces the export of aspartate leading to defect of the urea cycle cytosolic step where aspartate is condensed with citrulline to give argininosuccinate. Hypoglycemia and hyperammonemia observed during the metabolic crises of the patients thus reflect altered metabolism and not actual liver insufficiency. Consequently they are completely reversible provided intensive nutritional care restores the metabolism towards a fed state, with concomitant inhibition of β oxidation and gluconeogenesis. These metabolic crises are expected to be less frequent and less severe with increasing age because the period of time when glycogen breakdown suffices for the maintenance of blood glucose progressively increases [39]. That spontaneous improvement with age has been observed with the patients with UQCRB or CYC1 mutations [12, 13].
Figure 5. Intermediary metabolic pathways in prolonged fasting.
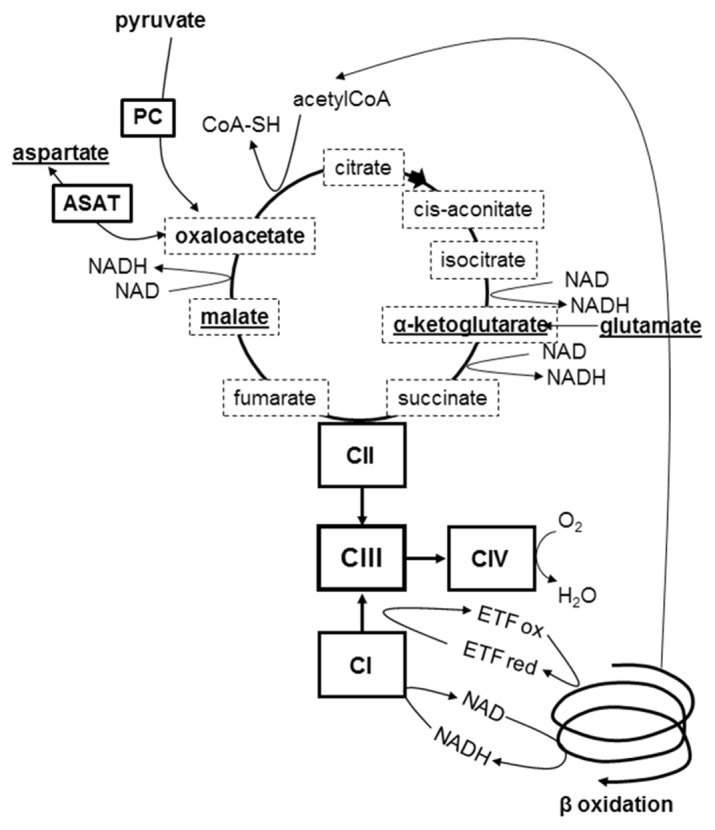
Main substrates of gluconeogenesis are show in bold, those involved in the malate-aspartate shuttle are underlined; Krebs intermediates are framed with a dotted line; PC= pyruvate carboxylase; ASAT= aspartate aminotransferase; CI= respiratory complex I, CII= complex II, CIII= complex III, CIV= complex IV; ETF ox and ETF red= electron transfer flavoprotein oxidized and reduced state. During prolonged fasting a defective complex III is bound to be overloaded by the convergent increased electrons fluxes from activated gluconeogenesis and β oxidation. Redox potential increases, resulting in decreased flux in the Krebs cycle oxidoreduction steps. Defect in gluconeogenic substrates induces hypoglycemia, defect in aspartate essential for argininosuccinate synthesis induces hyperammonemia and insufficient entry of acetylCoA into Krebs cycle induces ketosis. Graphical abstract
Comparison of the clinical presentation of the patient herein reported disclosed a pattern common to several other cases of complex III defect, including the first two patients with LYRM7 mutations [18], and the patients with UQCRB [12], CYC1 [13] or UQCRC2 [14] mutations (Table 3). All these genes but LYRM7 encode complex III structural subunits without tissue-specific isoform. The sole clinical difference among these diverse complex III defects was the occurrence of encephalopathy in the patients with LYRM7 mutations [18, 19]. The possibility to maintain brain functions despite the presence of a profound complex III defect is demonstrated by the patients with mutations in UQCRB [12] or CYC1 [13], two of whom, followed by one of us, have now reached adulthood with normal brain functions.
Table 3.
Genetic complex III defects sharing their clinical pattern
| Sign/symptom | UQCRB[12] | CYC1[13] | UQCRC2[14] | LYRM7£[18] |
|---|---|---|---|---|
|
| ||||
| Age at onset | 8 | 5/28 | NN/NN/NN | 20/6 |
|
| ||||
| Present age | 18 | 3/21 | 5/4/1.5 | 2.3*/1.2* |
|
| ||||
| Metabolic crises | +£ | +/+£ | +/+/+££ | +/+ |
| Severe acidosis | + | +/+ | +/+/+ | +/+ |
| Hyperlactatemia | + | +/+ | +/+/+ | +/+ |
| Hypoglycemia | + | ±/+ | +/+/+ | ?/+ |
| Hyperammonemia | ± | +/+ | +/+/? | +/? |
| Ketosis | + | +/+ | +/+/? | ?/? |
|
| ||||
| Liver failure | ± | 0/± | 0/0/0 | 0/0 |
|
| ||||
| Brain function | N | N | N/N/N | PR$/PR$$ |
|
| ||||
| Heart function | N | N | N | N |
|
| ||||
| Complex III | ||||
| Liver | 6% | 4%/? | ? | ?/0% |
| Skeletal Muscle | ? | 24%/45% | ? | 22%/? |
| Fibroblasts | 12% | 25%/24% | 50%/?/? | ?/26% |
0= absence of the sign; N=Normal function; Age at onset is expressed in months or as NN for neonatal period; present age is expressed in years, *=age at death; frequency of acute metabolic crises was reported as ~ four times a year from 0.5 to 4 years of age, progressively declining to less than one per year during adolescence (£) or as frequent and decreasing with age (££);severe acidosis=pH dropping to ~7.0 or below; ± liver failure= factor V<40% only observed during one acute metabolic crisis; PR=Psychomotor Regression, ($)=neurological development normal up to 20 months of age, severe psychomotor regression with loss of head control, spastic tetraparesis, marked demyelination and vacuolization of the white matter appearing after an acute metabolic decompensation with pH 6.78; ($$)=neurological development normal up to 6 months of age, severe psychomotor regression with loss of contact, profound axial hypotonia and peripheral hypertonia following a 3 day-acute metabolic crisis with coma.
During the preparation of this manuscript, 7 additional patients with LYRM7 mutations were identified based on the presence of a distinct multifocal leukoencephalopathy [19].
Metabolic crises were not reported in these patients who presented however with one or several episodes of subacute encephalopathy, during which metabolic parameters are not mentioned.
The mechanism underlying the encephalopathy of patients with LYRM7 mutations is essential. Indeed it could not be avoided if it were due to the role of LYRM7 in brain whereas it would be at least partially prevented by appropriate intensive metabolic treatment if it were a consequence of the severe metabolic disturbances due the liver defect. The latter mechanism is suggested by the initially normal neurological status of the patients and by the occurrence of encephalopathy as subacute episodes of deterioration, often triggered by febrile illness [18, 19]. The severe metabolic episodes (with blood pH dropping to 6.78 or prolonged coma) observed in our patient and in the first one reported [18] would also fit with a toxic encephalopathy. The patients most recently reported presented however with a different clinical pattern despite similar or identical LYRM7 mutations [19]. They did not present with severe metabolic disturbances, in accordance with their later onset and prolonged survival, but had significant encephalopathy with distinct leukoencephalopathy. However, because of the potential therapy, it would be important to evaluate these patients metabolic status during one of their episodes of subacute encephalopathy before definitely excluding a toxic component to their brain damage.
If the encephalopathy is essentially due to the role of LYRM7 in brain, the genotype/phenotype relationship of LYRM7 mutations would resemble that of BCS1L and TTC19, encoding other complex III assembly factors, where encephalopathy is either constant (TTC19) or very frequent (BCS1L) [40, 41]. In both cases, encephalopathy occurs in the absence of acute metabolic crises. Most importantly, TTC19 mutations spare liver function whereas severe BCS1L mutations induce generalized liver dysfunction, progressing towards cirrhosis. Part of these differences might be explained by the fact that, in contrast to structural subunits, assembly factors are expected to have other functions in addition to their role OXPHOS assembly. Indeed both BCS1L and TTC19 proteins have been involved in other protein-protein interactions [15, 42]. Mutations of UQCC2 [16] and UQCC3 [17], both encoding complex III assembly factor and associated with human disease, have each been reported only once in very young patients preventing to reliably ascertain their phenotype. In conclusion: profound complex III defect in liver induces severe lactic acidosis, ketosis, hypoglycemia, and hyperammonemia that occur after prolonged fasting and may induce irreversible brain damage in the absence of intensive metabolic care.
Supplementary Material
Acknowledgments
This work was supported by grants from the FRM (Fondation pour la Recherche Médicale) grant DPM20121125550 to AL; from the Association Française contre les Myopathies (AFM) to AL and Association contre les Maladies Mitochondriales (AMMi) to AL; German Bundesministerium für Bildung und Forschung (BMBF) through funding of the E-Rare project GENOMIT (01GM1207) (TM, HP); German Network for mitochondrial disorders (mitoNET, 01GM1113C) (TM, HP).
List of Abbreviations
- OXPHOS
oxidative phosphorylation pathway
- mtDNA
mitochondrial DNA
- respiratory complex III
ubiquinol cytochrome c oxidoreductase (EC 1.10.2.2)
Footnotes
The authors declare the absence of conflict of interest
Authors contribution
Laura S Kremer participated to the study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Caroline L’hermitte- Stead participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Pierre Lesimple participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Mylène Gilleron participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Sandrine Filaut participated to the acquisition of data and analysis and interpretation of data
Claude Jardel participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Tobias B Haack participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Tim M Strom participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Thomas Meitinger participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Hatem Azzouz participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Neji Tebib participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Hélène Ogier de Baulny participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Guy Touati participated to the acquisition of data; analysis and interpretation of data; drafting of the manuscript; and critical revision of the manuscript
Holger Prokisch participated to the study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; statistic4al analysis; and obtained funding.
Anne Lombès participated to the study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; statistical analysis; and obtained funding.
References
Author names in bold designate shared co-first authorship.
- 1.DiMauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004;1658:80–88. doi: 10.1016/j.bbabio.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 2.Auré K, Jardel C, Lombès A. Mitochondrial diseases: molecular mechanisms, clinical presentations and diagnosis investigations. Ann Pathol. 2005;25:270–281. doi: 10.1016/s0242-6498(05)80131-2. [DOI] [PubMed] [Google Scholar]
- 3.Labarthe F, Dobbelaere D, Devisme L, De Muret A, Jardel C, Taanman JW, et al. Clinical, biochemical and morphological features of hepatocerebral syndrome with mitochondrial DNA depletion due to deoxyguanosine kinase deficiency. J Hepatol. 2005;43:333–341. doi: 10.1016/j.jhep.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 4.Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D’Adamo P, Calvo S, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 5.Sarzi E, Goffart S, Serre V, Chretien D, Slama A, Munnich A, et al. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann Neurol. 2007;62:579–587. doi: 10.1002/ana.21207. [DOI] [PubMed] [Google Scholar]
- 6.Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706–712. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 7.Coenen MJ, Antonicka H, Ugalde C, Sasarman F, Rossi R, Heister JG, et al. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med. 2004;351:2080–2086. doi: 10.1056/NEJMoa041878. [DOI] [PubMed] [Google Scholar]
- 8.Zeharia A, Shaag A, Pappo O, Mager-Heckel AM, Saada A, Beinat M, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet. 2009;85:401–407. doi: 10.1016/j.ajhg.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vedrenne V, Galmiche L, Chretien D, de Lonlay P, Munnich A, Rotig A. Mutation in the mitochondrial translation elongation factor EFTs results in severe infantile liver failure. J Hepatol. 2012;56:294–297. doi: 10.1016/j.jhep.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 10.de Lonlay P, Valnot I, Barrientos A, Gorbatyuk M, Tzagoloff A, Taanman JW, et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat Genet. 2001;29:57–60. doi: 10.1038/ng706. [DOI] [PubMed] [Google Scholar]
- 11.Visapaa I, Fellman V, Vesa J, Dasvarma A, Hutton JL, Kumar V, et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet. 2002;71:863–876. doi: 10.1086/342773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haut S, Brivet M, Touati G, Rustin P, Lebon S, Garcia-Cazorla A, et al. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Hum Genet. 2003;113:118–122. doi: 10.1007/s00439-003-0946-0. [DOI] [PubMed] [Google Scholar]
- 13.Gaignard P, Menezes M, Schiff M, Bayot A, Rak M, Ogier de Baulny H, et al. Mutations in CYC1, encoding cytochrome c1 subunit of respiratory chain complex III, cause insulin-responsive hyperglycemia. Am J Hum Genet. 2013;93:384–389. doi: 10.1016/j.ajhg.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyake N, Yano S, Sakai C, Hatakeyama H, Matsushima Y, Shiina M, et al. Mitochondrial complex III deficiency caused by a homozygous UQCRC2 mutation presenting with neonatal-onset recurrent metabolic decompensation. Hum Mutat. 2013;34:446–452. doi: 10.1002/humu.22257. [DOI] [PubMed] [Google Scholar]
- 15.Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, Benna C, et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet. 2011;43:259–263. doi: 10.1038/ng.761. [DOI] [PubMed] [Google Scholar]
- 16.Tucker EJ, Wanschers BF, Szklarczyk R, Mountford HS, Wijeyeratne XW, van den Brand MA, et al. Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 2013;9:e1004034. doi: 10.1371/journal.pgen.1004034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wanschers BF, Szklarczyk R, van den Brand MA, Jonckheere A, Suijskens J, Smeets R, et al. A mutation in the human CBP4 ortholog UQCC3 impairs complex III assembly, activity and cytochrome b stability. Hum Mol Genet. 2014;23:6356–6365. doi: 10.1093/hmg/ddu357. [DOI] [PubMed] [Google Scholar]
- 18.Invernizzi F, Tigano M, Dallabona C, Donnini C, Ferrero I, Cremonte M, et al. A homozygous mutation in LYRM7/MZM1L associated with early onset encephalopathy, lactic acidosis, and severe reduction of mitochondrial complex III activity. Hum Mutat. 2013;34:1619–1622. doi: 10.1002/humu.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dallabona C, Abbink TE, Carrozzo R, Torraco A, Legati A, van Berkel CG, et al. LYRM7 mutations cause a multifocal cavitating leukoencephalopathy with distinct MRI appearance. Brain. 2016;139:782–794. doi: 10.1093/brain/awv392. [DOI] [PubMed] [Google Scholar]
- 20.Meunier B, Fisher N, Ransac S, Mazat JP, Brasseur G. Respiratory complex III dysfunction in humans and the use of yeast as a model organism to study mitochondrial myopathy and associated diseases. Biochim Biophys Acta. 2013;1827:1346–1361. doi: 10.1016/j.bbabio.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Barel O, Shorer Z, Flusser H, Ofir R, Narkis G, Finer G, et al. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am J Hum Genet. 2008;82:1211–1216. doi: 10.1016/j.ajhg.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auré K, Mamchaoui K, Frachon P, Butler-Browne GS, Lombès A, Mouly V. Impact on oxidative phosphorylation of immortalization with the telomerase gene. Neuromuscul Disord. 2007;17:368–375. doi: 10.1016/j.nmd.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 23.Klement P, Nijtmans LG, Van den Bogert C, Houstek J. Analysis of oxidative phosphorylation complexes in cultured human fibroblasts and amniocytes by blue-native-electrophoresis using mitoplasts isolated with the help of digitonin. Anal Biochem. 1995;231:218–224. doi: 10.1006/abio.1995.1523. [DOI] [PubMed] [Google Scholar]
- 24.Medja F, Allouche S, Frachon P, Jardel C, Malgat M, de Camaret BM, et al. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion. 2009;9:331–339. doi: 10.1016/j.mito.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 26.Legros F, Chatzoglou E, Frachon P, Ogier de Baulny H, Laforêt P, Jardel C, et al. Molecular consequences of novel mutations in the human cytochrome b gene. Eur J Hum Genet. 2001;9:510–518. doi: 10.1038/sj.ejhg.5200678. [DOI] [PubMed] [Google Scholar]
- 27.Barthélémy C, Ogier de Baulny H, Diaz J, Cheval MA, Frachon P, Romero N, et al. Late-onset mitochondrial DNA depletion: DNA copy number, multiple deletions, and compensation. Ann Neurol. 2001;49:607–617. [PubMed] [Google Scholar]
- 28.Haack TB, Haberberger B, Frisch EM, Wieland T, Iuso A, Gorza M, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet. 2012;49:277–283. doi: 10.1136/jmedgenet-2012-100846. [DOI] [PubMed] [Google Scholar]
- 29.Kornblum C, Nicholls TJ, Haack TB, Scholer S, Peeva V, Danhauser K, et al. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat Genet. 2013;45:214–219. doi: 10.1038/ng.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baruffini E, Dallabona C, Invernizzi F, Yarham JW, Melchionda L, Blakely EL, et al. MTO1 mutations are associated with hypertrophic cardiomyopathy and lactic acidosis and cause respiratory chain deficiency in humans and yeast. Hum Mutat. 2013;34:1501–1509. doi: 10.1002/humu.22393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danhauser K, Iuso A, Haack TB, Freisinger P, Brockmann K, Mayr JA, et al. Cellular rescue-assay aids verification of causative DNA-variants in mitochondrial complex I deficiency. Mol Genet Metab. 2011;103:161–166. doi: 10.1016/j.ymgme.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 32.Baker MJ, Palmer CS, Stojanovski D. Mitochondrial protein quality control in health and disease. Br J Pharmacol. 2014;171:1870–1889. doi: 10.1111/bph.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zara V, Conte L, Trumpower BL. Biogenesis of the yeast cytochrome bc1 complex. Biochim Biophys Acta. 2009;1793:89–96. doi: 10.1016/j.bbamcr.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Elstner M, Andreoli C, Klopstock T, Meitinger T, Prokisch H. The mitochondrial proteome database: MitoP2. Methods Enzymol. 2009;457:3–20. doi: 10.1016/S0076-6879(09)05001-0. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez E, Lobo T, Fox JL, Zeviani M, Winge DR, Fernandez-Vizarra E. LYRM7/MZM1L is a UQCRFS1 chaperone involved in the last steps of mitochondrial Complex III assembly in human cells. Biochim Biophys Acta. 2013;1827:285–293. doi: 10.1016/j.bbabio.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghezzi D, Baruffini E, Haack TB, Invernizzi F, Melchionda L, Dallabona C, et al. Mutations of the Mitochondrial-tRNA Modifier MTO1 Cause Hypertrophic Cardiomyopathy and Lactic Acidosis. Am J Hum Genet. 2012 doi: 10.1016/j.ajhg.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Umeda N, Suzuki T, Yukawa M, Ohya Y, Shindo H, Watanabe K, et al. Mitochondria-specific RNA-modifying enzymes responsible for the biosynthesis of the wobble base in mitochondrial tRNAs Implications for the molecular pathogenesis of human mitochondrial diseases. J Biol Chem. 2005;280:1613–1624. doi: 10.1074/jbc.M409306200. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Li R, Lin X, Guan MX. Isolation and characterization of the putative nuclear modifier gene MTO1 involved in the pathogenesis of deafness-associated mitochondrial 12 S rRNA A1555G mutation. J Biol Chem. 2002;277:27256–27264. doi: 10.1074/jbc.M203267200. [DOI] [PubMed] [Google Scholar]
- 39.Bonnefont JP, Specola NB, Vassault A, Lombes A, Ogier H, de Klerk JB, et al. The fasting test in paediatrics: application to the diagnosis of pathological hypo- and hyperketotic states. Eur J Pediatr. 1990;150:80–85. doi: 10.1007/BF02072043. [DOI] [PubMed] [Google Scholar]
- 40.Hinson JT, Fantin VR, Schonberger J, Breivik N, Siem G, McDonough B, et al. Missense mutations in the BCS1L gene as a cause of the Bjornstad syndrome. N Engl J Med. 2007;356:809–819. doi: 10.1056/NEJMoa055262. [DOI] [PubMed] [Google Scholar]
- 41.Fernandez-Vizarra E, Bugiani M, Goffrini P, Carrara F, Farina L, Procopio E, et al. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum Mol Genet. 2007;16:1241–1252. doi: 10.1093/hmg/ddm072. [DOI] [PubMed] [Google Scholar]
- 42.Wagener N, Neupert W. Bcs1, a AAA protein of the mitochondria with a role in the biogenesis of the respiratory chain. Journal of structural biology. 2012;179:121–125. doi: 10.1016/j.jsb.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 43.Osawa T, Ito K, Inanaga H, Nureki O, Tomita K, Numata T. Conserved cysteine residues of GidA are essential for biogenesis of 5-carboxymethylaminomethyluridine at tRNA anticodon. Structure. 2009;17:713–724. doi: 10.1016/j.str.2009.03.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.