

ABSTRACT
The highly conserved cytoplasmic tail of influenza virus glycoprotein hemagglutinin (HA) contains three cysteines, posttranslationally modified by covalently bound fatty acids. While viral HA acylation is crucial in virus replication, its physico-chemical role is unknown. We used virus-like particles (VLP) to study the effect of acylation on morphology, protein incorporation, lipid composition, and membrane fusion. Deacylation interrupted HA-M1 interactions since deacylated mutant HA failed to incorporate an M1 layer within spheroidal VLP, and filamentous particles incorporated increased numbers of neuraminidase (NA). While HA acylation did not influence VLP shape, lipid composition, or HA lateral spacing, acylation significantly affected envelope curvature. Compared to wild-type HA, deacylated HA is correlated with released particles with flat envelope curvature in the absence of the matrix (M1) protein layer. The spontaneous curvature of palmitate was calculated by molecular dynamic simulations and was found to be comparable to the curvature values derived from VLP size distributions. Cell-cell fusion assays show a strain-independent failure of fusion pore enlargement among H2 (A/Japan/305/57), H3 (A/Aichi/2/68), and H3 (A/Udorn/72) viruses. In contradistinction, acylation made no difference in the low-pH-dependent fusion of isolated VLPs to liposomes: fusion pores formed and expanded, as demonstrated by the presence of complete fusion products observed using cryo-electron tomography (cryo-ET). We propose that the primary mechanism of action of acylation is to control membrane curvature and to modify HA's interaction with M1 protein, while the stunting of fusion by deacylated HA acting in isolation may be balanced by other viral proteins which help lower the energetic barrier to pore expansion.
IMPORTANCE Influenza A virus is an airborne pathogen causing seasonal epidemics and occasional pandemics. Hemagglutinin (HA), a glycoprotein abundant on the virion surface, is important in both influenza A virus assembly and entry. HA is modified by acylation whose removal abrogates viral replication. Here, we used cryo-electron tomography to obtain three-dimensional images to elucidate a role for HA acylation in VLP assembly. Our data indicate that HA acylation contributes to the capability of HA to bend membranes and to HA's interaction with the M1 scaffold protein during virus assembly. Furthermore, our data on VLP and, by hypothesis, virus suggests that HA acylation, while not critical to fusion pore formation, contributes to pore expansion in a target-dependent fashion.
KEYWORDS: cryo-electron microscopy, electron microscopy, membrane fusion, palmitoylation, protein acylation
INTRODUCTION
Influenza A virus is an enveloped virus which assembles spheroidal or filamentous virions on the plasma membrane via a process called budding (1). At least four influenza virus membrane-associated proteins, hemagglutinin (HA), neuraminidase (NA), matrix 1 (M1), and the matrix 2 (M2) ion channel, orchestrate the plasma membrane remodeling to assemble new virions (2). HA and NA, both serving as the main antigens, stud the virion surface. HA binds to sialic acid moieties on host membrane proteins and, upon acidification in the late endosomes, drives membrane fusion. HA is a homotrimer, synthesized as a precursor H0, which becomes fusogenic only after proteolytic cleavage that results in two proteins connected by disulfide bonds, HA1 and HA2 (3). Both HA and NA interact with M1 protein via their cytoplasmic tails (CTs) (4, 5). M1, which electrostatically binds to negatively charged lipids (6) and assembles in a helical array beneath the inner monolayer of the plasma membrane (7), is an important determinant of virion morphology (8, 9) and provides a platform for viral genome incorporation (10). The negative-sense RNA genome is segmented into eight physically independent viral ribonucleoprotein (vRNP) complexes which have an average diameter of ∼14 nm and a length ranging from ∼24 to ∼110 nm (11). The budding process is terminated by the formation of a constricted neck at the plasma membrane and subsequent M2 protein-dependent membrane scission (12). To prevent reentry into the producing cell and promote virion release, NA concentrates at the rear tip of the budding virion where cleavage of sialic acid moieties on the surface of the plasma membrane occurs (7, 13). The resulting virus particles have pleomorphic geometry, with a predominance of spheroidal and filamentous shapes (8). Effects of glycoproteins on the viral envelope curvature have been reviewed recently (2).
Spheroidal and filamentous virions are able to infect cells and enter via receptor-mediated clathrin-dependent endocytosis (14) and macropinocytosis (15), respectively. To accomplish vRNP release into the cytoplasm, both endosomes and macropinocytosomes must be acidified to pH ∼5.0 to trigger HA trimer conformational changes that induce fusion between viral and endosomal membranes. HA-mediated membrane fusion has been extensively studied in reconstituted systems, usually free of other influenza virus proteins except HA, employing various biophysical methods such as content and lipid fluorescent indicators and electrophysiology. Membrane fusion occurs through several intermediates and requires overcoming a substantial energy barrier as the two membranes approach each other (16, 17). Initially, the two membranes must be tightly apposed, and, subsequently, the endosomal membrane integrity is distorted by the insertion of HA fusion peptides resulting in the merger of the outer monolayers of the two bilayers; this stage is called hemifusion (18, 19), whose structure has been recently characterized by cryo-electron tomography (cryo-ET) (20). In this stage, only lipid indicators can enter the target membrane. In a subsequent step a small transient fusion pore is formed, which repeatedly opens and closes, a process called “flicker,” and ultimately remains open. Diameters of a flickering pore are in the range of nanometers (21, 22); and the pore is permeable to small aqueous indicators, but it is too small to allow release of vRNPs. Fusion pore enlargement, a process whose mechanism is not entirely elucidated, seems to depend on numerous factors, including the number and density of HA trimers in a fusogenic state (23–25). Although the minimal number of HA trimers capable of opening a small pore, evaluated experimentally and theoretically, is around three (26, 27), additional HA trimers in a fusogenic conformation might be required to promote pore enlargement (25). Moreover, in the context of an intact virus, other factors influencing membrane fusion should be considered. The M2 ion channel plays an indirect but important role in destabilization and detachment of the matrix layer beneath the membrane, which is also an important requirement for pore enlargement (28, 29). Interestingly, a recent study showed that influenza virions are not permeable to protons unless subjected to trypsin treatment (30). Increased H+ and K+ concentrations (31) inside the virion interior result in weakening interactions between M1 proteins and vRNPs (32). Recent findings highlight that a gradual, rather than a steep, influenza virus acidification leads to successful vRNP delivery into the cytoplasm; this process occurs in at least two steps (29, 31).
Both the HA and M2 proteins are acylated, and a plethora of studies pinpoint this posttranslational modification as crucial in the influenza virus A replication cycle for both assembly and fusion (33). Two cysteines present within the cytoplasmic tail (CT) sequence and one cysteine in the transmembrane domain (TM) sequence of HA are S acylated by two palmitates (CT cysteines) and one stearic or palmitic acid (TM cysteine), respectively (34, 35). The role of HA acylation has been studied extensively since these cysteines are conserved at their respective positions to a greater extent than other residues among all otherwise highly variable HA subtypes. Hence, the cytoplasmic tail cysteine acylation is likely crucial in the influenza virus replication cycle (reviewed in reference 33). HA CT deacylation or complete removal of the CT results in reduced M1 incorporation and, consequently, changes in virus morphology and infectivity (36, 37). Removal of HA acylation results in decreased M1 and vRNP incorporation into assembling virions of A/Udorn/72 (H3N2); the observed decreases were rescued by M1 protein from the A/WSN/33 (WSN) virus (38). With respect to the role of HA acylation in membrane fusion, although all studies are consistent with a lack of effect of HA acylation on hemifusion, contrasting data have been reported regarding the role of HA acylation in fusion pore enlargement (reviewed in reference 33). Acylation of H1 (A/USSR/77) (39), H2 (A/Japan/305/57) (40), and H7 (A/FPV/Ro/34) (41) in contrast to that of H3 (A/Aichi/2/68) (42) and H3 (A/Udorn/72) (38) contributes to pore enlargement. One study employing conductance measurements of fusion pores induced by H3 (A/Udorn/72) showed that palmitoyl group removal reduced fusion pore flickering but, compared to wild-type (WT) HA, did not influence pore enlargement kinetics (43). Several studies also tested which of the three acyl groups have the most profound function in pore enlargement and indicated that the palmitoyl group most distal from the TM is crucial (37, 38, 44).
To study the role of HA acylation in influenza virus-like particle (VLP) assembly and membrane fusion, we have mutated two or all three cysteines on the HA CT in the HA proteins from three different strains. We compared the release and morphology of wild-type VLPs with VLPs containing depalmitoylated HA, both by biochemistry and by cryo-ET. We used a fluorescent cell-red blood cell (RBC) fusion assay, cryo-ET, and model simulation to investigate the roles of acylation in influenza VLP morphology and membrane fusion. The data indicate that HA acylation modulates virus envelope curvature, influencing HA-mediated membrane fusion and influenza VLP assembly. Atomistic simulation of the spontaneous curvature of palmitic acid was consistent with a curvature modulating role for HA acylation.
RESULTS
HA acylation does not inhibit release of influenza VLP proteins.
To express completely or partially deacylated HA (A/Aichi/2/68), we mutated the cysteines present in the CT of HA. Codons for two cysteines (Cys 555 and Cys 562) or for all three cysteines (Cys 555, Cys 562, and Cys 565) were changed to codons that coded for a MAC (Met 555 and Ala 562) or MAY (Met 555, Ala 562, and Tyr 565) mutant.
Following transfection of both 293T and Cos7 cells with MAC and MAY, trafficking to the plasma membrane comparable to that of wild-type HA was observed (data not shown).
To assess the role of HA acylation on incorporation of M1, NA, and M2 proteins into VLPs, we cotransfected 293T cells with plasmids containing NA, M1, M2, and either HA or MAY (yielding HANAM1M2 or MAYNAM1M2, respectively). We opted for plasmid-driven VLPs since attempts to substitute all cysteines in HA (A/WSN/33) and produce a virus using a reverse genetic system (45) resulted in a reverted virus that recovered the most distant cysteine, as revealed by virus genome sequencing (Table 1). The expression and release of proteins were analyzed using a bicinchoninic acid assay and Western blotting (Fig. 1A). Both HANAM1M2 and MAYNAM1M2 preparations, following sucrose cushion purification, yielded 30 to 50 μl of VLPs with total protein concentrations of ∼2 mg/ml. Although the levels of released HA/MAY, M1, and M2 protein from HANAM1M2 and MAYNAM1M2 transfected cells were similar, NA release in MAYNAM1M2 transfected cells was slightly but significantly increased (P = 0.008; n = 4, t test) compared to the level of NA released from HANAM1M2 transfected cells (Fig. 1A). The HA/NA and MAY/NA ratios, 1.1 and 0.8, were significantly different (P = 0.006; t test).
TABLE 1.
Preparation of recombinant A/WSN/33 virus carrying MAY mutationa
| Sample no. | No. of HA U/ml at 24 h | CPE (%) at 24 h | No. of HA U/ml at 48 h | CPE (%) at 48 h | No. of HA U/ml at 72 h | CPE (%) at 72 h | Sequence at 72 h |
|---|---|---|---|---|---|---|---|
| 1 | 80 | 0 | 320 | 90 | 640 | 100 | MAY+MAC |
| 2 | 0 | 0 | 320 | 40 | 320 | 45 | MAY+MAC |
| 3 | 0 | 0 | 160 | 70 | 320 | 100 | MAY+MAC |
| 4 | 0 | 0 | 160 | 50 | 320 | 70 | MAY+MAC |
| 5 | 0 | 0 | 0 | 10 | 0 | 15 | MAY+MAC |
| 6 | 80 | 0 | 320 | 20 | 640 | 60 | MAC |
| 7 | 80 | 0 | 320 | 80 | 1280 | 85 | MAY+MAC |
| 8 | 80 | 0 | 320 | 80 | 1280 | 80 | MAY+MAC |
The table shows sequence changes and virus titer during recovery of A/WSN/33 virus prepared by reverse genetics. 293T cells were transfected using sets of plasmids used for generation of the recombinant virus for a duration of 72 h. The viral titer (HA U/ml) was measured, and the HA sequence was obtained at 48 and at 72 h posttransfection. At 72 h posttransfection the rest of media was collected and used to infect MDCK cells. Virus titer (HA U/ml) and the HA sequence were again obtained at 72 h postinfection.
FIG 1.

Release of influenza virus proteins from HANAM1M2 and MAYNAM1M2 transfected 293T cells. (A) SDS-PAGE under reducing conditions followed by Western blotting of lysates and supernatants from 293T cells transfected with plasmids containing HA (A/Aichi/2/68), NA (A/Singapore/1/57), M1 (A/Hong Kong/8/68), and M2 (synthetic) and harvested at 48 h posttransfection. (B) Bar plot showing the averaged (n = 4) and lysate-normalized relative intensities of the Western blot bands. Relative M1 release levels into the supernatant were similar in HANAM1M2 and MAYNAM1M2 transfected cells. NA release to the supernatant was increased in MAYNAM1M2 transfected cells.
Spheroidal particles containing deacylated HA are impaired in M1 layer incorporation and are larger in diameter.
We further investigated the role of HA acylation on VLP morphology using cryo-ET (Fig. 2A and B). Filamentous (19%) and spheroidal (81%) particles were similarly represented in both HANAM1M2 (n = 62) and MAYNAM1M2 (n = 57) samples (Fig. 2C). HA and NA glycoproteins can be distinguished from each other by their shape as resolved in cryo-ET; HA proteins appear as narrow spikes with an obvious stem connected to the surface of the particle, while NA proteins, in contrast, show a large globular domain connected to the particle via a thin stem, which is hardly visible (11). We observed both HA and NA spikes on all three types of particles, indicating that neither filamentous nor spheroidal particles were devoid of either of the two glycoproteins (Fig. 2A and B). HANAM1M2 filamentous particles had NA clusters localized at the tip of the VLP as reported before (8), whereas MAYNAM1M2 filamentous particles contained, overall, more NA glycoproteins, and NA clusters were also present on the cylindrical surface of the filamentous particle. Importantly, 52% (n = 29) of filamentous MAYNAM1M2 particles contained only NA, an observation consistent with the increased release of NA detected by Western blotting. All observed HANAM1M2 and MAYNAM1M2 filamentous VLPs contained an electron-dense layer formed presumably by interior membrane-associated M1 protein (Fig. 2A and B). The average diameter, 51 ± 4 nm (standard deviation [SD]), was the same for both HANAM1M2 and MAYNAM1M2 filamentous VLPs. More than half (62%, n = 50) of the analyzed spheroidal particles isolated from HANAM1M2 transfected cells contained the M1 layer. In contrast, a majority (91%, n = 46) of the spheroidal particles isolated from the MAYNAM1M2 transfected cells did not show the M1 layer underneath the membrane (Fig. 2C). HA spheroidal particles without an M1 layer were larger (90 ± 39 nm [SD]; n = 24) than those containing M1 (63 ± 14 nm [SD], n = 30; P = 0.003; unequal variance t test) and more variable in diameter (P = 0.001; Levene's test for equality of variance using median). Spheroidal particles without M1 containing HA were significantly smaller (∼1.3-fold) in diameter than MAY spheroidal particles without M1 (113 ± 40 nm [SD], n = 45; P = 0.026; t test) and displayed no difference in diameter variability (P = 0.66; Levene's test for equality of variance using median). The distributions of the aspect ratios (length/width) of HANAM1M2 and MAYNAM1M2 were similar (P = 0.856; Kolmogorov-Smirnov test), indicating that the NA-M1 interaction can rescue the weakened depalmitoylated HA-M1 interaction necessary for filamentous VLP formation (Fig. 2D). To investigate further, we isolated particles from supernatants of cells transfected with only MAY, M1, and M2. MAYM1M2 particles were predominantly spheroidal without an M1 layer; filamentous particle was observed only once (data not shown). Thus, HA acylation plays a role in HA-modulated membrane curvature and M1 protein incorporation into spheroidally shaped particles. The filamentous particles containing depalmitoylated HA are formed only if NA is also present.
FIG 2.

Morphology of HANAM1M2 and MAYNAM1M2 particles by cryo-ET. Slices of tomograms were reconstructed from a tilt series collected using a Krios microscope at 300 kV with a Volta phase plate and nominal defocus of −0.5 μm. The slices capture spheroidal particles with and without an M1 layer and three examples of filamentous particles, isolated from supernatants of the HANAM1M2 (A) and MAYNAM1M2 (B) transfected 293T cells (scale bar, 50 nm). The M1 layer is present underneath the membrane of filamentous VLPs and in some spheroidal particles in both HANAM1M2 and MAYNAM1M2 samples. Examples of regions containing HA or NA spikes are surrounded by a black rectangle, and magnified views of the respective regions are on the right (scale bar, 5 nm). MAYNAM1M2 filamentous particles incorporate more NA spikes than HANAM1M2 filamentous particles. The MAYNAM1M2 filamentous particle in the bottom image contains only NA spikes. Two representative tomograms were deposited into the wwPDB under accession numbers EMD-8843 and EMD-8844. (C) Column plot showing percentage representation of filamentous and spheroidal particles with and without the M1 layer for HANAM1M2 and MAYNAM1M2 isolates. (D) A normalized cumulative distribution function (nCDF) plot shows that the HANAM1M2 and MAYNAM1M2 particles have similar distributions of aspect ratios (P = 0.856, Kolmogorov-Smirnov test).
Neither HA acylation nor the presence of the matrix layer influences glycoprotein spacing on filamentous and spheroidal particles.
We next analyzed whether HA acylation influences the distance between glycoproteins in the presence or absence of the M1 layer. Glycoprotein (HA and NA or MAY and NA) spacing on the surface of the filamentous and spheroidal VLPs, with and without matrix layer, was analyzed either in longitudinal tomographic slices (Fig. 2) using a line scan (Materials and Methods) or in the transverse tomographic slices through the protruding spikes (Fig. 3A) using radially averaged power spectrum (PS) (Table 2). There were no statistically significant differences between group mean spacing as determined by one-way analysis of variance (ANOVA) (F5,114 = 0.985, P = 0.43). The average spacing over all species (filamentous and spheroidal, HA and MAY) is 8.33 ± 0.21 nm (mean ± 95% confidence). The average spacing between glycoproteins calculated from PS (Fig. 3B) over all species is 8.74 ± 0.43 nm (mean ± 95% confidence); the difference between the spacing determined using the two methods was not significantly different from zero (0.41 ± 0.48 nm for the difference ± 95% confidence). The average spacing for the surface glycoproteins over all species and methods is 8.54 ± 0.48 nm (mean ± 95% confidence), corresponding to a surface glycoprotein density of 0.016 glycoproteins/nm2 assuming hexagonal packing. Thus, HA acylation does not influence the spacing of the surface glycoproteins in either the presence or absence of the M1 layer.
FIG 3.
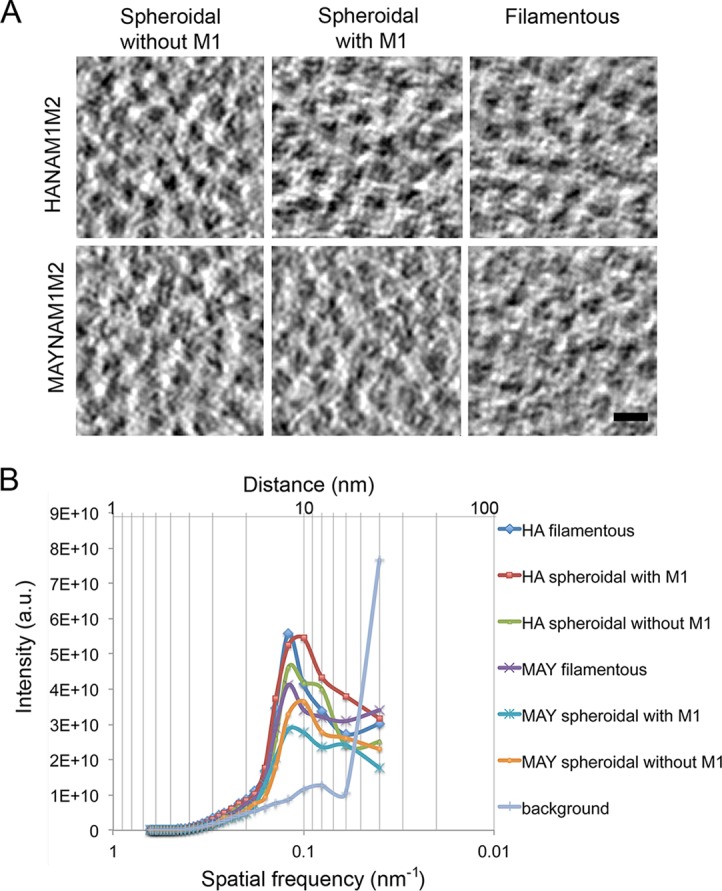
Glycoprotein spacing of HA- and MAY-containing particles. (A) Representative slices of a tomogram showing the electron densities of the glycoproteins of the filamentous particles, spheroidal particles with and without an M1 layer, and a vitreous ice background. Scale bar, 10 nm. (B) Radially averaged power spectra showing high-order glycoprotein distances for HANAM1M2 and MAYNAM1M2 particles. a.u., arbitrary units.
TABLE 2.
Summary of glycoprotein spacing measured by power spectrum analysis and line plot
| Particle type | Top-view measurement (PS)a |
Side-view measurement (avg ± SD [nm])b | ||
|---|---|---|---|---|
| Peak (nm) | μ fit (1/nm) | Sigma fit (1/nm) | ||
| Spherical with HA | ||||
| Without M1 | 9.46 | 0.11 | 0.02 | 8.3 ± 1.4 |
| With M1 | 8.90 | 0.11 | 0.03 | 8.8 ± 1.2 |
| Filamentous with HA | 8.36 | 0.12 | 0.02 | 8.2 ± 1.1 |
| Spherical with MAY | ||||
| Without M1 | 9.17 | 0.11 | 0.02 | 8.0 ± 1.0 |
| With M1 | 8.73 | 0.11 | 0.03 | 8.5 ± 1.4 |
| Filamentous with MAY | 8.18 | 0.12 | 0.02 | 8.2 ± 0.8 |
Values were obtained by power spectrum analysis from transverse slices of glycoproteins on the tops of the particles. Mean spatial frequency and sigma values were obtained from modified two-term Lorentzian-Gaussian fit of the power spectrum for filamentous particles and for spheroidal particles with and without an M1 layer containing either HA or MAY. Glycoprotein spacing at the peak of the curve is shown.
Mean and standard deviation (n = 20) of glycoprotein spacing obtained from density line scans on horizontal slices of the glycoprotein spikes.
The difference in HANA and MAYNA particle radii implies that acylation imposes large negative spontaneous curvature.
HA spheroidal particles without an M1 layer were larger (90 ± 39 nm [SD], n = 24) than those containing M1 (63 ± 14 nm [SD], n = 30; P = 0.003, unequal variance t test) and more variable in diameter (P = 0.001; Levene's test for equality of variance using median). Interestingly, HA-containing spheroidal particles without M1 were significantly smaller (∼1.3-fold) in diameter than MAY-containing spheroidal particles without M1 (113 ± 40 nm [SD], n = 45; P = 0.026, t test), with no difference in diameter variability (P = 0.66; Levene's test for equality of variance using median) (Fig. 4A). The size differences suggested that palmitoylation has an impact on particle curvature.
FIG 4.

HA acylation affects curvature of the released particles. (A) Representative slices of tomograms showing MAYNA (no M1), HANA (no M1), and HANAM1M2 spheroidal particles. Respective average radii, standard deviations, and numbers of measurements are shown below the images. (B) Peak normalized probability density functions (PDF) for the three species, HA with and without M1 and MAY without M1, calculated using the truncated Gaussian parameters identified from the global fit of the cumulative distributions of VLP radii. HA and MAY differ in their most probable size (peak of the PDF), and the distribution width is significantly greater in the absence of M1. (C) Spontaneous curvature of palmitic acid and palmitoylation on short alpha-helical peptide sequences computed from simulation. Extrapolation of the data to the 100% area fraction of palmitic acid yields an estimate of the spontaneous curvature of palmitic acid. Shown for reference (red point on the right curvature axis) is the same value estimated from the change in average size of the VLPs with HA compared to that with MAY.
We used a simple model describing the effect of HA palmitoylation on curvature, which compares the size of the particles with and without palmitoylation and then infers what the spontaneous curvature of the membrane monolayers must be to account for the observed size difference. To normalize the radius distribution, peak normalized probability density functions (PDF) for the three species, HA with and without M1 and MAY without M1, were calculated using the truncated Gaussian parameters identified from the global fit of the cumulative distributions of VLP radii (Fig. 4B). In the model, palmitoylation affects only the VLP's inner monolayer (membrane monolayer facing the interior of the VLP), which, according to the established sign convention, has negative curvature.
A patch of VLP surface with area A, bending modulus kc, radius RVLP, and membrane monolayer spontaneous curvatures, c0(outer) and c0(inner), has Helfrich/Canham (46, 47) curvature energy:
Minimizing the energy (per unit area) relative to RVLP yields:
Comparing two systems with the same value of c0(outer) but with different c0(inner) and RVLP values, we find:
where RVLP(MAY) and RVLP(HA) are the most probable radii estimated from the global truncated Gaussian fit (50.9 ± 0.8 and 30.0 ±0.2 nm, respectively [value ± 95% confidence]). A VLP with a 30-nm radius having a glycosylated protein density of 0.016 per nm2 will contain ∼150 HA trimers (assuming HA/NA = 5:1), each with nine palmitoylated cysteines, with an effective area fraction of the palmitoylated groups fpalm of 3.6%, assuming the palmitoyl group has an area of 0.3 nm2, as it does in simulation of the free acid. The estimate of the effective spontaneous curvature of palmitoylation depends on the value of c0(inner; MAY):
Solving for c0(palm) yields:
The second term on the right side is −1.52 nm−1, itself a very large negative spontaneous curvature. This value serves as a reasonable lower bound for the effect as it would be unlikely for c0(inner; MAY) to be positive, given that even without palmitoylation it forms small VLPs. An upper estimate of the effect is to assume that c0(outer) is zero, requiring all the curvature energetics to occur in the inner monolayer, in which case c0(palm) is −1.59 nm−1. This value represents a very high negative spontaneous curvature, much higher than the −0.34 of the canonical negative-curvature lipid used for biophysical studies, dioleoylphosphatidylethanolamine (DOPE) (48).
Simulations of palmitic acid indicate large negative spontaneous curvature.
To estimate the effect of palmitoylation of HA on membrane spontaneous curvature, two simple model systems were simulated. The first is protonated palmitic acid residues mixed in POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine). The second is the relative curvature stress of palmitoylated HA and MAY sequences of short alpha-helical subunits. These model systems have several attractive features: the simulations are computationally inexpensive, the signal is simple to interpret and has excellent statistics, and the physicochemical effect of the palmitic acid head group “size” relative to its tail entropy is captured. The spontaneous curvature as a function of mole fraction is plotted in Fig. 4C, with a line computed for pure palmitate and the palmitoylated peptide indicated separately. Extrapolating the curve to 100% palmitic acid yields an estimate of the spontaneous curvature of the lipid: −1.1 nm−1. Although this value is 31% lower in magnitude than that estimated from the experimental VLP size distribution, nevertheless, both estimates yield extremely high inferred spontaneous curvatures for palmitoylation.
The effect of palmitoylation on short alpha-helical peptide sequences, indicated by circles in Fig. 4C, agrees qualitatively with the palmitate simulations. Alpha-helical secondary structure places the palmitoyl groups on opposite sides of the helix, so that two orientations are consistent with the palmitoyl chains in the bilayer. These two configurations were both simulated. The spontaneous curvature of palmitoylation was estimated by subtracting the value of c0 from simulations of unpalmitoylated peptide from that of the palmitoylated, adjusting for the “wedge” effect of the peptide itself. These raw values for curvature induction (−0.077 and −0.041 nm−1 at 7% fraction palmitoyl) were then added to the c0 of POPC to be plotted in Fig. 4C.
Lipid mass spectrometry analysis does not reveal major differences between the lipid composition of HANAM1M2 and MAYNAM1M2 particles.
To investigate whether HA acylation influences lipids present in the VLP envelope, the lipid compositions of HANAM1M2, MAYNAM1M2 particles and X31 virions were analyzed (Fig. 5A to C). Lipid concentration (in nanomoles) was normalized to protein concentration determined by bicinchoninic acid assay prior to the lipid analysis. Both VLP preparations showed similar levels of cholesterol (HANAM1M2, 39% ± 4.5%; MAYNAM1M2, 43% ± 7% [average ± difference of two preparations divided by 2]). The levels of cholesterol were lower in the VLPs than in X31 virus grown in 293T cells (51%). Levels of sphingomyelin were similar in both HANAM1M2 (8% ± 1.5%) and MAYNAM1M2 (9% ± 1.5%) particles but lower than those found in the X31 virus grown in 293T cells (29%). The ratio between saturated and unsaturated lipids was maintained at around 1:2 in all analyzed preparations (Fig. 5D). Thus, HA acylation does not seem to dramatically affect the lipid composition of the VLP envelope. However, VLPs seem to incorporate less cholesterol and sphingomyelin in the envelope than the X31 virus grown in 293T cells.
FIG 5.

Lipid mass spectrometry analysis of HANAM1M2 and MAYNAM1M2 particles. Relative distribution of cholesterol (chol) and phospholipids in isolated and sucrose cushion-purified X31 virus grown on 293T cells (A) and HANAM1M2 (B) and MAYNAM1M2 (C) particles isolated from supernatants of the transfected 293T cells. The mean percentages and the differences between the two values divided by 2 were calculated from two independent analyses (except for X31 virus, for which only one preparation was analyzed). (D) Relative distributions of saturated and unsaturated acyl chains found in phospholipids. LPC, lysophosphatidylcholine; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PS, phosphatidylserine; SM, sphingomyelin.
Deacylated HA of three different influenza virus strains forms an arrested fusion pore unable to expand.
To investigate the fusion properties of deacylated HA, we transfected Cos7 cells with plasmids containing either HA, MAC, or MAY. The transfected cells were incubated with human RBCs labeled with the lipid probe PKH26 and one aqueous probe of either carboxyfluorescein or fluorescein isothiocyanate (FITC)-dextran (10 kDa). Acylation does not prevent hemifusion because the lipid probe PKH26 transferred from RBCs to HA, MAC, and MAY transfected cells following pH 4.9 acidification (Fig. 6A). We observed carboxyfluorescein mixing between RBCs and HA, MAY, or MAC transfected cells (Fig. 6B). However, FITC–10-kDa dextran was transferred only to HA and MAC transfected cells, consistent with the previously proposed hypothesis that the completely depalmitoylated HA can induce only a fusion pore that is too small to support transfer of molecules equal to or larger than 10-kDa dextran (Fig. 6C). We further tested several conditions, prolonging the pH 4.9 exposure time to up to 20 min, followed by incubation at pH 7.0 up to 60 min at 37°C. In all cases MAY-induced fusion pores were not permeable to 10-kDa dextran (Fig. 6D).
FIG 6.

Cell-RBC fusion assay monitoring lipid and content mixing upon fusion mediated by HA, MAC, or MAY. Cos7 cells were transfected 24 h prior to fusion with either HA, MAC, or MAY (Aichi [H3N2]), incubated with RBCs labeled with PKH26 lipid mixing indicator (red) and with either anionic FITC-dextran (10 kDa) or a carboxyfluorescein aqueous indicator (green). The fusion was triggered by exposure to citrate buffer with pH 4.9 for 2 min at 37°C, followed by 20 min at pH 7 prior to imaging and counting. (A) Set of representative fluorescent micrographs showing the fusion pairs of RBCs and transfected Cos7 cells. Hemifused cell-RBC pairs contain only red indicator. Fully fused cell-RBC pairs containing both red and green indicators are indicated by white arrows. MAY is not able to induce fusion pores supporting permeation of FITC–10-kDa dextran. Bar plots represent counted fusion events (green cells) containing either carboxyfluorescein (B) or FITC–10-kDa dextran (C) normalized to hemifusion events (red cells labeled with PKH26) driven by HA, MAC, and MAY. (D) The fusion was triggered by exposure to citrate buffer with pH 5 for between 2 and 20 min, followed by 20 to 60 min at pH 7 at 37°C prior to imaging and counting. (E) Bar plot of fusion events normalized to hemifusion events driven by HA and MAY from three different influenza virus strains, Aichi (H3N2), Udorn (H3N2), and WSN (H1N2).
Since the role of HA acylation seems to vary among HA proteins from different influenza A viruses, we tested the fusogenic properties of HAs from A/Udorn/72 (H3N2) and A/WSN/33 (H1N1). MAY from both A/Udorn/72 (H3N2) and A/WSN/33 (H1N1) supported hemifusion and the formation of fusion pores, which were not permeable to 10-kDa dextran (Fig. 6E). These data further support the hypothesis that HA acylation has a role in fusion pore enlargement.
Deacylated HA forms a fusion pore able to expand in the presence of NA, M1, and M2 proteins.
To investigate the role of HA-mediated membrane fusion in a system that more closely mimics influenza virus fusion, we modified the cell-RBC fusion assay. In this assay, the labeled RBCs may bind and fuse with the budding HANAM1M2 filamentous VLPs rather than or in addition to HA-studded regions of the cell membrane. We used 293T cells instead of Cos7 cells because Cos7 cells did not form HANAM1M2 VLPs upon transfection with HA (data not shown). To verify that most of the established contacts are between RBCs and budding VLPs rather than between RBCs and plasma membrane, we analyzed the binding pairs of HANAM1M2 transfected 293T cells and RBCs using thin-section electron microscopy (EM) (Fig. 7A). In 70% of all cases (n = 40), we observed long budding filamentous VLPs near RBC membrane, and in 30% of all cases the RBCs and 293T membranes were in direct contact.
FIG 7.

Cell-RBC fusion assay monitoring lipid and content mixing upon fusion mediated by HA or MAY in the presence of other influenza virus proteins. (A) Thin-section EM of chemically fixed and EPON-embedded 293T cells expressing MAY, NA, M1, and M2 proteins and incubated with RBCs for 20 min at 37°C. RBCs bind to budding filamentous VLPs rather than to the surfaces of the cells. Magnified views show a strong connection between HAMAY present on the VLP and the RBC membrane. (B) Bar plot of the HA- or MAY-induced fusion monitoring lipid (PKH26) and content (FITC–10-kDa dextran) mixing in the presence or absence of NA, M1, and M2 proteins. (C) Bar plot of the MAY-induced fusion monitoring lipid (PKH26) and content (FITC–10-kDa dextran) mixing in the presence of NA only, M2 only, or NA, M1, and M2 proteins together. Coexpression of M1 and M2 proteins enhances pore enlargement of the MAY-driven fusion pore.
The number of MAY-induced fusion pores that support 10-kDa dextran content mixing in the 293T-RBC fusion assay was approximately 3-fold lower (P = 0.011, t test; 5 independent assays) (Fig. 7B) than that in HA-induced fusion pores similarly observed in Cos7 cells (Fig. 6). Interestingly, the coexpression of NA, M1, and M2 proteins slightly increased (2-fold), but not significantly (P = 0.06, t test), the number of MAY-induced fusion pores permeable to 10-kDa dextran. However, the MAYNAM1M2 ability to support 10-kDa dextran mixing was only 1.6-fold lower than that of HANAM1M2, and the difference was not statistically significant (P = 0.06, t test). To investigate whether the increase in dextran mixing in MAYNAM1M2 is specific to one of the coexpressed proteins, we cotransfected 293T cells with MAY and NA, with MAY, NA, and M1, or with MAY, NA, M1, and M2 genes. 293T cells forming both MAYNAM1 and MAYNAM1M2 VLPs showed slight, but not significant, increases (∼1.5-fold) in the number of pores supporting 10-kDa dextran penetration (Fig. 7C). These data indicate that HA deacylation inhibits fusion pore opening but that opening is partially rescued by the presence of NA, M1, and M2 proteins.
Isolated MAYNAM1M2 particles form complete fusion products with liposomes.
Cryo-ET was recently used to structurally characterize the influenza A VLP-driven hemifusion intermediates (20) and the influenza A virus-driven fusion intermediates (49–51). To investigate the role of HA acylation in HA-mediated membrane fusion in a system that more closely mimics influenza virus fusion, we used cryo-ET. Mixtures of isolated HANAM1M2 or MAYNAM1M2 VLPs and liposomes (42 mol% POPC, 13 mol% POPE, 40 mol% cholesterol, and 5 mol% of total gangliosides [GA]; POPE is 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) were incubated for 20 to 30 min and subsequently subjected to pH 5 for 2 to 5 min prior to being plunged into liquid ethane. Based on the cell-RBC fusion assay where we observed dramatic inhibition of expansion of the deacylated HA-induced fusion pores, we expected to observe an increased number of fusion pores with inner diameters of less than 4.6 nm (hydrodynamic diameter of FITC–10-kDa dextran used in the cell-RBC assay). Cryo-ET revealed the presence of large compartments in both HANAM1M2 (n = 8 observable ratio R = 0.8; five tomograms) and MAYNAM1M2 (n = 38 and R = 1.46; 15 tomograms) samples. The large compartments with electron-dense disassembled M1 protein and more sparsely distributed glycoproteins are characteristic of complete fusion. Evaluating the fusion probability as a function of either a discrete, N (N = 0 to 10), or continuous, <N> (<N> = 0 to 10), proportionality was consistent with no significant differences between the fusion probability of MAYNAM1M2 and HANAM1M2. These data suggest that a large fraction of the fusion pores induced by either acylated or deacylated HA underwent expansion (Fig. 8). In the MAYNAM1M2 sample, several hemifusion diaphragms with an average diameter of 10 ± 3 nm (n = 22) were often present between the completely fused compartment and unfused liposomes, whereas in the HANAM1M2 sample the hemifusion diaphragms were present between unfused VLPs and unfused liposomes (Fig. 8). Fusion pores, which by definition have an electron-lucent interior, could not be unambiguously determined in this data set. Thus, in this fusion system of liposomes to VLPs having protein composition close to that of the native virus, HA deacylation did not have an observable effect on fusion pore expansion.
FIG 8.
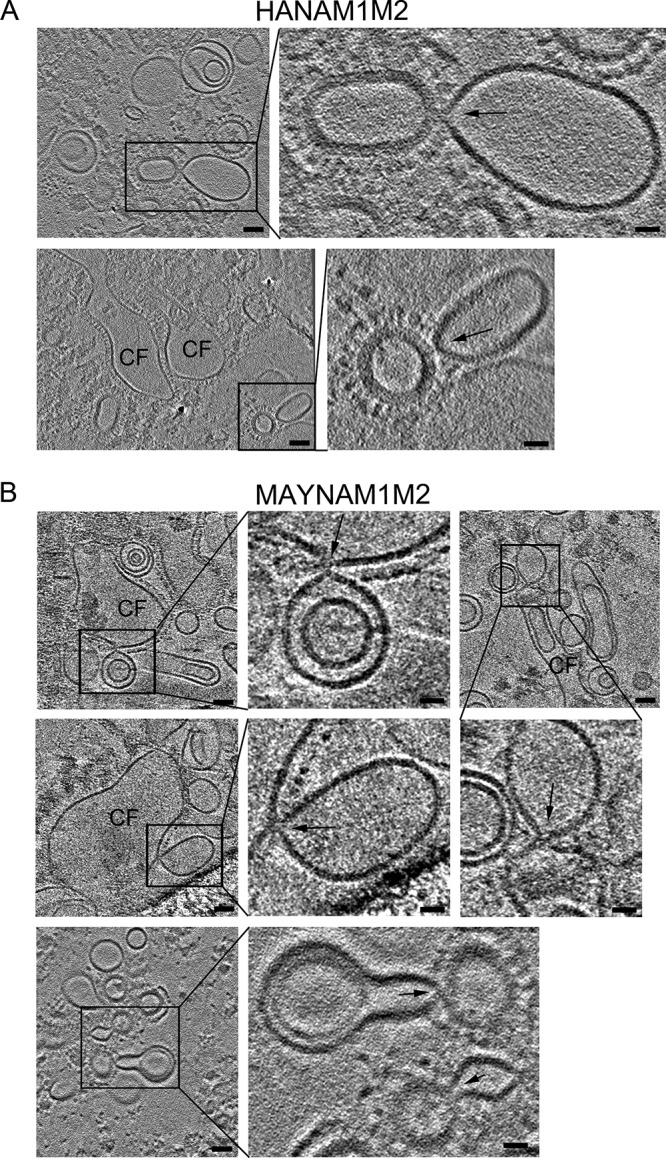
Cryo-ET of MAYNAM1M2 and liposome fusion events. HANAM1M2 or MAYNAM1M2 particles were mixed with liposomes containing 5 mol% total gangliosides, incubated for 20 to 30 min at neutral pH, and subjected to low pH for 2 min at 37°C prior to being plunged into liquid ethane. (A) Tomogram slices (3 nm thick) showing HANAM1M2 fusion events. The tomograms were calculated by a weighted back-projection method from a tilt series acquired on a Krios microscope at a defocus of −0.5 μm with a Volta phase plate. (B) Tomogram slices (3 nm thick) showing MAYNAM1M2 fusion events. Tomograms were calculated by a weighted back-projection method using a simultaneous iteration reconstruction technique (SIRT)-like filter from a tilt series acquired on a Krios microscope at a defocus of −5 μm, except for the slices of the tomogram on the bottom, which were reconstructed as described for panel A. The slices capture complete fusion (CF) compartments and fusion events formed between liposomes and VLPs, marked by black rectangles (scale bar, 50 nm). Magnified areas of the region of interest surrounded by the rectangle are on the right or bottom, as indicated by black lines (scale bar, 20 nm). Black arrows mark the contact between liposomes and VLPs. Note that the fusion intermediates in panel B are between complete fusion compartments and liposomes, and in panel A they are between an unfused VLP and unfused liposome. The bottom image in panel B shows a rare event where unfused MAYNAM1M2 spheroidal particles containing M1 protein form a hemifusion diaphragm with liposomes. Two representative tomograms were deposited in the wwPDB under accession numbers EMD-8845 and EMD-8846.
DISCUSSION
Our data indicate that the removal of HA terminal acylation affects influenza virus VLP assembly and HA-mediated membrane fusion in a cell-RBC fusion assay. Findings in this study suggest that some apparent discrepancies in the field are likely caused by differences in fusion assays and HA expression systems rather than by different HA subtypes. We propose that a role of HA acylation to inhibit fusion pore expansion in cell-RBC fusion assays relates to the impact of acylation on membrane curvature, regardless of which HA subtype is used. For assembly, we propose that acylation of HA increases its contribution to overall bilayer curvature, facilitating the incorporation and assembly of the M1 layer, which maximizes envelope curvature. Consistent with our proposal, molecular dynamics shows that insertion of palmitic acid into the inner envelope monolayer increases monolayer negative spontaneous curvature.
HA palmitoylation has a profound effect on M1 protein incorporation. The release of influenza virus proteins, assessed by Western blotting, was similar in both HANAM1M2 and MAYNAM1M2 transfected cells, except for that of NA, whose release was increased in MAYNAM1M2 transfected cells. Consistently, cryo-ET revealed that filamentous MAYNAM1M2 VLPs incorporated more NA than HANAM1M2 filamentous VLPs and that spheroidal MAYNAM1M2 particles rarely contained an M1 layer. These data are in agreement with a previous study where a deacylated HA-SSS mutant (with three cysteines changed to serines) failed to incorporate M1 into H3N2 Udorn influenza virions (38). Thus, our data indicate that the M1-MAY protein interaction is weaker than the M1–acylated-HA interaction, perhaps due to steric changes or exposure of the deacylated CT within the cytoplasm.
A previous study showed that HA expression induces ruffling of the plasma membrane and shedding of HA containing spheroidal particles, suggesting that HA is able to alter membrane curvature (8). To address whether HA acylation has a role in modulating membrane curvature, we compared the diameters of released spheroidal VLPs containing either HANA or MAYNA but with no M1. This approach also has the advantage of eliminating possible contributions of the cellular cytoskeleton. Membrane curvature J is inversely proportional to the radius R (J = 1/R). Interestingly, we found that spheroidal MAY VLPs were larger than spheroidal HA VLPs, indicating that HA acylation can influence HA-induced membrane curvature. Although NA also contributes to the curvature, we presumed that the NA contribution to the curvature is equal in both MAYNA and HANA particles (ratio of HA/MAY to NA, = 5:1). HANA and MAYNA vesicles, with an average radius of 45 nm and 56.5 nm, have curvatures of 0.022 nm−1 and 0.018 nm−1, respectively. If we assume that the only difference between HA WT and MAY is the presence or absence of acyl groups, the curvature difference can be directly attributed to the nine acyl groups present on a single HA trimer. Thus, acylation may contribute globally to an 0.004 nm−1 increase in HA-induced positive curvature by affecting the inner monolayer curvature. Based on average HA and MAY spacing and an HA/NA ratio of 5:1, approximately 340 HA and 530 MAY trimers are required to transform a flat membrane into a sphere in the absence of an M1 layer. Since the location of the acyl groups is in the inner VLP monolayer, formally the acylated cytoplasmic tail itself has a more negative spontaneous curvature. That the acylated CT induces negative spontaneous curvature is supported further by the results of molecular dynamics simulations on palmitic acid. The simulated spontaneous curvature (−1.1 nm−1) agrees with that inferred from the VLP distribution (−1.62 nm−1) in the sense that both values indicate an extremely high negative curvature propensity. Note here that the spontaneous curvature of palmitoylation extracted from the VLP distribution is for the effect of an individual palmitoylation, rather than its effect on the entire bilayer (0.004 nm−1).
It is conceivable that acylation contributes to both the position and depth of CT insertion into the membrane, which may cause local membrane distortion. It has been shown that C16 alkylated peptides of the transmembrane domain and CT have a greater tendency within a micelle to form oligomers of higher order (52). Thus, acylation may be responsible for arrangements and spacing of HA molecules on the surface of the virions. A molecular dynamics study suggests that palmitoylation may influence rotation of the protein with respect to the bilayer (53). To address whether the spatial distribution of the deacylated HA is different from that of the WT HA, we measured the glycoprotein spacing in filamentous and spheroidal particles with and without the M1 layer. The glycoprotein spacings were similar in all instances, 8.54 ± 0.48 nm (mean ± 95% confidence), indicating that neither HA acylation nor the M1 layer influences the distance between glycoproteins. Thus, our results support the hypothesis that HA acylation does not influence glycoprotein spacing.
To elucidate whether acylation influences VLP lipid composition, membrane samples obtained from HANAM1M2 and MAYNAM1M2 isolated particles were analyzed using mass spectrometry. We found that the relative levels of cholesterol and sphingolipids were similar regardless of HA acylation. These data are consistent with lipid analysis performed on influenza A virus A/FPV/Ro/34 (H7N1), where both deacylated HA mutant virus and wild-type virus had similar lipid compositions (41). Interestingly, the VLP envelope contained less (∼40%) cholesterol and sphingomyelin than X31 influenza A virus grown and isolated from 293T cells, perhaps indicating differences between transfection and infection. The relative cholesterol level in X31 influenza A virus envelope was similar to the relative cholesterol level found in A/FPV/Ro/34 (H7N1) (54).
To test the fusogenic properties of MAY, we performed an RBC-cell fusion assay where hemifusion and fusion are monitored as lipid and content mixing, respectively. As previously shown, we found that MAY-induced fusion pores were able to induce hemifusion and a small fusion pore supporting transfer of small molecules such as carboxyfluorescein (38). However, in contrast to data shown when Udorn MAY was used (38, 43), the Aichi MAY used in our study was not able to support content mixing of larger indicators such as 10-kDa dextran. Initially, we hypothesized that this discrepancy is due to a unique single amino acid difference (Ser 348) present in the fusion peptide of the Udorn HA. However, introduction of Ser mutation into the fusion peptide of the Aichi HA did not rescue pore enlargement. In addition, in our setup neither the Udorn MAY nor additionally tested WSN or H1N1 MAY could induce a fusion pore that would support content mixing of 10-kDa dextran. It is possible that the discrepancy is caused by the difference in expression systems: plasmid-driven expression used in our study versus recombinant simian virus 40 (SV40)-driven expression used in the study by Melikyan et al. (43) where the background SV40 infection may have altered membrane properties. Furthermore, we found that A/Udorn/72 (H3N2) and A/WSN/33 (H1N1) deacylated HA proteins are also impaired in fusion pore expansion. Thus, the role of HA acylation in fusion pore expansion is not strain dependent.
In the context of the whole virus there are additional factors that play a role in fusion pore enlargement: M1 matrix layer disassembly upon acidification facilitated by the M2 proton channel. Fusion pore expansion driven solely by HA might not recapitulate fusion pore expansion driven by the virus. Clearly, our data show that HA acylation is important in M1 protein recruitment and assembly. Conversely, we hypothesize that HA acylation may influence M1 layer disassembly that is necessary for fusion pore formation and expansion. M1 disassembly occurs in two independent steps, and the M1 layer must first undergo a conformational change prior to detachment from the membrane (28, 29, 31). We showed that the presence of other influenza virus proteins (NA, M1, and M2) diminished the impact of HA acylation on fusion pore expansion in a cell-RBC fusion assay. To further study this in a system closer to virus-mediated fusion, we used cryo-ET to structurally characterize fusion pores between HANAM1M2 or MAYNAM1M2 VLPs and liposomes as in our recent study (20). We observed numerous, large, postfusion compartments with dispersed influenza virus spikes and often associated electron-dense material, presumably disassembled M1 protein, indicating that a large fraction of fusion pores had fully expanded. Using the observed numbers of hemifusion and complete fusion structures and models relating the observables with transitive intermediates, we found no evidence for differences in the probabilities of fusion between MAYNAM1M2 and HANAM1M2. In addition, the large complete fusion compartments in MAYNAM1M2 samples often showed secondary fusion events, which appeared to be at the hemifusion stage. We believe that this is due to the presence of more MAY containing spheroidal particles without the M1 layer, which would otherwise have had to be disassembled prior to formation of a completely fused compartment. Detected MAY-induced hemifusion diaphragms had diameters consistent with hemifusion diaphragms found in wild-type HA (9 ± 2 nm)- and G1S mutant (9 ± 3 nm)-driven fusion published in our recent study (20). Some of the fusion events were similar to those described by Calder and Rosenthal as fusion pores in their recent study (49) or similar to those described by Gui et al. as target membrane-pinching deformations at liposome-virus contact sites (50). However, due to the presence of an electron-dense material between the MAYNAM1M2 VLPs and liposomes, we classified these events as hemifusion diaphragms/stalks, as in our previous study (20). The observation of multiple complete fusion compartments suggests that HA deacylation in VLP-mediated fusion does not have a dramatic role in fusion pore expansion in VLP-liposome fusion, in contrast to the role observed in a cell-RBC fusion assay. Thus, HA-mediated fusion in a cell-RBC fusion assay may not recapitulate all aspects of viral fusion, likely due to a lack of additional viral proteins and target membrane proteins and different geometry and dimensions of the fusing compartments.
It is plausible that fusion pore expansion driven by HA in cell-RBC fusion requires additional negative curvature exerted by palmitoyls on the inner monolayer of the fusion pore, which is not needed in virus fusion due to the factors listed above. We propose that in the fusion of two flat membranes (cell-RBC fusion), the acylated CT and fusion peptide act from opposite sites, both decreasing the local spontaneous curvature of the inner and outer monolayers, respectively. By convention, the inner monolayer of the fusion pore is facing a fusion pore lumen, and the outer monolayer is facing the extracellular space. Consistently, it has been shown that the fusion peptide promotes negative spontaneous curvature of the outer monolayer of the fusion pore (55). This hypothesis is supported by a continuum analysis, which calculated that fusion pores with negative spontaneous curvature in both monolayers lower their total energy by expansion (56). However, in the virus fusion scenario where the viral envelope is already highly curved, the subsequent release of membrane bending energy upon membrane fusion may contribute to pore expansion without an additional requirement for negative curvature exerted by palmitoyl. This hypothesis is supported by calculations on the kinetics of model fusion pore dilatation where fusion pores are shown to widen more rapidly from curved membranes than from flat membranes (57).
In conclusion, our study indicates that acylation increases the tendency of HA to bend membranes and interact with M1 protein. It is plausible that both HA-M1 interactions and high curvature induced by HA are needed for M1 layer formation. Finally, the influenza virus proteins studied in this work could be hierarchically classified based on their propensity to bend membranes, with highly membrane bending M1 protein followed by NA, then by acylated HA, and finally by moderately membrane bending HA after deacylation. It is of further interest to elucidate how these proteins cooperate in membrane bending in virus assembly and membrane fusion.
MATERIALS AND METHODS
Plasmids.
HA (A/Aichi/2/68) and NA (A/Singapore/1/57) DNA sequences cloned into eukaryotic expression vector pCAGGS (58) were a kind gift from H. D. Klenk, University of Marburg. The HA (A/Udorn/72) DNA sequence of HA (A/Udorn/72) was a kind gift from Fredrick S. Cohen, Rush University. Sequences of M1 (A/Hong Kong/8/68) and M2 (synthetic) were cloned into pCAGGS. Nucleotide mutations resulting in MAC and MAY mutants in HA (A/Aichi/2/68) and a MAY mutant in HA (A/WSN/33) and HA (A/Udorn/72) were introduced by a QuikChange II XL site-directed mutagenesis kit (Agilent). Codons for two cysteines (Cys 555 and Cys 562) or for all three cysteines (Cys 555, Cys 562, and Cys 565) where changed to codons that coded for the MAC (Met 555 and Ala 562) or MAY (Met 555, Ala 562, and Tyr 565) mutant.
Cells.
293T human embryonic kidney cells, Cos7 African green monkey kidney fibroblast-like cells and Madin-Darby canine kidney (MDCK) cells were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), l-glutamine, and Primocin.
Antibodies.
Polyclonal rabbit serum was raised against a peptide homologous with a 25-amino-acid sequence at the C terminus of HA1 [A/X31 (H3N2)]. Polyclonal goat serum raised against NA [A/Singapore/1/57 (H2N2)] was obtained from BEI Resources (Manassas, VA). Monoclonal mouse anti-M1 antibody (59) was a gift of J. W. Yewdell (National Institutes of Health, Bethesda, MD). Monoclonal mouse anti-M2 (14C2) was purchased from Abcam (Cambridge, MA). Antibodies against influenza virus proteins were diluted 1:2,000 or 1:1,000 (mouse anti-M2) for Western blotting. Donkey anti-goat, goat anti-mouse, and goat anti-rabbit alkaline phosphatase conjugates (Invitrogen) were used at a dilution of 1:8,000.
Production of MAY A/WSN virus by reverse genetics.
Plasmids for A/WSN/33 reverse genetics (pTM-PolI-WSN-PB2-PB1-PA-NP-M-NS, pTM-PolI-WSN-HA-NA, and pC-PolII-WSN-PB2-PB1-PA) were obtained as a gift from Y. Kawaoka's group (45). A QuikChange II XL site-directed mutagenesis kit (Agilent) was used to create site-directed mutations in three acylation sites of influenza virus hemagglutinin (Cys 555, Cys 562, and Cys 565) located in the protein C terminus. The HA mutants A/WSN/33 HA-MAY and A/WSN/33 HA-MAC were created. The pTM and pC plasmids required for reverse genetics were transfected into 293T cells according to a published protocol (45). After 48 h of transfection, cytopathic effects were monitored, and hemagglutinin assays were performed to confirm the presence of viruses. The supernatant containing viruses was then transferred to MDCK cells for virus amplification. In each step, the supernatant was collected, and viral RNAs were purified using a Qiagen QIAamp viral RNA minikit. cDNAs of viruses were created using an Invitrogen Super Script III reverse transcriptase kit. PCRs with oligonucleotides specific for HAs from different influenza virus strains were carried out in a Bio-Rad MJ Mini PCR machine. PCR products were purified using a Qiagen MiniElute PCR Purification kit and subjected to 2% Tris-acetate-EDTA (TAE) gel electrophoresis for product observation. The purified PCR products were sent for sequencing, and sequence results were analyzed in Sequencher, version 4.9 (Gene Codes Corporation, Ann Arbor, MI) and the Vector NTI Advance 11 program (Invitrogen).
VLP preparation.
293T cells were seeded in a 10-cm-diameter tissue culture dish (3.2 × 106 cells/dish) and grown at 37°C and 5% CO2 in DMEM–10% FCS supplemented with l-glutamine and Primocin. Twenty-four hours later, cells were transfected with the appropriate plasmids using Trans-IT LT1 (Mirus) according to the manufacturer's instructions. VLPs were produced using the following quantities: 2.5 of μg pCAGGS-HA, 0.5 μg of pCAGGS-NA, and either 1 μg of pCAGGS-M (coding for both M1 and M2) or 1 μg of pCAGGS-M1 and 0.25 μg of pCAGGS-M2. At 5 h posttransfection, the medium was replaced with serum-free SFM4MegaVir, and exogenous bacterial neuraminidase from Clostridium perfringens (Sigma-Aldrich) was added at a final concentration of 25 mU/ml. At 48 h tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (5 μg/ml) was directly added to the medium in the culture dish and incubated at 37°C for 15 min. Trypsin was inactivated by the addition of soybean trypsin inhibitor (0.1 mg/ml); supernatant was collected and clarified at 4°C by centrifugation at 1,000 × g for 10 min. Subsequently, the supernatant was layered onto a 3-ml 30% (wt/vol) sucrose-KHE (100 mM KCl, 10 mM HEPES, 1 mM EDTA, pH 7.4) cushion and centrifuged at 200,000 × g for 2 h at 4°C in a Beckman Optima centrifuge using a 50.2 Ti rotor (Beckman Coulter, Fullerton). The pellet was resuspended in KHE buffer, loaded onto an OptiPrep 10 to 30% continuous gradient, and centrifuged at 200,000 × g for 2 h at 4°C in a tabletop Beckman Optima centrifuge using a swing-out TLS-55 rotor. The VLP band was collected, washed in KHE buffer, and centrifuged again at 130,000 × g for 30 min at 4°C using a TLA 100.3 rotor. The pellet was resuspended in KHE buffer.
SDS-PAGE and Western blotting.
To prepare the cell lysates for Western blot analysis, transfected cells were lysed in 750 μl of lysis buffer (50 mM Tris-Cl, pH 8.0), 1% sodium dodecyl sulfate [SDS], and protease inhibitor complex [Roche], homogenized by passage three times through a 500-μl Hamilton syringe, and sonicated using a rod sonicator (Sonifier 250; Branson) set to 15 cycles, output 4, and 40% duty cycle, Samples were centrifuged at 18,000 × g for 10 min at 4°C and the supernatant was mixed with 4× lithium dodecyl sulfate (LDS) loading buffer (NuPAGE) and dithiothreitol (final concentration, 100 mM). Lysates and supernatants were analyzed under reducing conditions on 4 to 12% SDS-polyacrylamide gels (NuPAGE) and transferred to polyvinylidene difluoride membranes (Invitrogen) using a Fastblot 44 semidry system (Biometra, Gottingen). Membranes with proteins were blocked in 5% low-fat milk in Tris-buffered saline and 1% Tween 20 (TBS-T) and then incubated for 1 h with appropriate antibodies diluted in TBS-T. Membranes were incubated with enhanced chemifluorescent (ECF) substrate (Amersham) for 5 min and scanned with an FLA 3000 scanner (Fujifilm) using a 473-nm laser and 580-nm detection filter. Protein densities were analyzed using a gel macro implemented in FIJI imaging software.
RBC labeling with hydrophilic (content) and hydrophobic (lipid) fluorescent indicators.
Human red blood cells (RBCs) were spun at 700 × g for 8 min at 4°C and subsequently washed five times with phosphate-buffered saline (PBS) without Ca2+ and Mg2+ to remove serum. The RBC pellet was hemolyzed with hemolysis buffer (PBS without Ca2+ and Mg2+ plus H2O, 1:1) and incubated on ice for 2 to 15 min with an aqueous fluorescent indicator [5-(and 6-) carboxy-2′,7′-dichloro-fluorescein; Molecular Probes], anionic FITC–10-kDa dextran, or anionic FITC–500-kDa dextran (Invitrogen). The RBC membrane was sealed according to Ellens et al. (60) and spun at 300 × g for 4 min at 4°C to remove any unincorporated fluorescent indicator. The RBC membrane was labeled with PKH26 (Sigma).
Cell-RBC fusion assay.
Cos7 cells were seeded in a two-well Lab-Tek chambered cover glass (Nunc) at a density of 3.5 × 104 cells per well and grown at 37°C and 5% CO2 in DMEM–10% FCS supplemented with l-glutamine and Primocin. Twenty-four hours later, cells were transfected with 2.5 μg of pCAGGS-HA using Trans-IT LT1 (Mirus) according to the manufacturer's instructions. At 1 day posttransfection the cells were washed twice with PBS (pH 7.4) and treated for 4 min at room temperature (RT) with TPCK-treated trypsin (10 μg/ml) and exogenous bacterial neuraminidase from Clostridium perfringens (0.2 mg/ml). Subsequently, the RBCs labeled with fluorescent content and lipid indicators were added to transfected Cos7 cells and incubated for 20 min at RT. The Cos7 RBC-cell pairs were washed three times with PBS (pH 7.4) and incubated for 2 min with acidified PBS (pH 4.9) at 37°C and an additional 20 min with PBS (pH 7.4) at 37°C. Cell pairs were imaged using an Axiovert 25 (Zeiss) inverted microscope equipped with 20× Zeiss objective (numerical aperture [NA], 0.8) and filter sets 45 and 42 (Zeiss). Cell-RBC pairs showing content and lipid indicator mixing were counted. Fusion was evaluated using the number of cell pairs exhibiting both content and lipid indicator mixing and presented as a number ratio.
To study the effect of NA, M1, and M2 on HA-induced fusion pores, the cell-RBC fusion assay was modified accordingly: 293T cells were seeded in a 6-cm-diameter tissue culture dish (1 × 106 cells/dish) and grown at 37°C and in 5% CO2 and DMEM–10% FCS supplemented with l-glutamine and Primocin. Twenty-four hours later, cells were transfected with appropriate plasmids (2.5 μg of pCAGGS-HA, 0.5 μg of pCAGGS-NA, 1 μg of pCAGGS-M1, and 0.25 μg of pCAGGS-M2) using Trans-IT LT1 (Mirus) according to the manufacturer's instructions. At 1 day posttransfection the cells were washed twice with PBS (pH 7.4) and detached from the dish using TPCK-treated trypsin (10 μg/ml) and exogenous bacterial neuraminidase from Clostridium perfringens (0.2 mg/ml) for 4 min. 293T cell suspensions were mixed with RBCs labeled with content and membrane fluorescent indicators, incubated on a rotator for 20 min at RT, and plated for 1 h at RT in Lab-Tek chambers (Nunc) coated with poly-l-lysine (0.5 mg/ml). Subsequently, the 293T-RBC cell pairs were incubated for 2 min with acidified PBS (pH 4.9) at 37°C and an additional 20 min with PBS (pH 7.4) at 37°C. Cell pairs were imaged using an Axiovert 25 (Zeiss) inverted microscope and counted as described above.
Sample preparation for thin-section electron microscopy.
For thin-section electron microscopy, 293T-RBC pairs were fixed for 1 h at room temperature with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) and then fixed for 30 min at 4°C with OsO4 in 0.1 M cacodylate buffer (pH 7.2). Specimens were subsequently dehydrated with ethanol, stained en bloc for 2 h at 4°C with 6% uranyl acetate in 70% ethanol, and further dehydrated with 80%, 90%, 95%, and finally with 100% ethanol. To detach the cells, the dish was partially dissolved by propylene oxide. The cells were further embedded in epoxy resin (Embed-812; EMS) polymerized for 24 h at 60°C. Sections (50 nm) were obtained using a Leica EM UC7 microtome equipped with a diamond knife (Diatome) and poststained with 2% lead citrate in water.
MS lipid analysis.
Two independent preparations of HANAM1M2 and MAYNAM1M2 and one preparation of X31 influenza A virus grown on 293T cells in KHE buffer were processed by the Metabolomics Core, Washington University School of Medicine, for lipid analysis. A protein precipitation method was used to extract lipids from each sample in the presence of internal standards. The final reconstituted solutions were used for mass spectrometry (MS) analysis of phospholipids. In order to improve MS sensitivity of free cholesterol, a small aliquot of the reconstituted solutions was further derivatized by nicotinic acid to convert free cholesterol into ester. Sample analysis was performed with a Shimadzu 10A high-performance liquid chromatography (HPLC) system coupled to a TSQ Quantum Ultra triple quadrupole mass spectrometer operated in selective reaction monitoring (SRM) mode under positive electrospray ionization (ESI+). Different analytical HPLC columns and mobile phases were employed for different analytes to achieve optimal sensitivity and separation. Data processing was conducted using Xcalibur software (Thermo). Analyte concentration was calculated as the concentration of a corresponding internal standard multiplied by the peak area ratio of the analyte to the internal standard.
Liposome preparation.
POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine), cholesterol, and total gangliosides (GA) (Avanti Polar Lipids) were dissolved in chloroform-methanol (2:1) at a final concentration of 10 mM containing 42 mol% POPC, 13 mol% POPE, 40 mol% cholesterol, and 5 mol% GA. The solvent was removed by evaporation, first using a stream of argon gas and, second, a high vacuum for 1 h. The lipid film was resuspended in KHE buffer, processed using 21 freeze-thaw cycles, and extruded at room temperature through 100-nm polycarbonate (PC) membrane (Whatman) using a mini-extruder set (Avanti Polar Lipids). Liposomes were stored at 4°C and used within 1 to 5 days.
Cryo-ET of VLPs and VLP-liposome mixtures.
For structural analysis of HANAM1M2 and MAYNAM1M2 VLPs, 3 μl of VLPs containing 10-nm protein A colloidal gold nanoparticles was applied to C-flat grids (CF-2/1-2C-50; Protochips) maintained at 25°C and 100% relative humidity, and grids were vitrified by being plunged into liquid ethane using a Vitrobot (FEI). For structural analysis of fusion pores, 7 μl of MAYNAM1M2 VLPs and 5 μl of liposomes (10 mM) were incubated for 20 to 30 min at 37°C. Membrane fusion was triggered by the addition of 17 mM citric acid in 20 mM HEPES to achieve a final pH of 5.0 and incubated at 37°C for 2 to 5 min. VLP-liposome mixtures containing 10-nm protein A colloidal gold nanoparticles were applied to C-flat grids (CF-2/1-2C-50; Protochips) which were plunged into liquid ethane using a Vitrobot (FEI) instrument as described above. A cryo-electron tilt series of vitrified samples was collected either with a transmission electron microscope T20 (FEI) equipped with a K2 Summit camera (Gatan) using the counting mode or with a Krios (FEI) equipped with a Falcon II camera (FEI). Tomographic tilt ranges were typically acquired from +50° to −50° with an angular increment of 3° using −8-μm defocus, 0.387 nm/pixel, and a total electron dose below 80 e−/Å2 using the T20 microscope. Tilt series collected using the Krios microscope (FEI) were typically acquired from +65° to −65° with an angular increment of 2° using either −5-μm defocus with a 70-μm aperture or −0.5-μm defocus with the Volta phase plate, 0.368 nm/pixel, and a total dose of 80 e−/Å2. Tilt series were reconstructed using weighted back projection, and the tilt series collected on the T20 was reconstructed using the simultaneous iterative reconstruction technique (SIRT)-like filter in the IMOD software suite (61). In total, 21 tomograms of HANAM1M2 particles, 24 tomograms of MAYNAM1M2 VLPs, 15 tomograms of MAYNAM1M2 VLPs-liposomes incubated at pH 5, and 5 tomograms of HANAM1M2 VLPs-liposomes incubated at pH 5 were used for analyses. All statistical tests were done either in Microsoft Excel or MathWorks MatLab. The cumulative distributions of VLP radii were first individually fit to a left-truncated Gaussian distribution. However, since the cutoff values, truncated mean parameters (μ) for HA with and without M1, and truncated standard deviations (σ) for HA and MAY without M1 were not statistically different (individually fitted parameters within the 95% confidence bounds), the data were fit using a global fitting function that allowed for a reduction in the number of fitting parameters. The global fitting results for the entire data set (HA with and without M1 and MAY without M1) are as follows (fitted value [± 95% confidence bounds]): cutoff c = 20.8 nm (20.0, 21.7), μHA = 30.0 nm (29.8, 30.2), μMAY = 50.9 nm (50.0, 51.7), σ with M1 (σwM1) = 4.6 nm (4.2, 4.9), and σ without M1 (σwoM1) = 22.2 nm (21.4, 23.1).
Glycoprotein spacing analysis.
For measurements from the top, two-dimensional slices (thickness, 0.774 nm) capturing glycoproteins on the surface of the VLPs were extracted from tomograms as MRC files in IMOD Slicer and converted to TIF using mrc2tif IMOD script. Tomograms were reconstructed from tilt series collected on a T20 microscope with −8-μm defocus. Images were cropped to 50 by 50 nm2 in FIJI with a pixel size of 0.774 nm/pixel and tapered in the MatLab image-processing toolbox. The calculated power spectra of images (HA filamentous, n = 23; MAY filamentous, n = 14; HA spheroidal with M1, n = 12; MAY spheroidal with M1, n = 5; HA spheroidal without M1, n = 8; MAY spheroidal without M1, n = 4; background, n = 20) were averaged in MatLab. A radially averaged power spectrum was calculated from the averaged power spectrum using the MatLab function rotavg written by Bruno Olshausen (University of California, Berkeley [https://redwood.berkeley.edu/bruno/npb261b/lab2/rotavg.m]). The power spectra were fit in MatLab using a mixture model composed of a modified Lorentzian and a Gaussian fit [A/(1 + (x × 6 × B)3C) + D × normpdf (x, μ, σ)] to obtain convergence on all fits. For measurements from the side, two-dimensional slices (thickness, 0.774 nm) capturing the cross-section of glycoproteins on the surface of the VLPs were extracted from tomograms as MRC files in IMOD Slicer and converted to TIF using mrc2tif IMOD script. Plot line and peak-to-peak measurements were done in FIJI.
Computation of palmitate spontaneous curvature.
Palmitic acid spontaneous curvature was computed from molecular dynamics simulations of small patches of lipid bilayers. Systems were assembled using the CHARMM-GUI online protocol (62). Eight systems were simulated: one system with 100 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) molecules per monolayer; three systems with 90:20, 80:40, 70:60 POPC/palmitic acid molecules per monolayer; and two pairs of simulations with palmitoylated and unpalmitoylated short alpha-helical units with segments of the HA sequence (ACQRGNIRCNICI) in two orientations. The pure palmitate simulations approximate 0%, 10%, 20%, and 30% palmitic acid by surface area. Peptide systems included three helices per leaflet, initially equally spaced, on a hexagonal bilayer patch, whereas lipid-only simulations were square. The systems were each run for at least 80 ns, total, using either the CHARMM (63) or NAMD (64) molecular dynamics package for the pure lipid or lipid peptide simulations, respectively. The C36 lipid force field (65) was used for all simulations. Standard simulation parameters were used: the particle mesh Ewald (66) algorithm was used to compute long-ranged electrostatics, the SHAKE (67) algorithm constrained bonds to hydrogen atoms, and a 1-fs time step was used for the leapfrog dynamics integrator. One atmosphere of normal pressure was applied, and the system was maintained at zero surface tension using the Langevin piston algorithm with a 20 ps−1 collision frequency (68). The temperature was controlled to 310 K. The spontaneous curvature is computed from the lateral pressure profile (69) as:
where kc is the bending modulus (70), Lc is the height of the simulation cell, and pT(z)is the tangential pressure profile, available from standard molecular dynamics packages, as well as a developmental version of CHARMM. The flexibility of the force field does not allow the palmitic acid lipids to deprotonate; thus, they are effectively constrained to be neutral.
Computation of fusion probabilities from the numbers of hemifusion and complete fusion structures.
The fusion probability can be inferred using a model that relates the observable hemifusion and complete fusion structures. The model assumes a proportional relationship between the number of hemifusion structures that transition to complete fusion and the number of observed complete fusion structures. Let Hobserved and Fobserved be the observed number of hemifusion and complete fusion structures and Htransition be an unknown number of hemifusion structures that transitioned into complete fusion structures. The probability of observing a hemifusion structure, PHF, for unknown Htransition is PHF = Hobserved/(Hobserved + Htransition) and the probability of fusion, PF, is PF = 1 − PHF. Next, Htransition is expressed in terms of the observable, complete fusion structures, through the relationship Htransition= N · Fobserved, where N is an unknown proportionality constant that may be discrete or continuously distributed. For discrete N, PF = 1 − [1/(1 + NR)], where R is the ratio Fobserved/Hobserved. If N is a Poisson-distributed random variable, characterized by a Poisson average, <N>, the probability of fusion can be expressed as follows:
R can be derived either from the observed sums over all tomograms or from the individual counts for each preparation. When individual tomograms are evaluated, zero examples for either observable may occur. In these cases, PF, which may be 0 or 1, is adjusted according to the number of hemifusion and complete fusion structures observed, n, using the relationships 1/(4n) and 1 − 1/4n for probabilities 0 and 1. The dependence of the probability function PF on either N or <N> is then compared; similar probability functions are evidence for no change in probability between compared data sets.
Accession number(s).
Four tomograms were deposited in the Worldwide Protein Data Bank (wwPDB) under accession codes EMD-8843, EMD-8844, EMD-8845, and EMD-8846.
ACKNOWLEDGMENTS
We are grateful to Hans-Dieter Klenk (Marburg, Germany) for pCAGGS plasmids encoding HA (A/Aichi/2/68) and NA (A/Singapore/1/57) DNA sequences and to Fredrick S. Cohen (Chicago, IL) for the pCAGGS plasmid encoding HA (A/Udorn/72) DNA sequence. We thank J. W. Yewdell (Bethesda, MD) for mouse-anti-M1 antibody and BEI Resources (Manassas, VA) for polyclonal goat serum raised against NA. We thank Yoshihiro Kawaoka (Madison, WI) for kindly providing an A/WSN/33 reverse genetics plasmid set. We thank Jane E. Farrington for assistance with electron microscopy. We are grateful to Elizabeth Fischer for access to the Rocky Mountain Laboratories EM facility and the Krios microscope. We thank Hui Jiang for performing the lipid analysis. The NIH Biowulf high-performance computing resource was used for the simulation work. NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign.
This work was supported by the Division of Intramural Research of the Intramural Program of the NIH.
REFERENCES
- 1.Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chlanda P, Zimmerberg J. 2016. Protein-lipid interactions critical to replication of the influenza A virus. FEBS Lett 590:1940–1954. doi: 10.1002/1873-3468.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 4.Ali A, Avalos RT, Ponimaskin E, Nayak DP. 2000. Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J Virol 74:8709–8719. doi: 10.1128/JVI.74.18.8709-8719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enami M, Enami K. 1996. Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J Virol 70:6653–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruigrok RW, Barge A, Durrer P, Brunner J, Ma K, Whittaker GR. 2000. Membrane interaction of influenza virus M1 protein. Virology 267:289–298. doi: 10.1006/viro.1999.0134. [DOI] [PubMed] [Google Scholar]
- 7.Calder LJ, Wasilewski S, Berriman JA, Rosenthal PB. 2010. Structural organization of a filamentous influenza A virus. Proc Natl Acad Sci U S A 107:10685–10690. doi: 10.1073/pnas.1002123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chlanda P, Schraidt O, Kummer S, Riches J, Oberwinkler H, Prinz S, Krausslich HG, Briggs JA. 2015. Structural analysis of the roles of influenza A virus membrane-associated proteins in assembly and morphology. J Virol 89:8957–8966. doi: 10.1128/JVI.00592-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts PC, Lamb RA, Compans RW. 1998. The M1 and M2 proteins of influenza A virus are important determinants in filamentous particle formation. Virology 240:127–137. doi: 10.1006/viro.1997.8916. [DOI] [PubMed] [Google Scholar]
- 10.Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, Digard P. 2007. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J Gen Virol 88:2280–2290. doi: 10.1099/vir.0.82809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC. 2006. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci U S A 103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossman JS, Jing X, Leser GP, Lamb RA. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murti KG, Webster RG. 1986. Distribution of hemagglutinin and neuraminidase on influenza virions as revealed by immunoelectron microscopy. Virology 149:36–43. doi: 10.1016/0042-6822(86)90084-X. [DOI] [PubMed] [Google Scholar]
- 14.Chen C, Zhuang X. 2008. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc Natl Acad Sci U S A 105:11790–11795. doi: 10.1073/pnas.0803711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossman JS, Leser GP, Lamb RA. 2012. Filamentous influenza virus enters cells via macropinocytosis. J Virol 86:10950–10960. doi: 10.1128/JVI.05992-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chernomordik LV, Frolov VA, Leikina E, Bronk P, Zimmerberg J. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol 140:1369–1382. doi: 10.1083/jcb.140.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chernomordik LV, Kozlov MM. 2008. Mechanics of membrane fusion. Nat Struct Mol Biol 15:675–683. doi: 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chernomordik LV, Leikina E, Frolov V, Bronk P, Zimmerberg J. 1997. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J Cell Biol 136:81–93. doi: 10.1083/jcb.136.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuzmin PI, Zimmerberg J, Chizmadzhev YA, Cohen FS. 2001. A quantitative model for membrane fusion based on low-energy intermediates. Proc Natl Acad Sci U S A 98:7235–7240. doi: 10.1073/pnas.121191898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chlanda P, Mekhedov E, Waters H, Schwartz CL, Fischer ER, Ryham RJ, Cohen FS, Blank PS, Zimmerberg J. 2016. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat Microbiol 1:16050. doi: 10.1038/nmicrobiol.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frolov VA, Dunina-Barkovskaya AY, Samsonov AV, Zimmerberg J. 2003. Membrane permeability changes at early stages of influenza hemagglutinin-mediated fusion. Biophys J 85:1725–1733. doi: 10.1016/S0006-3495(03)74602-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melikyan GB, Niles WD, Peeples ME, Cohen FS. 1993. Influenza hemagglutinin-mediated fusion pores connecting cells to planar membranes: flickering to final expansion. J Gen Physiol 102:1131–1149. doi: 10.1085/jgp.102.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clague MJ, Schoch C, Blumenthal R. 1991. Delay time for influenza virus hemagglutinin-induced membrane fusion depends on hemagglutinin surface density. J Virol 65:2402–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gunther-Ausborn S, Schoen P, Bartoldus I, Wilschut J, Stegmann T. 2000. Role of hemagglutinin surface density in the initial stages of influenza virus fusion: lack of evidence for cooperativity. J Virol 74:2714–2720. doi: 10.1128/JVI.74.6.2714-2720.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leikina E, Mittal A, Cho MS, Melikov K, Kozlov MM, Chernomordik LV. 2004. Influenza hemagglutinins outside of the contact zone are necessary for fusion pore expansion. J Biol Chem 279:26526–26532. doi: 10.1074/jbc.M401883200. [DOI] [PubMed] [Google Scholar]
- 26.Ivanovic T, Choi JL, Whelan SP, van Oijen AM, Harrison SC. 2013. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife 2:e00333. doi: 10.7554/eLife.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danieli T, Pelletier SL, Henis YI, White JM. 1996. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol 133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fontana J, Steven AC. 2013. At low pH, influenza virus matrix protein M1 undergoes a conformational change prior to dissociating from the membrane. J Virol 87:5621–5628. doi: 10.1128/JVI.00276-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S, Sieben C, Ludwig K, Hofer CT, Chiantia S, Herrmann A, Eghiaian F, Schaap IA. 2014. pH-controlled two-step uncoating of influenza virus. Biophys J 106:1447–1456. doi: 10.1016/j.bpj.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhirnov OP, Manykin AA, Rossman JS, Klenk HD. 2016. Intravirion cohesion of matrix protein M1 with ribonucleocapsid is a prerequisite of influenza virus infectivity. Virology 492:187–196. doi: 10.1016/j.virol.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 31.Stauffer S, Feng Y, Nebioglu F, Heilig R, Picotti P, Helenius A. 2014. Stepwise priming by acidic pH and high K+ is required for efficient uncoating of influenza A virus cores after penetration. J Virol 88:13029–13046. doi: 10.1128/JVI.01430-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamb RA, Zebedee SL, Richardson CD. 1985. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell 40:627–633. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- 33.Veit M, Serebryakova MV, Kordyukova LV. 2013. Palmitoylation of influenza virus proteins. Biochem Soc Trans 41:50–55. doi: 10.1042/BST20120210. [DOI] [PubMed] [Google Scholar]
- 34.Kordyukova LV, Ksenofontov AL, Serebryakova MV, Ovchinnikova TV, Fedorova NV, Ivanova VT, Baratova LA. 2004. Influenza A hemagglutinin C-terminal anchoring peptide: identification and mass spectrometric study. Protein Pept Lett 11:385–391. doi: 10.2174/0929866043406850. [DOI] [PubMed] [Google Scholar]
- 35.Kordyukova LV, Serebryakova MV, Baratova LA, Veit M. 2008. S acylation of the hemagglutinin of influenza viruses: mass spectrometry reveals site-specific attachment of stearic acid to a transmembrane cysteine. J Virol 82:9288–9292. doi: 10.1128/JVI.00704-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen BJ, Leser GP, Morita E, Lamb RA. 2007. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol 81:7111–7123. doi: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin H, Leser GP, Lamb RA. 1994. The influenza virus hemagglutinin cytoplasmic tail is not essential for virus assembly or infectivity. EMBO J 13:5504–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen BJ, Takeda M, Lamb RA. 2005. Influenza virus hemagglutinin (H3 subtype) requires palmitoylation of its cytoplasmic tail for assembly: M1 proteins of two subtypes differ in their ability to support assembly. J Virol 79:13673–13684. doi: 10.1128/JVI.79.21.13673-13684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakai T, Ohuchi R, Ohuchi M. 2002. Fatty acids on the A/USSR/77 influenza virus hemagglutinin facilitate the transition from hemifusion to fusion pore formation. J Virol 76:4603–4611. doi: 10.1128/JVI.76.9.4603-4611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naeve CW, Williams D. 1990. Fatty acids on the A/Japan/305/57 influenza virus hemagglutinin have a role in membrane fusion. EMBO J 9:3857–3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagner R, Herwig A, Azzouz N, Klenk HD. 2005. Acylation-mediated membrane anchoring of avian influenza virus hemagglutinin is essential for fusion pore formation and virus infectivity. J Virol 79:6449–6458. doi: 10.1128/JVI.79.10.6449-6458.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinhauer DA, Wharton SA, Wiley DC, Skehel JJ. 1991. Deacylation of the hemagglutinin of influenza A/Aichi/2/68 has no effect on membrane fusion properties. Virology 184:445–448. doi: 10.1016/0042-6822(91)90867-B. [DOI] [PubMed] [Google Scholar]
- 43.Melikyan GB, Jin H, Lamb RA, Cohen FS. 1997. The role of the cytoplasmic tail region of influenza virus hemagglutinin in formation and growth of fusion pores. Virology 235:118–128. doi: 10.1006/viro.1997.8686. [DOI] [PubMed] [Google Scholar]
- 44.Zurcher T, Luo G, Palese P. 1994. Mutations at palmitylation sites of the influenza virus hemagglutinin affect virus formation. J Virol 68:5748–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neumann G, Fujii K, Kino Y, Kawaoka Y. 2005. An improved reverse genetics system for influenza A virus generation and its implications for vaccine production. Proc Natl Acad Sci U S A 102:16825–16829. doi: 10.1073/pnas.0505587102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canham PB. 1970. The minimum energy of bending as a possible explanation of the biconcave shape of the human red blood cell. J Theor Biol 26:61–81. doi: 10.1016/S0022-5193(70)80032-7. [DOI] [PubMed] [Google Scholar]
- 47.Helfrich W. 1973. Elastic properties of lipid bilayers: theory and possible experiments. Z Naturforsch C 28:693–703. doi: 10.1515/znc-1973-11-1209. [DOI] [PubMed] [Google Scholar]
- 48.Rand RP, Fuller NL, Gruner SM, Parsegian VA. 1990. Membrane curvature, lipid segregation, and structural transitions for phospholipids under dual-solvent stress. Biochemistry 29:76–87. doi: 10.1021/bi00453a010. [DOI] [PubMed] [Google Scholar]
- 49.Calder LJ, Rosenthal PB. 2016. Cryomicroscopy provides structural snapshots of influenza virus membrane fusion. Nat Struct Mol Biol 23:853–858. doi: 10.1038/nsmb.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gui L, Ebner JL, Mileant A, Williams JA, Lee KK. 2016. Visualization and sequencing of membrane remodeling leading to influenza virus fusion. J Virol 90:6948–6962. doi: 10.1128/JVI.00240-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee KK. 2010. Architecture of a nascent viral fusion pore. EMBO J 29:1299–1311. doi: 10.1038/emboj.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mineev KS, Lyukmanova EN, Krabben L, Serebryakova MV, Shulepko MA, Arseniev AS, Kordyukova LV, Veit M. 2013. Structural investigation of influenza virus hemagglutinin membrane-anchoring peptide. Protein Eng Des Sel 26:547–552. doi: 10.1093/protein/gzt034. [DOI] [PubMed] [Google Scholar]
- 53.Lakkaraju AK, Abrami L, Lemmin T, Blaskovic S, Kunz B, Kihara A, Dal Peraro M, van der Goot FG. 2012. Palmitoylated calnexin is a key component of the ribosome-translocon complex. EMBO J 31:1823–1835. doi: 10.1038/emboj.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerl MJ, Sampaio JL, Urban S, Kalvodova L, Verbavatz JM, Binnington B, Lindemann D, Lingwood CA, Shevchenko A, Schroeder C, Simons K. 2012. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J Cell Biol 196:213–221. doi: 10.1083/jcb.201108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Epand RM. 2003. Fusion peptides and the mechanism of viral fusion. Biochim Biophys Acta 1614:116–121. doi: 10.1016/S0005-2736(03)00169-X. [DOI] [PubMed] [Google Scholar]
- 56.Ryham RJ, Ward MA, Cohen FS. 2013. Teardrop shapes minimize bending energy of fusion pores connecting planar bilayers. Phys Rev E Stat Nonlin Soft Matter Phys 88:062701. doi: 10.1103/PhysRevE.88.062701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chizmadzhev YA, Cohen FS, Shcherbakov A, Zimmerberg J. 1995. Membrane mechanics can account for fusion pore dilation in stages. Biophys J 69:2489–2500. doi: 10.1016/S0006-3495(95)80119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 59.Yewdell JW, Frank E, Gerhard W. 1981. Expression of influenza A virus internal antigens on the surface of infected P815 cells. J Immunol 126:1814–1819. [PubMed] [Google Scholar]
- 60.Ellens H, Doxsey S, Glenn JS, White JM. 1989. Delivery of macromolecules into cells expressing a viral membrane fusion protein. Methods Cell Biol 31:155–178. doi: 10.1016/S0091-679X(08)61607-6. [DOI] [PubMed] [Google Scholar]
- 61.Kremer JR, Mastronarde DN, McIntosh JR. 1996. Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 62.Jo S, Kim T, Iyer VG, Im W. 2008. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 63.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. 2009. CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. 2010. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: an N · log(N) method for Ewald sums in large systems. J Chem Phys 98:10089. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 67.Andersen HC. 1983. Rattle: a velocity version of the shake algorithm for molecular-dynamics calculations. J Comput Phys 52:24–34. doi: 10.1016/0021-9991(83)90014-1. [DOI] [Google Scholar]
- 68.Feller SE, Zhang YH, Pastor RW, Brooks BR. 1995. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys 103:4613. doi: 10.1063/1.470648. [DOI] [Google Scholar]
- 69.Szleifer I, Kramer D, Benshaul A, Gelbart WM, Safran SA. 1990. Molecular theory of curvature elasticity in surfactant films. J Chem Phys 92:6800–6817. doi: 10.1063/1.458267. [DOI] [Google Scholar]
- 70.Bouvrais H, Meleard P, Pott T, Jensen KJ, Brask J, Ipsen JH. 2008. Softening of POPC membranes by magainin. Biophys Chem 137:7–12. doi: 10.1016/j.bpc.2008.06.004. [DOI] [PubMed] [Google Scholar]