

ABSTRACT
The initial goal of this study was to reexamine the requirement of UL21 for herpes simplex virus 1 (HSV-1) replication. Previous studies suggested that UL21 is dispensable for replication in cell cultures, but a recent report on HSV-2 challenges those findings. As was done for the HSV-2 study, a UL21-null virus was made and propagated on complementing cells to discourage selection of compensating mutations. This HSV-1 mutant was able to replicate in noncomplementing cells, even at a low multiplicity of infection (MOI), though a reduction in titer was observed. Also, increased proportions of empty capsids were observed in the cytoplasm, suggesting a role for UL21 in preventing their exit from the nucleus. Surprisingly, passage of the null mutant resulted in rapid outgrowth of syncytial (Syn) variants. This was unexpected because UL21 has been shown to be required for the Syn phenotype. However, earlier experiments made use of only the A855V syncytial mutant of glycoprotein B (gB), and the Syn phenotype can also be produced by substitutions in glycoprotein K (gK), UL20, and UL24. Sequencing of the syncytial variants revealed mutations in the gK locus, but UL21 was shown to be dispensable for UL20Syn and UL24Syn. To test whether UL21 is needed only for the A855V mutant, additional gBSyn derivatives were examined in the context of the null virus, and all produced lytic rather than syncytial sites of infection. Thus, UL21 is required only for the gBSyn phenotype. This is the first example of a differential requirement for a viral protein across the four syn loci.
IMPORTANCE UL21 is conserved among alphaherpesviruses, but its role is poorly understood. This study shows that HSV-1 can replicate without UL21, although the virus titers are greatly reduced. The null virus had greater proportions of empty (DNA-less) capsids in the cytoplasm of infected cells, suggesting that UL21 may play a role in retaining them in the nucleus. This is consistent with reports showing UL21 to be capsid associated and localized to the nuclei of infected cells. UL21 also appears to be needed for viral membrane activities. It was found to be required for virus-mediated cell fusion, but only for mutants that harbor syncytial mutations in gB (not variants of gK, UL20, or UL24). The machinery needed for syncytial formation is similar to that needed for direct spread of the virus through cell junctions, and these studies show that UL21 is required for cell-to-cell spread even in the absence of syncytial mutations.
KEYWORDS: glycoproteins, HSV-1, syncytia
INTRODUCTION
Herpesviruses are large, double-stranded DNA viruses that cause diseases ranging from relatively benign cold sores to life-threatening conditions, such as encephalitis. Their genomes are encased within a proteinaceous capsid, which is further surrounded by a lipid envelope that is embedded with a multitude of virally encoded membrane proteins. The region between the envelope and the capsid contains many additional proteins, collectively known as the tegument, and while the functions of many of these are undefined, they are in a prime location to interact with the cytoplasmic tails of the envelope proteins, one another, or the surface of the capsid (reviewed in reference 1). Our laboratory has been investigating the tegument proteins UL11, UL16, and UL21 of herpes simplex virus 1 (HSV-1), all of which come together to form a complex on the cytoplasmic tail of glycoprotein E (gE) to control its function (2). The focus of the experiments described here was UL21.
Homologs of UL21 are found within all the alphaherpesviruses, but not very much is known about their function (3, 4). Previous studies have shown that UL21 binds directly to UL16 (5, 6), and this interaction induces UL16 to bind to UL11 (2). It has also been shown that UL21 is associated with capsids and may influence genome packaging (7–10). Another study has shown that the protein is nucleotidylated (11). UL21 has been suggested to have two functional domains (6), and the structures of these have been solved by X-ray crystallography (4, 12). The N-terminal domain has a novel “sail-like” structure, while the C-terminal domain has an α-helical structure that resembles a dragonfly and can bind to RNA (4, 12). Unfortunately, these structures are unique and offer little insight into the function of UL21.
To gain insight into the function of UL21, several groups of investigators have made and characterized null mutants. Most of these studies used HSV-1 and porcine pseudorabies virus (PRV). The results suggested that UL21 is not essential, although the mutant viruses did have replication defects and produced small plaques (5, 7, 8, 13–16). For HSV-1, a delay in expression of certain viral transcripts has been observed, particularly for immediate early genes (15), and in some studies, the growth defect can be overcome by increasing the amount of virus used to start the infection (16). In the case of PRV, the absence of UL21 is associated with DNA packaging defects as well as with attenuated virulence in mice (5, 7, 8, 14).
While all previous studies with HSV-1 and PRV have suggested UL21 to be dispensable for replication in cell culture, a recent study argues that this tegument protein is essential for HSV-2 (3). This is surprising, because the UL21 proteins of HSV-1 and HSV-2 are 84% identical and 90% similar. The HSV-2 deletion mutant was made in Escherichia coli by “recombineering” a copy of the viral genome carried in a bacterial artificial chromosome (BAC), and the mutant was propagated in complementing cells prior to elucidation of its phenotype in noncomplementing cells. In contrast, complementing cells were not used to propagate the HSV-1 mutants, regardless of whether they were produced by homologous recombination in mammalian cells (13) or by recombineering (15). Thus, it was possible that the previous studies of HSV-1 had selected variants that could replicate in the absence of UL21. In the experiments described here, BAC recombineering and a complementing cell line were used to make and characterize a deletion mutant of HSV-1. The results definitively show that UL21 is not essential for HSV-1 replication in cell culture.
Unexpectedly, the UL21-null mutant was found to have a propensity for promoting the outgrowth of syncytial (Syn) variants within very few passages in noncomplementing cells. Syncytial viruses have a selective advantage because they are capable of spreading rapidly by fusing infected cells with adjacent uninfected cells. The responsible mutations are known to arise in any of four syn genes, namely, those that encode the viral fusogen gB, the fusion modulatory membrane proteins gK and UL20, and the highly basic, nucleus-associated protein UL24 (reviewed in reference 17). The locations of the numerous syn mutations that arose in our studies were mapped, and detailed investigations revealed that UL21 is required for only one of the four syn loci.
RESULTS
Generation of a UL21 deletion virus.
Mutants lacking UL21 expression have been made for HSV-1 and PRV both by recombination in mammalian cells, which requires several rounds of plaque purification (7, 8, 13, 14, 16), and, more recently, by recombineering methods for E. coli that utilize BACs carrying the viral genome (3, 15). However, unlike what was done for HSV-2 (3), all these mutants were propagated without the use of complementing cell lines to prevent the selection of compensating mutations. In this study, a UL21 deletion virus named ΔUL21 was made with the KOS genome maintained in a BAC. We used a two-step BAC recombineering protocol (18) that left only the stop codon (TAA) in place, which was verified by DNA sequencing. This deletion is not expected to interfere with expression of the upstream UL20 or downstream UL22 (gH) gene. While available KOS sequences are not annotated with regulatory elements, the annotated strain 17 sequence (NCBI accession no. NC_001806.2) does not predict any regulatory elements of surrounding genes to be located within the UL21 coding sequence. The UL20 locus and surrounding regions were subsequently amplified, sequenced, and determined to be of the wild type (WT). A repaired virus in which the removed sequences were restored to the original locus was also made and is referred to as ΔUL21Rep.
To produce a virus stock, ΔUL21 BAC DNA was transfected into Vero cells, and while infectious virus was produced following transfection, the resulting virus titers were quite low. With the hope of identifying a UL21-complementing cell line, we infected G5 cells, which are a derivative of Vero cells that contain a segment of the viral genome spanning UL16 to UL21 (19). This cell line has been useful for complementation of the capsid proteins VP5 (UL19) and VP23 (UL18) (19), the tegument protein UL16 (20), and the membrane protein UL20 (21). Since the UL21 gene is at one end of the viral fragment present in G5 cells, we needed to make sure that it could be expressed. The UL21 protein was indeed detected when G5 cells were infected with the ΔUL21 virus (Fig. 1), and similar to the case for UL16 (20), this protein was present in low quantity and was thus immunoprecipitated prior to Western blotting to enhance detection.
FIG 1.
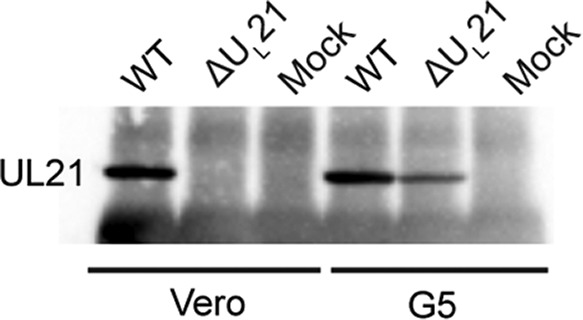
Infected G5 cells express UL21. Confluent monolayers of Vero or G5 cells were either infected with wild-type or ΔUL21 virus or not infected (mock). At 18 h postinfection, cells were lysed, and proteins were immunoprecipitated with an antibody against UL21. The proteins were then separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with an antibody against UL21.
Replication of ΔUL21 virus.
Having identified a putative complementing cell line, we transfected the cells with ΔUL21 BAC DNA to produce a virus stock for subsequent experiments while reducing selective pressure for variants that can grow without UL21. The chance of recombination with the cellular copy of UL21 to create a wild-type virus was expected to be low, and two lines of evidence confirmed this to be the case. First, the resulting G5-made stock of ΔUL21 virus produced uniformly small plaques on Vero cells (Fig. 2A and B). These were 1/3 the size of the wild-type plaques, whereas there was no difference on complementing cells. Second, the G5-made stock was used to infect fresh G5 cells, and the virions were harvested 48 h later, thereby providing an extended opportunity for recombination. Viral DNAs were extracted and analyzed by a PCR using primers flanking the UL21 locus. A single product was produced from the ΔUL21 template, and it was shorter than that from the wild type, as expected (data not shown).
FIG 2.

UL21 is not essential for HSV-1 replication but is needed for cell-to-cell spread. (A) Plaques of the wild-type or UL21-null mutant virus were developed on Vero or G5 cells, and representative images are shown. The areas of 50 plaques per virus type were measured using ImageJ, and the average ± standard deviation is indicated below each image, with the mean value for the wild-type sample on either cell type set to 100%. (B) Individual plaque measurements are shown. The data were graphed using a logarithmic y axis to allow for better visualization of the distribution of data points. Each circle represents an individual area measurement, and error bars show means ± standard errors of the means (SEM). (C and D) Confluent monolayers of Vero or G5 cells were infected at either a low (0.1) (C) or high (10.0) (D) MOI. At approximately 6-h intervals postinfection, total virus (cells and medium) was harvested and titrated on Vero cells. The experiment was done in duplicate, and average values are shown. (E) Vero cells seeded onto glass coverslips were infected with the WT or ΔUL21 virus at an MOI of 0.001 in the presence (+IgG) or absence (−IgG) of 5 mg/ml of human IgG to neutralize any free virus released into the medium. At 18 or 30 h postinfection (hpi), cells were fixed with paraformaldehyde, and foci of infection were immunostained with an antibody against the major capsid protein VP5 (red). Nuclei were stained with DAPI (blue). A representative image from each infection is shown. (F) The sizes of 20 plaques from the images used for the experiment described for panel E were measured. For ease of comparison, the resulting plaque area measurements were normalized, with the mean value for plaque size resulting from wild-type infection at 18 h postinfection without IgG overlay (WT, 18 hpi, −IgG) set to 1.
The ability of the deletion virus to produce plaques on Vero cells suggested that UL21 is not needed for HSV-1, but it also provided evidence that the genes surrounding the UL21 locus are functional. In particular, disruptions in the expression of any of the upstream, essential genes UL15 (22), UL17 (23), UL18 (19), UL19 (19), and UL20 (24) are known to render the virus unable to make plaques on Vero cells. Similarly, disrupted expression of the downstream locus, UL22, which encodes an essential entry glycoprotein (gH), would also render the ΔUL21 virus noninfectious on noncomplementing cells (25).
To more thoroughly assess whether UL21 is necessary for HSV-1 in cell culture, the replication kinetics of the null virus were examined. For this purpose, the G5-grown virus was used to infect G5 or Vero cells at a low (0.1) multiplicity of infection (MOI). Total virus was collected at approximately 6-h intervals following infection, and the titers of infectious virus were measured with plaque assays on Vero cells. As the growth curve clearly shows (Fig. 2C), the deletion virus was able to replicate on the noncomplementing cell line (red line), indicating that UL21 is not essential for replication of HSV-1, although there was a clear reduction in titer. In contrast, the repaired virus replicated like the wild type on Vero cells, suggesting that there are no other mutations that account for the defect. One-way analysis of variance (ANOVA) comparisons with the growth of the wild-type virus on Vero cells as the control condition revealed a significant difference only for the deletion virus grown on Vero cells. All other curves showed nonsignificant variations.
Although the deletion virus was clearly capable of replicating in the absence of UL21, its growth lagged behind that of the wild-type and repaired viruses. On examining the defect at 36 h postinfection (approximately the peak for ΔUL21 virus growth in Vero cells), the deletion virus had an approximately 2-log reduction in titer compared to that of the WT (Fig. 2C). This replication defect was less severe, but was not fully overcome, when an MOI of 10 was used (Fig. 2D). Moreover, the rates of increase for the mutant at the early time points were obviously different, with a slower rise when the MOI was low. These data suggest two distinct roles for UL21. First, it is needed for the production of infectious virions, since use of an MOI in which virtually every Vero cell would be infected still resulted in titers lower than those of the wild type. Second, the trend toward lower titers when fewer cells were infected (low MOI) suggested that the deletion virus may have a defect in cell-to-cell spread.
Analysis of cell-to-cell spread.
UL21 is part of a complex on the cytoplasmic tail of gE (2), which is a known mediator of cell-to-cell spread (26). To test the hypothesis that UL21 is needed for this mode of spreading, we infected Vero cells at a very low MOI (0.001) and added neutralizing serum to the culture medium. This limits cell-free spread, and plaques can form only when viruses spread directly from an infected cell to an uninfected cell. To visualize and measure foci of infection, we used an immunofluorescence-based approach to stain the major capsid protein VP5 and used DAPI (4′,6-diamidino-2-phenylindole) to visualize the nuclei of all cells. Preliminary experiments showed that 5 mg/ml of pooled human IgG mixed with the virus prior to infection resulted in no infected cells, even after incubation of the cultures for 2 days (data not shown).
To assay spreading defects, cells were infected for an hour and then washed, and neutralizing serum was added to the cultures. Controls were identical but received no IgG. The cultures were stained and analyzed after 18 and 30 h of incubation (Fig. 2E and F). At the earlier time point, the wild-type virus produced smaller plaques in the presence of neutralizing antibody than in its absence, as expected. Moreover, distal foci with single infected cells could be seen in the absence of IgG (an example is shown in the top left panel of Fig. 2E), but these sites of cell-free spread were not observed in the presence of neutralizing serum. By the later time point, cell-to-cell spread had enabled the wild-type foci to expand despite the presence of the neutralizing antibody, and their sizes had become indistinguishable from those of foci produced without IgG present (Fig. 2F).
For the ΔUL21 virus, the sites of infection at the early time point were the same size as those of the wild type when it was limited by neutralizing IgG. However, while the mutant foci did increase in size over time in the absence of neutralizing antibody, their expansion did not keep up with that of the wild type. Thus, by 30 h postinfection, their foci were only 1/8 the size of the wild-type foci. When a methylcellulose overlay was used to keep cell-free virions concentrated near the cells in the monolayer during a 3-day plaque assay (Fig. 2A and B), the progeny ΔUL21 virus was better able to infect nearby cells, and as noted earlier, the plaque size was 1/3 that of the wild type. However, in the cell-to-cell spread assay, neutralizing antibodies prevented the foci from increasing in size (Fig. 2F), even after 40 h (not shown), although on G5 cells the mutant behaved like the wild type (not shown). The continuing expansion of the wild-type foci in the presence of IgG enabled them to become 20 times larger than those of the mutant at 30 h. Thus, it seems clear that UL21 is needed for the mechanism of cell-to-cell spread.
Electron microscopy of cells infected with ΔUL21 virus.
With hopes of gaining insight into why the ΔUL21 virus produces fewer infectious virions, we used electron microscopy to examine infected cells. We were motivated by previous reports on PRV, some of which showed DNA packaging aberrations when UL21 was absent (7, 8), although others did not observe any ultrastructural changes (5). For HSV-1, there are no reports of DNA packaging defects for UL21 deletion viruses, but since others did not use complementing cell lines to produce their mutant viruses, we decided to examine infected Vero cells and G5 cells by transmission electron microscopy. Cells were infected at an MOI of 10 to ensure that virtually all would be producing virions, and they were analyzed 24 h later.
For the wild-type virus grown on Vero cells (Fig. 3A and B) or G5 cells (data not shown), particles at various maturation states were seen, with empty and DNA-filled capsids detected in the nucleus and the cytoplasm. Enveloped capsids were also detected in both the cytoplasm and the nucleus. The deletion virus grown on Vero cells exhibited a similar range of maturation states (Fig. 3C to E), but there were noticeably fewer particles in the nucleus, cytoplasm, and extracellular spaces, and there appeared to be a greater number of empty capsids. Growth in G5 cells appeared to compensate for this (Fig. 3F). To test these initial impressions, many more sections were examined, and the total numbers of DNA-containing and empty capsids present anywhere in the cell (cytoplasm or nucleus) were counted. Because ΔUL21 virus-infected cells produced fewer particles than the wild-type virus-infected controls, it was necessary to examine twice as many sections in order to obtain a number of capsids equal to that for the wild type (Table 1). The results of this survey revealed that 17.8% of the capsids produced by the WT were empty, compared to 31.4% for the mutant, which represents a statistically significant difference (Fig. 3G). As expected, replication of the deletion virus on G5 cells reduced the number of empty capsids to wild-type levels (Table 1 and Fig. 3G).
FIG 3.

Morphology of capsids produced by the UL21 deletion virus. (A to F) Vero or G5 cells were infected at an MOI of 10 with either wild-type or ΔUL21 virus. The samples were fixed and processed for electron microscopy at 24 h postinfection, and representative images are shown. The black arrowheads (A, C, and F) mark mature extracellular particles. The red asterisk (A) marks a rare enveloped extracellular empty capsid. The black arrows (B, D, and E) indicate complete, DNA-filled capsids, whereas the red arrows (B, D, and E) denote empty capsids. The green arrowhead (C) marks a partially wrapped capsid in the cytoplasm, and the white arrowheads (A and C) show wrapped capsids in the cytoplasm. Where applicable, the nucleus (Nuc) is indicated. (G) The percentage of empty capsids within each total capsid population, regardless of subcellular position, was plotted for the wild-type and deletion viruses on Vero or G5 cells, and the error bars indicate means and SEM. One-way ANOVA revealed significant differences in the percentages of empty capsids among the groups, and the relevant P values (from unpaired t tests) are shown. (H to J) For analysis of the subcellular locations of empty capsids produced by the wild-type and mutant viruses, a subset of 20 independent sections, all having the nuclei and cytoplasm in view, were assessed as described for panel G and are explained in the text.
TABLE 1.
Summary of sections used for quantitation of empty capsidsa
| Virus strain (cell line) | No. of sections | Total no. of capsids | No. of empty capsids | % empty capsids |
|---|---|---|---|---|
| WT (Vero) | 31 | 695 | 124 | 17.8 |
| ΔUL21 (Vero) | 58 | 692 | 217 | 31.3 |
| WT (G5) | 17 | 240 | 36 | 15 |
| ΔUL21 (G5) | 24 | 252 | 35 | 13.8 |
Total numbers of capsids, regardless of their positions within cells.
For a positional analysis of the empty capsids, 20 independent sections of WT- or ΔUL21 virus-infected Vero cells were analyzed that had both the nucleus and the cytoplasm within view (Table 2). Ignoring their DNA content, the capsids were similarly distributed, with approximately 2/3 of them in the nucleus and 1/3 in the cytoplasm, with no significant differences for the wild type and the mutant (Fig. 3H). Ignoring their cellular positions, there were about twice as many empty capsids for the mutant in these 20 sections (Table 2 and Fig. 3I), similar to what was found in the previous analysis of many more sections and equal numbers of capsids (Fig. 3G), indicating that we had a representative sampling. Next, the nucleus and cytoplasm were assessed separately for the proportions of empty capsids. One-way ANOVA comparisons revealed a significant increase (from ∼20% to ∼60%) in the proportion of empty capsids in the cytoplasm for the ΔUL21 virus relative to the wild-type control, but no significant difference was observed for the nucleus (Fig. 3J). Growth of the mutant on G5 cells restored the proportion of empty capsids in the cytoplasm back to wild-type levels (data not shown). It is not clear what UL21 does to limit the appearance of empty capsids in the cytoplasm (see Discussion).
TABLE 2.
Summary of sections used for positional analysis of empty capsidsa
| Virus strain (cell line) | Total no. of capsids | No. of empty capsids | % empty capsids |
|---|---|---|---|
| WT (Vero) | 430 | 83 | 19.3 |
| ΔUL21 (Vero) | 229 | 67 | 29.3 |
Twenty sections of cells with both the nucleus and the cytoplasm in view were selected for each virus strain.
Propensity of ΔUL21 virus to give rise to syncytial variants.
In the course of this study, we noticed that very prominent, large plaques occasionally appeared among the small plaques produced by the ΔUL21 virus (Fig. 4A). Microscopy revealed that these were syncytia and not the result of several smaller plaques coalescing. Because of our interest in the mechanism of syncytium formation, several distinct variants were purified for further characterization. For this purpose, ΔUL21 BAC DNA was transfected into several cultures of G5 cells so that independently arising syncytia could be identified. The viruses produced from these transfections were used to infect Vero cells, and the progeny from those cells were then used to infect fresh monolayers of Vero cells. Virus propagation continued until a syncytial variant was noticed, typically within one or two passages, at which point the syncytium was picked with a pipet tip, transferred to fresh Vero cells, and subjected to three rounds of plaque purification from a 0.5% agarose overlay. Viral DNAs were extracted from the cloned viruses, and the four known syn loci—those encoding gB, gK, UL20, and UL24—were amplified and sequenced.
FIG 4.

Passage of ΔUL21 virus leads to outgrowth of gKSyn variants. (A) Representative image of very large plaques obtained during an early passage of ΔUL21 virus. The plaque marked with a star was found to be syncytial when viewed under a microscope. (B) The three novel gK variants were recreated in the wild-type KOS.BAC genome, and the DNAs were transfected into Vero cells. The resultant cytopathic effect (syncytium formation) is shown. (C) Two gKSyn alleles were built into the ΔUL21 background, and the DNAs were transfected into Vero cells. The resultant cytopathic effect (syncytium formation) is shown.
It was difficult to predict what we would find. UL21 forms a complex on the cytoplasmic tail of gE, along with UL11 and UL16, and all the members of this complex are required for the fusogenic phenotype of the gB A855V mutant (2). Hence, we did not anticipate finding any gB mutations among the ΔUL21 variants. However, it was recently reported that UL11 is also required to achieve fusion in the gK syncytial background (27), and thus we did not expect to find mutations in its coding sequence either. There have been no studies addressing whether UL21 and its binding partners are required for the UL20 or UL24 Syn phenotype. To our surprise, sequencing revealed that all the syncytial variants encoded substitutions within gK, and there were no changes to gB, UL20, or UL24. As expected, all lacked the UL21 coding sequence as well.
There were four different gK substitutions: A40V (GCG → GUA), T60I (ACC → ATC), L118Q (CTA → CAA), and G167D (GGC → GAC). While the A40V mutant (also known as the syn20 mutant) is a well-described Syn variant (28, 29), the other substitutions have not been reported. To verify that these mutations were solely responsible for the Syn phenotype, we built them into the WT KOS background (i.e., with UL21 present). All the mutants were immediately syncytial upon transfection (Fig. 4B), confirming that they represent novel gK syn alleles. To verify that UL21 is not required, two of the alleles (for the A40V and L118Q mutants) were separately built into the ΔUL21 BAC. The recombinants were immediately and exclusively syncytial upon transfection into Vero cells (Fig. 4C). These results also show that expression of UL20 must not be affected by removal of UL21, because UL20 expression is required for the gK Syn (A40V) phenotype (21).
Requirement of UL21 for the gB Syn phenotype.
To our knowledge, there are no examples of a differential requirement for an accessory protein among the four syn loci. Having found that UL21 is not required for syncytium formation with any of the gK syn alleles, we began to question our claim that this protein is required for the gB Syn phenotype (2). Only a single Syn mutant (A855V) (Fig. 5A) was used in the previous experiments, and complementing (G5) cells were not used. We reported that removal of the UL21 coding sequence resulted in loss of the syncytial phenotype and, instead, that small lytic plaques and occasional mixed sites of limited fusion were visible.
FIG 5.

gB syncytial mutants require UL21 for fusion. (A) Representation of the gB cytoplasmic tail, which is predicted to contain helical domains. The syncytial variants made for this study are shown in red above the amino acid sequence, with an arrow indicating the location of the truncation to make the gBΔ28 mutation. With the exception of the LL871/872AA mutant, all the other mutants had single amino acid substitutions. (Adapted from references 30 and 31 with permission.) (B) The fusion levels in Vero (V) or G5 (G) cells were measured with a quantitative luciferase-based assay. Virus stocks of UL21-null mutants were prepared in G5 cells for this assay. The experiment was done in duplicate, and the results are a representative sample, with triplicate wells for each sample. (C) Results of fusion assay with the gBSyn/ΔUL21 viruses. The mock sample indicates cells that were transfected with reporter plasmids and cocultured but not infected. Background levels of fusion activity obtained for the wild-type virus for each cell type were subtracted from the mean values for all the samples. The experiment was done in duplicate, and the results are a representative sample, with triplicate wells for each sample.
To more thoroughly investigate the UL21 requirement, 11 additional, well-known gB syncytial mutants were made within the context of the ΔUL21 virus (Fig. 5A). Ten of these have amino acid substitutions which cluster in two regions of the cytoplasmic tail (30, 31), either in close proximity to the membrane or at a more central location (near the A855V mutation). An additional mutant (gBΔ28) was made that lacks the last 28 amino acids of gB (32). Virus stocks for each of the 12 mutants were prepared on G5 cells.
To quantitate the fusogenic activity of the mutants, a split-reporter assay was used (33, 34). Briefly, plasmids RLuc8(1–7) and RLuc8(8–11), each encoding a portion of green fluorescent protein (GFP) fused to a portion of Renilla luciferase, were separately transfected into two different cultures of Vero cells. At 12 h posttransfection, the two cell populations were detached and reseeded together at a 1:1 ratio. Infection with a fusogenic virus causes cytoplasmic mixing, which allows the products of each plasmid to associate and become functional. Preliminary experiments showed that no luciferase activity was produced in (i) untransfected Vero or G5 cells, (ii) cells transfected with only RLuc8(1–7), or (iii) cells transfected with RLuc8(1–7) and then infected with a syncytial virus; however, cells cotransfected with both plasmids produced high levels of luciferase activity, which were not increased upon infection with a fusogenic virus (data not shown). Cocultured cells in which one population received RLuc8(1–7) and the other received RLuc8(8–11) did not show luciferase activity unless the cells were infected (Fig. 5B). While all infected samples exhibited some level of fusion, those for the ΔUL21 and wild-type viruses were the lowest and represented background syncytia containing two or three nuclei. As expected, the gK Syn mutant used in this assay exhibited very high levels of fusion activity whether UL21 was present or not (Fig. 5B, compare the bars for gK L118Q/WT [V] and gK L118Q/ΔUL21 [V]). Both of these viruses were much more robust than the gB A855V mutant in Vero cells (∼2.5 times more luciferase activity), which matches expectations based on the sizes of the syncytia observed in cell cultures. All of the viruses used in these experiments exhibited higher-than-expected fusion activities on G5 cells, but it is not clear which one or which combination of viral genes (UL16, UL17, UL18, UL19, UL20, or UL21) present in these complementing cells was responsible. It seems likely that at least one of these viral proteins is normally limiting for the phenotypes of both gB and gK Syn variants.
Having validated the fusion assay, we then analyzed the collection of gB Syn mutants. Consistent with our previous work (2), the gB A855V mutant exhibited a large reduction in fusion activity in the absence of UL21, and similar results were obtained with gBΔ28 (Fig. 5B). In both cases, the remaining fusion activity was equivalent to the background level of the wild-type virus in Vero cells. To make it easier to appreciate the effects of eliminating UL21, the background fusion level for the wild-type virus in Vero or G5 cells was subtracted from the data obtained with the other gB mutants in those two cell types (Fig. 5C). On Vero cells, all the viruses lacking UL21 exhibited virtually no fusion activity above the background, but on G5 cells, all showed far greater levels of fusion than those of the wild type. The percentages of reduction in luciferase activity, as well as the P values obtained by unpaired t tests, are shown in Table 3. From these data, it can be concluded definitively that UL21 is required for fusion in the context of a gB syncytial mutation, whereas it is not necessary for fusion in the background of a gK syncytial mutant.
TABLE 3.
Summary of changes in luciferase activity of viruses with gBSyn cytoplasmic domain substitutions in the absence of UL21
| gBSyn cytoplasmic domain substitution in ΔUL21 background | % reduction in luciferase activity in absence of UL21a | P valueb (Vero vs G5 cells) |
|---|---|---|
| R796C | 81.8 | <0.0001 |
| R800W | 98.8 | 0.0004 |
| P805A | 74.7 | 0.0002 |
| T813I | 83.7 | 0.0048 |
| L817H | 86.7 | <0.0001 |
| S854F | 87.8 | <0.0001 |
| A855V | 81.7 | 0.0034 |
| E857D | 84.6 | <0.0001 |
| R858H | 87.8 | 0.0006 |
| LL871/872AA | 77.3 | <0.0001 |
| A874P | 82.2 | 0.0003 |
| Δ28 | 80.5 | <0.0001 |
Calculated as follows: % reduction = [(luciferase activity on G5 cells − luciferase activity on Vero cells)/luciferase activity on G5 cells] × 100.
Determined by unpaired t test.
UL20 and UL24 syncytial phenotypes do not require UL21.
Syncytial forms of HSV-1 can also arise from mutations in either the polytopic membrane protein UL20 (24) or the highly basic nucleus-associated protein UL24 (35, 36). In the case of both of these proteins, multiple mutations are capable of leading to the syncytial phenotype. To assess the requirement of UL21 for these Syn mutants, we chose two syncytial mutations for each locus to test whether the absence of UL21 leads to a loss of fusion. For UL20, we used the syncytial substitutions R209A (37) and F222A (38), as these have been described as ones that are not associated with loss of viral titer in an otherwise wild-type background. For UL24, the syncytial substitutions were the E99A/K101A combination of changes and the singular point mutation G121A (39). The UL24 G121A mutation, similar to the UL20 syn mutations, causes a purely syncytial phenotype that is not associated with a loss of virus titer (39); however, the combination mutation occurs in a putative functional domain of UL24 (40), so this change is accompanied by attenuated growth in cell culture (39). Each of the aforementioned mutations was built into the ΔUL21 background, and the resulting phenotypes are shown (Fig. 6). Upon transfection, each of these viruses was immediately syncytial, with syncytia of the same size as those for the corresponding substitution in an otherwise wild-type background (i.e., with UL21 present), leading us to conclude that the presence of UL21 is not essential to achieve a fusogenic phenotype. This result highlights the fact that of the four syn loci, only syncytial mutations arising in the gB locus require the presence of the tegument protein UL21 for full fusogenic capacity, whereas syncytial mutations in gK, UL20, and UL24 are fully fusogenic even in viruses lacking UL21.
FIG 6.

Viruses with syncytial mutations in UL20 or UL24 do not require UL21. Syncytial mutations in UL20 or UL24 were built into the ΔUL21 BAC, and the resulting viruses were transfected into Vero cells. Representative images are shown.
DISCUSSION
Discrepancy between HSV-1 and HSV-2 regarding the essentiality of UL21.
Use of a complementing cell line to eliminate the selection of HSV-1 variants that can replicate without UL21 enabled us to show that this tegument protein is not essential for the KOS strain in cell culture. Nevertheless, UL21 is important, because in its absence virus titers are impaired when growth is assessed at both low (0.1) and high (10.0) MOIs. It is difficult to explain why different results were found for HSV-2, in which UL21 seems to be essential (3). The homologs of these viruses have 84% amino acid identity and share other common properties, such as nuclear and cytoplasmic localization and the promotion of timely viral transcription (3, 6, 9, 13, 15).
One noteworthy difference in experimental design is that the HSV-2 study used a cell line of murine origin (L cells), while our studies made use of primate (Vero) cells, and it is possible that the essentiality of UL21 is cell type dependent. Moreover, Vero cells (and Vero-derived G5 cells) are deficient in interferon production (41), whereas L cells are interferon competent (42). Thus, it is possible that UL21 has some function in immune system modulation that renders it essential in the context of an innate immune response and dispensable when antiviral signaling is absent. Arguing against this notion, previous studies of HSV-1-null mutants used a variety of cell types (11, 13), including those capable of producing interferon in response to infection; however, none made use of a complementing cell line.
It is possible that the HSV-2 study represents an “outlier” in the sense that there has been only one such study, using a single strain (3). For HSV-1, multiple strains have been examined (13, 15, 16), with the consistent finding that UL21 is not necessary for replication in cell culture. On the other hand, all of the laboratory strains were passaged extensively in cell culture prior to the construction of the BACs that carry their genomes, and this makes it difficult to know whether mutations have arisen that enable all the HSV-1 strains to be less dependent upon UL21 in cell culture. Thus, the strain of HSV-2 used in the previous study may be more similar to clinical isolates than the HSV-1 laboratory strains. In any case, further investigation of this discrepancy is warranted. For example, it should be possible to use clustered regularly interspaced short palindromic repeat (CRISPR) technology to make UL21-null mutants of clinical isolates to ascertain their dependence on this protein.
Relevance of empty capsids observed in the absence of UL21.
It is clear from both the virus replication and electron microscopy experiments that UL21 is needed for efficient virus production. The null mutant produced lower titers, and there were fewer capsids to be found in the cytoplasm and nuclei of infected cells. Moreover, based on the examination of ∼100 thin sections, it is clear that the ΔUL21 virus produced more empty capsids than the wild type, in accordance with a study of PRV (8); however, for HSV-1, these empty capsids were found primarily in the cytoplasm. Although there are conflicting reports on a role for UL21 in DNA packaging for PRV (5, 7, 8), this step in capsid assembly did not seem to be affected in our ΔUL21 virus, because empty capsids did not accumulate in the nucleus. Indeed, we did not observe an increase of any type of capsid in the nucleus, suggesting that UL21 does not play a role in nuclear egress, in contrast to what was reported for HSV-2 (3).
It was not surprising to find capsid defects with our null virus because UL21 is well known to localize to the nucleus (6, 9, 13) and to be capsid associated (9). The mechanism by which empty capsids arise in the cytoplasm warrants further investigation, and there are at least two possible explanations. (i) UL21 may be part of a regulatory mechanism for retaining capsids in the nucleus until they receive DNA, and its disruption would allow empty capsids to be transported into the cytoplasm. (ii) UL21 may be part of a mechanism that protects DNA-filled capsids, and in its absence, something may trigger the loss of DNA when the capsids arrive in the cytoplasm. Neither of these hypotheses explains why there are fewer capsids overall for the null virus. Perhaps empty capsids produced in the absence of UL21 are degraded in the cytoplasm, and if so, our analysis greatly underestimates the magnitude of the defect. Consistent with this conjecture, empty capsids did not seem to be enveloped in an efficient manner, as they were rare in the visible extracellular virions for both the mutant and the wild type; however, a thorough analysis of envelopment was not done.
Whatever function UL21 provides for capsids, it appears not to require UL16, even though this binding partner is in the nucleus (43, 44), bound to capsids (10, 44), and packaged into virions less efficiently in the absence of UL21 (10; data not shown). That is, studies of a UL16-null mutant did not reveal an accumulation of empty capsids. Instead, capsid envelopment in the cytoplasm was inefficient, and multicapsid virions were observed (20). Neither of these phenotypes was observed with the ΔUL21 virus.
Roles for UL21 in cell-to-cell spread and the Syn phenotype.
Wild-type HSV-1 can infect susceptible cells by either of two mechanisms. In cell-free spread, viruses released from an infected cell attach to an uninfected cell and enter via fusion at the plasma membrane or following endocytosis (1). In cell-to-cell spread, viruses move through lateral junctions to the uninfected cell, bypassing apical release and neutralizing antibodies (45). Here we report that foci of infection do not enlarge for the UL21-null virus when neutralizing antibody is present in the culture medium, strongly suggesting that this protein is needed for cell-to-cell spread, and thus that the ΔUL21 virus relies primarily on cell-free spread.
Syncytial mutations enable a third mechanism of virus spreading. Cells infected with these mutants fuse their plasma membranes with those of surrounding uninfected cells, allowing capsids to rapidly transfer viral DNA to many nuclei at once (17). Viruses with replication defects can gain a spreading advantage by becoming syncytial, and in the case of the ΔUL21 virus, syncytial variants would be strongly favored due to the restrictions imposed by reduced titers and the cell-to-cell spreading defect. Syn mutants are interesting because the viral proteins required for cell fusion are similar to those required for cell-to-cell spread, as shown here for UL21, but there are other well-known examples, such as gE (2, 46, 47).
Syn mutants can arise from particular alterations in the viral proteins gB, gK, UL20, and UL24, and the phenotypes for each of these require the virus entry machinery, composed of gB, gD, gH, and gL (17). However, a number of accessory proteins are also required, and these have been identified mostly by accident or by trial-and-error deletions of various genes within the context of a particular Syn mutant. For example, our studies have shown that gBSyn mutants become lytic in the absence of UL21. The list of accessory proteins is long and also includes the glycoproteins gE, gI, and gM (2, 46), the membrane proteins UL11 (2) and UL45 (48), and the tegument proteins UL16 (2), UL21 (2), and UL51 (49). Unfortunately, most of the accessory proteins have been examined only in the context of one syn gene, and in reading the literature, there seems to be an assumption that an accessory protein needed for one should also be needed for the others. Supporting this idea, a recent study showed that UL11 and gM, known accessory proteins for gBSyn-mediated fusion, are also required for fusion in a gKSyn background (27). Thus, we were initially perplexed when syncytial variants arose upon passage of the ΔUL21 virus, especially since UL21 is known to participate with UL11 in an interaction network. Thus, UL21 is the first described example of an accessory protein that is differentially required across the syncytial backgrounds. Learning that it is required only for gBSyn mutants does not provide insight about its function in cell fusion (or cell-to-cell spread); however, it shows the need for more studies of accessory proteins within multiple syn backgrounds.
In retrospect, it is easy to understand why all of the Syn variants we obtained had alterations in gK. The two largest targets in terms of numbers of previously described, distinct Syn substitutions are gB (50) and gK (28), and only a few Syn variants have been reported for UL20 and UL24. Because UL21 is required for gBSyn variants, the most likely target in the ΔUL21 virus became gK. What is unclear is whether all of the many different gKSyn variants can tolerate the absence of UL21, because there is a functional and physical relationship between gB and gK. While gK is a Syn target (28, 29, 51, 52), it is also an accessory protein for gBSyn-mediated fusion. It directly interacts with gB via residues in its N terminus (53), and deletion of gK in the context of a gBSyn mutant renders the virus lytic (54). Thus, it is conceivable that some gKSyn variants actually depend on UL21.
Our discovery of three novel gKSyn variants suggests that others may be found. This polytopic protein crosses the membrane four times (28, 55). Several Syn substitutions (including A40V, A40T, and D99N) have already been described for extracellular domain I and the short extracellular portion predicted between transmembrane passes two and three (C243S) (55, 56), and two of the novel mutations described here, T60I and L118Q, are located within the first and largest extracellular domain. At least two substitutions (L304P and R310L) in a transmembrane domain have been reported, but no syncytial mutations have been reported to exist within the cytoplasmic domains (55, 56). That has changed with the novel G167D mutant reported here, which is predicted to lie within cytoplasmic domain II of the protein (55).
MATERIALS AND METHODS
Cell lines.
Vero cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 5% bovine calf serum (BCS; HyClone), 5% fetal bovine serum (FBS; HyClone), and penicillin-streptomycin (pen/strep; Gibco). G5 cells (19) were a gift from Prashant Desai (Johns Hopkins University) and were maintained in DMEM containing 5% BCS, 5% FBS, and 1 mg/ml G418 (Gibco). Infected cells were maintained in DMEM containing 2% FBS, 25 mM HEPES, glutamine (0.3 μg/ml), and pen/strep.
Viruses.
The KOS strain of HSV-1 was used in this study, and site-specific mutations were made in a copy of the genome that is contained within a BAC (57). Mutagenesis took place in E. coli as described in detail elsewhere (18). In brief, the method has two steps and utilizes the galactokinase (galK) gene as a selectable marker. In the first step, galK is inserted into a target region of the genome, while the second step removes the galK sequence, leaving the mutation of interest. Clones were screened by HindIII digestion to ensure that gross deletions had not occurred, and putative mutants were validated by PCR amplification of the target gene and sequencing.
The null virus, ΔUL21, lacks the open reading frame but retains the stop codon (TAA). The step I forward primer had the sequence 5′-AGC GTT GCC CTC CAG TTT CTG TTG TCG GTG TTC CCC CAT ACC CAC GCC CAC ATC CAC CGT AGG GGG CCT CTG GGC CGT GTC ACT TCG CCG CCC GCG CCT GTT GAC AAT TAA TCA TCG CGA, while the step I reverse primer had the sequence 5′-GCC GCT CCC CCA GCC CCT CTT TGT TTT CCC TTC CCC CCC GGA GAG GCG TCC ATT GAC ACA CAA GGG TGT GGT AGC GAC ATA CGT TTA TTG GGG TCT GTT ATC AGC ACT GTC CTG CTC CTT. Underlined nucleotides are complementary to the galK sequence. The deletion virus was generated with the step II double-stranded oligonucleotide 5′-CCC CCA TAC CCA CGC CCA CAT CCA CCG TAG GGG GCC TCT GGG CCG TGT CAC GTC GCC GCC CGC GTA ACA GAC CCC AAT AAA CGT ATG TCG CTA CCA CAC CCT TGT GTG TCA ATG GAC GCC TCT CC.
The repaired virus had the missing UL21 sequence inserted back at its normal location. For this purpose, we used the forward primer 5′-CCC CCA TAC CCA CGC CCA CAT CCA CCG TAG GGG GCC TCT GGG CCG TGT CAC GTC GCC GCC CGC GAT GGA GCT TAG CTA CGC CAC CAC CAT GCA C and the reverse primer 5′-GGA GAG GCG TCC ATT GAC ACA CAA GGG TGT GGT AGC GAC ATA CGT TTA TTG GGG TCT GTT ACA CAG ACT GTC CGT GTT GGG AGC GAG C (underlined nucleotides are complementary to UL21). The PCR assay amplified the UL21 sequence along with flanking homology arms to allow for recombination via recombineering in E. coli, and the positive clones were sequence verified.
BAC DNA for transfection was isolated from inoculated E. coli cultures by use of a NucleoBond BAC 100 kit (Macherey-Nagel). For all viruses, the DNA was transfected into Vero or G5 cells by use of Lipofectamine 2000 (Invitrogen), and once the virus had spread through the monolayer, the cells and medium were frozen and thawed three times to prepare what is termed the P0 or transfection stock. These were propagated to make the first-passage or P1 virus stock by inoculating Vero or G5 cells at a low multiplicity of infection (MOI). All viruses were titrated by plaque assay on Vero or G5 cells. Images of cytopathic effects (CPE) were taken with a Nikon Eclipse TS100 inverted microscope or an Olympus IX73 inverted microscope.
Immunoprecipitation of UL21.
UL21 is present at low levels in HSV-1-infected G5 cells, and thus its detection necessitated concentration of the protein by immunoprecipitation. The method described has been utilized previously (20, 58). Cells were infected at an MOI of 5 and lysed with 1% NP-40 at 18 h postinfection. Lysates were clarified by centrifugation before being incubated with rabbit anti-UL21 serum (6) with rocking for 1 h at 4°C. Protein G-agarose beads (Roche) were added, and rocking was continued for another 4 h at 4°C. The beads were washed 3 times with phosphate-buffered saline (PBS) and resuspended in sample buffer prior to protein resolution by SDS-PAGE. The proteins were transferred to a nitrocellulose membrane, and UL21 was visualized with rabbit anti-UL21 serum (6) and True-Blot horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody (eBioscience).
Measurement of virus replication kinetics.
P1 virus stocks were used at both low (0.1) and high (1.0) MOIs to infect confluent monolayers of Vero or G5 cells. The infections were done in 6-well plates and allowed to proceed for 1 h at 37°C, at which point the cells were rinsed with a low-pH citric acid buffer (135 mM NaCl, 10 mM KCl, 40 mM citric acid, pH 3.0, 1% FBS in PBS) for 1 min, followed by neutralization of the low pH with a DMEM rinse. The wells were then overlaid with 1 ml of infection medium per well. Total virus (cells and supernatant) was collected at 0, 6, 12, 18, 24, 36, and 48 h postinfection, and cellular virions were released by 3 cycles of freeze-thawing. Titers were determined by plaque assay on Vero cells. For the plaque assays, confluent monolayers of Vero cells were infected with 400 μl/well of serially diluted virus. The infection was allowed to proceed for 1 h at 37°C, after which the virus was aspirated, and the cells were rinsed with DMEM once before being overlaid with a solution containing 0.5% methylcellulose to limit diffusion of released virions within the culture supernatant. At 3 days postinfection, the methylcellulose was removed, and infected cells were fixed and stained with a solution containing 10% formaldehyde and 0.5% crystal violet. Resultant plaques were counted to determine the titer, and the plates were imaged using a Bio-Rad ChemiDoc MP imaging system with a white light conversion screen. To determine plaque area, the images were analyzed using ImageJ. All images were taken using the same magnification, and a global scale was set using the known diameter of the well. The area of outlined plaques was determined in square millimeters.
Cell-to-cell spreading assay.
Vero or G5 cells were seeded onto glass coverslips in 6-well dishes 24 h prior to infection with WT or ΔUL21 virus at an MOI of 0.001. The infection was allowed to proceed for 1 h at 37°C, after which the cells were rinsed once with culture medium and overlaid with medium containing 5 mg/ml of pooled human IgG (Equitech-Bio) to neutralize any viruses released from the infected cells. At 18 and 30 h postinfection, cells were rinsed once with PBS, fixed with 3.7% paraformaldehyde for 10 min at room temperature, and processed for immunofluorescence assay as described previously (59). Sites of infection were visualized by using a rabbit polyclonal antibody against the major capsid protein, VP5, at a dilution of 1:500 and an Alexa Fluor 568-conjugated goat anti-rabbit antibody (Life Technologies) at a dilution of 1:1,000. Nuclei were stained with DAPI (Molecular Probes). Fluorescence images were taken using an Olympus IX73 inverted microscope, and plaque areas were measured using ImageJ. For measurement, a global scale was set by using a 100-μm scale bar, and the areas of outlined plaques were reported in square micrometers.
Electron microscopy.
Samples were prepared as detailed previously (20). Vero or G5 cells were seeded onto 60-mm Permanox-treated plates (Nalge Nunc International) 24 h prior to infection with WT or ΔUL21 virus at an MOI of 10. At 24 h postinfection, samples were rinsed once with warm PBS and then fixed for 1 h with cold fixation buffer (0.5% [vol/vol] glutaraldehyde, 4.0% [wt/vol] paraformaldehyde in 0.1 M sodium cacodylate). The fixed samples were further processed, stained, and sectioned in the Pennsylvania State College of Medicine Microscopy Imaging Facility, and images were taken using a JEOL JEM-1400 digital capture transmission electron microscope.
Plaque purification of syncytial variants.
The ΔUL21 BAC was first transfected into G5 cells by use of Lipofectamine 2000 in two 6-well plates (for a total of 12 transfected wells), seeded at a density of 0.5 × 106 cells per well. The resultant G5 cell-derived transfection stocks were harvested and maintained in subsequent infections as 12 individual virus stocks that were passaged independently on Vero cells, which were monitored daily for CPE. Cultures in which all the plaques were lytic were harvested, virus stocks were prepared, and these were used to infect a fresh monolayer of Vero cells. If a syncytial variant was identified, it was picked by use of a pipet tip, transferred to a tube containing 2 ml of DMEM, and used to infect a fresh monolayer (100-mm dish) of Vero cells at 37°C with rocking at 15-min intervals. After 1 h, cells were rinsed with DMEM and overlaid with a 0.5% agarose mixture consisting of 1% agarose (SeaPlaque; Lonza) mixed at a 1:1 ratio with 2× infection medium (DMEM with 4% FBS, 50 mM HEPES, 0.6 μg/ml glutamine, and pen/strep). CPE was allowed to develop, and subsequent syncytia were picked and used to infect another Vero cell monolayer. This process was repeated for 3 rounds of purification, until the sites of infection were 100% syncytial, and virus stocks were prepared by freeze-thawing.
Sequencing of viral DNAs.
DNAs were isolated from the ΔUL21 syncytial variants by use of an Invitrogen PureLink viral RNA/DNA kit and 200 to 400 μl of culture supernatant for each virus. To amplify and sequence the syn loci from these viral DNAs, the following primers were used: for UL20, 5′-CGG GCC CGG ACC CAA AAA GGC GGG GG and 5′-GCG TGG GCA TTG GGG CGT AGG CGT; for UL24, 5′-CGC GCG AAC CCA GGG CCA CCA and 5′-TGG CGT GGG GAA ACC CGG GGG C; for the gB tail, 5′-GGG GGG GGG GGG AAT CGG CAC TG and 5′-GCG CGG TCG GCA AGG TGG TGA TG; and for gK, 5′-CTC CTA CAG CTA GTC CCC GTT CGC and 5′-TGG GTT GGT CTT GGT AAC GGG ACG. The genes were amplified by PCR using TaKaRa GXL DNA polymerase (Clontech), and sequencing was done by Eurofins Genomics.
RLuc8 cell fusion assay.
To quantify the fusion of syncytial mutants, a split-reporter assay was used (33, 34). Plasmids RLuc8(1–7) and RLuc8(8–11) (60, 61) were a generous gift of Zene Matsuda (Institute of Medical Science, University of Tokyo), and they encode the two portions of a split GFP-luciferase chimeric protein which becomes functional if both portions are present in the same cell. Briefly, cells were seeded at a density of 0.35 × 106 cells per well in a 6-well plate and transfected with 1 μg of either RLuc8(1–7) or RLuc8(8–11) by use of Lipofectamine 2000 (7 μl/well). Positive-control wells were cotransfected with 1 μg of each plasmid. At 12 h posttransfection, the cells were detached by use of trypsin and cocultured at a 1:1 ratio in white 96-well plates (Greiner). At 12 h postreseeding, the cells were infected at an MOI of 0.3 for 1 h at 37°C, rinsed once with DMEM, and overlaid with 65 μl of medium (DMEM without phenol red, 2% FBS, 25 mM HEPES, 0.3 μg/ml glutamine, and pen/strep) containing the Renilla luciferase substrate EnduRen (Promega). Luminescence readings were taken at 24 h postinfection by using a BioTek Synergy H1M plate reader with the temperature set to 37°C and the gain set to 150.
ACKNOWLEDGMENTS
We thank our coworker Carol B. Wilson for technical help and encouragement, Roland Myers (Pennsylvania State College of Medicine Microscopy Imaging Facility) for his expertise and technical skills, and Anne Stanley and Suja Rani Maddukuri (Macromolecular Synthesis Core Facility, Pennsylvania State University College of Medicine) for generating the oligonucleotide primers used in this study. We also thank Gary Cohen (University of Pennsylvania) and Zene Matsuda (Institute of Medical Science, University of Tokyo) for their generosity in providing the necessary reagents to complete this study.
This work and authors A.S., E.M., P.C., and J.C. were supported by NIH grant R01AI071286 to J.W.W. J.S. and D.Z. were supported in part by NIH training grant T32CA060395.
REFERENCES
- 1.Roizman B, Knipe D, Whitley R. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Han J, Chadha P, Starkey JL, Wills JW. 2012. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc Natl Acad Sci U S A 109:19798–19803. doi: 10.1073/pnas.1212900109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le Sage V, Jung M, Alter JD, Wills EG, Johnston SM, Kawaguchi Y, Baines JD, Banfield BW. 2013. The herpes simplex virus 2 UL21 protein is essential for virus propagation. J Virol 87:5904–5915. doi: 10.1128/JVI.03489-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Metrick CM, Chadha P, Heldwein EE. 2015. The unusual fold of herpes simplex virus 1 UL21, a multifunctional tegument protein. J Virol 89:2979–2984. doi: 10.1128/JVI.03516-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klupp BG, Bottcher S, Granzow H, Kopp M, Mettenleiter TC. 2005. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J Virol 79:1510–1522. doi: 10.1128/JVI.79.3.1510-1522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harper AL, Meckes DG, Marsh J, Ward MD, Yeh PC, Baird NL, Wilson CB, Semmes OJ, Wills JW. 2010. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J Virol 84:2963–2971. doi: 10.1128/JVI.02015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Wind N, Wagenaar F, Pol J, Kimman T, Berns A. 1992. The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J Virol 66:7096–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagenaar F, Pol JM, de Wind N, Kimman TG. 2001. Deletion of the UL21 gene in pseudorabies virus results in the formation of DNA-deprived capsids: an electron microscopy study. Vet Res 32:47–54. doi: 10.1051/vetres:2001108. [DOI] [PubMed] [Google Scholar]
- 9.Takakuwa H, Goshima F, Koshizuka T, Murata T, Daikoku T, Nishiyama Y. 2001. Herpes simplex virus encodes a virion-associated protein which promotes long cellular processes in over-expressing cells. Genes Cells 6:955–966. doi: 10.1046/j.1365-2443.2001.00475.x. [DOI] [PubMed] [Google Scholar]
- 10.Meckes DG, Marsh JA, Wills JW. 2010. Complex mechanisms for the packaging of the UL16 tegument protein into herpes simplex virus. Virology 398:208–213. doi: 10.1016/j.virol.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaho JA, Mitchell C, Roizman B. 1994. An amino acid sequence shared by the herpes simplex virus 1 alpha regulatory proteins 0, 4, 22, and 27 predicts the nucleotidylylation of the UL21, UL31, UL47, and UL49 gene products. J Biol Chem 269:17401–17410. [PubMed] [Google Scholar]
- 12.Metrick CM, Heldwein EE. 2016. Novel structure and unexpected RNA-binding ability of the C-terminal domain of herpes simplex virus 1 tegument protein UL21. J Virol 90:5759–5769. doi: 10.1128/JVI.00475-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baines JD, Koyama AH, Huang T, Roizman B. 1994. The UL21 gene products of herpes simplex virus 1 are dispensable for growth in cultured cells. J Virol 68:2929–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klupp BG, Lomniczi B, Visser N, Fuchs W, Mettenleiter TC. 1995. Mutations affecting the UL21 gene contribute to avirulence of pseudorabies virus vaccine strain Bartha. Virology 212:466–473. doi: 10.1006/viro.1995.1504. [DOI] [PubMed] [Google Scholar]
- 15.Mbong EF, Woodley L, Frost E, Baines JD, Duffy C. 2012. Deletion of UL21 causes a delay in the early stages of the herpes simplex virus 1 replication cycle. J Virol 86:7003–7007. doi: 10.1128/JVI.00411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muto Y, Goshima F, Ushijima Y, Kimura H, Nishiyama Y. 2012. Generation and characterization of UL21-null herpes simplex virus type 1. Front Microbiol 3:394. doi: 10.3389/fmicb.2012.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ambrosini AE, Enquist LW. 2015. Cell-fusion events induced by alpha-herpesviruses. Future Virol 10:185–200. doi: 10.2217/fvl.14.100. [DOI] [Google Scholar]
- 18.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desai P, DeLuca NA, Glorioso JC, Person S. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J Virol 67:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Starkey JL, Han J, Chadha P, Marsh JA, Wills JW. 2014. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J Virol 88:110–119. doi: 10.1128/JVI.02555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster TP, Melancon JM, Baines JD, Kousoulas KG. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J Virol 78:5347–5357. doi: 10.1128/JVI.78.10.5347-5357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baines JD, Cunningham C, Nalwanga D, Davison AJ. 1997. The UL15 gene of herpes simplex virus type 1 contains within its second exon a novel open reading frame that is translated in frame with the UL15 gene product. J Virol 71:2666–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salmon B, Cunningham C, Davison AJ, Harris WJ, Baines JD. 1998. The herpes simplex virus 1 UL17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J Virol 72:3779–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baines JD, Ward PL, Campadelli-Fiume G, Roizman B. 1991. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J Virol 65:6414–6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desai PJ, Schaffer PA, Minson AC. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1—evidence that gH is essential for virion infectivity. J Gen Virol 69:1147–1156. doi: 10.1099/0022-1317-69-6-1147. [DOI] [PubMed] [Google Scholar]
- 26.Dingwell KS, Brunetti CR, Hendricks RL, Tang Q, Tang M, Rainbow AJ, Johnson DC. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J Virol 68:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim IJ, Chouljenko VN, Walker JD, Kousoulas KG. 2013. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J Virol 87:8029–8037. doi: 10.1128/JVI.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Debroy C, Pederson N, Person S. 1985. Nucleotide sequence of a herpes simplex virus type 1 gene that causes cell fusion. Virology 145:36–48. doi: 10.1016/0042-6822(85)90199-0. [DOI] [PubMed] [Google Scholar]
- 29.Bond VC, Person S. 1984. Fine structure physical map locations of alterations that affect cell fusion in herpes simplex virus type 1. Virology 132:368–376. doi: 10.1016/0042-6822(84)90042-4. [DOI] [PubMed] [Google Scholar]
- 30.Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J Virol 86:8171–8184. doi: 10.1128/JVI.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogalin HB, Heldwein EE. 2015. Interplay between the herpes simplex virus 1 gB cytodomain and the gH cytotail during cell-cell fusion. J Virol 89:12262–12272. doi: 10.1128/JVI.02391-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baghian A, Huang L, Newman S, Jayachandra S, Kousoulas KG. 1993. Truncation of the carboxy-terminal 28 amino acids of glycoprotein B specified by herpes simplex virus type 1 mutant amb1511-7 causes extensive cell fusion. J Virol 67:2396–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saw WT, Matsuda Z, Eisenberg RJ, Cohen GH, Atanasiu D. 2015. Using a split luciferase assay (SLA) to measure the kinetics of cell-cell fusion mediated by herpes simplex virus glycoproteins. Methods 90:68–75. doi: 10.1016/j.ymeth.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Atanasiu D, Saw WT, Gallagher JR, Hannah BP, Matsuda Z, Whitbeck JC, Cohen GH, Eisenberg RJ. 2013. Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J Virol 87:11332–11345. doi: 10.1128/JVI.01700-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tognon M, Guandalini R, Romanelli MG, Manservigi R, Trevisani B. 1991. Phenotypic and genotypic characterization of locus Syn 5 in herpes simplex virus 1. Virus Res 18:135–150. doi: 10.1016/0168-1702(91)90014-M. [DOI] [PubMed] [Google Scholar]
- 36.Pearson A, Coen DM. 2002. Identification, localization, and regulation of expression of the UL24 protein of herpes simplex virus type 1. J Virol 76:10821–10828. doi: 10.1128/JVI.76.21.10821-10828.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melancon JM, Foster TP, Kousoulas KG. 2004. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J Virol 78:7329–7343. doi: 10.1128/JVI.78.14.7329-7343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charles AS, Chouljenko VN, Jambunathan N, Subramanian R, Mottram P, Kousoulas KG. 2014. Phenylalanine residues at the carboxyl terminus of the herpes simplex virus 1 UL20 membrane protein regulate cytoplasmic virion envelopment and infectious virus production. J Virol 88:7618–7627. doi: 10.1128/JVI.00657-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bertrand L, Leiva-Torres GA, Hyjazie H, Pearson A. 2010. Conserved residues in the UL24 protein of herpes simplex virus 1 are important for dispersal of the nucleolar protein nucleolin. J Virol 84:109–118. doi: 10.1128/JVI.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knizewski L, Kinch L, Grishin NV, Rychlewski L, Ginalski K. 2006. Human herpesvirus 1 UL24 gene encodes a potential PD-(D/E)XK endonuclease. J Virol 80:2575–2577. doi: 10.1128/JVI.80.5.2575-2577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desmyter J, Melnick JL, Rawls WE. 1968. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J Virol 2:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wagner RR. 1963. The interferons: cellular inhibitors of viral infection. Annu Rev Microbiol 17:285–296. doi: 10.1146/annurev.mi.17.100163.001441. [DOI] [PubMed] [Google Scholar]
- 43.Nalwanga D, Rempel S, Roizman B, Baines JD. 1996. The UL16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 226:236–242. doi: 10.1006/viro.1996.0651. [DOI] [PubMed] [Google Scholar]
- 44.Meckes DG, Wills JW. 2007. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J Virol 81:13028–13036. doi: 10.1128/JVI.01306-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson DC, Huber MT. 2002. Directed egress of animal viruses promotes cell-to-cell spread. J Virol 76:1–8. doi: 10.1128/JVI.76.1.1-8.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis-Poynter N, Bell S, Minson T, Browne H. 1994. Analysis of the contributions of herpes simplex virus type 1 membrane proteins to the induction of cell-cell fusion. J Virol 68:7586–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balan P, Davis-Poynter N, Bell S, Atkinson H, Browne H, Minson T. 1994. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J Gen Virol 75:1245–1258. doi: 10.1099/0022-1317-75-6-1245. [DOI] [PubMed] [Google Scholar]
- 48.Haanes EJ, Nelson CM, Soule CL, Goodman JL. 1994. The UL45 gene product is required for herpes simplex virus type 1 glycoprotein B-induced fusion. J Virol 68:5825–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roller RJ, Haugo AC, Yang K, Baines JD. 2014. The herpes simplex virus 1 UL51 gene product has cell type-specific functions in cell-to-cell spread. J Virol 88:4058–4068. doi: 10.1128/JVI.03707-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gage PJ, Levine M, Glorioso JC. 1993. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J Virol 67:2191–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruyechan WT, Morse LS, Knipe DM, Roizman B. 1979. Molecular genetics of herpes simplex virus. II. Mapping of the major viral glycoproteins and of the genetic loci specifying the social behavior of infected cells. J Virol 29:677–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hutchinson L, Goldsmith K, Snoddy D, Ghosh H, Graham FL, Johnson DC. 1992. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J Virol 66:5603–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chouljenko VN, Iyer AV, Chowdhury S, Kim J, Kousoulas KG. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J Virol 84:8596–8606. doi: 10.1128/JVI.00298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Melancon JM, Luna RE, Foster TP, Kousoulas KG. 2005. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J Virol 79:299–313. doi: 10.1128/JVI.79.1.299-313.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Foster TP, Alvarez X, Kousoulas KG. 2003. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): the UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J Virol 77:499–510. doi: 10.1128/JVI.77.1.499-510.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dolter KE, Ramaswamy R, Holland TC. 1994. Syncytial mutations in the herpes simplex virus type 1 gK (UL53) gene occur in two distinct domains. J Virol 68:8277–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gierasch WW, Zimmerman DL, Ward SL, VanHeyningen TK, Romine JD, Leib DA. 2006. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods 135:197–206. doi: 10.1016/j.jviromet.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 58.Yeh PC, Han J, Chadha P, Meckes DG, Ward MD, Semmes OJ, Wills JW. 2011. Direct and specific binding of the UL16 tegument protein of herpes simplex virus to the cytoplasmic tail of glycoprotein E. J Virol 85:9425–9436. doi: 10.1128/JVI.05178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chadha P, Han J, Starkey JL, Wills JW. 2012. Regulated interaction of tegument proteins UL16 and UL11 from herpes simplex virus. J Virol 86:11886–11898. doi: 10.1128/JVI.01879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813–820. doi: 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 61.Kondo N, Miyauchi K, Meng F, Iwamoto A, Matsuda Z. 2010. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J Biol Chem 285:14681–14688. doi: 10.1074/jbc.M109.067090. [DOI] [PMC free article] [PubMed] [Google Scholar]