

ABSTRACT
There is an urgent need for therapeutic development to combat infections caused by Rift Valley fever virus (RVFV), which causes devastating disease in both humans and animals. In an effort to repurpose drugs for RVFV treatment, our previous studies screened a library of FDA-approved drugs. The most promising candidate identified was the hepatocellular and renal cell carcinoma drug sorafenib. Mechanism-of-action studies indicated that sorafenib targeted a late stage in virus infection and caused a buildup of virions within cells. In addition, small interfering RNA (siRNA) knockdown studies suggested that nonclassical targets of sorafenib are important for the propagation of RVFV. Here we extend our previous findings to identify the mechanism by which sorafenib inhibits the release of RVFV virions from the cell. Confocal microscopy imaging revealed that glycoprotein Gn colocalizes and accumulates within the endoplasmic reticulum (ER) and the transport of Gn from the Golgi complex to the host cell membrane is reduced. Transmission electron microscopy demonstrated that sorafenib caused virions to be present inside large vacuoles inside the cells. p97/valosin-containing protein (VCP), which is involved in membrane remodeling in the secretory pathway and a known target of sorafenib, was found to be important for RVFV egress. Knockdown of VCP resulted in decreased RVFV replication, reduced Gn Golgi complex localization, and increased Gn ER accumulation. The intracellular accumulation of RVFV virions was also observed in cells transfected with siRNA targeting VCP. Collectively, these data indicate that sorafenib causes a disruption in viral egress by targeting VCP and the secretory pathway, resulting in a buildup of virions within dilated ER vesicles.
IMPORTANCE In humans, symptoms of RVFV infection mainly include a self-limiting febrile illness. However, in some cases, infected individuals can also experience hemorrhagic fever, neurological disorders, liver failure, and blindness, which could collectively be lethal. The ability of RVFV to expand geographically outside sub-Saharan Africa is of concern, particularly to the Americas, where native mosquito species are capable of virus transmission. Currently, there are no FDA-approved therapeutics to treat RVFV infection, and thus, there is an urgent need to understand the mechanisms by which the virus hijacks the host cell machinery to replicate. The significance of our research is in identifying the cellular target of sorafenib that inhibits RVFV propagation, so that this information can be used as a tool for the further development of therapeutics used to treat RVFV infection.
KEYWORDS: Rift Valley fever virus, VCP, egress, sorafenib
INTRODUCTION
Rift Valley fever virus (RVFV) is a member of the genus Phlebovirus in the family Bunyaviridae. RVFV is the causative agent of Rift Valley fever (RVF), an arboviral zoonosis that can cause severe disease in humans and livestock. In ruminants, the disease is characterized by a spontaneous abortion rate of 40 to 100%, as well as an increased incidence of mortality among neonates (1). Humans may contract the virus through a mosquito bite or through exposure to infected animal carcasses or body fluid. Human infections are generally asymptomatic or characterized by a mild febrile illness. However, a subset of infected people can progress to severe disease, with symptoms ranging from flu-like symptoms to hepatitis, maculoretinitis, encephalitis, and fatal hemorrhagic fever, which can result in death (2). RVFV has caused outbreaks of RVF throughout Africa and has recently expanded into Madagascar and parts of the Middle East (3). Studies with native mosquitoes in the Americas have demonstrated the potential of virus autochthonous transmission, and thus, the expansion to the Americas remains a concern. In addition, deliberate aerosol dissemination of the virus could enable widespread transmission of the disease and is thus considered a viable threat. Therefore, RVFV is classified as a category A priority pathogen by the National Institute of Allergy and Infectious Diseases and ranked as the third most dangerous animal threat by the National Veterinary Stockpile (4). Currently, there are no licensed antiviral treatments or vaccines to prevent or treat RVFV infection, and the U.S. population remains vulnerable to natural disease outbreaks. Further research into the virulence mechanisms of the virus is necessary to develop safe and effective drugs against this disease.
The RVFV genome is comprised of small (S), medium (M), and large (L) segments, each of which is encapsidated by the nucleoprotein (N). Once encapsidated, the segments are referred to as ribonucleoproteins (RNPs). The L segment encodes the viral RNA-dependent RNA polymerase (RdRp) or L protein (5). The genomic sense S segment encodes the N protein, while the antigenomic S segment encodes a nonstructural protein, NSs, a virulence factor known to modulate the host immune response (6). The M segment encodes the major structural glycoproteins Gn and Gc as well as two nonstructural proteins, NSm1 and NSm2, which are involved in the suppression of cellular apoptosis (7, 8). A characteristic feature of bunyaviruses is that viral particle assembly occurs at the Golgi complex, from which virions have been shown to bud within Golgi vesicles and transport to the host cell plasma membrane for egress (9).
Previous studies conducted by our group demonstrated the utility of an FDA-approved hepatocellular and renal cell carcinoma drug, sorafenib, to inhibit RVFV replication. Studies suggested that sorafenib targets RVFV in at least two stages in the viral life cycle, viral RNA synthesis (viral genome replication and/or viral transcription) and viral egress from the host cell, resulting in an intracellular buildup of virions. Sorafenib was first discovered as a Raf kinase inhibitor. However, our results showed that the loss of Raf does not result in inhibition of RVFV (10), suggesting that an additional protein(s) is targeted by sorafenib in order to decrease viral replication/egress. Here, we investigate the mechanism by which sorafenib inhibits RVFV replication.
RESULTS
Sorafenib treatment causes the inhibition of viral egress.
The process of RVFV virion egress consists of virion packaging and budding into the Golgi complex lumen followed by transport to the cell surface to exit the host cell (11). In order to provide further support for the finding that sorafenib inhibits viral egress, an assay that analyzes the ability of the viral glycoprotein Gn to reach the cell surface at increasing times postinfection was performed (Fig. 1A). This assay has been optimized and used before in the study of RVFV egress (12, 13). All cells were fixed at 12 h postinfection (hpi) and analyzed for the levels of Gn present on the cell surface. Infected cells treated with the solvent control (dimethyl sulfoxide [DMSO]) displayed 67% surface Gn staining, indicating normal viral egress (Fig. 1B and F). In contrast, the Gn staining was significantly reduced to 11% in cells treated with sorafenib immediately following infection (Fig. 1C and F). Similarly, when sorafenib was added to the cells at 8 hpi (Fig. 1D), before the majority of viral egress is thought to occur, the levels of surface Gn were still significantly lower than those for the DMSO-treated control cells at 37% surface Gn staining (Fig. 1F). However, when sorafenib was added at 10 hpi (Fig. 1E), after the viral egress stage had already begun, the treatment was no longer able to inhibit viral Gn trafficking to the cell surface, resulting in a surface staining of 70% (Fig. 1F), which was comparable to that for the DMSO-treated control cells.
FIG 1.

Sorafenib treatment causes the inhibition of viral egress. (A) Schematic of the treatments used for the cells whose images appear in panels B to E. (B to E) Vero cells were infected with MP12 at an MOI of 10 and either treated with DMSO immediately following infection (B), treated with sorafenib immediately following infection (C), treated with sorafenib at 8 h postinfection (D), or treated with sorafenib at 10 h postinfection (E). (F) Cells were fixed, left unpermeabilized, and stained for virus glycoprotein Gn on the cell surface at 12 h postinfection. The percentage of cells that stained positive for Gn expression was determined by high-content quantitative image-based analysis (see Materials and Methods). The means and standard deviations from three biological replicates are plotted. *, P < 0.05.
In order to corroborate these findings, the intracellular distribution and levels of Gn were analyzed. To address this, an experiment similar to that whose results are presented Fig. 1 was conducted (Fig. 2A), but one condition was altered. After cells were fixed at 12 hpi, they were permeabilized before probing for Gn. This allowed the detection of both intracellular and surface viral Gn, providing an indication of whether or not sorafenib affects Gn protein production and/or its distribution. When cells were treated with DMSO (Fig. 2B), total Gn staining appeared punctate and perinuclear, which could indicate its colocalization with the site of budding, the Golgi complex. In addition, quantitation of the signal showed that 71% of total cells displayed Gn staining (Fig. 2F). When cells were treated with sorafenib immediately postinfection, a more diffuse phenotype was observed (Fig. 2C) and what appeared to be a slight drop in the level of Gn staining to 55% was detected; however, this decrease was not significant (P > 0.05) (Fig. 2F). Similar results were obtained when cells were treated before the viral egress stage at 8 hpi (Fig. 2D), with diffuse Gn staining appearing in 75% of cells (Fig. 2F), comparable to the levels in the DMSO-treated control cells. When cells were treated with sorafenib at 12 hpi, after the majority of viral egress had begun (Fig. 2E), 75% of the cells were positive for total Gn, a finding which was again similar to that for the DMSO-treated control cells (Fig. 2F). The same experiment was conducted to determine the total levels of N protein, and no significant change in total protein levels with sorafenib treatment was observed (Fig. 2G). Collectively, these results reaffirm the hypothesis that sorafenib impedes the viral egress stage of the RVFV life cycle, while it produces no significant change in the level of viral protein production at 12 hpi.
FIG 2.

Sorafenib treatment does not affect intracellular Gn protein levels. (A) Schematic of the treatments used for the cells whose images appear in panels B to E. (B to E) Vero cells were infected with MP12 at an MOI of 10 and either treated with DMSO immediately following infection (B), treated with sorafenib immediately following infection (C), treated with sorafenib at 8 h postinfection (D), or treated with sorafenib at 10 h postinfection (E). (F) Cells were fixed, permeabilized, and stained for virus glycoprotein Gn both inside the cell and on the cell surface at 12 h postinfection. The percentage of cells that stained positive for Gn expression was determined by high-content quantitative image-based analysis (see Materials and Methods). The means and standard deviations from three biological replicates are plotted. (G) Cells were fixed, permeabilized, and stained for viral N protein both inside the cell and on the cell surface at 12 h postinfection. The percentage of cells that stained positive for N expression was determined by high-content quantitative image-based analysis (see Materials and Methods). The means and standard deviations from three biological replicates are plotted.
Sorafenib treatment of infected cells generates large intracellular vacuoles containing virus.
Previous work published in the literature has shown that sorafenib causes the fragmentation of organelles within the secretory pathway, which includes the endoplasmic reticulum (ER) and the Golgi complex (14). Since we know that sorafenib causes an intracellular buildup of virions (10), we wanted to obtain a general visualization of infected cells that were treated with sorafenib compared to that of cells treated with DMSO. Images obtained by transmission electron microscopy (TEM) allowed us to view the physical state of the organelles and the localization of the virions under the respective conditions. As expected, when cells were treated with DMSO, we observed several small vesicles containing virus particles, which we could reasonably deduce were Golgi vesicles containing virions being transported toward the plasma membrane to be exocytosed (Fig. 3A). After sorafenib treatment, large intracellular vacuoles containing virions were observed (Fig. 3B). It has been reported in the literature that sorafenib alters critical organelles of the secretory pathway, as exemplified by the fragmentation of the Golgi complex (14) and the appearance of dilated ER lumens, indicative of ER stress (15). Thus, we can hypothesize that the vacuoles observed could be a dilated ER or Golgi complex. Another possibility is that they might be the result of membranes that fuse together and that are unable to further vesiculate. These results indicate that in the presence of sorafenib, RVFV is trapped within these large intracellular vacuoles, which correlates with the observed decrease in viral egress.
FIG 3.

Sorafenib treatment of infected cells generates large intracellular vacuoles containing virus. Vero cells were infected with MP12 at an MOI of 10 and treated with DMSO (A) or sorafenib (B). Samples were prepared and analyzed by transmission electron microscopy. Arrowheads indicate virions.
Sorafenib treatment prevents Gn from colocalizing with the Golgi complex and increases Gn colocalization with the ER.
Since a defect in egress was observed after sorafenib treatment, confocal microscopy was used to determine whether sorafenib affects the colocalization of Gn and potentially affects virus budding into the Golgi complex. Vero (Fig. 4A) and Huh7 (Fig. 5A) cells that were infected and treated with DMSO displayed a normal pattern of Gn distribution, where the Gn signal colocalized with the signal from the Golgi complex marker trans-Golgi network protein 46 (TGN46), indicated by the yellow signal in the merged panels. In contrast, sorafenib treatment caused the cells to display a diffuse Gn staining pattern, with Gn no longer colocalizing with the Golgi complex (Fig. 4B and 5B). Additionally, the morphology of the Golgi complex was altered with sorafenib treatment, displaying a more fragmented appearance, as has been previously observed in studies published in the literature (14). Thus, the aberrance in viral egress caused by sorafenib could be the result of the virions not being retained in the Golgi complex.
FIG 4.
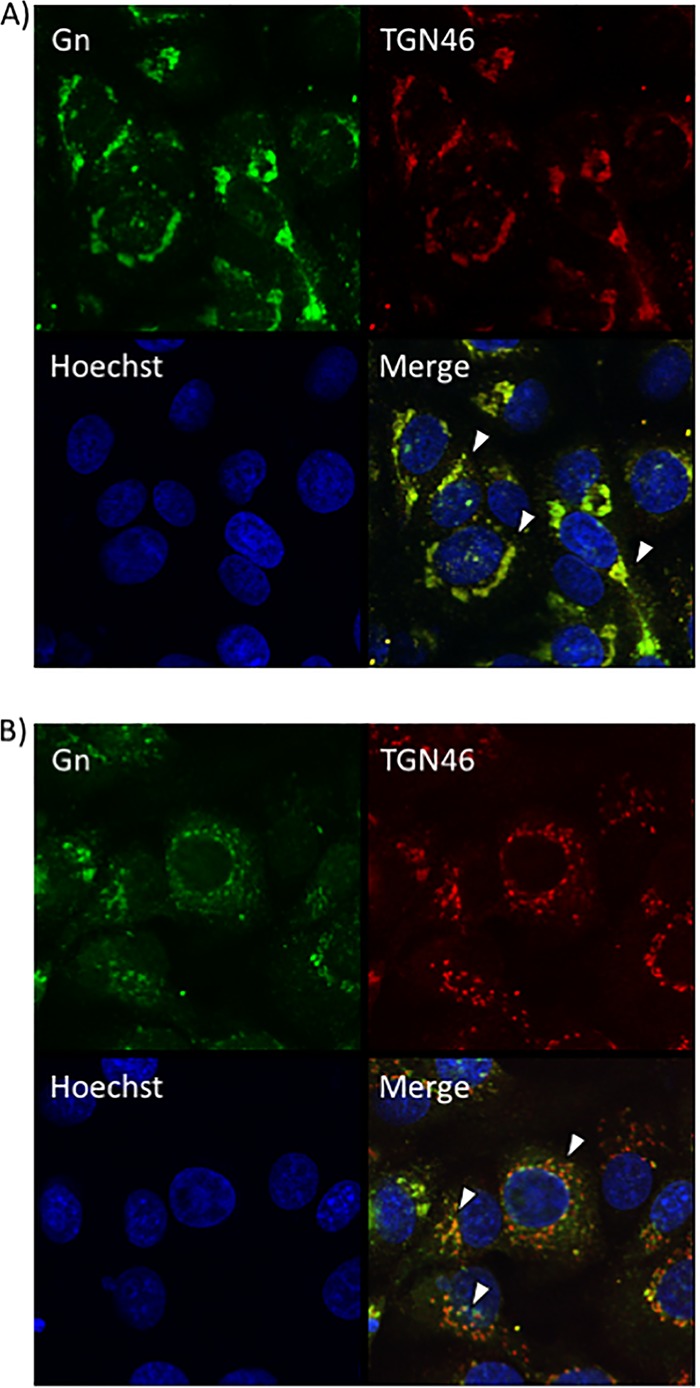
Sorafenib treatment prevents Gn from colocalizing with the Golgi complex in Vero cells. Vero cells were infected with MP12 at an MOI of 10 and either treated with DMSO immediately following infection (A) or treated with sorafenib immediately following infection (B). Cells were fixed, permeabilized, and stained for Gn, trans-Golgi network protein 46 (TGN46), and Hoechst at 12 h postinfection. Arrowheads indicate areas of TGN46 and Gn colocalization (A) or areas that lack TGN46 and Gn colocalization (B).
FIG 5.
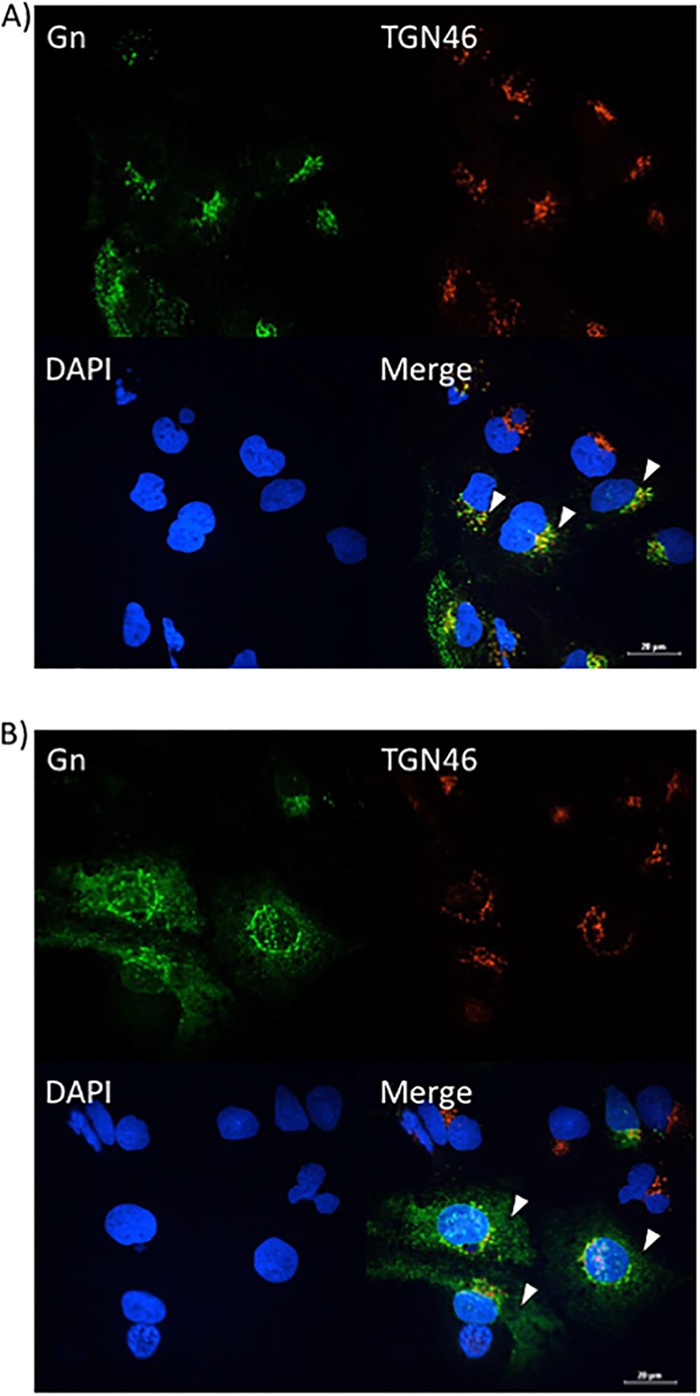
Sorafenib treatment prevents Gn from colocalizing with the Golgi complex in Huh7 cells. Huh7 cells were infected with MP12 at an MOI of 10 and either treated with DMSO immediately following infection (A) or treated with sorafenib immediately following infection (B). Cells were fixed, permeabilized, and stained for Gn, trans-Golgi network protein 46 (TGN46), and Hoechst at 12 h postinfection. Arrowheads indicate areas of TGN46 and Gn colocalization (A) or areas that lack TGN46 and Gn colocalization (B).
Given that sorafenib treatment prevented Gn from colocalizing with the Golgi complex, we hypothesized that it was relocated to another location within the secretory pathway. To test this hypothesis, Gn localization to the ER and/or autophagosomes was analyzed using calnexin and p62 as markers of ER and autophagosomes, respectively. Infected Vero (Fig. 6A) and Huh7 (Fig. 7A) cells treated with DMSO displayed the characteristic perinuclear Gn staining of RVFV-infected cells. There was a low level of colocalization with Gn and the ER marker calnexin (Fig. 6A and 7A), which is not surprising, as Gn is translated on the rough ER. However, sorafenib-treated cells exhibited a far greater level of colocalization with the ER, as indicated by the prevalence of yellow staining compared to that for DMSO-treated control cells (Fig. 6B and 7B). Gn colocalization with p62 was not observed (data not shown), suggesting that Gn does not relocate to autophagosomes following sorafenib treatment. So far, our overall data support the idea that in the presence of sorafenib, the assembly of RVFV virions occurs in the Golgi complex but prevents them from egressing by redirecting them to the ER after assembly, where they remain.
FIG 6.
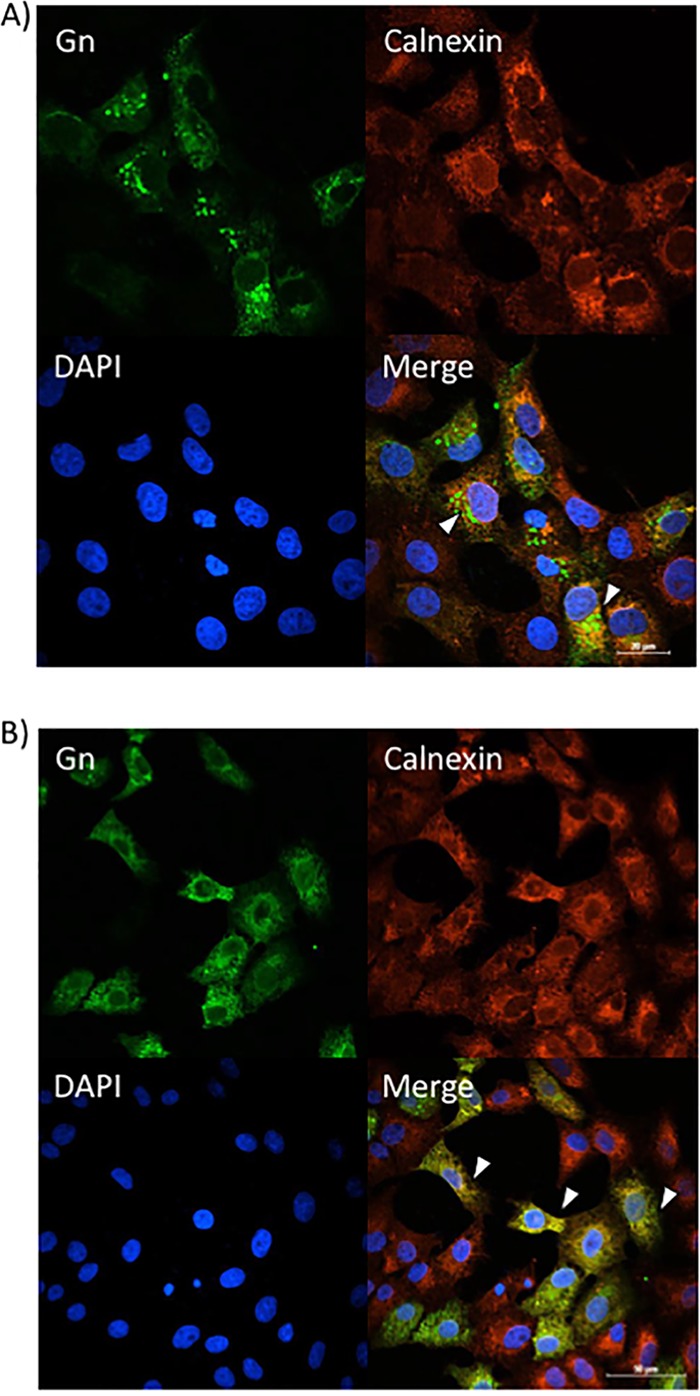
Sorafenib treatment increases Gn colocalization with the ER in Vero cells. Vero cells were infected with MP12 at an MOI of 10 and either treated with DMSO immediately following infection (A) or treated with sorafenib immediately following infection (B). Cells were fixed, permeabilized, and stained for Gn, the ER marker calnexin, and DAPI at 12 h postinfection. Arrowheads indicate areas that lack calnexin and Gn colocalization (A) or areas of calnexin and Gn colocalization (B).
FIG 7.
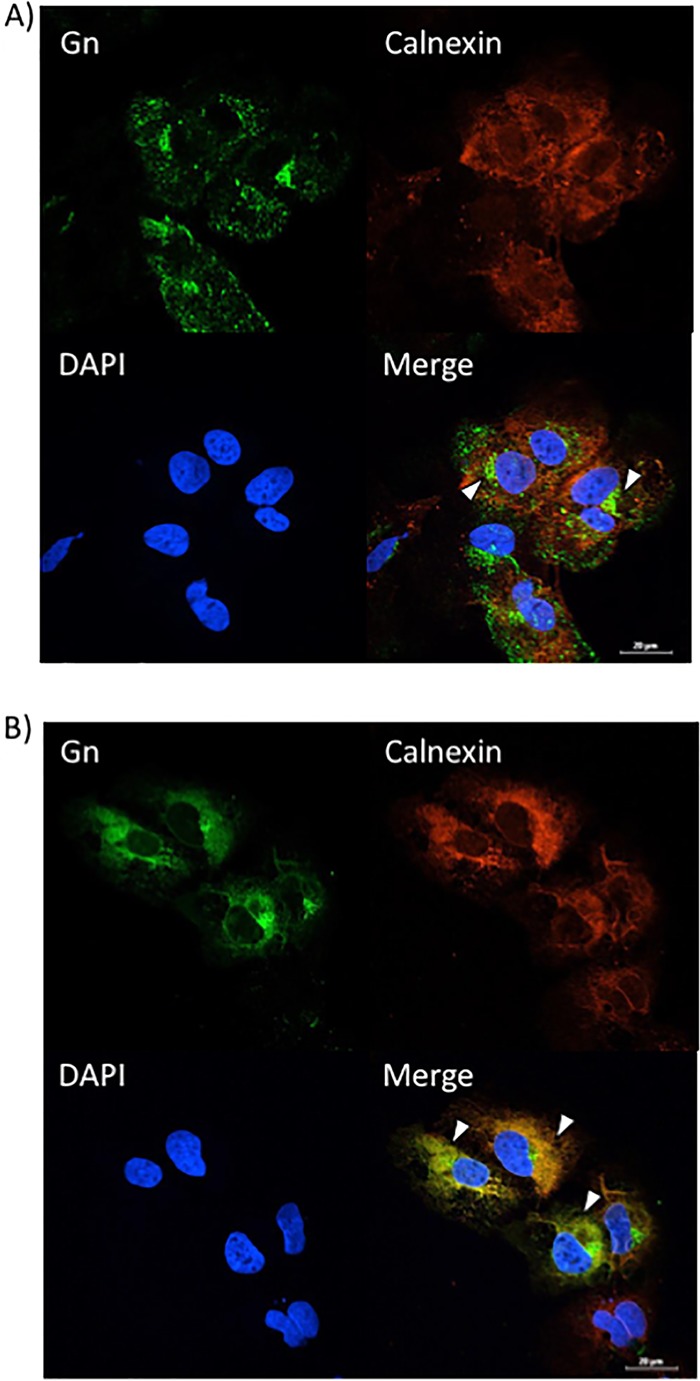
Sorafenib treatment increases Gn colocalization with the ER in Huh7 cells. Huh7 cells were infected with MP12 at an MOI of 10 and either treated with DMSO immediately following infection (A) or treated with sorafenib immediately following infection (B). Cells were fixed, permeabilized, and stained for Gn, the ER marker calnexin, and DAPI at 12 h postinfection. Arrowheads indicate areas that lack calnexin and Gn colocalization (A) or areas of calnexin and Gn colocalization (B).
siRNA knockdown of VCP causes a reduction in the amount of RVFV released into the extracellular medium.
The classical target of sorafenib, Raf, was shown to not be important for RVFV replication in our previously published research (10). Therefore, we carried out an in-depth literature search to identify a candidate sorafenib target protein which would induce the effects on the secretory pathway that were observed, leading to the subsequent block in viral egress. One potential target of sorafenib is valosin-containing protein (VCP), a hexameric AAA+ ATPase (ATPases are associated with diverse cellular activities) also known as p97. It has been shown that sorafenib prevents the tyrosine phosphorylation of VCP, inducing ER stress; fragmentation of the Golgi complex; and disruption of the cellular secretory pathway (14).
To determine whether VCP is important for viral egress, VCP expression was knocked down using small interfering RNA (siRNA), and this was followed by RVFV infection. As a control, a very closely related AAA+ ATPase called N-ethylmaleimide-sensitive fusion protein (NSF) (16) was also knocked down by siRNA. NSF is also linked to the secretory pathway and is involved in the heterotypic fusion between small transport vesicles and organelles (17). At 24 h postinfection, extracellular supernatants were harvested and viral titers were analyzed by plaque assay to determine the levels of virus that were able to egress the cell. In conjunction with harvest of the supernatant, protein lysates were collected from the cells and Western blot analysis was performed in order to determine the knockdown of the protein in question. Both VCP and NSF were successfully knocked down (Fig. 8A). Analysis of the viral titer of the extracellular supernatants revealed that the knockdown of VCP was able to significantly reduce the level of infectious virus that was released from the cell, while knockdown of NSF resulted in virus titers that were not significantly different from those for the controls (Fig. 8B). These results indicate that the loss of VCP inhibits RVFV replication.
FIG 8.

The loss of VCP decreases RVFV virion release. Huh7 cells were treated with either negative-control siRNA or siRNA targeting VCP, followed by infection with MP12 at an MOI of 0.1 for 24 h. (A) Protein levels were detected by Western blotting. (B) The level of virus in the cell supernatants was determined by plaque assay. siNSF, siRNA targeting NSF. The means and standard deviations from three biological replicates are plotted. *, P < 0.05. (C and D) Huh7 cells were treated with either negative-control siRNA (C) or siRNA targeting VCP (D), followed by infection with MP12 at an MOI of 0.1. Cells were fixed, permeabilized, and stained for Gn, VCP, and DAPI at 24 h postinfection. Arrowheads indicate areas displaying typical Gn staining (A) or areas with diffuse Gn staining (B).
siRNA knockdown of VCP causes Gn localization patterns similar to those caused by sorafenib treatment.
In order to determine the importance of VCP for RVFV egress, the staining patterns of Gn, VCP, and the organelle markers were characterized by confocal microscopy following siRNA knockdown of VCP. Negative-control siRNA (siNeg)-treated control cells displayed the typical morphology of Gn localization (Fig. 8C). VCP staining was present both in the nucleus and in the cytoplasm, as expected (18). siRNA knockdown of VCP resulted in a very diffuse staining of Gn throughout the cell (Fig. 8D). Also, the siRNA knockdown of VCP proved to be robust, as demonstrated by the very faint staining of VCP (Fig. 8D).
Since the knockdown of VCP resulted in Gn staining that was analogous to that after the alteration of Gn localization caused by sorafenib, we next wanted to determine whether VCP knockdown also affected Gn colocalization with the secretory pathway organelles in a manner similar to that achieved with sorafenib treatment. siNeg-transfected RVFV-infected cells again showed the distinctive Gn staining, indicating that Gn colocalized closely with the Golgi complex marker (TGN46), as shown by the yellow staining indicated by the white arrowheads in Fig. 9A. Conversely, in RVFV-infected cells transfected with siRNA targeting VCP (siVCP), the level of Gn colocalization with the Golgi complex (TGN46) was highly reduced, as indicated by the near absence of yellow staining (Fig. 9B). Furthermore, a low level of colocalization between Gn and the ER (calnexin) was observed in RVFV-infected cells transfected with siNeg (Fig. 10A). However, when RVFV-infected cells were transfected with siVCP, the cells displayed an increase in the level of colocalization between Gn and the ER (calnexin) (Fig. 10B). These results demonstrate that the loss of VCP results in Gn being relocalized to the ER. They also support the hypothesis that VCP plays a key role in the inhibition of viral egress caused by sorafenib.
FIG 9.
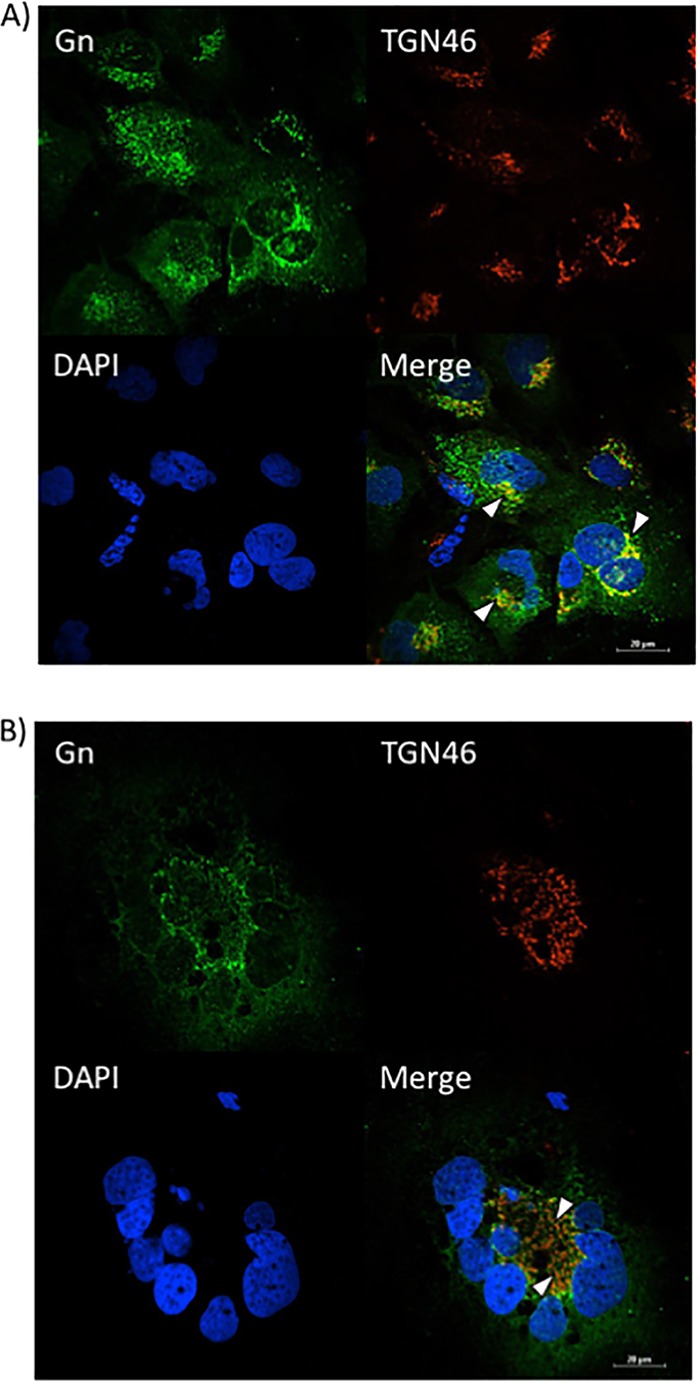
The loss of VCP prevents Gn from colocalizing with the Golgi complex. Huh7 cells were treated with either negative-control siRNA (A) or siRNA targeting VCP (B), followed by infection with MP12 at an MOI of 0.1. Cells were fixed, permeabilized, and stained for Gn, trans-Golgi network protein 46 (TGN46), and DAPI at 24 h postinfection. Arrowheads indicate areas of TGN46 and Gn colocalization (A) or areas that lack TGN46 and Gn colocalization (B).
FIG 10.
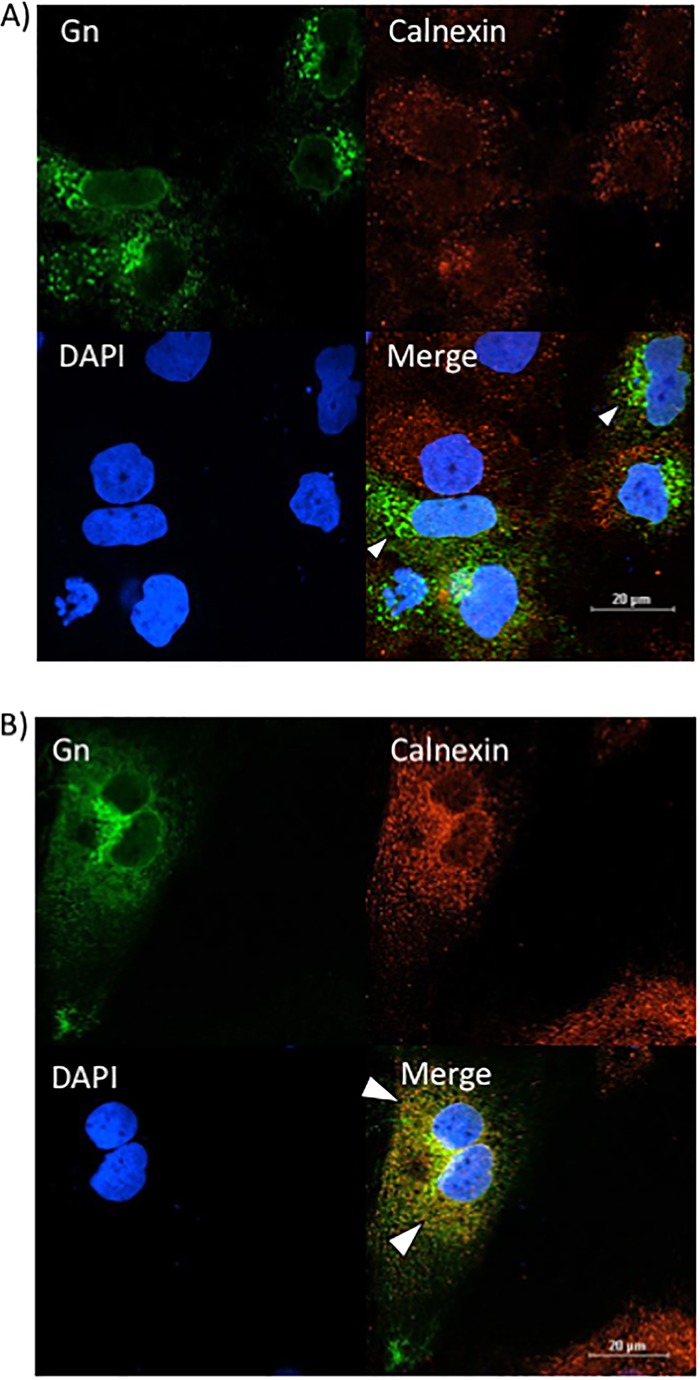
The loss of VCP increases Gn colocalization with the ER. Huh7 cells were treated with either negative-control siRNA (A) or siRNA targeting VCP (B), followed by infection with MP12 at an MOI of 0.1. Cells were fixed, permeabilized, and stained for Gn, the ER marker calnexin, and DAPI at 24 h postinfection. Arrowheads indicate areas that lack calnexin and Gn colocalization (A) or areas of calnexin and Gn colocalization (B).
siRNA knockdown of VCP causes the intracellular buildup of infectious virions comparable to that after sorafenib treatment.
When infected Huh7 cells were treated with sorafenib, we observed a block in viral egress that led to a buildup in infectious viral particles within the cell (10). To ensure that this effect was not cell type dependent, we performed the same experiment in Vero cells (Fig. 11C and D). Cells were infected and treated with sorafenib immediately following infection. At 24 hpi, extracellular supernatants were harvested and the remaining cells were lysed using repeated cycles of freezing and thawing in order to release the intact virions from within the cells. These extracts were analyzed by plaque assay, and the results were plotted as the total number of PFU in each fraction (Fig. 11C). These results were also plotted in terms of the percentage of intracellular virions in the total pool of extracellular and intracellular virions (Fig. 11D). The results show that cells treated with DMSO had about 40% of the virus pool retained within the cell. When the cells were treated with sorafenib, close to 100% of the virion pool was retained within the cell. We hypothesized that the loss of VCP would cause a similar intracellular accumulation of infectious virions through disruption of the secretory pathway. To test this hypothesis, Huh7 cells were subjected to transfection of either siVCP or siNeg prior to infection. At 24 hpi, the extracellular and intracellular fractions were harvested, analyzed by plaque assay, and plotted as described above (Fig. 11A and B). Similar to the findings for cells treated with sorafenib, siRNA knockdown of VCP caused a significant reduction in the amount of virus released by the cell (Fig. 11A). Furthermore, intracellular fraction data demonstrated a significant increase in the amount of infectious virus particles observed within the cell following knockdown of VCP compared to the amount seen in siNeg-transfected cells. The results showed that siNeg-transfected cells had about 50% of the virus pool retained within the cell. When the cells were transfected with siVCP, close to 100% of the virion pool was retained within the cell (Fig. 11B). These data demonstrate that the loss of VCP inhibits viral egress.
FIG 11.

The loss of VCP inhibits RVFV virion egress to a level comparable to that achieved with sorafenib treatment. (A and B) Huh7 cells were treated with either negative-control siRNA or siRNA targeting VCP, followed by infection with MP12 at an MOI of 0.1 for 24 h. (A) The intra- and extracellular infectivity per infection, as determined by plaque assay, is plotted. (B) Total infectivity (i.e., the sum of intra- and extracellular infectivity) per infection was determined. The percentage of intracellular infectivity relative to the total infectivity was plotted for siNeg- and siVCP-transfected cells. (C and D) Vero cells were treated with either DMSO or sorafenib, followed by infection with MP12 at an MOI of 0.1 for 24 h. Intracellular versus extracellular infectivity (C) and the percentage of intracellular infectivity (D) were plotted as mentioned above. The means and standard deviations from three biological replicates are plotted. *, P < 0.05.
DISCUSSION
This study is a follow-up to our group's work on screening FDA-approved drugs to find a therapeutic to combat RVFV infection. We found our top candidate, sorafenib, and our goal was to ascertain the mechanism by which sorafenib inhibits RVFV replication and to determine one of the host's cellular targets that RVFV hijacks to propagate itself. We discovered that sorafenib was capable of inhibiting RVFV replication by impeding viral egress while still allowing viral protein production and virion assembly to occur. Our data show that sorafenib causes virions to get trapped in large vesicles, which we can conclude are formed from dilated ER since viral Gn no longer colocalized with the site of budding, the Golgi complex, but, rather, colocalized with the ER.
A literature search revealed that sorafenib modulates the function of a protein called VCP, which is involved in vesiculation and membrane remodeling in the secretory pathway. Interestingly, our results show that the loss of VCP results in decreased Gn Golgi complex localization, increased Gn ER localization, and the increased accumulation of intracellular virions. VCP functions as a chaperone to control various cellular processes, primarily endoplasmic reticulum-associated protein degradation (ERAD), the ubiquitin-proteasome system (UPS), and ER and Golgi complex morphogenesis. VCP associates with a range of binding partners which determine its diverse activities. It is expected that the expression levels of the cofactors affect the function of VCP by competing for interaction and, thus, directing VCP to different protein machineries. When VCP interacts with a cofactor named p47, it regulates membrane fusion in ER and Golgi complex morphogenesis (19).
RVFV virions normally bud from the Golgi complex within small transport vesicles that travel toward the plasma membrane in order to get exocytosed. We observed that sorafenib treatment caused the virions to get trapped within large intracellular vacuoles unable to egress properly. The fusion of transport vesicles with their appropriate target membrane is caused by the binding of soluble vesicle N-ethylmaleimide-sensitive factor attachment (v-SNARE) proteins embedded in the membrane of the transport vesicle to the corresponding target-localized SNARE (t-SNARE) proteins in the target membrane. VCP utilizes ATP hydrolysis to break apart SNARE complexes within a restricted set of homotypic fusion pathways. This permits v-SNARE and t-SNARE proteins to be recycled for additional rounds of transport. For both Golgi complex reassembly and yeast ER fusion, it is believed that the most likely ATPase target is a t-SNARE–t-SNARE pairing (syntaxin 5 and Ufe1p, respectively) (20). Thus, in the context of RVFV infection, the loss of VCP could prevent SNARE complexes from breaking apart within homotypic fusion pathways, which would prevent Golgi vesicle formation.
NSF is a protein that is important in heterotypic vesicle formation, which includes ER-to-Golgi vesicle formation. The loss of NSF had no effect on RVFV replication or egress. VCP, on the other hand, has been shown to play a role in enabling homotypic vesicle formation (20), which includes Golgi vesicle formation. The loss of VCP or sorafenib-mediated inhibition of VCP resulted in virions not being able to bud from the Golgi complex properly in order to make their way to the plasma membrane to be exocytosed from the cell. Thus, we can deduce that VCP, which is necessary for Golgi vesicle formation, is utilized by the virus to egress from the Golgi complex. Gn colocalized primarily with the ER following sorafenib treatment and siRNA knockdown of VCP, suggesting that the intact heterotypic vesicle formation allows the virions to be shuttled back to the ER.
The loss of VCP resulted in an ∼1.5-log drop in viral titers. In comparison, sorafenib treatment resulted in a 2- to 3-log decrease in the amount of virus, depending on the cell type analyzed (10). These data suggest that sorafenib-mediated inhibition of VCP is not the only factor contributing to RVFV inhibition. Our previous work revealed that sorafenib also interferes with an earlier step in the virus life cycle, most likely viral transcription and/or genome replication (10). It is therefore likely that sorafenib inhibits a yet to be identified event/protein that suppresses viral RNA production. Work recently published in the literature has revealed that sorafenib can associate with heat shock proteins through their N termini in order to suppress their chaperone function (21). Chaperones are necessary to maintain the conformation of proteins so that they can preserve their function. If sorafenib-induced inhibition of heat shock proteins resulted in misfolded N protein or RdRp, this could inhibit the ribonucleoproteins from properly forming or could interfere with the functionality of the RdRp, thus causing a disruption in further steps of the virus life cycle. Interestingly, inhibition of heat shock proteins using the small molecular inhibitors 17-N-allylamino-17-demethoxygeldanamycin and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) reduces RVFV replication (22). This inhibition was determined to be directed at a postentry event, possibly influencing viral transcription or RNA replication. Hence, the interaction between chaperones and RVFV and the role that sorafenib plays in this interaction warrant further study.
Collectively, our data have resulted in our working model of sorafenib-mediated viral egress inhibition (Fig. 12). In the absence of sorafenib treatment (Fig. 12A), viral proteins are translated by ribosomes that are either free-floating in the cytoplasm or embedded in the ER. Gn and Gc are initially expressed as a precursor polypeptide, which is then cotranslationally cleaved, leading to the maturation of both glycoproteins. Once they have matured, Gn and Gc form oligomers which are transported from the ER to the Golgi apparatus due to the presence of a Golgi apparatus-targeting signal located within the Gn C-terminal sequence (9). Once the proteins are ready to move on to the Golgi complex, vesicles containing the viral proteins are able to form and are shuttled from the ER to the Golgi complex. Within the Golgi complex, the Gn and Gc oligomers organize into the virus envelope as cylindrical hollow spikes that cluster into an icosahedral lattice of 122 distinct capsomers. Published data have revealed that distinct regions of the Gn cytosolic tail are required for binding RdRp and N to package within the virion (5). Once the virions are formed, they are released into the lumen of the Golgi complex in virus-filled vesicles. These vesicles subsequently fuse with the plasma membrane, releasing mature virions into the extracellular medium (11).
FIG 12.
Working model of sorafenib effect on viral egress. (A) Normal RVFV egress; (B) disruption of egress following sorafenib treatment.
In the presence of sorafenib treatment, the localization of Gn is dramatically altered (Fig. 12B). The majority of Gn now colocalizes with the ER. The large vacuoles containing virus that were observed in the TEM images are hypothesized to be dilated ER. We also noted fragmentation of the Golgi complex, as visualized in Fig. 4B. Since we observed fully assembled virions in the TEM images (Fig. 3B) and also detected a buildup of infectious virus particles within the cell (10), we can infer that the virions are allowed to assemble properly with sorafenib treatment. This leads us to believe that the viral proteins are able to be transported from the ER to the Golgi complex within vesicles to be assembled into virions within the Golgi complex. However, since the majority of Gn is found to colocalize with the ER, we hypothesize that the assembled virions are prevented from egressing to the plasma membrane due to the inhibited function of VCP. Instead, the virions are then shuttled back to the ER, which is facilitated by NSF, and they remain in the ER.
MATERIALS AND METHODS
Cells, virus, and drug treatment.
Vero cells (ATCC CCL-81) were grown in Dulbecco's modified minimum essential medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin-streptomycin, and 1% l-glutamine. Huh7 cells (a kind gift from Charles M. Rice, Rockefeller University, New York, NY) were grown in DMEM containing 1% l-glutamine, 1% nonessential amino acids, 10% FBS, 1% penicillin-streptomycin, and 1% sodium pyruvate. The cells were maintained at 37°C in a humidified atmosphere with 5% CO2. Unless noted otherwise, all cells were plated at a density of 5.0 × 105 cells in 6-well plates, 2.5 × 105 cells in 12-well plates, and 1 × 104 cells in 96-well plates.
RVFV strain MP12 was rescued, and the titer was determined as previously described (10). Infections were performed under biosafety level 2 conditions. Cells were infected with virus at a multiplicity of infection (MOI) of 10 in drug treatment experiments and an MOI of 0.1 in siRNA knockdown experiments. Inocula were removed after 1 h, the cells were washed one time with 1× phosphate-buffered saline (PBS), and the medium was replaced with fresh medium. Infection was allowed to proceed for a specific duration of time, as indicated above for each experiment. Infectious virus was detected by plaque assay (23). The intracellular infectivity assay was performed as described in reference 10.
Sorafenib tosylate (S1040) was purchased from Selleckchem, made into 10 mM stocks dissolved in DMSO, and stored at −80°C. For treatment, the drug was used at a concentration of 10 μM in complete medium appropriate for the respective cells.
Immunofluorescence and antibodies.
At the end of the specified incubation time, virus-infected cells were fixed in 10% neutral buffered Formalin (Sigma, St. Louis, MO) for a minimum of 15 min. Briefly, Formalin-fixed cells were either untreated or, when necessary, permeabilized (to stain for the intracellular expression of RVFV Gn or N proteins) with 0.1% (vol/vol) Triton X-100 in PBS for 15 min at room temperature. The cells were washed three times with PBS, blocked in 3% bovine serum albumin in PBS and treated with the corresponding primary antibody at 37°C for 1 h, followed by three additional washes with 1× PBS and secondary antibody treatment. The mouse monoclonal antibodies 4D4 and R3-1D8-1-1a were used to detect RVFV glycoprotein (Gn) and RVFV nucleocapsid (N), respectively. All antibodies to viral antigens were purified from the hybridoma stocks at USAMRIID (available from BEI Resources). The rabbit monoclonal antibody ab109240 (Abcam) was used to detect VCP. The sheep polyclonal antibody AHP500GT (AbD Serotec) was used to detect TGN46. The rabbit polyclonal antibody sc11397 (Santa Cruz Biotechnology) was used to detect calnexin. The rabbit monoclonal antibody 3924S (Cell Signaling Technologies) was used to detect NSF. Cell nuclei were labeled with Hoechst 33342 (Life Technologies) at a 1:10,000 dilution or DAPI (4′,6-diamidino-2-phenylindole) at a 1:1,000 dilution. Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin secondary antibody, Alexa Fluor 568-conjugated goat anti-rabbit immunoglobulin, or Alexa Fluor 568-conjugated donkey anti-sheep immunoglobulin (1:500; Life Technologies) was used to visualize the primary antibodies.
High-content quantitative imaging data were acquired and analyzed on an Opera confocal reader (model 3842 [quadruple excitation high sensitivity] or model 5025; PerkinElmer, Waltham, MA) at two exposures using a 10× air objective. Analyses of the images were accomplished within the Opera or Columbus environment using standard Acapella scripts (24).
Transmission electron microscopy (TEM).
Vero cells were infected with MP12 at an MOI of 10, followed by treatment with sorafenib for 24 h. The cells were then fixed and processed as described in reference 25.
siRNA knockdowns.
Huh7 cells, plated at 120,000 cells per well in 12-well plates or 280,000 cells per well on coverslips in 6-well plates, were transfected with SMARTpool siRNA targeting VCP (10 nM; catalog number L-008727-00-0005; Dharmacon) or NSF (50 nM; catalog number L-009401-00-0005; Dharmacon) or negative-control siRNA (50 nM; catalog number D-001810-01-05; Dharmacon). All transfections were performed using the DharmaFECT 2 reagent (catalog number T-2002-02; Thermo Scientific). An untreated control transfection with the DharmaFECT 2 reagent alone was also performed. After 24 h, the transfection medium was replaced with complete medium, and the cells were cultured for an additional 48 h before infection. Protein lysates and extracellular medium supernatants were collected at 24 h postinfection (hpi). Protein expression was measured by Western blot analysis, and infectious titers were determined by plaque assay. Western blot assays were performed as described elsewhere (10). For confocal microscopy, coverslips were fixed at 24 hpi, processed, and imaged as mentioned earlier.
Statistics.
All quantifications are based on data obtained from triplicate samples unless indicated otherwise. Error bars in all figures indicate standard deviations. P values were calculated using an unpaired Student's t test. P values below 0.05 were considered significant.
ACKNOWLEDGMENTS
A.B., R.M., K.K., F.N., S.B., and K.K.-H. designed the experiments. A.B., R.M., C.P., K.K., and R.Z. performed the experiments. A.B., R.M., C.P., K.K., F.N., R.Z., S.B., and K.K.-H. wrote and edited the manuscript.
A.B. was supported by a fellowship from the Institutes of Infectious Animal Diseases. This material is based upon work supported by the U.S. Department of Homeland Security under cooperative agreement number DHS 2010-ST-061-AG0002. This work was funded through Defense Threat Reduction Agency (DTRA) grant HDTRA1-13-1-0006 to K.K.-H. The research described herein was also sponsored by DTRA, Joint Science and Technology Office for Chem Bio Defense (JSTO-CBM.THRV.01.10.RD.19 to S.B.), and the National Interagency Confederation for Biomedical Research (award number 132103643 to R.M. and K.K.).
The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official polices, either expressed or implied, of the U.S. Department of Homeland Security. DTRA did not have any role in the design of the study or the collection, analysis, and interpretation of data or in the writing of the manuscript. The opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army.
REFERENCES
- 1.Mansfield KL, Banyard AC, McElhinney L, Johnson N, Horton DL, Hernández-Triana LM, Fooks AR. 2015. Rift Valley fever virus: a review of diagnosis and vaccination, and implications for emergence in Europe. Vaccine 33:5520–5531. doi: 10.1016/j.vaccine.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 2.Bird BH, Ksiazek TG, Nichol ST, MacLachlan NJ. 2009. Rift Valley fever virus. J Am Vet Med Assoc 234:883–893. doi: 10.2460/javma.234.7.883. [DOI] [PubMed] [Google Scholar]
- 3.Chevalier V, Pépin M, Plée L, Lancelot R. 2010. Rift Valley fever—a threat for Europe? Euro Surveill 15:19506 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19506. [PubMed] [Google Scholar]
- 4.Rolin AI, Berrang-Ford L, Kulkarni MA. 2013. The risk of Rift Valley fever virus introduction and establishment in the United States and European Union. Emerg Microbes Infect 2:e81. doi: 10.1038/emi.2013.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piper ME, Sorenson DR, Gerrard SR. 2011. Efficient cellular release of Rift Valley fever virus requires genomic RNA. PLoS One 6:e18070. doi: 10.1371/journal.pone.0018070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pepin M, Bouloy M, Bird BH, Kemp A, Paweska J. 2010. Rift Valley fever virus (Bunyaviridae: Phlebovirus): an update on pathogenesis, molecular epidemiology, vectors, diagnostics and prevention. Vet Res 41:61. doi: 10.1051/vetres/2010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Won S, Ikegami T, Peters CJ, Makino S. 2007. NSm protein of Rift Valley fever virus suppresses virus-induced apoptosis. J Virol 81:13335–13345. doi: 10.1128/JVI.01238-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wichgers Schreur PJ, Kortekaas J. 2016. Single-molecule FISH reveals non-selective packaging of Rift Valley fever virus genome segments. PLoS Pathog 12:e1005800. doi: 10.1371/journal.ppat.1005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carnec X, Ermonval M, Kreher F, Flamand M, Bouloy M. 2014. Role of the cytosolic tails of Rift Valley fever virus envelope glycoproteins in viral morphogenesis. Virology 448:1–14. doi: 10.1016/j.virol.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 10.Benedict A, Bansal N, Senina S, Hooper I, Lundberg L, de la Fuente C, Narayanan A, Gutting B, Kehn-Hall K. 2015. Repurposing FDA-approved drugs as therapeutics to treat Rift Valley fever virus infection. Front Microbiol 6:676. doi: 10.3389/fmicb.2015.00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spiegel M, Plegge T, Pöhlmann S. 2016. The role of phlebovirus glycoproteins in viral entry, assembly and release. Viruses 8:E202. doi: 10.3390/v8070202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mudhasani R, Kota KP, Retterer C, Tran JP, Tritsch SR, Zamani R, Whitehouse CA, Bavari S. 2015. High-content image-based screening of a signal transduction pathway inhibitor small-molecule library against highly pathogenic RNA viruses. J Biomol Screen 20:141–152. doi: 10.1177/1087057114556253. [DOI] [PubMed] [Google Scholar]
- 13.Mudhasani R, Tran JP, Retterer C, Radoshitzky SR, Kota KP, Altamura LA, Smith JM, Packard BZ, Kuhn JH, Costantino J, Garrison AR, Schmaljohn CS, Huang I-C, Farzan M, Bavari S. 2013. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J Virol 87:8451–8464. doi: 10.1128/JVI.03382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yi P, Higa A, Taouji S, Bexiga MG, Marza E, Arma D, Castain C, Le Bail B, Simpson JC, Rosenbaum J, Balabaud C, Bioulac-Sage P, Blanc J-F, Chevet E. 2012. Sorafenib-mediated targeting of the AAA+ ATPase p97/VCP leads to disruption of the secretory pathway, endoplasmic reticulum stress, and hepatocellular cancer cell death. Mol Cancer Ther 11:2610–2620. doi: 10.1158/1535-7163.MCT-12-0516. [DOI] [PubMed] [Google Scholar]
- 15.Shi Y-H, Ding Z-B, Zhou J, Hui B, Shi G-M, Ke A-W, Wang X-Y, Dai Z, Peng Y-F, Gu C-Y, Qiu S-J, Fan J. 2011. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 7:1159–1172. doi: 10.4161/auto.7.10.16818. [DOI] [PubMed] [Google Scholar]
- 16.Kano F, Kondo H, Yamamoto A, Kaneko Y, Uchiyama K, Hosokawa N, Nagata K, Murata M. 2005. NSF/SNAPs and p97/p47/VCIP135 are sequentially required for cell cycle-dependent reformation of the ER network. Genes Cells 10:989–999. doi: 10.1111/j.1365-2443.2005.00894.x. [DOI] [PubMed] [Google Scholar]
- 17.Nichols BJ, Ungermann C, Pelham HRB, Wickner WT, Haas A. 1997. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature 387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- 18.Totsukawa G, Kaneko Y, Uchiyama K, Toh H, Tamura K, Kondo H. 2011. VCIP135 deubiquitinase and its binding protein, WAC, in p97ATPase-mediated membrane fusion. EMBO J 30:3581–3593. doi: 10.1038/emboj.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shih Y-T, Hsueh Y-P. 2016. VCP and ATL1 regulate endoplasmic reticulum and protein synthesis for dendritic spine formation. Nat Commun 7:11020. doi: 10.1038/ncomms11020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodman PG. 2003. p97, a protein coping with multiple identities. J Cell Sci 116:4283–4290. doi: 10.1242/jcs.00817. [DOI] [PubMed] [Google Scholar]
- 21.Booth L, Shuch B, Albers T, Roberts JL, Tavallai M, Proniuk S, Zukiwski A, Wang D, Chen C-S, Bottaro D, Ecroyd H, Lebedyeva IO, Dent P. 2016. Multi-kinase inhibitors can associate with heat shock proteins through their NH2-termini by which they suppress chaperone function. Oncotarget 7:12975–12996. doi: 10.18632/oncotarget.7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nuss JE, Kehn-Hall K, Benedict A, Costantino J, Ward M, Peyser BD, Retterer CJ, Tressler LE, Wanner LM, McGovern HF, Zaidi A, Anthony SM, Kota KP, Bavari S, Hakami RM. 2014. Multi-faceted proteomic characterization of host protein complement of Rift Valley fever virus virions and identification of specific heat shock proteins, including HSP90, as important viral host factors. PLoS One 9:e93483. doi: 10.1371/journal.pone.0093483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baer A, Kehn-Hall K. 2014. Viral concentration determination through plaque assays: using traditional and novel overlay systems. J Vis Exp 2014:e52065. doi: 10.3791/52065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mudhasani R, Kota KP, Retterer C, Tran JP, Whitehouse CA, Bavari S. 2014. High content image-based screening of a protease inhibitor library reveals compounds broadly active against Rift Valley fever virus and other highly pathogenic RNA viruses. PLoS Negl Trop Dis 8:e3095. doi: 10.1371/journal.pntd.0003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiang C-Y, Ulrich RL, Ulrich MP, Eaton B, Ojeda JF, Lane DJ, Kota KP, Kenny TA, Ladner JT, Dickson SP, Kuehl K, Raychaudhuri R, Sun M, Bavari S, Wolcott MJ, Covell D, Panchal RG. 2015. Characterization of the murine macrophage response to infection with virulent and avirulent Burkholderia species. BMC Microbiol 15:259. doi: 10.1186/s12866-015-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]