

ABSTRACT
Endoglin is part of the TGF-β receptor complex and has a crucial role in fibrogenesis and angiogenesis. It is also an important protein for tumor growth, survival, and cancer cell metastasis. In a previous study, we have shown that hepatitis C virus (HCV) infection induces epithelial-mesenchymal transition (EMT) state and cancer stem-like cell (CSC) properties in human hepatocytes. Our array data suggested that endoglin (CD105) mRNA is significantly upregulated in HCV-associated CSCs. In this study, we have observed increased endoglin expression on the cell surface of an HCV core-expressing hepatocellular carcinoma (HepG2) cell line or immortalized human hepatocytes (IHH) and activation of its downstream signaling molecules. The status of phospho-SMAD1/5 and the expression of inhibitor of DNA binding protein 1 (ID1) were upregulated in HCV-infected cells or viral core gene-transfected cells. Additionally, we observed upregulation of endoglin/ID1 mRNA expression in chronic HCV patient liver biopsy samples. CSC generation by HCV core protein was dependent on the endoglin signaling pathway using activin receptor-like kinase 1 (ALK1) Fc blocking peptide and endoglin small interfering RNA (siRNA). Further, follow-up from in vitro analysis suggested that the antiapoptosis Bcl2 protein, proliferation-related cyclin D1 protein, and CSC-associated Hes1, Notch1, Nanog, and Sox2 proteins are enhanced during infection or ectopic expression of HCV core protein.
IMPORTANCE Endoglin plays a crucial role in fibrogenesis and angiogenesis and is an important protein for tumor growth, survival, and cancer cell metastasis. Endoglin enhances ALK1-SMAD1/5 signaling in different cell types, leading to increased proliferation and migration responses. We have observed endoglin expression on the HCV core-expressing cell surface of human hepatocyte origin and activation of phospho-SMAD1/5 and ID1 downstream signaling molecules. ID1 protein plays a role in CSC properties, and we found that this pathway is important for antiapoptotic and cell proliferation signaling. Blocking of endoglin-ALK1-SMAD1/5 might be a good candidate for therapy for liver cancer stem cells together with liver cirrhosis.
KEYWORDS: HCV, HCC, core, endoglin, angiogenesis, hepatitis C virus
INTRODUCTION
Hepatitis C virus (HCV) often causes persistent infection and disease progression to hepatocellular carcinoma (HCC). A major global research effort has illuminated many aspects of the HCV life cycle, facilitating the development of effective direct-acting antiviral (DAA) regimens with high cure rates. However, many hurdles remain. Cure of HCV viremia by DAA treatment does not prevent risk of reinfection with the same or different genotypes and may not reverse virus-associated pathology. Further, the high cost of DAA treatment results in restricted access. Additionally, the largely asymptomatic nature of infection facilitates continued transmission in at-risk groups and resource-constrained settings due to limited surveillance. Approximately 50% of HCV-infected patients in the United States are not aware that they are infected (1), and even those cured of viremia remain at a significantly elevated risk for HCC (2–6). Increased intrahepatic endoglin and transforming growth factor-β1 (TGF-β1) expression is significantly associated with progressive hepatic fibrosis in chronic HCV infection (7). This raises the question of how the endoglin level rises and which cell types generate it following HCV infection.
Endoglin is an ∼180-kDa integral membrane-bound glycoprotein and is found on many cell surfaces. It forms homodimers and consists of a large extracellular domain, a hydrophobic transmembrane domain, and a short cytoplasmic tail. This receptor binds to a large variety of extra- and intracellular binding partners and modulates numerous cellular properties, including morphology, migration, endocytic vesicular transport, microtubular structures, and functionality of focal adhesion proteins. Endoglin plays a crucial role in TGF-β signaling (8, 9) and is enhanced upon TGF-β stimulation and exposure to hypoxic conditions, such as those found in tumors (10). Endoglin is an accessory coreceptor for TGF-β, a pleotropic protein involved in cytokine-regulating cellular proliferation, differentiation, migration, and adhesion. Endoglin plays a role in fibrosis involving hepatic stellate cells (HSCs) (11). Endoglin is also expressed in renal cell carcinoma and is predictive of increased tumorigenic potential and disease staging (12, 13). Endoglin is highly expressed by activated endothelial cells and has a crucial role in angiogenesis. Endoglin knockout animals die in utero because of defects in the vascular system (14). Endoglin expression is upregulated in various cancers (8) and correlates with the development of metastatic disease (15).
In our previous studies, we have shown that HCV infection of hepatocytes induces an epithelial-mesenchymal transition (EMT) state and cancer stem-like cell (CSC) properties (16, 17). HCV core protein has many intriguing properties, including immortalization of primary human hepatocytes (PHH) (18) and induction of EMT (19, 20) and CSCs (17). Our CSC array result shows that endoglin is upregulated (∼850-fold) in sphere-forming cells compared to primary hepatocytes (17). In HCV patients with fibrosis, the expression of intrahepatic and circulating endoglin is higher than that in patients with early fibrosis and normal liver (7). Inhibitor of DNA binding protein 1 (ID1) proteins, which are downstream molecules of the endoglin and SMAD1/5 pathway, are important for generation of cancer stem cells (21). In this study, we examined endoglin expression on the cell surface of HCV core protein-expressing hepatocytes. We also determined the downstream signaling pathway of endoglin for cell proliferation, antiapoptosis, and CSC markers. Our results suggest that endoglin and TGF-β1 regulation may lead to HCV-associated liver disease progression.
RESULTS
HCV core protein induces endoglin expression on cell surfaces.
We have previously shown by cancer stem cell-specific PCR array analysis that endoglin is upregulated in sphere-forming cells of HCV-infected primary human hepatocytes (17). HepG2 cells have previously been observed not to express endoglin on the cell surface (11). We determined whether endoglin is upregulated in HCV core-transfected HepG2 cells and immortalized human hepatocytes (IHH) which were generated by stable transfection of the HCV core genomic region into primary human hepatocytes. Interestingly, HepG2 cells stably expressing HCV core displayed a strong expression of endoglin on the cell surface by immunofluorescence, in contrast to a weak expression level on parental cells. IHH are expected to display endoglin on the cell surface for transfection of the HCV core gene. We verified the relationship between HCV core and endoglin with IHH antisense core-expressing cells. Endoglin expression was decreased on cell surface of antisense core-expressing IHH compared to parental IHH as determined by immunofluorescence (Fig. 1A).
FIG 1.

Endoglin expression on HCV core- or anticore-expressing hepatocyte surface. Endoglin expression on HCV core- or anticore-expressing hepatocytes is shown. (A) Immunofluorescence for endoglin on the HepG2 cell surface (upper left), HepG2 core 1a (upper right), IHH (lower left), and IHH antisense core (lower right). Photomicrographs taken at a magnification of ×20 are shown. Enlarged insets of cells marked with short arrows are shown in the upper left corners. (B) Results from qRT-PCR analysis for endoglin expression in core-transfected or HCV genotype 2a-infected Huh7.5 cells (panel B).
Huh7.5 cells are an appropriate cell line for HCV infection. We could not detect endoglin expression or any significant change in Huh7.5 cells before or after introduction of the core gene or infection with HCV2a by immunofluorescence. For this, we analyzed endoglin expression by quantitative reverse transcription-PCR (qRT-PCR) in transfected or infected Huh7.5 cells (Fig. 1B). These results suggested that HCV core protein expression upregulates endoglin expression in Huh7.5 cells. The results from our multiple cell lines of human hepatocyte origin suggest that the enhancement of endoglin by HCV is not a cell line-specific effect.
HCV core protein activates downstream of the endoglin/ALK1 pathway.
Endoglin promotes signaling mechanisms for cancer stem cell generation by activating the ALK1 receptor and SMAD 1/5 phosphorylation (Fig. 2A). We analyzed the HCV core-mediated effect on hepatocytes by examining the status of SMAD phosphorylation in mock-transfected and stably HCV core 1a-transfected HepG2 cells. Phospho-SMAD1/5 (Ser463/465) was induced in HepG2 core 1a-transfected cells compared to in parental HepG2 cells (Fig. 2B). We further verified whether HCV core protein induces the endoglin-mediated SMAD1/5 signaling pathway using IHH or IHH transfected with an antisense core gene. Our observations suggested that the phospho-SMAD1/5 (Ser463/465) expression was higher in IHH than in IHH transfected with the antisense core gene. Interestingly, PHH may not express detectable SMAD1 and are stimulated upon HCV core protein expression. Subsequently, we examined expression of ID1, which is one of the downstream molecules of SMAD1/5 and acts as transcription factor, by Western blotting for protein expression and densitometric scanning. ID1 was upregulated in HepG2 core 1a-transfected compared to parental HepG2 cells and was downregulated in antisense core-transfected IHH compared to IHH (Fig. 2C).
FIG 2.

HCV core 1a protein promotes SMAD1/5 phosphorylation and ID1. (A) Schematic representation of the downstream portion of the endoglin-mediated signaling pathway. (B). Phospho-SMAD1/5 (S463/465) status in HepG2 cells, HepG2 core 1a-transfected cells, primary human hepatocytes (PHH), IHH, and IHH antisense core-transfected cells cell lines as determined by Western blotting. Densitometric scanning of the Western blot results is shown on the right. Actin was used as an internal control for comparison of protein load in each lane. (C) ID1 protein expression in these cells was analyzed similarly. (D) HCV core 1a protein expression in transfected cells was analyzed by Western blotting. Densitometric scanning of the Western blot results is shown on the right. The negative-control HepG2 value was arbitrarily considered to be 1 for comparison of core expression in transfected cells. (E) Core protein expression was separately analyzed by ELISA.
The HCV core protein expression status in transfected HepG2 cells was analyzed by Western blotting (Fig. 2D) and enzyme-linked immunosorbent assay (ELISA). HCV core protein expression was detected in transiently HCV core-transfected cells by Western blotting and ELISA. We have used different transfection reagents in the past. However, the HCV core genomic region from the H77 clone always displays lower expression than JFH1 core. This could be due to the differences or sequence variations between HCV clones used.
Similar enhanced p-SMAD1/5 and ID1 expression was observed in HCV genotype 2a-infected Huh7.5 cells (Fig. 3A and B). Densitometric scanning results suggested that HCV infection induces SMAD1/5 phosphorylation and ID1 to approximately 6-fold-higher levels than in mock-treated parent Huh7.5 cells. Together, our results suggested that HCV core expression promotes SMAD1/5 phosphorylation and ID1 expression in hepatocytes.
FIG 3.

HCV infection induces SMAD1/5 phosphorylation and ID1 expression. (A and B) P-SMAD1/5 (S463/S465) (A) and ID1 (B) expression status in Huh7.5 cells and HCV 2a (JFH1)-infected Huh7.5 cells. The actin expression status was determined and included as an internal control for comparison of protein expression. Densitometric scanning of Western blot results is shown on the right. Values are means from three independent experiments ± SD. Statistical significance was analyzed using the two-tailed Student t test: *, P < 0.05; **P < 0.01. (C and D) qRT-PCR data from nine liver biopsy RNAs for endoglin (C) and ID1 (D). Results from control liver RNAs (n = 2) are shown for comparison.
Endoglin and ID1 enhancement in chronically HCV-infected patient liver.
We examined whether liver biopsy samples from chronically infected HCV patients have a higher expression of endoglin or ID1 at the mRNA level. RNAs from archived liver biopsy samples from nine patients with chronic HCV genotype 1a or 1b infection with a viral titer between 3.9 ×104 and 2.8 ×106 IU/ml were used (before treatment) to investigate the status of endoglin and ID1 by qRT-PCR. Endoglin was upregulated (2.7- to 34-fold) in all infected liver specimens compared to control liver RNA (Fig. 3A). ID1, which is downstream of the endoglin/ALK pathway, was also upregulated (1.6- to 23-fold) in seven out of nine samples (Fig. 3D). The status of endoglin or ID1 was not related to the genotype of the infecting HCV, liver fibrosis stage, or viral load in blood.
ALK1-Fc acts downstream of the endoglin/ALK pathway, impairing SMAD1/5 phosphorylation and ID1 expression in HCV core-expressing cells.
To investigate the role of ALK1, we examined the status of SMAD1/5 phosphorylation and ID1 expression in HepG2 cells transiently transfected with the HCV core genomic region from genotype 1a using the peptide inhibitor ALK1-Fc. Phospho-SMAD1/5 (Ser463/465) and ID1 expression was induced in HepG2 cells transiently transfected with HCV core 1a and was downregulated upon treatment with AKL1-Fc (Fig. 4A and B). Together, the results show that ALK1-Fc inhibited induction of both SMAD1/5 phosphorylation and ID1 expression, indicating a role of endoglin in regulating signaling pathways for HCV core protein.
FIG 4.
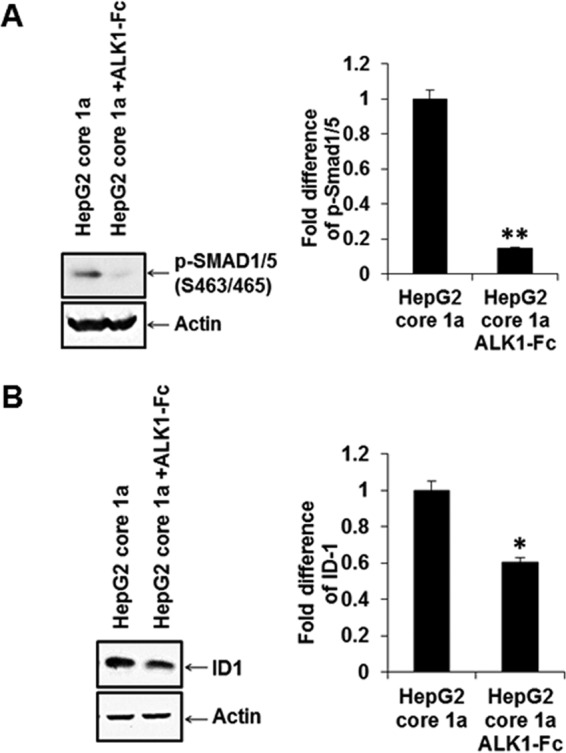
HCV core-induced SMAD1/5 phosphorylation and ID1 increase are inhibited by ALK1-Fc. HCV core from genotype 1a transfected into HepG2 cells was analyzed by Western blotting for induction of SMAD1/5 phosphorylation (A), ID-1 upregulation (B), and inhibition by ALK1-Fc. Actin expression status was determined and included as an internal control for comparison of protein expression and densitometric scanning. Statistical significance was analyzed using the two-tailed Student t test: *, P < 0.05; **P < 0.01.
HCV infection acts downstream of ID1-related antiapoptosis, cell proliferation, and CSC marker proteins.
We focused on the ID1 family of proteins, which are induced by the endoglin-ALK1-SMAD1/5 pathways. ID1 is related to antiapoptosis, cell cycle progression, and cancer stem cell-associated properties (21). We analyzed the protein expression status of an antiapoptosis-related protein (Bcl-2) and cancer stem cell property-associated proteins (N-cadherin, Notch1, Nanog, and Sox2) in Huh7.5 cells infected with HCV by Western blotting (Fig. 5A and B). Cell cycle regulatory proteins (cyclins E and D1) and cancer stem cell property-related protein (HES1) in Huh7.5 cells infected with HCV were separately analyzed (Fig. 5C). Interestingly, the antiapoptosis marker protein (Bcl-2) and cancer stem cell marker (Hes1) were upregulated in HCV-infected Huh7.5 cells compared with parental Huh7.5 control cells.
FIG 5.

HCV infection alters the ID1 downstream antiapoptosis Bcl-2 protein and enhances cell proliferation and CSC marker proteins. (A and B) Infection of Huh7.5 cells with cell culture-grown HCV genotype 2a (clone JFH1) enhanced Bcl2 (A) and CSC marker proteins (N-cadherin, Notch1, Nanog, and Sox2) (B) as determined by Western blotting. (C) Cell cycle progression markers (cyclin E and cyclin D1) and a Notch signaling marker (Hes1) were examined similarly from infected cells by Western blotting. Densitometric scanning for comparison of protein status is shown on the right. Statistical significance was analyzed using the two-tailed Student t test: *, P < 0.05; **P < 0.01.
ALK1-Fc and endoglin small interfering RNA (siRNA) act downstream of the endoglin/ALK pathway, impairing SMAD1/5 phosphorylation and ID1 expression in HCV-infected cells.
To investigate the role of ALK1-Fc, we examined the phosphorylation status of SMAD1/5 and ID1 and Bcl-2 expression in Huh7.5 cells infected with HCV using peptide inhibitor ALK1Fc and Western blotting for protein expression status (Fig. 6A and B). Phospho-SMAD1/5 (Ser463/465) and ID1 expression was downregulated by AKL1-Fc in Huh7.5 cells infected with HCV compared to parental Huh7.5 cells. The antiapoptosis marker Bcl-2 and cancer stem cell-associated proteins (Notch1 and Hes1) were also downregulated following ALK1-Fc treatment of Huh7.5 cells infected with HCV.
FIG 6.

Inhibitory role of ALK1 peptide on downstream ID1 signaling molecules in HCV-infected cells. Phosphorylation status of SMAD1/5 and ID1 (A) and anti-apoptotic protein Bcl2 and stem cell-associated proteins Notch1 and Hes1 (B) was inhibited by ALK1 peptide. Densitometric scanning for comparison of protein status is shown on the right. Statistical significance was analyzed using the two-tailed Student t test: *, P < 0.05; **P < 0.01.
Similar inhibition of p-SMAD1/5, ID1, or Bcl2 was noted in Huh7.5 cells infected with HCV2a after treatment of cells with endoglin-specific siRNA (Fig. 7A). ID1 and pSMAD1/5 expression was also observed using endoglin-specific siRNA in core-transfected Huh7.5 cells (Fig. 7B). These results suggested that the endoglin/ALK pathway may be related to HCV-induced hepatoma cell viability and cancer stem cell properties.
FIG 7.

Endoglin-specific siRNA inhibits downstream ID1 signaling molecules in HCV-infected or core-transfected cells. The P-SMAD1/5, ID1, or Bcl2 status was altered upon treatment of HCV-infected (A) or core-transfected (B) cells with endoglin siRNA. Actin was detected and used as an internal control for densitometric scanning and comparison of protein status (shown on the right). Statistical significance was analyzed using the two-tailed Student t test: *, P < 0.05; **P < 0.01.
DISCUSSION
Endoglin expression and dysregulation have been shown in a number of cell types (22). Endoglin can be detected in HCV-infected serum (7, 23). In this study, we observed endoglin expression and activation of phospho-SMAD1/5 and ID1 downstream signaling molecules in HCV core-expressing cells of human hepatocyte origin (Fig. 8). Endoglin is not detected on HepG2 and Hep3B cell surfaces (11). Since ID1 protein plays a role in CSC properties, we further examined whether CSC generation by HCV core protein is dependent on the endoglin signaling pathway, using ALK1-Fc blocking peptide and endoglin siRNA, and found that this pathway is important for antiapoptotic and cell proliferation signaling. Additionally, we observed upregulated mRNA expression of endoglin/ID1 in chronically HCV infected patient liver.
FIG 8.
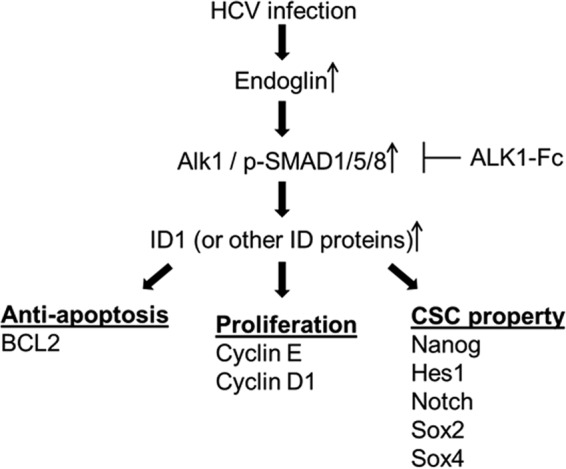
Schematic diagram showing the observed changes induced by HCV in endoglin-associated cell signaling pathways.
Overexpression of endoglin leads to increased SMAD1/SMAD5 phosphorylation (22). Endoglin enhances ALK1/SMAD1/5 signaling in different cell types (24), leading to increased proliferation and migration responses. The characteristic activation phase of angiogenesis is negatively affected upon endoglin reduction (25–27). On the other hand, ALK5/SMAD3 signaling, which inhibits proliferation and migration (characteristics of the resolution phase of angiogenesis), is blocked by endoglin (26, 28, 29). Blocking of endoglin-ALK1-SMAD1/5 might be a good candidate for therapy of liver cancer stem cells together with liver cirrhosis.
HCC, a typical hypervascular tumor, is the most common hepatic malignancy worldwide. Approximately 80% of HCC patients have pathology associated with liver cirrhosis (30). The expression of endoglin at the mRNA and protein levels was higher in tumor tissues than in normal liver but was lower than in nontumor tissues with cirrhosis (31). High vascularity could be one of the reasons for the poor prognosis (32). Most malignant tumors, as well as HCCs, have developed efficient strategies to promote fast blood vessel growth for supply of nutrients and oxygen. Angiogenesis is a highly regulated, complex process modulated by many intersecting pathways, including vascular endothelial growth factor (VEGF), TGF-β, and endoglin (33), angiopoietins (34), Notch (35), and integrins (36). By modulating TGF-β signaling, endoglin plays a crucial role in angiogenesis and tumor growth and could be linked to HCC (37) and other cancers (38–41). Endoglin is an angiogenesis marker in breast cancer (42), malignant melanoma (43), non-small-cell lung cancer (44), and colorectal carcinoma (45). These findings have shown the usefulness of targeting endoglin in antiangiogenic therapy of cancer (46, 47).
In the pathogenesis of liver fibrosis, TGF-β is the most potent fibrogenic cytokine. It induces fibrosis through multiple mechanisms, including direct activation of HSCs, stimulation of ECM production, and prompting the synthesis of tissue inhibitors of matrix metalloproteases (TIMPs), thereby inhibiting ECM degradation (48). Endoglin is a marker of angiogenesis in HCC (49, 50) and might be appropriate target for antiangiogenesis therapy in HCC. Understanding the underlying mechanisms associated with alterations in endoglin expression will be of interest in establishing new therapeutic modalities for chronic liver disease. Intrahepatic and circulating levels of endoglin are elevated in patients with chronic hepatitis C infection, liver cirrhosis, and HCC (7, 31, 37). Our results suggest that endoglin and TGF-β1 regulation by HCV core in hepatocytes may initiate as a molecular basis for development of HCV-associated liver disease progression. Endoglin has been used as a target in clinical trials for development of therapy in fibrosis/cirrhosis. Therefore, our results suggest the potential for use of endoglin as an additional target for HCV-associated HCC.
MATERIALS AND METHODS
Liver biopsy specimens and RNA isolation.
Archived liver biopsy samples from nine patients with chronic HCV infection were kindly provided by Adrian M. Di Bisceglie (Saint Louis University, MO). The biopsy specimens were obtained from patients infected with HCV genotype 1a (n = 5) or 1b (n = 4), and liver fibrosis stages were 1 (n = 3) and 2 (n = 6). Specimens were collected from subjects with their written consent, and the human studies protocol was approved by the Saint Louis University Institutional Review Board. RNA was prepared from liver specimens using TRIzol (Invitrogen), as previously described (17). Commercially available control liver RNAs (Clontech, Mountain View, CA; Lonza, Allendale, NJ) were included in experiments for comparison.
Cells, virus, and transfection.
Cells of human hepatocyte origin (Huh7.5 and HepG2) were used. Immortalized human hepatocytes (IHH) were generated by transfection of a plasmid DNA expressing the HCV core genomic region of genotype 1a (GenBank accession number M62321) into primary human hepatocytes under the control of a cytomegalovirus (CMV) promoter (51). Inhibition of core protein expression in IHH (antisense core IHH) was done as previously described (52). For infection, Huh7.5 cells were treated with cell culture-grown HCV genotype 2a, clone JFH1 (multiplicity of infection [MOI] of 0.2), in a minimum volume of medium and incubated for 3 to 5 days. Infected cells were lysed and processed for either RNA isolation or protein lysate preparation. For transient expression, HepG2 cells were transfected with pBabe HCV core plasmid DNA using jetPrime reagent (Polyplus-transfection, NY). Huh7.5 cells were transfected with endoglin siRNA using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific).
RNA isolation and qRT-PCR.
Total RNA was isolated using a Qiagen RNeasy minikit (Qiagen, CA). RNA was quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized using random hexamers and a Superscript III reverse transcriptase kit (Invitrogen, CA). Quantitative reverse transcription-PCR (qRT-PCR) was performed with cDNA for quantitation using TaqMan gene expression PCR master mix and 6-carboxyfluorescein (FAM)-MGB probes for ENG (assay ID, Hs00923996_m1) and ID1 (assay ID, Hs03676575_s1) (Thermo Fisher Scientific). A FAM-MGB probe for 18S rRNA (assay ID, Hs03928985_g1) was used as an endogenous control. The relative gene expression levels were normalized with housekeeping gene 18S rRNA level by using the 2−ΔΔCT formula (ΔΔCT = ΔCT for sample − ΔCT for untreated control).
Immunofluorescence.
Cells were stained with antiendoglin antibody (Santa Cruz, CA) in blocking buffer for 1 h at 4°C, washed with phosphate-buffered saline (PBS), and incubated for 1 h with Alexa 488-conjugated anti-mouse IgG (Molecular Probes, CA). The cells were fixed with 3.7% formaldehyde, stained with 1 μg/ml DAPI (4′,6′-diamidino-2-phenylindole) in PBS, and washed with PBS. Images were captured with a fluorescence microscope (Leica, model DMI4000B).
Western blot analysis.
Cells were lysed in sample-reducing buffer and subjected to SDS-PAGE, and separated proteins were transferred onto a nitrocellulose membrane (Bio-Rad Laboratories). The blot was blocked with 5% skim milk and incubated with a primary antibody, followed by treatment with a secondary antibody conjugated to horseradish peroxidase (Bio-Rad Laboratories). The membrane was probed with antibodies to phospho-Smad1/5, Smad1, Hes1, and Notch1 (Cell Signaling), ID1, Bcl-2, cyclin E, and cyclin D1 (Santa Cruz), and N-cadherin, Notch, Nanog, and Sox2 (Cell Signaling), and HCV core protein was detected by a specific rabbit antiserum, followed by treatment with a secondary antibody conjugated with horseradish peroxidase. The membrane was reprobed with actin as an internal control. The protein bands were detected with SuperSignal West Pico ECL reagents (Pierce). Densitometric scanning of Western blots was performed by ImageJ software (NIH).
HCV core ELISA.
Cells were lysed in Triton X lysis buffer and subjected to an HCV ELISA test (Ortho Clinical Diagnostics, Tokyo, Japan).
Statistical analysis.
The results are presented as means ± standard deviations (SD). Data were analyzed by Student's t test with a two-tailed distribution in Microsoft Excel. A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This work was supported by research grant DK113645 from the National Institutes of Health and by the Presidential and Liver Center Research Funds of Saint Louis University.
We thank Adrian M. Di Bisceglie for providing clinical archived specimens.
REFERENCES
- 1.Litwin AH, Smith BD, Drainoni ML, McKee D, Gifford AL, Koppelman E, Christiansen CL, Weinbaum CM, Southern WN. 2012. Primary care-based interventions are associated with increases in hepatitis C virus testing for patients at risk. Dig Liver Dis 44:497–503. doi: 10.1016/j.dld.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 2.van der Meer AJ, Veldt BJ, Feld JJ, Wedemeyer H, Dufour JF, Lammert F, Duarte-Rojo A, Heathcote EJ, Manns MP, Kuske L, Zeuzem S, Hofmann WP, de Knegt RJ, Hansen BE, Janssen HL. 2012. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 308:2584–2593. doi: 10.1001/jama.2012.144878. [DOI] [PubMed] [Google Scholar]
- 3.Aleman S, Rahbin N, Weiland O, Davidsdottir L, Hedenstierna M, Rose N, Verbaan H, Stal P, Carlsson T, Norrgren H, Ekbom A, Granath F, Hultcrantz R. 2013. A risk for hepatocellular carcinoma persists long-term after sustained virologic response in patients with hepatitis C-associated liver cirrhosis. Clin Infect Dis 57:230–236. doi: 10.1093/cid/cit234. [DOI] [PubMed] [Google Scholar]
- 4.Hoshida Y, Fuchs BC, Bardeesy N, Baumert TF, Chung RT. 2014. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J Hepatol 61:S79–90. doi: 10.1016/j.jhep.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reig M, Marino Z, Perello C, Inarrairaegui M, Ribeiro A, Lens S, Diaz A, Vilana R, Darnell A, Varela M, Sangro B, Calleja JL, Forns X, Bruix J. 2016. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J Hepatol 65:719–726. doi: 10.1016/j.jhep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Conti F, Buonfiglioli F, Scuteri A, Crespi C, Bolondi L, Caraceni P, Foschi FG, Lenzi M, Mazzella G, Verucchi G, Andreone P, Brillanti S. 2016. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J Hepatol 65:727–733. doi: 10.1016/j.jhep.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 7.Clemente M, Nunez O, Lorente R, Rincon D, Matilla A, Salcedo M, Catalina MV, Ripoll C, Iacono OL, Banares R, Clemente G, Garcia-Monzon C. 2006. Increased intrahepatic and circulating levels of endoglin, a TGF-beta1 co-receptor, in patients with chronic hepatitis C virus infection: relationship to histological and serum markers of hepatic fibrosis. J Viral Hepat 13:625–632. doi: 10.1111/j.1365-2893.2006.00733.x. [DOI] [PubMed] [Google Scholar]
- 8.Duff SE, Li C, Garland JM, Kumar S. 2003. CD105 is important for angiogenesis: evidence and potential applications. FASEB J 17:984–992. doi: 10.1096/fj.02-0634rev. [DOI] [PubMed] [Google Scholar]
- 9.Bernabeu C, Lopez-Novoa JM, Quintanilla M. 2009. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim Biophys Acta 1792:954–973. doi: 10.1016/j.bbadis.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C. 2002. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J Biol Chem 277:43799–43808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- 11.Meurer SK, Tihaa L, Borkham-Kamphorst E, Weiskirchen R. 2011. Expression and functional analysis of endoglin in isolated liver cells and its involvement in fibrogenic Smad signalling. Cell Signal 23:683–699. doi: 10.1016/j.cellsig.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. 2008. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J 22:3696–3705. doi: 10.1096/fj.08-102590. [DOI] [PubMed] [Google Scholar]
- 13.Khan MI, Czarnecka AM, Duchnowska R, Kukwa W, Szczylik C. 2014. Metastasis-initiating cells in renal cancer. Curr Signal Transduct Ther 8:240–246. doi: 10.2174/1574362409666140206222431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, Charlton R, Parums DV, Jowett T, Marchuk DA, Burn J, Diamond AG. 2000. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol 217:42–53. doi: 10.1006/dbio.1999.9534. [DOI] [PubMed] [Google Scholar]
- 15.Romani AA, Borghetti AF, Del Rio P, Sianesi M, Soliani P. 2006. The risk of developing metastatic disease in colorectal cancer is related to CD105-positive vessel count. J Surg Oncol 93:446–455. doi: 10.1002/jso.20456. [DOI] [PubMed] [Google Scholar]
- 16.Bose SK, Meyer K, Di Bisceglie AM, Ray RB, Ray R. 2012. Hepatitis C virus induces epithelial-mesenchymal transition in primary human hepatocytes. J Virol 86:13621–13628. doi: 10.1128/JVI.02016-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwon YC, Bose SK, Steele R, Meyer K, Di Bisceglie AM, Ray RB, Ray R. 2015. Promotion of cancer stem-like cell properties in hepatitis C virus-infected hepatocytes. J Virol 89:11549–11556. doi: 10.1128/JVI.01946-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon YC, Ray RB, Ray R. 2014. Hepatitis C virus infection: establishment of chronicity and liver disease progression. EXCLI J 13:977–996. [PMC free article] [PubMed] [Google Scholar]
- 19.Akkari L, Gregoire D, Floc'h N, Moreau M, Hernandez C, Simonin Y, Rosenberg AR, Lassus P, Hibner U. 2012. Hepatitis C viral protein NS5A induces EMT and participates in oncogenic transformation of primary hepatocyte precursors. J Hepatol 57:1021–1028. doi: 10.1016/j.jhep.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 20.Quan H, Zhou F, Nie D, Chen Q, Cai X, Shan X, Zhou Z, Chen K, Huang A, Li S, Tang N. 2014. Hepatitis C virus core protein epigenetically silences SFRP1 and enhances HCC aggressiveness by inducing epithelial-mesenchymal transition. Oncogene 33:2826–2835. doi: 10.1038/onc.2013.225. [DOI] [PubMed] [Google Scholar]
- 21.Lasorella A, Benezra R, Iavarone A. 2014. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer 14:77–91. doi: 10.1038/nrc3638. [DOI] [PubMed] [Google Scholar]
- 22.Meurer SK, Alsamman M, Scholten D, Weiskirchen R. 2014. Endoglin in liver fibrogenesis: bridging basic science and clinical practice. World J Biol Chem 5:180–203. doi: 10.4331/wjbc.v5.i2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rath T, Hage L, Kugler M, Menendez Menendez K, Zachoval R, Naehrlich L, Schulz R, Roderfeld M, Roeb E. 2013. Serum proteome profiling identifies novel and powerful markers of cystic fibrosis liver disease. PLoS One 8:e58955. doi: 10.1371/journal.pone.0058955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Velasco S, Alvarez-Munoz P, Pericacho M, Dijke PT, Bernabeu C, Lopez-Novoa JM, Rodriguez-Barbero A. 2008. L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J Cell Sci 121:913–919. doi: 10.1242/jcs.023283. [DOI] [PubMed] [Google Scholar]
- 25.Tian H, Mythreye K, Golzio C, Katsanis N, Blobe GC. 2012. Endoglin mediates fibronectin/alpha5beta1 integrin and TGF-beta pathway crosstalk in endothelial cells. EMBO J 31:3885–3900. doi: 10.1038/emboj.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P. 2004. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J 23:4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jerkic M, Rodriguez-Barbero A, Prieto M, Toporsian M, Pericacho M, Rivas-Elena JV, Obreo J, Wang A, Perez-Barriocanal F, Arevalo M, Bernabeu C, Letarte M, Lopez-Novoa JM. 2006. Reduced angiogenic responses in adult Endoglin heterozygous mice. Cardiovasc Res 69:845–854. doi: 10.1016/j.cardiores.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 28.Lee NY, Ray B, How T, Blobe GC. 2008. Endoglin promotes transforming growth factor beta-mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J Biol Chem 283:32527–32533. doi: 10.1074/jbc.M803059200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. 2002. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J 21:1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikai I, Yamaoka Y, Yamamoto Y, Ozaki N, Sakai Y, Satoh S, Shinkura N, Yamamoto M. 1998. Surgical intervention for patients with stage IV-A hepatocellular carcinoma without lymph node metastasis: proposal as a standard therapy. Ann Surg 227:433–439. doi: 10.1097/00000658-199803000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu D, Zhuang L, Sun X, Chen J, Yao Y, Meng K, Ding Y. 2007. Particular distribution and expression pattern of endoglin (CD105) in the liver of patients with hepatocellular carcinoma. BMC Cancer 7:122. doi: 10.1186/1471-2407-7-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung GE, Lee JH, Yoon JH, Myung SJ, Lee K, Jang JJ, Lee JM, Kim SH, Suh KS, Kim YJ, Lee HS. 2012. Prognostic implications of tumor vascularity and its relationship to cytokeratin 19 expression in patients with hepatocellular carcinoma. Abdom Imaging 37:439–446. doi: 10.1007/s00261-011-9756-3. [DOI] [PubMed] [Google Scholar]
- 33.Bellone G, Gramigni C, Vizio B, Mauri FA, Prati A, Solerio D, Dughera L, Ruffini E, Gasparri G, Camandona M. 2010. Abnormal expression of Endoglin and its receptor complex (TGF-beta1 and TGF-beta receptor II) as early angiogenic switch indicator in premalignant lesions of the colon mucosa. Int J Oncol 37:1153–1165. doi: 10.3892/ijo_00000767. [DOI] [PubMed] [Google Scholar]
- 34.Fagiani E, Christofori G. 2013. Angiopoietins in angiogenesis. Cancer Lett 328:18–26. doi: 10.1016/j.canlet.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 35.Benedito R, Hellstrom M. 2013. Notch as a hub for signaling in angiogenesis. Exp Cell Res 319:1281–1288. doi: 10.1016/j.yexcr.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Eklund L, Saharinen P. 2013. Angiopoietin signaling in the vasculature. Exp Cell Res 319:1271–1280. doi: 10.1016/j.yexcr.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 37.Yagmur E, Rizk M, Stanzel S, Hellerbrand C, Lammert F, Trautwein C, Wasmuth HE, Gressner AM. 2007. Elevation of endoglin (CD105) concentrations in serum of patients with liver cirrhosis and carcinoma. Eur J Gastroenterol Hepatol 19:755–761. doi: 10.1097/MEG.0b013e3282202bea. [DOI] [PubMed] [Google Scholar]
- 38.Blanco FJ, Bernabeu C. 2011. Alternative splicing factor or splicing factor-2 plays a key role in intron retention of the endoglin gene during endothelial senescence. Aging Cell 10:896–907. doi: 10.1111/j.1474-9726.2011.00727.x. [DOI] [PubMed] [Google Scholar]
- 39.Bellone G, Solerio D, Chiusa L, Brondino G, Carbone A, Prati A, Scirelli T, Camandona M, Palestro G, Dei Poli M. 2007. Transforming growth factor-beta binding receptor endoglin (CD105) expression in esophageal cancer and in adjacent nontumorous esophagus as prognostic predictor of recurrence. Ann Surg Oncol 14:3232–3242. doi: 10.1245/s10434-007-9528-z. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S, Ghellal A, Li C, Byrne G, Haboubi N, Wang JM, Bundred N. 1999. Breast carcinoma: vascular density determined using CD105 antibody correlates with tumor prognosis. Cancer Res 59:856–861. [PubMed] [Google Scholar]
- 41.Yu JX, Zhang XT, Liao YQ, Zhang QY, Chen H, Lin M, Kumar S. 2003. Relationship between expression of CD105 and growth factors in malignant tumors of gastrointestinal tract and its significance. World J Gastroenterol 9:2866–2869. doi: 10.3748/wjg.v9.i12.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beresford MJ, Harris AL, Ah-See M, Daley F, Padhani AR, Makris A. 2006. The relationship of the neo-angiogenic marker, endoglin, with response to neoadjuvant chemotherapy in breast cancer. Br J Cancer 95:1683–1688. doi: 10.1038/sj.bjc.6603491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodey B, Bodey B Jr, Siegel SE, Kaiser HE. 1998. Immunocytochemical detection of endoglin is indicative of angiogenesis in malignant melanoma. Anticancer Res 18:2701–2710. [PubMed] [Google Scholar]
- 44.Tanaka F, Otake Y, Yanagihara K, Kawano Y, Miyahara R, Li M, Yamada T, Hanaoka N, Inui K, Wada H. 2001. Evaluation of angiogenesis in non-small cell lung cancer: comparison between anti-CD34 antibody and anti-CD105 antibody. Clin Cancer Res 7:3410–3415. [PubMed] [Google Scholar]
- 45.Li C, Gardy R, Seon BK, Duff SE, Abdalla S, Renehan A, O'Dwyer ST, Haboubi N, Kumar S. 2003. Both high intratumoral microvessel density determined using CD105 antibody and elevated plasma levels of CD105 in colorectal cancer patients correlate with poor prognosis. Br J Cancer 88:1424–1431. doi: 10.1038/sj.bjc.6600874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carmeliet P, Jain RK. 2000. Angiogenesis in cancer and other diseases. Nature 407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka F, Otake Y, Yanagihara K, Kawano Y, Miyahara R, Li M, Ishikawa S, Wada H. 2003. Correlation between apoptotic index and angiogenesis in non-small cell lung cancer: comparison between CD105 and CD34 as a marker of angiogenesis. Lung Cancer 39:289–296. doi: 10.1016/S0169-5002(02)00534-2. [DOI] [PubMed] [Google Scholar]
- 48.Brenner DA. 2009. Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc 120:361–368. [PMC free article] [PubMed] [Google Scholar]
- 49.Yao Y, Pan Y, Chen J, Sun X, Qiu Y, Ding Y. 2007. Endoglin (CD105) expression in angiogenesis of primary hepatocellular carcinomas: analysis using tissue microarrays and comparisons with CD34 and VEGF. Ann Clin Lab Sci 37:39–48. [PubMed] [Google Scholar]
- 50.Ho JW, Poon RT, Sun CK, Xue WC, Fan ST. 2005. Clinicopathological and prognostic implications of endoglin (CD105) expression in hepatocellular carcinoma and its adjacent non-tumorous liver. World J Gastroenterol 11:176–181. doi: 10.3748/wjg.v11.i2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ray RB, Meyer K, Ray R. 2000. Hepatitis C virus core protein promotes immortalization of primary human hepatocytes. Virology 271:197–204. doi: 10.1006/viro.2000.0295. [DOI] [PubMed] [Google Scholar]
- 52.Basu A, Meyer K, Ray RB, Ray R. 2002. Hepatitis C virus core protein is necessary for the maintenance of immortalized human hepatocytes. Virology 298:53–62. doi: 10.1006/viro.2002.1460. [DOI] [PubMed] [Google Scholar]