

ABSTRACT
The use of overlapping open reading frames (ORFs) to synthesize more than one unique protein from a single mRNA has been described for several viruses. Segment 11 of the rotavirus genome encodes two nonstructural proteins, NSP5 and NSP6. The NSP6 ORF is present in the vast majority of rotavirus strains, and therefore the NSP6 protein would be expected to have a function in viral replication. However, there is no direct evidence of its function or requirement in the viral replication cycle yet. Here, taking advantage of a recently established plasmid-only-based reverse genetics system that allows rescue of recombinant rotaviruses entirely from cloned cDNAs, we generated NSP6-deficient viruses to directly address its significance in the viral replication cycle. Viable recombinant NSP6-deficient viruses could be engineered. Single-step growth curves and plaque formation of the NSP6-deficient viruses confirmed that NSP6 expression is of limited significance for RVA replication in cell culture, although the NSP6 protein seemed to promote efficient virus growth.
IMPORTANCE Rotavirus is one of the most important pathogens of severe diarrhea in young children worldwide. The rotavirus genome, consisting of 11 segments of double-stranded RNA, encodes six structural proteins (VP1 to VP4, VP6, and VP7) and six nonstructural proteins (NSP1 to NSP6). Although specific functions have been ascribed to each of the 12 viral proteins, the role of NSP6 in the viral replication cycle remains unknown. In this study, we demonstrated that the NSP6 protein is not essential for viral replication in cell culture by using a recently developed plasmid-only-based reverse genetics system. This reverse genetics approach will be successfully applied to answer questions of great interest regarding the roles of rotaviral proteins in replication and pathogenicity, which can hardly be addressed by conventional approaches.
KEYWORDS: rotavirus, reverse genetics, NSP6, viral replication
INTRODUCTION
Group A rotavirus (RVA), a member of the family Reoviridae, is the leading pathogen of severe diarrhea in young children worldwide, being responsible for an estimated 215,000 deaths annually (1). The RVA virion encapsidates a genome of 11 segments of double-stranded RNA (dsRNA), which encode 12 proteins, six structural proteins (VP1 to VP4, VP6, and VP7), and six nonstructural proteins (NSP1 to NSP6) (2). All of the gene segments are monocistronic with the exception of segment 11, which encodes two nonstructural proteins, NSP5 and NSP6, in overlapping open reading frames (ORFs) (2). The NSP6 protein (12 kDa) is expressed from a +1 alternative ORF in segment 11, and its coding sequence lies entirely within that of NSP5 (3). This small protein (92 amino acids) is the least studied among the 12 RVA proteins (4). In infected cells, NSP6 has been reported to be localized to viroplasms, sites of viral genome replication, and to mitochondria via its different conserved sequences (3–8). The NSP6 protein has been shown to interact with the NSP5 protein in viroplasms (7). In common with most of the other RVA proteins found in viroplasms, NSP6 is an RNA binding protein exhibiting binding activities toward both single-stranded RNA (ssRNA) and dsRNA without sequence specificity (4, 9). Sequence analysis of segment 11 from various RVA strains has revealed that few cell culture-adapted strains lack a 12-kDa NSP6 protein (2). That is, human strain 512C (G3P[9]) (10) does not have the start codon of NSP6, while lapine strain Alabama (G3P[14]) (11) and porcine strain OSU (G5P[7]) (12) possess truncated NSP6 ORFs. This, together with its low level of expression in infected cells (3), has suggested that the NSP6 protein plays a nonessential regulatory role in the viral replication cycle (3, 7, 13). However, direct evidence of its role or requirement in viral replication has remained to be obtained.
A reverse genetics technology to engineer infectious viruses that contain specific genomic sequence modifications from cloned cDNAs is a powerful tool for investigating virus replication and pathogenicity. For RVA, partial plasmid-based reverse genetics systems that require a helper virus have been established (14–18). Although these reverse genetics systems have been exploited to generate novel recombinant RVAs possessing a cDNA-derived gene segment, the development of a versatile entirely plasmid-based reverse genetics system for RVA has lagged significantly behind that for other RNA viruses, including other members of the Reoviridae (19–25). Recently, a long-awaited entirely plasmid-based reverse genetics system has been established (26). In this system, recombinant RVAs are rescued following transfection of cells constitutively expressing T7 RNA polymerase (BHK-T7 cells) with 14 plasmids (11 RVA cDNA plasmids for 11 gene segments, flanked by the T7 RNA polymerase promoter and hepatitis delta virus [HDV] ribozyme sequences, in combination with three expression plasmids encoding the Nelson Bay orthoreovirus fusion-associated small transmembrane [FAST] protein and the two subunits of the vaccinia virus [VV] capping enzyme). In this study, we took advantage of this technology to generate NSP6-deficient viruses to directly address the significance of the NSP6 protein in viral replication.
RESULTS AND DISCUSSION
Generation of a mutant SA11 virus with a silenced NSP6 ORF.
To determine the significance of the NSP6 protein in the viral replication cycle, we employed an entirely plasmid-based reverse genetics system (26) to generate recombinant RVAs capable or incapable of expressing the NSP6 protein. Genetic manipulation was performed in a T7-driven plasmid, pT7-NSP5SA11 (26), encoding the full-length segment 11 (NSP5/6 genes) of the SA11 virus (Fig. 1A). To generate plasmid pT7-NSP5SA11-delNSP6 for the rescue of the NSP6 ORF-silenced virus, the start codon and three downstream AUG codons in the NSP6 ORF were disrupted by introducing a silent mutation (AUG to ACG) at nucleotide positions 80 to 82, 152 to 154, 257 to 259, and 326 to 328, respectively (Fig. 1A). With these manipulations, the amino acid sequence of the overlapping NSP5 was not affected. Viable recombinant SA11 viruses capable or incapable of expressing the NSP6 protein were rescued from BHK-T7 cells transfected with 12 plasmids (11 rescue T7 plasmids for 11 gene segments in combination with the FAST-expressing plasmid), which contain the wild-type or NSP6 ORF-silenced segment 11 (rSA11 and rSA11-delNSP6, respectively) (Fig. 1B). For the rescue of rSA11 and rSA11-delNSP6 viruses, cotransfection of expression plasmids encoding the two subunits of the VV capping enzyme was not absolutely required. Therefore, we did not employ these two expression plasmids throughout this study. PAGE analysis of viral genomic dsRNAs extracted from the rescued viruses demonstrated that rSA11 and rSA11-delNSP6 exhibited the expected migration patterns that are indistinguishable from that of the native SA11 virus (Fig. 1B). To confirm that the rescued viruses were generated from the cloned cDNAs, pT7/VP4SA11-ΔPstI (15) was used as the segment 4 (VP4 gene) rescue T7 plasmid, in which a unique PstI site was destroyed in the VP4 gene of SA11 by the introduction of two silent mutations (T to C and A to C at nucleotide positions 1365 and 1368, respectively) (Fig. 1C). The full-length VP4 genes (2,362 bp) of native SA11, rSA11, and rSA11-delNSP6 were amplified by reverse transcription-PCR (RT-PCR). The amplified VP4 gene derived from native SA11 was digested to produce 1,368- and 994-bp fragments, whereas the VP4 segments from rSA11 and rSA11-delNSP6 were not digested (Fig. 1D). The full-length segment 11 (667 bp) of rSA11 and rSA11-delNSP6 were amplified by RT-PCR. Direct sequencing of the RT-PCR products confirmed the presence of the expected AUG-to-ACG mutations on segment 11 of mutant rSA11-delNSP6 and the absence of unwanted mutations on segment 11 of both rescued viruses (data not shown). The genetic stability of the introduced mutations remained unchanged after five passages in MA104 cells (data not shown). Thus, these results demonstrated that the rSA11 and rSA11-delNSP6 viruses originated from the cloned cDNAs, and viable and genetically stable RVAs with the silenced NSP6 ORF could be generated by means of reverse genetics.
FIG 1.

Generation of the rSA11-delNSP6 virus incapable of expressing the NSP6 protein. (A) Schematic presentation of the plasmids encoding segment 11 (NSP5/6 genes), used for rescue of the wild-type (wt) rSA11 and mutant rSA11-delNSP6 viruses (pT7-NSP5SA11 and pT7-NSP5SA11-delNSP6, respectively). To generate plasmid pT7-NSP5SA11-delNSP6, the start codon and three AUG codons in the NSP6 ORF of SA11 were disrupted by replacing them with ACG codons. The arrowheads indicate the ACG mutation sites. UTR, untranslated region; aa, amino acid. (B) PAGE of viral genomic dsRNAs extracted from the native SA11, rSA11, and rSA11-delNSP6 viruses. Lane 1, dsRNAs from the native SA11; lanes 2 and 3, dsRNAs from the rescued rSA11 (lane 2) and rSA11-delNSP6 (lane 3) viruses. The numbers on the left indicate the order of the genomic dsRNA segments of the native SA11. (C) Rescued rSA11 and rSA11-delNSP6 contain a signature mutation in their VP4 genes as a gene marker. Nucleotide substitutions (T to C and A to C at nucleotide positions 1365 and 1368, respectively) were introduced to eliminate a unique PstI site in the VP4 gene (15). (D) The VP4 gene RT-PCR products were digested with PstI, followed by separation in a 1.2% agarose gel. Native SA11 (lanes 1 and 2), rSA11 (lanes 3 and 4), and rSA11-delNSP6 (lanes 5 and 6) are shown. The 2,362-bp fragments (lanes 1, 3, and 5) were digested with PstI (lanes 2, 4, and 6). M, 1-kb ladder markers. (E) Expression of the NSP6 protein and other RVA proteins in MA104 cells infected with rSA11 or rSA11-delNSP6. Whole-cell lysates of infected cells were analyzed by immunoblotting using anti-NSP6 antiserum, anti-NSP5 antiserum, anti-VP6 antiserum, or anti-β-actin monoclonal antibody. The anti-β-actin antibody was used as a loading control. Mock (lane 1), rSA11 (lane 2), and rSA11-delNSP6 (lane 3) virus infection is shown.
To confirm that the AUG-to-ACG mutations in segment 11 of rSA11-delNSP6 prevent NSP6 expression, immunoblotting analysis was performed to assess the expression of NSP6 using NSP6-specific antiserum in MA104 cells infected with the rescued viruses. The expression of the NSP6 protein was detected in cells infected with wild-type rSA11 but not in cells infected with mutant rSA11-delNSP6 (Fig. 1E). Immunoblotting with other RVA-specific antisera demonstrated equivalent expression levels of other RVA proteins (NSP5 and VP6) in cells infected with the wild-type and mutant viruses, suggesting a similar level of infection by rSA11 and rSA11-delNSP6 (Fig. 1E). Thus, it was demonstrated that the mutant rSA11-delNSP6 virus was viable and did not express the NSP6 protein.
Growth properties of the NSP6-deficient virus in cell culture.
To determine whether the NSP6 protein influences RVA infectivity in cell culture, single-step growth curves of rSA11 and rSA11-delNSP6 were determined after infection of MA104 cells at a high multiplicity of infection (MOI) of 5 PFU/cell. The growth curves demonstrated that the replication of the mutant rSA11-delNSP6 was slower (by about 8-fold) than that of the wild-type rSA11 during the replication cycle but that comparable end titers were reached (Fig. 2A). We then examined the plaque sizes in CV-1 cells for these viruses by measuring the mean diameter of each of 25 plaques in two independent assays (Fig. 2B). The virus growth titers correlated well with the sizes of the plaques formed. That is, the mutant rSA11-delNSP6 formed smaller-sized plaques (diameter, 1.39 ± 0.21 mm) than those of the wild-type rSA11 (diameter, 3.43 ± 0.35 mm). Therefore, the results confirmed that the NSP6 protein is not essential for viral replication in cell culture, although the NSP6 protein appeared to promote efficient virus growth.
FIG 2.
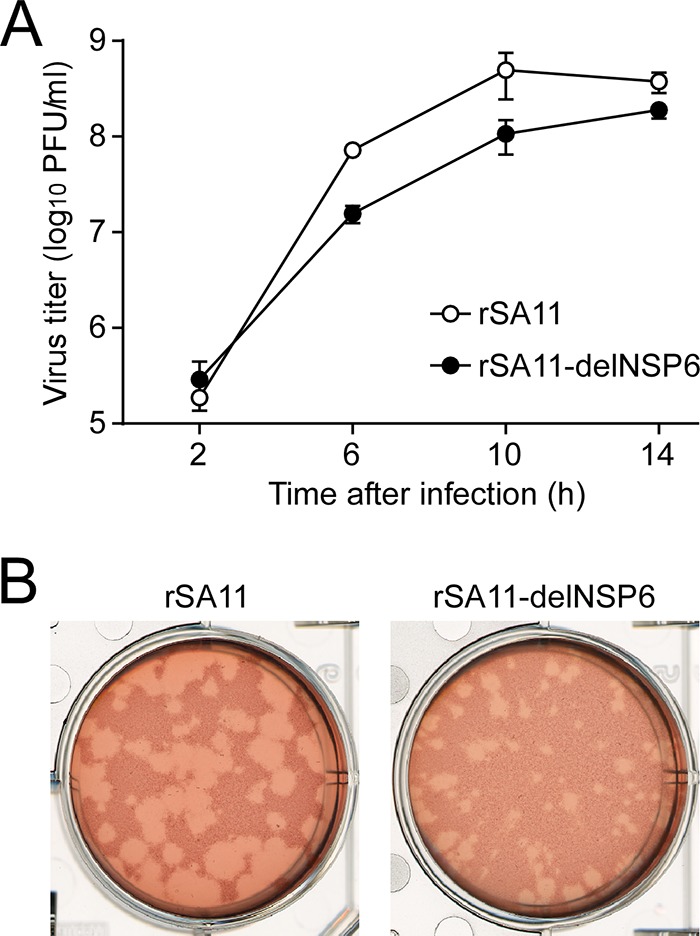
Infectivity of the rSA11-delNSP6 virus lacking NSP6 expression. (A) Single-step growth curves of rSA11 and rSA11-delNSP6. MA104 cells were infected with rSA11 or rSA11-delNSP6 at an MOI of 5 and then incubated for various times. Virus titers in the cultures were determined by plaque assay. The data shown are the mean viral titers and standard deviations (SDs) from three independent cell cultures. (B) Plaque formation by rSA11 and rSA11-delNSP6. rSA11 or rSA11-delNSP6 was directly plated onto CV-1 cells for plaque formation. The experiment was repeated three times with similar results, and representative results are shown.
Generation of recombinant SA11-based monoreassortant viruses possessing a KU-derived segment 11 capable or incapable of expressing the NSP6 protein.
In order to determine whether the effect of deletion of NSP6 expression differs among RVA strains, we attempted to rescue recombinant monoreassortant viruses that possess segment 11 from the human KU virus and the other 10 segments from the SA11 virus. To construct rescue T7 plasmid pT7/NSP5KU, encoding the authentic segment 11 of KU, the full-length cDNA of segment 11 was amplified by RT-PCR from viral genomic dsRNA of KU. The amplified cDNA of KU segment 11 was inserted into a T7-driven plasmid, pX8dT (Fig. 3A). To generate plasmid pT7/NSP5KU-delNSP6 for the rescue of the NSP6 ORF-silenced monoreassortant virus, the start codon and five downstream AUG codons in the NSP6 ORF were disrupted by introduction of a silent ACG mutation at nucleotide positions 80 to 82, 140 to 142, 158 to 160, 257 to 259, 272 to 274, and 278 to 280, respectively, without an effect of the amino acid sequence of the overlapping NSP5 (Fig. 3A). Viable recombinant SA11-based monoreassortant viruses possessing KU-derived segment 11, capable or incapable of expressing the NSP6 protein, were rescued from the cells transfected with 12 plasmids (11 rescue T7 plasmids for 11 gene segments in combination with the FAST-expressing plasmid), which contain the wild-type or NSP6 ORF-silenced KU segment 11 (rSA11-s11KU and rSA11-s11KU-delNSP6, respectively) (Fig. 3B). PAGE analysis of viral dsRNAs showed that segment 11 of the rescued monoreassortant viruses migrated to the same position as the corresponding segment 11 of the native KU virus, with an SA11 backbone (Fig. 3B). The authenticity and genetic stability of mutations in segment 11 of the rescued monoreassortant viruses was confirmed by nucleotide sequence analysis using extracted viral dsRNAs (data not shown). To confirm that the mutant rSA11-s11KU-delNSP6 virus did not express the NSP6 protein, as intended, immunoblotting analysis using NSP6-specific antiserum was carried out. The expression of the NSP6 protein was detected in MA104 cells infected with wild-type rSA11-s11KU but not in cells infected with mutant rSA11-s11KU-delNSP6 (Fig. 3C). Immunoblotting with other RVA-specific antisera demonstrated equivalent expression levels of other RVA proteins (NSP5 and VP6) in cells infected with the wild-type and mutant viruses, suggesting a similar level of infection by rSA11-s11KU and rSA11-s11KU-delNSP6 (Fig. 3C). Thus, the mutant rSA11-s11KU-delNSP6, which did not express the NSP6 protein, was viable, demonstrating that RVAs that lack NSP6 expression can be rescued by means of reverse genetics without strain dependence.
FIG 3.

Generation of recombinant SA11-based monoreassortant viruses having a KU-derived segment 11 capable or incapable of NSP6 expression. (A) Schematic presentation of the plasmids encoding KU segment 11 (NSP5/6 genes) for rescue of the wild-type rSA11-s11KU and mutant rSA11-s11KU-delNSP6 viruses (pT7/NSP5KU and pT7/NSP5KU-delNSP6, respectively). To generate plasmid pT7/NSP5KU-delNSP6, the start codon and five AUG codons in the NSP6 ORF of KU were disrupted by replacing them with ACG codons. The arrowheads indicate the ACG mutation sites. (B) PAGE of viral dsRNAs extracted from rSA11, rSA11-s11KU, rSA11-s11KU-delNSP6, and native KU. Lane 1, dsRNAs from rSA11; lanes 2 and 3, rSA11-s11KU (lane 2) and rSA11-s11KU-delNSP6 (lane 3); lane 4, native KU. The numbers on the left indicate the order of the genomic dsRNA segments of rSA11. (C) Expression of the NSP6 protein and other RVA proteins in MA104 cells infected with rSA11-s11KU or rSA11-s11KU-delNSP6. Whole-cell lysates of infected cells were analyzed by immunoblotting using anti-NSP6 antiserum, anti-NSP5 antiserum, anti-VP6 antiserum, or anti-β-actin monoclonal antibody. Shown are mock (lane 1), rSA11-s11KU (lane 2), and rSA11-s11KU-delNSP6 (lane 3) infection.
Growth properties of the NSP6-deficient monoreassortant virus in cell culture.
To confirm the significance of the NSP6 protein in viral replication, MA104 cells were infected with the rSA11-s11KU and rSA11-s11KU-delNSP6 monoreassortant viruses at a high MOI of 5 PFU/cell to determine their single-step growth curves. The growth curves showed that both viruses reached 108 PFU/ml at 14 h after infection, although the mutant rSA11-s11KU-delNSP6 grew somewhat less (<10-fold lower titers) than the wild-type rSA11-s11KU (Fig. 4A), and the titers were almost the same as those seen with the rSA11 and rSA11-delNSP6 viruses (Fig. 2A). We then determined the plaque sizes in CV-1 cells for these monoreassortant viruses by measuring the mean diameter of each of 25 plaques in two independent assays (Fig. 4B). A reduction in the size of plaques formed by the mutant rSA11-s11KU-delNSP6 (diameter, 1.38 ± 0.49 mm) compared to that of the wild-type rSA11-s11KU (diameter, 3.38 ± 0.42 mm) was observed, which was compatible with the reduction seen with the mutant rSA11-delNSP6 compared to that of the wild-type rSA11 (Fig. 2B). Based on these results, we conclude that the NSP6 protein is dispensable for viral replication in cell culture, although the NSP6 protein appeared to promote efficient virus growth.
FIG 4.
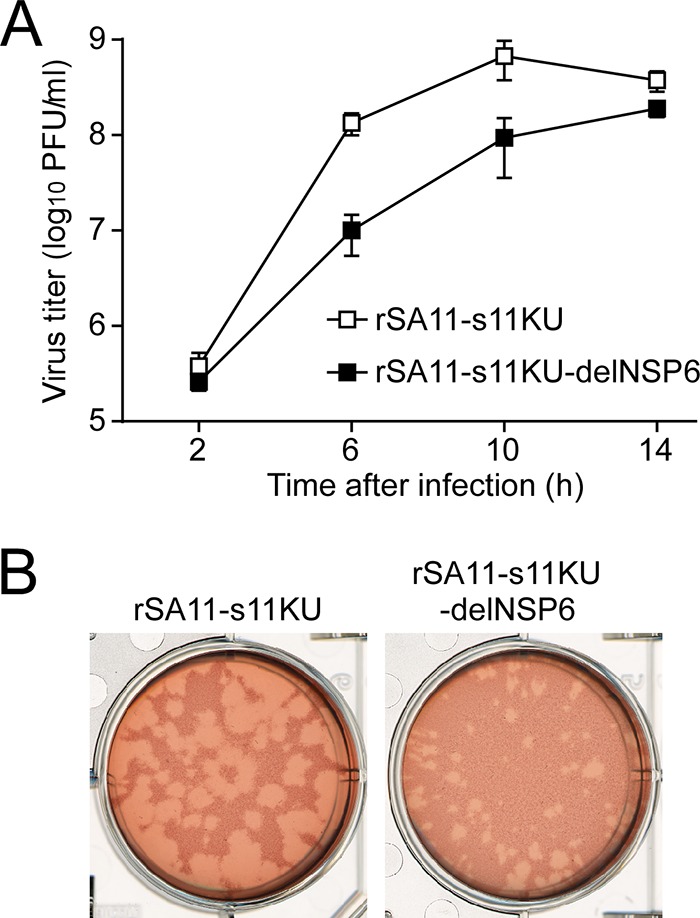
Infectivity of the rSA11-s11KU-delNSP6 monoreassortant virus lacking NSP6 expression. (A) Single-step growth curves of rSA11-s11KU and rSA11-s11KU-delNSP6. MA104 cells were infected with rSA11-s11KU or rSA11-s11KU-delNSP6 at an MOI of 5 and then incubated for various times. Virus titers in the cultures were determined by plaque assay. The data shown are the mean viral titers and SDs for three independent cell cultures. (B) Plaque formation by rSA11-s11KU and rSA11-s11KU-delNSP6. rSA11-s11KU or rSA11-s11KU-delNSP6 was directly plated onto CV-1 cells for plaque formation. The experiment was repeated three times with similar results, and representative results are shown.
Growth properties of NSP6-deficient viruses in Vero E6 cells.
As RVAs actively suppress induction and signaling of type I interferons (IFN), i.e., IFN-α and IFN-β (27–29), we wondered whether the reduction in replication of the NSP6-deficient viruses in cell culture is due to IFN-α/β effects. To address this question, we infected Vero E6 cells that lack the IFN-α/β genes (30–32) with the rSA11 and rSA11-delNSP6 viruses at a high MOI of 5 PFU/cell to determine their single-step growth curves in IFN-α/β-deficient cells. The growth curves showed that the mutant rSA11-delNSP6 replicated less efficiently than the wild-type rSA11 (Fig. 5A), as seen for IFN-α/β-competent MA104 and CV-1 cells (33, 34) (Fig. 2A and 4A). The same tendency was found between the mutant rSA11-s11KU-delNSP6 and the wild-type rSA11-s11KU (Fig. 5B), suggesting that the reduced viral replication of the NSP6-deficient viruses is type I IFN independent, thereby pointing to a potentially different pathway.
FIG 5.

Single-step growth curves of the NSP6-deficient viruses in IFN-α/β-deficient Vero E6 cells. Vero E6 cells were infected with rSA11 or rSA11-delNSP6 (A) or with rSA11-s11KU or rSA11-s11KU-delNSP6 (B) at an MOI of 5 and then incubated for various times. The virus titers in the cultures were determined by plaque assay. The data shown are the mean viral titers and SDs from three independent cell cultures.
In the present study, we exploited a recently developed plasmid-based reverse genetics system to determine the significance of the NSP6 protein in the viral replication cycle. The generated NSP6-deficient viruses were viable and genetically stable. Single-step growth curves and plaque formation of the NSP6-deficient viruses confirmed that the NSP6 protein is not essential for viral replication in cell culture. Thus, the finding of few cell culture-adapted strains possessing defective or truncated NSP6 ORFs as well as the present results showed that the NSP6 protein is not essential for viral replication, at least in cell culture. However, the presence of the NSP6 ORF is highly conserved among RVA isolates from natural infections, suggesting an important but unknown role of the NSP6 protein in the viral replication cycle. In fact, the NSP6-deficient viruses in this study showed somewhat impaired growth properties in cell culture. Therefore, NSP6 expression is of limited significance for RVA replication in cell culture, but it may be important for viral replication in vivo where there are much more complex situations compared to those in cultured cells. Further studies using animal models will be important to confirm the role(s) of the NSP6 protein in viral replication and pathogenicity.
An entirely plasmid-based reverse genetics system can now be used for studying the mechanisms of RVA replication and pathogenesis. Here, we employed this approach to generate recombinant RVAs with engineered segment 11 to determine the significance of the NSP6 protein, since the current partial plasmid-based reverse genetics system has been successfully applied to only 3 of the 11 gene segments (VP4, NSP2, and NSP3 genes) (23). Two efficacious and safe live attenuated RVA vaccines (Rotarix and RotaTeq) are currently available; however, the molecular bases for the attenuation of these vaccine strains remain unknown. Therefore, it is expected that this reverse genetics approach will contribute to our understanding of the molecular bases of RVA replication and pathogenicity, as well as to the development of novel next-generation RVA vaccines with known attenuating mutations.
MATERIALS AND METHODS
Cells and viruses.
Baby hamster kidney cell lines, BHK/T7-9 (35) and BHK-T7/P5 (26), stably expressing T7 RNA polymerase (BHK-T7 cells) were cultured in Dulbecco's modified Eagle medium (DMEM) (Nissui) supplemented with 5% fetal calf serum (FCS) (Gibco) (complete medium) in the presence of 600 ng/ml hygromycin (BHK/T7-9) (Invitrogen) or 4 μg/ml puromycin (BHK-T7/P5) (Sigma-Aldrich). Monkey kidney cell lines, MA104, CV-1, and Vero E6 (CRL-1586; American Type Culture Collection), were cultured in complete medium. Simian RVA strain SA11-L2 (SA11) (G3P[2]) (36) and human RVA strain KU (G1P[8]) (37) were propagated as described previously (15). Briefly, the SA11 and KU viruses were pretreated with trypsin (type IX, from porcine pancreas) (10 μg/ml; Sigma-Aldrich) and then propagated in MA104 cells in Eagle's minimum essential medium (Nissui) without FCS (incomplete medium) but containing trypsin (1 μg/ml).
Construction of rescue T7 plasmids encoding full-length segment 11 (NSP5/6 genes).
To generate a construct for the rescue of the NSP6 ORF-silenced SA11 virus, T7-driven plasmid pT7-NSP5SA11 (26) was modified using a QuikChange II site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions. To generate plasmid pT7-NSP5SA11-delNSP6, the start codon and three downstream AUG codons in the NSP6 ORF of SA11 were disrupted (AUG to ACG) at nucleotide positions 80 to 82, 152 to 154, 257 to 259, and 326 to 328 in pT7-NSP5SA11. The coding sequence of the overlapping NSP5 ORF was not affected.
To construct T7-driven plasmid pT7/NSP5KU, which encodes the authentic segment 11 of the KU virus, the cDNA was amplified by RT-PCR from its genomic dsRNA with ReverTra Ace reverse transcriptase (Toyobo), PrimeStar HS DNA polymerase (TaKaRa Bio), and specific primers. As described previously (15), the forward primer contains the T7 RNA polymerase promoter sequence and a sequence corresponding to the 5′ terminus of the viral gene. The primer sequences will be provided on request. After digestion with restriction enzymes, the cDNA was ligated into the corresponding restriction enzyme site of T7 expression plasmid pX8dT (38). The constructed plasmid, pT7/NSP5KU, contains the full-length segment 11 cDNA of KU (GenBank/EMBL/DDBJ accession no. LC309019), flanked by the T7 RNA promoter and HDV ribozyme sequences, followed by the T7 RNA polymerase terminator sequence. To generate a construct for the rescue of the SA11-based monoreassortant virus having KU segment 11 incapable of NSP6 expression, pT7/NSP5KU was modified using a QuikChange II site-directed mutagenesis kit as described above. To generate plasmid pT7/NSP5KU-delNSP6, the start codon and five downstream AUG codons in the NSP6 ORF were disrupted (AUG to ACG) at nucleotide positions 80 to 82, 140 to 142, 158 to 160, 257 to 259, 272 to 274, and 278 to 280 in pT7/NSP5KU. The coding sequence of the overlapping NSP5 ORF was not affected. The nucleotide sequences of all of the constructed plasmids were sequenced to ensure the presence of the intended mutations and the absence of unwanted mutations.
An entirely plasmid-based reverse genetics system.
Recombinant RVAs entirely derived from cloned cDNAs were generated with a plasmid-only-based reverse genetics system (26), with some modifications. Briefly, monolayers of BHK-T7 cells in 6-well plates (Falcon) were cotransfected with 11 rescue T7 plasmids representing the cloned cDNAs of RVA 11 gene segments and FAST-expressing plasmids pT7-VP1SA11 (0.75 μg), pT7-VP2SA11 (0.75 μg), pT7-VP3SA11 (0.75 μg), pT7/VP4SA11-ΔPstI (0.75 μg) (15), pT7-VP6SA11 (0.75 μg), pT7-VP7SA11 (0.75 μg), pT7-NSP1SA11 (0.75 μg), pT7-NSP2SA11 (0.75 μg), pT7-NSP3SA11 (0.75 μg), pT7-NSP4SA11 (0.75 μg), and pCAG-FAST (0.02 μg), in combination with 0.75 μg of pT7-NSP5SA11, pT7-NSP5SA11-delNSP6, pT7/NSP5KU, or pT7/NSP5KU-delNSP6. Following 1 day of incubation, the transfected BHK-T7 cells were washed with incomplete medium and then cocultured with overlaid CV-1 cells for 3 days in incomplete medium containing trypsin (0.3 μg/ml). After incubation, the cultures were subjected to two cycles of freezing and thawing and then treated with trypsin (10 μg/ml) for RVA activation, followed by inoculation on MA104 cells. After 1 or 2 days of incubation, recombinant RVAs were rescued and then plaque purified in CV-1 cells as described previously (39).
Electrophoretic analysis of viral genomic dsRNAs.
Viral genomic dsRNAs were extracted from cell cultures using a QIAamp viral RNA minikit (Qiagen). The extracted viral dsRNAs were used for PAGE analysis. The dsRNAs were electrophoresed in a 10% polyacrylamide gel for 16 h at 20 mA at room temperature, followed by silver staining (15) to determine the genomic dsRNA profiles.
Generation of RVA-specific antisera.
Rabbit polyclonal anti-NSP6 serum was generated by BioGate. Two RVA-seronegative rabbits were immunized subcutaneously five times with mixed synthetic peptides (corresponding to 1-MNRLLQRQLFLENLLVGTN-19 of KU NSP6 and 74-HQHNHDLQVMSDAIKWISP-92 of SA11 NSP6) conjugated to keyhole limpet hemocyanin.
Rabbit polyclonal anti-NSP5 serum was generated by immunizing rabbits with the recombinant NSP5 protein. To construct an NSP5 expression plasmid, the full-length NSP5 ORF of SA11 was amplified by RT-PCR with specific primers containing restriction enzyme sites. The sequences of the primers will be provided on request. After digestion with restriction enzymes, the resultant PCR product was ligated into the corresponding site of the pGEX6P-3 vector (GE Healthcare) to yield plasmid pGEX-NSP5 expressing the entire NSP5 as a fusion protein with glutathione S-transferase (GST) at the N terminus. Escherichia coli XL1-Blue cells were transformed with pGEX-NSP5. The GST-NSP5 fusion protein was purified by glutathione Sepharose 4B column chromatography (GE Healthcare) as described previously (40). Two RVA-seronegative rabbits were immunized subcutaneously five times with the purified GST-NSP5 protein using standard procedures (Medical and Biological Laboratories).
Rabbit polyclonal anti-VP6 serum was generated by Medical and Biological Laboratories. Two RVA-seronegative rabbits were immunized subcutaneously five times with mixed synthetic peptides (corresponding to 134-EYIENWNLQNRRQRTGF-150 and 366-NWTDLITNYSPSREDNL-382 of SA11 VP6) conjugated to keyhole limpet hemocyanin.
The antisera were evaluated for sensitivity and specificity by enzyme-linked immunosorbent assay (ELISA) and immunoblotting against their respective RVA antigens and whole-cell lysates of RVA-infected cells.
Immunoblotting.
Immunoblotting was performed as described previously (41, 42). Briefly, monolayers of MA104 cells in 12-well plates (Falcon) were infected with trypsin-pretreated recombinant RVAs at an MOI of 3. Sixteen hours after infection, the infected cells were washed twice with phosphate-buffered saline (PBS) and then lysed in SDS-PAGE sample buffer (63 mM Tris-HCl [pH 6.8], 2% SDS, 5% sucrose, 5% 2-mercaptoethanol, and 0.002% bromophenol blue). Protein expression was assessed by SDS-PAGE (15%) and immunoblotting using RVA-specific polyclonal antiserum (anti-NSP6, -NSP5, or -VP6) or β-actin-specific monoclonal antiserum (Sigma-Aldrich). The antigen-antibody complexes were detected using a 1-step Ultra TMB blotting solution system (Thermo Fisher Scientific).
Single-step virus replication.
Monolayers of MA104 and Vero E6 cells in 12-well plates (Falcon) were infected in triplicate with trypsin-pretreated recombinant RVAs at an MOI of 5, washed twice with incomplete medium, and then incubated for various times in incomplete medium without trypsin. The infected cells were frozen and thawed twice before determination of virus titers by plaque assay.
Plaque assay.
The plaque assays were performed as described previously (43). Briefly, monolayers of CV-1 cells were infected with trypsin-pretreated recombinant RVAs, washed twice with incomplete medium, and then cultured with trypsin (1 μg/ml) in primary overlay medium (0.7% agarose). After 2 days, the infected cells were stained with secondary overlay medium containing 0.7% agarose and 0.005% neutral red (Sigma-Aldrich). Plaque sizes were determined by measuring the mean diameters of 25 plaques in two independent assays.
Accession number(s).
The sequence determined in the course of this work was deposited in GenBank/EMBL/DDBJ under accession number LC309019.
ACKNOWLEDGMENTS
We thank Chihiro Yamashiro, Naoko Nagasawa, and Misa Onishi for their technical assistance.
This study was supported in part by the MEXT-Supported Program for the Research Program on Emerging and Re-emerging Infectious Diseases of the Japan Agency for Medical Research and Development, AMED (K.T. and S.K.), and JSPS KAKENHI grant number 15K08505 (S.K.).
We have no conflicts of interest to declare.
REFERENCES
- 1.Tate JE, Burton AH, Boschi-Pinto C, Parashar UD, World Health Organization-Coordinated Global Rotavirus Surveillance Network. 2016. Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis 62(Suppl 2):S96–S105. doi: 10.1093/cid/civ1013. [DOI] [PubMed] [Google Scholar]
- 2.Estes MK, Greenberg HB. 2013. Rotaviruses, p 1347–1401. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Mattion NM, Mitchell DB, Both GW, Estes MK. 1991. Expression of rotavirus proteins encoded by alternative open reading frames of genome segment 11. Virology 181:295–304. doi: 10.1016/0042-6822(91)90495-W. [DOI] [PubMed] [Google Scholar]
- 4.Rainsford EW, McCrae MA. 2007. Characterization of the NSP6 protein product of rotavirus gene 11. Virus Res 130:193–201. doi: 10.1016/j.virusres.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 5.González RA, Torres-Vega MA, López S, Arias CF. 1998. In vivo interactions among rotavirus nonstructural proteins. Arch Virol 143:981–996. doi: 10.1007/s007050050347. [DOI] [PubMed] [Google Scholar]
- 6.Holloway G, Johnson RI, Kang Y, Dang VT, Stojanovski D, Coulson BS. 2015. Rotavirus NSP6 localizes to mitochondria via a predicted N-terminal α-helix. J Gen Virol 96:3519–3524. doi: 10.1099/jgv.0.000294. [DOI] [PubMed] [Google Scholar]
- 7.Torres-Vega MA, González RA, Duarte M, Poncet D, López S, Arias CF. 2000. The C-terminal domain of rotavirus NSP5 is essential for its multimerization, hyperphosphorylation and interaction with NSP6. J Gen Virol 81:821–830. doi: 10.1099/0022-1317-81-3-821. [DOI] [PubMed] [Google Scholar]
- 8.Viskovska M, Anish R, Hu L, Chow DC, Hurwitz AM, Brown NG, Palzkill T, Estes MK, Prasad BV. 2014. Probing the sites of interactions of rotaviral proteins involved in replication. J Virol 88:12866–12881. doi: 10.1128/JVI.02251-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patton JT. 2001. Rotavirus RNA replication and gene expression. Novartis Found Symp 238:64–81. doi: 10.1002/0470846534.ch5. [DOI] [PubMed] [Google Scholar]
- 10.Wu H, Taniguchi K, Urasawa T, Urasawa S. 1998. Serological and genomic characterization of human rotaviruses detected in China. J Med Virol 55:168–176. doi:. [DOI] [PubMed] [Google Scholar]
- 11.Gorziglia M, Nishikawa K, Fukuhara N. 1989. Evidence of duplication in super short segment 11 of rabbit rotavirus Alabama strain. Virology 170:587–590. doi: 10.1016/0042-6822(89)90453-4. [DOI] [PubMed] [Google Scholar]
- 12.González SA, Burrone OR. 1989. Porcine OSU rotavirus segment 11 sequence shows common features with the viral gene of human origin. Nucleic Acids Res 17:6402. doi: 10.1093/nar/17.15.6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.López T, Rojas M, Ayala-Bretón C, López S, Arias CF. 2005. Reduced expression of the NSP5 gene has a pleiotropic effect on virus replication. J Gen Virol 86:1609–1617. doi: 10.1099/vir.0.80827-0. [DOI] [PubMed] [Google Scholar]
- 14.Johne R, Reetz J, Kaufer BB, Trojnar E. 2015. Generation of an avian-mammalian rotavirus reassortant by using a helper virus-dependent reverse genetics system. J Virol 90:1439–1443. doi: 10.1128/JVI.02730-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komoto S, Sasaki J, Taniguchi K. 2006. Reverse genetics system for introduction of site-specific mutations into the double-stranded RNA genome of infectious rotavirus. Proc Natl Acad Sci U S A 103:4646–4651. doi: 10.1073/pnas.0509385103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navarro A, Trask SD, Patton JT. 2013. Generation of genetically stable recombinant rotaviruses containing novel genome rearrangements and heterologous sequences by reverse genetics. J Virol 87:6211–6220. doi: 10.1128/JVI.00413-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trask SD, Taraporewala ZF, Boehme KW, Dermody TS, Patton JT. 2010. Dual selection mechanisms drive efficient single-gene reverse genetics for rotavirus. Proc Natl Acad Sci U S A 107:18652–18657. doi: 10.1073/pnas.1011948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Troupin C, Dehée A, Schnuriger A, Vende P, Poncet D, Garbarg-Chenon A. 2010. Rearranged genomic RNA segments offer a new approach to the reverse genetics of rotaviruses. J Virol 84:6711–6719. doi: 10.1128/JVI.00547-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyce M, Celma CC, Roy P. 2008. Development of reverse genetics systems for bluetongue virus: recovery of infectious virus from synthetic RNA transcripts. J Virol 82:8339–8348. doi: 10.1128/JVI.00808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaname Y, Celma CC, Kanai Y, Roy P. 2013. Recovery of African horse sickness virus from synthetic RNA. J Gen Virol 94:2259–2265. doi: 10.1099/vir.0.055905-0. [DOI] [PubMed] [Google Scholar]
- 21.Kawagishi T, Kanai Y, Tani H, Shimojima M, Saijo M, Matsuura Y, Kobayashi T. 2016. Reverse genetics for fusogenic bat-borne orthoreovirus associated with acute respiratory tract infections in humans: role of outer capsid protein σC in viral replication and pathogenesis. PLoS Pathog 12:e1005455. doi: 10.1371/journal.ppat.1005455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi T, Antar AAR, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komoto S, Taniguchi K. 2013. Genetic engineering of rotaviruses by reverse genetics. Microbiol Immunol 57:479–486. [DOI] [PubMed] [Google Scholar]
- 24.Matsuo E, Saeki K, Roy P, Kawano J. 2015. Development of reverse genetics for Ibaraki virus to produce viable VP6-tagged IBAV. FEBS Open Biol 5:445–453. doi: 10.1016/j.fob.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taniguchi K, Komoto S. 2012. Genetics and reverse genetics of rotavirus. Curr Opin Virol 2:399–407. doi: 10.1016/j.coviro.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Kanai Y, Komoto S, Kawagishi T, Nouda R, Nagasawa N, Onishi M, Matsuura Y, Taniguchi K, Kobayashi T. 2017. Plasmid-based reverse genetics for rotavirus allows manipulation of the viral genome and development of efficient gene-transduction systems. Proc Natl Acad Sci U S A 114:2349–2354. doi: 10.1073/pnas.1618424114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holloway G, Coulson BS. 2013. Innate cellular responses to rotavirus infection. J Gen Virol 94:1151–1160. doi: 10.1099/vir.0.051276-0. [DOI] [PubMed] [Google Scholar]
- 28.López S, Sánchez-Tacuba L, Moreno J, Arias CF. 2016. Rotavirus strategies against the innate antiviral system. Annu Rev Virol 3:591–609. doi: 10.1146/annurev-virology-110615-042152. [DOI] [PubMed] [Google Scholar]
- 29.Morelli M, Ogden KM, Patton JT. 2015. Silencing the alarms: innate immune antagonism by rotavirus NSP1 and VP3. Virology 479–480:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD. 1988. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci U S A 85:5259–5263. doi: 10.1073/pnas.85.14.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosca JD, Pitha PM. 1986. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol Cell Biol 6:2279–2283. doi: 10.1128/MCB.6.6.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osada N, Kohara A, Yamaji T, Hirayama N, Kasai F, Sekizuka T, Kuroda M, Hanada K. 2014. The genome landscape of the African green monkey kidney-derived Vero cell line. DNA Res 21:673–683. doi: 10.1093/dnares/dsu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKimm-Breschkin JL, Holmes IH. 1982. Conditions required for induction of interferon by rotaviruses and for their sensitivity to its action. Infect Immun 36:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thacore HR, Lin HY, Davis PJ, Schoenl M. 1990. Effect of protein kinase C inhibitors on interferon-beta production by viral and nonviral inducers. J Gen Virol 71:2833–2839. doi: 10.1099/0022-1317-71-12-2833. [DOI] [PubMed] [Google Scholar]
- 35.Ito N, Takayama-Ito M, Yamada K, Hosokawa J, Sugiyama M, Minamoto N. 2003. Improved recovery of rabies virus from cloned cDNA using a vaccinia virus-free reverse genetics system. Microbiol Immunol 47:613–617. doi: 10.1111/j.1348-0421.2003.tb03424.x. [DOI] [PubMed] [Google Scholar]
- 36.Taniguchi K, Nishikawa K, Kobayashi N, Urasawa T, Wu H, Gorziglia M, Urasawa S. 1994. Differences in plaque size and VP4 sequence found in SA11 virus clones having simian authentic VP4. Virology 198:325–330. doi: 10.1006/viro.1994.1035. [DOI] [PubMed] [Google Scholar]
- 37.Urasawa S, Urasawa T, Taniguchi K, Chiba S. 1984. Serotype determination of human rotavirus isolates and antibody prevalence in pediatric population in Hokkaido, Japan. Arch Virol 81:1–12. doi: 10.1007/BF01309292. [DOI] [PubMed] [Google Scholar]
- 38.Schnell MJ, Mebatsion T, Conzelmann KK. 1994. Infectious rabies viruses from cloned cDNA. EMBO J 13:4195–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taniguchi K, Morita Y, Urasawa T, Urasawa S. 1987. Cross-reactive neutralization epitopes on VP3 of human rotavirus: analysis with monoclonal antibodies and antigenic variants. J Virol 61:1726–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komoto S, Kinomoto M, Ibrahim MS, Zhong Q, Auwanit W, Ayuthaya PI, Otake T, Mori H, Oishi I, Kurosu T, Takahashi H, Mukai T, Ikuta K. 2001. Low or no antibody responses to human immunodeficiency virus type 1 Nef in infected carriers with subtype E, in contrast to subtype B that showed antibodies preferentially recognizing subtype-specific Nef epitopes. Vaccine 19:3019–3032. doi: 10.1016/S0264-410X(00)00444-8. [DOI] [PubMed] [Google Scholar]
- 41.Komoto S, Kinomoto M, Horikoshi H, Shiraga M, Kurosu T, Mukai T, Auwanit W, Otake T, Oishi I, Ikuta K. 2002. Ability to induce p53 and caspase-mediated apoptosis in primary CD4+ T cells is variable among primary isolates of human immunodeficiency virus type 1. AIDS Res Hum Retrovir 18:435–446. doi: 10.1089/088922202753614209. [DOI] [PubMed] [Google Scholar]
- 42.Komoto S, Wakuda M, Ide T, Niimi G, Maeno Y, Higo-Moriguchi K, Taniguchi K. 2011. Modification of the trypsin cleavage site of rotavirus VP4 to a furin-sensitive form does not enhance replication efficiency. J Gen Virol 92:2914–2921. doi: 10.1099/vir.0.033886-0. [DOI] [PubMed] [Google Scholar]
- 43.Urasawa S, Urasawa T, Taniguchi K. 1982. Three human rotavirus serotypes demonstrated by plaque neutralization of isolated strains. Infect Immun 38:781–784. [DOI] [PMC free article] [PubMed] [Google Scholar]