

ABSTRACT
The K15P membrane protein of Kaposi's sarcoma-associated herpesvirus (KSHV) interacts with multiple cellular signaling pathways and is thought to play key roles in KSHV-associated endothelial cell angiogenesis, regulation of B-cell receptor (BCR) signaling, and the survival, activation, and proliferation of BCR-negative primary effusion lymphoma (PEL) cells. Although full-length K15P is ∼45 kDa, numerous lower-molecular-weight forms of the protein exist as a result of differential splicing and poorly characterized posttranslational processing. K15P has been reported to localize to numerous subcellular organelles in heterologous expression studies, but there are limited data concerning the sorting of K15P in KSHV-infected cells. The relationships between the various molecular weight forms of K15P, their subcellular distribution, and how these may differ in latent and lytic KSHV infections are poorly understood. Here we report that a cDNA encoding a full-length, ∼45-kDa K15P reporter protein is expressed as an ∼23- to 24-kDa species that colocalizes with the trans-Golgi network (TGN) marker TGN46 in KSHV-infected PEL cells. Following lytic reactivation by sodium butyrate, the levels of the ∼23- to 24-kDa protein diminish, and the full-length, ∼45-kDa K15P protein accumulates. This is accompanied by apparent fragmentation of the TGN and redistribution of K15P to a dispersed peripheral location. Similar results were seen when lytic reactivation was stimulated by the KSHV protein replication and transcription activator (RTA) and during spontaneous reactivation. We speculate that expression of different molecular weight forms of K15P in distinct cellular locations reflects the alternative demands placed upon the protein in the latent and lytic phases.
IMPORTANCE The K15P protein of Kaposi's sarcoma-associated herpesvirus (KSHV) is thought to play key roles in disease, including KSHV-associated angiogenesis and the survival and growth of primary effusion lymphoma (PEL) cells. The protein exists in multiple molecular weight forms, and its intracellular trafficking is poorly understood. Here we demonstrate that the molecular weight form of a reporter K15P molecule and its intracellular distribution change when KSHV switches from its latent (quiescent) phase to the lytic, infectious state. We speculate that expression of different molecular weight forms of K15P in distinct cellular locations reflects the alternative demands placed upon the protein in the viral latent and lytic stages.
KEYWORDS: K15P, Kaposi's sarcoma-associated herpesvirus, latent and lytic infection, subcellular localization
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiological agent of AIDS-associated and endemic Kaposi's sarcoma (KS) (1), a highly angiogenic and invasive tumor of endothelial origin. KSHV is also associated with the B-cell lymphoproliferative disorders primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) (2–8). K15, a nonstructural membrane-associated protein encoded by 8 alternatively spliced exons located at the “right end” of the long unique region of the KSHV genome (9, 10), is thought to play important roles in the progression of KS, PEL, and MCD (2, 8, 11, 12).
K15 exists as two major alleles, K15P (predominant) and K15M (minor) (13–16); for both K15P and K15M, the 8-exon message encodes an ∼45-kDa protein with 12 predicted membrane-spanning domains and a C-terminal cytoplasmic tail. The tail recruits several cellular proteins (10, 17–19), enabling K15 to influence numerous signaling pathways (15–17, 20, 21; reviewed in references 2 and 11). In KSHV-infected endothelial cells, this appears to result in increased angiogenesis and invasiveness (22, 23). In B cells, the major reservoir of KSHV in healthy seropositive hosts (24, 25), K15 is thought to downregulate signaling through the B-cell receptor (BCR) (9, 21, 26), which may help to maintain KSHV latency or suppress apoptosis (2, 24). When KSHV-infected B cells transform into primary effusion lymphomas (27), BCR expression is lost, giving a situation that is normally incompatible with survival (28, 29). In these BCR-negative PEL cells, K15 (and another KSHV-encoded protein, K1) may act redundantly to provide survival, activation, and proliferation functions (30), playing a role similar to that of latent membrane protein 2A (LMP2A) of Epstein-Barr virus (EBV) (10, 11, 30, 31).
Consistent with roles in both KSHV latency and lytic replication, the K15 transcript is expressed in a population of mostly latent PEL cells, and transcript levels are induced upon lytic reactivation (9, 10, 13, 16, 17, 32–35). Furthermore, in the PEL cell line BCBL-1, the phorbol ester tetradecanoyl phorbol acetate (TPA) and the KSHV lytic switch protein replication and transcription activator (RTA) (36) both activate the K15 promoter (35). In endothelia, it remains unclear whether the role of K15 in angiogenesis reflects its function in latently infected cells or in those undergoing lytic gene expression (22, 37, 38).
The predicted product of the 8-exon K15P open reading frame (ORF) is a polypeptide of ∼45 kDa, and K15P migrates at this expected molecular mass when expressed by in vitro translation (18) in heterologous hosts, such as COS or HEK293 cells (9, 17, 20), and when immunoprecipitated from KSHV-infected endothelial cells (23). However, in KSHV-infected BC3 and JSC-1 PEL cells, the major form of K15P is 23 to 24 kDa (18). Similarly, for primary human umbilical vein endothelial cells and the endothelial cell line IE7, K15P runs as ∼35-kDa and ∼24-kDa species (18). In all cases, the smaller forms of K15P correspond to the carboxy-terminal portion of the molecule, since they have been detected by Western blotting with antibodies against the cytoplasmic tail (18, 20). The ∼35-kDa and ∼24-kDa forms of K15P do not necessarily arise from alternative splicing of the 8-exon K15P transcript (10, 20), since they are expressed even by the 8-exon full-length K15P cDNA (18). Removal of the initiation codon from the start of the 8-exon K15P ORF abolishes production of the lower-molecular-weight forms of K15P (18); this implies that they are not generated by translational initiation at internal start codons in the spliced mRNA but instead suggests that the full-length, ∼45-kDa K15P protein is a precursor to the lower-molecular-weight species. It has been suggested that ∼45-kDa K15P might be processed by multiple cleavages at internal signal sequences (18); however, signal peptidase is unlikely to be the processing enzyme, since it is believed to cleave only once, following a single amino-terminal signal sequence (39, 40).
Despite the ability of the full-length K15P cDNA to generate lower-molecular-weight forms of the K15P protein, it is also clear that differential splicing of the K15P transcript does occur (9, 20). In addition to the 8-exon full-length transcript, an abundant K15P mRNA with the capacity to encode an ∼36-kDa form of K15P is expressed in unstimulated PEL cells (10). Moreover, several less abundant splice variants can give rise to K15P proteins of 21 kDa, 26 kDa, 33 kDa, 34 kDa, and 35 kDa (20). Like full-length, ∼45-kDa K15P, a subpopulation of the lower-molecular-weight isoforms are found localized to lipid rafts, consistent with the possibility that they also assemble into signaling complexes (20). Indeed, it has been suggested that the lower-molecular-weight forms of the protein might serve to modulate the activity of full-length K15P (9, 20). Nevertheless, the full-length 8-exon K15P cDNA is sufficient to provide the BCR-like activity that supports survival, activation, and proliferation of BCR-negative human B cells (30), and it can also drive capillary tube formation through the phospholipase Cγ1 (PLCγ1)-calcineurin-NFAT pathway in a human umbilical vein endothelial cell model of angiogenesis (22).
Another poorly understood aspect of the biology of the K15P protein concerns its intracellular localization. Transient-transfection studies with HeLa, COS, and 293 cells have been interpreted to suggest that K15P localizes to the endoplasmic reticulum (ER) (18), the Golgi apparatus and the plasma membrane (9), the mitochondria (16), or other, undefined dispersed punctate structures (10, 41). Similar studies have reported a dispersed punctate or lysosomal location for K15M (15, 16). The subcellular localization of K15 in KSHV-infected endothelial or B/PEL cells is the most relevant to its biological function, and in KSHV-infected endothelial cells, endogenously expressed K15P exhibits a dispersed punctate intracellular distribution (23) resembling that seen in butyrate-induced KSHV bacterial artificial chromosome (BAC)-transformed 293 cells (17), though K15P colocalization with organellar markers was not examined in the previous studies. The K15 protein has been reported to localize to endosomes in transfected B cells (26) and to be expressed in PEL cells (8, 16, 18, 30), but the intracellular distribution of K15 in KSHV-infected B or PEL cells has not been reported.
In this study, we examined the expressed forms and intracellular distribution of a K15P reporter molecule in a KSHV-infected PEL cell line and tested the response of the protein to lytic reactivation of KSHV. We found that the full-length 8-exon K15P cDNA is expressed predominantly as an ∼23- to 24-kDa protein, as reported by others (18), and substantially colocalizes with a marker of the trans-Golgi network (TGN). Upon lytic reactivation by sodium butyrate treatment, levels of the ∼23- to 24-kDa protein diminish, and the full-length, ∼45-kDa protein accumulates. This is accompanied by redistribution of K15P to a dispersed peripheral location and apparent fragmentation of the TGN. A similar K15P relocalization is seen when lytic reactivation is stimulated by transfection with the lytic activator RTA and during spontaneous lytic reactivation of KSHV in untreated PEL cells.
RESULTS
Expression of HA-tagged full-length K15P cDNA in KSHV-infected PEL cells.
We prepared a recombinant lentivirus capable of expressing the 8-exon K15P cDNA carboxy-terminally tagged with two copies of the influenza virus hemagglutinin epitope (K15P-HA). In this lentivirus, the K15P-HA gene is transcribed on a bicistronic message located 5′ to an internal ribosome entry site (IRES) sequence (42, 43) and a neomycin resistance marker; this helps to ensure stable K15P expression when cells are maintained in the presence of G418. The lentiviruses were used to prepare K15P-HA-expressing versions of the latently KSHV-infected PEL cell line BCBL-1 (44) (termed BCBL-1-HA) and, for comparison, the KSHV-negative Burkitt's lymphoma cell line BJAB (45) (termed BJAB-HA).
The full-length 8-exon K15P cDNA is predicted to encode a polypeptide of ∼45 kDa. Following preparation of cell extracts and Western blotting for the HA tag, BCBL-1-HA and BJAB-HA cells were found to express an anti-HA-reactive doublet band at ∼23 to 24 kDa that was absent from parental BCBL-1 and BJAB cells (Fig. 1A). Similar results were previously reported for expression of the full-length K15P cDNA in HeLa and endothelial cells, in which the predominant K15P product was ∼23 kDa (18). Our data suggest that in PEL cells, as in the other cell types mentioned, an ∼23/24-kDa C-terminally derived form of K15P can arise from the full-length K15P ORF in the absence of alternative splicing. Since we observed identical results for BCBL-1-HA and BJAB-HA cells (Fig. 1A), we concluded that this form of K15P can be generated in B-cell lymphomas in the absence of KSHV infection. We similarly used recombinant lentiviruses to prepare a K15P-HA-expressing clone of the KSHV/EBV-infected PEL cell line JSC-1 (46, 47), and also a BCBL-1-derived cell line expressing a hexahistidine-tagged form of K15P. In both cases, we observed results similar to those described here (data not shown).
FIG 1.
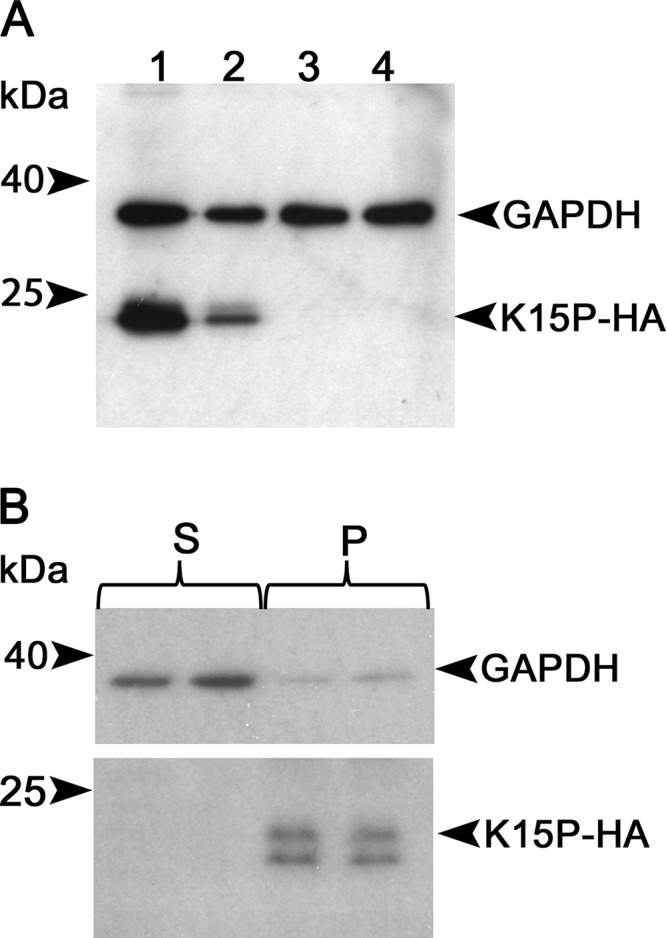
Expression of the K15P 8-exon cDNA in PEL cells. (A) RIPA extracts of cells were subjected to denaturing SDS-PAGE and Western blotted with anti-HA and anti-GAPDH antibodies (loading control), as indicated at right. Lanes: 1, BCBL-1-HA; 2, BJAB-HA; 3, BCBL-1; 4, BJAB. Positions of molecular size markers are indicated at left. (B) A PNS was prepared from BCBL-1-HA cells, and duplicate samples were subjected to centrifugation at 50,000 × g to prepare pellet (P) or supernatant (S) fractions. After SDS-PAGE and Western blotting, the membrane was probed exactly as described for panel A.
Brinkmann and colleagues reported that PEL cells can express ∼21- to 23-kDa forms of K15P that are not associated with cellular membranes (20). To test whether the ∼23- to 24-kDa K15P-HA doublet we observed was membrane bound, we prepared a postnuclear supernatant (PNS) from BCBL-1-HA cells and then collected a membranous organelle-enriched 50,000 × g pellet and soluble supernatant. The ∼23- to 24-kDa forms of K15P-HA were present in the pellet fraction, consistent with their membrane localization, and absent from the supernatant, which was enriched in the soluble cytosolic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Fig. 1B). The higher resolution of the two lower-molecular-weight species of K15P-HA in Fig. 1B compared to that in Fig. 1A reflects a longer time of gel electrophoresis and differing salt/detergent conditions in the extracts.
K15P-HA does not colocalize with lysosomal or mitochondrial markers following density gradient centrifugation.
Although K15P appears to be capable of influencing numerous signaling pathways in KSHV-infected endothelial cells, B cells, and PEL cells (2, 11, 15–17, 20–25), there is no known extracellular ligand that regulates K15P activity, and it is unclear whether the protein localizes to the cell surface or intracellular organelles (9, 10, 16, 18, 41). To examine the cellular localization of K15P in the context of KSHV-infected PEL cells, we prepared a BCBL-1-HA cell PNS and centrifuged it through an iodixanol density gradient. Fractions were then collected and Western blotted for HA-tagged K15P and organellar markers. As shown in Fig. 2, the K15P-HA distribution was distinct from those of markers of the mitochondria (second mitochondrion-derived activator of caspase [SMAC]) and lysosomes (cathepsin B [CB]), suggesting that K15P-HA was not resident in those organelles. However, these density gradient fractionation conditions could not separate K15P-HA-containing organelles from the endoplasmic reticulum (protein disulfide isomerase [PDI]), the Golgi apparatus (p58K) (48), and the plasma membrane (Na+/K+ ATPase) (Fig. 2).
FIG 2.

K15P-HA does not colocalize with lysosomal or mitochondrial markers in a density fractionation gradient. The PNS from BCBL-1-HA cells was loaded at the top of a 2.5% to 30% iodixanol gradient and subjected to ultracentrifugation and then SDS-PAGE and Western blotting of fractions from top (fraction 1) to bottom (fraction 24). Blots were probed with antibodies against K15P-HA (HA), the endoplasmic reticulum (PDI), the Golgi apparatus (p58K), the plasma membrane (Na/K [Na+/K+ ATPase]), mitochondria (SMAC), and lysosomes (CB [cathepsin B]), as indicated at right. Positions of molecular size markers are indicated at left.
K15P-HA staining overlaps the trans-Golgi network but not the ER, mitochondria, or plasma membrane.
We further investigated the intracellular location of K15P-HA by using immunocytochemistry. Since PEL cells are small and round, with a narrow volume of cytoplasm surrounding the nucleus, we used cytospinning to flatten the cells prior to fixation and optically sectioned them by using confocal microscopy. Following DAPI (4′,6-diamidino-2-phenylindole) staining and immunostaining, we observed HA immunoreactivity in a compact juxtanuclear location in BCBL-1-HA cells; anti-HA staining was not seen in control BCBL-1 cells under identical conditions (Fig. 3, top row). Fields of BCBL-1-HA and control BCBL-1 cells were then stained for antigens of the plasma membrane (Fig. 3A and B), mitochondria (Fig. 3C and D), trans-Golgi network (Fig. 3E and F), and endoplasmic reticulum (Fig. 3G and H). We observed substantial colocalization of K15P-HA immunoreactivity with the trans-Golgi network antigen TGN46 (Fig. 3E) but not with any other organellar markers tested.
FIG 3.

Localization of K15P-HA in BCBL-1-HA cells. The top row shows BCBL-1 (left) and BCBL-1-HA (right) cells stained with DAPI (blue) and anti-HA antibody (green). Insets show higher-magnification images of cells from similar fields. The lower panels show merged images of fields of BCBL-1-HA (A, C, E, and G) and control BCBL-1 (B, D, F, and H) cells stained with DAPI (blue) and immunostained for K15P-HA (green) and an organellar marker (red). Organellar antigens label the plasma membrane (Na+/K+ ATPase) (A and B), mitochondria (SMAC) (C and D), the trans-Golgi network (TGN46) (E and F), and the endoplasmic reticulum (calnexin) (G and H). For each BCBL-1-HA merged image (A, C, E, and G), the individual green and red channels are shown to the left. Bars, 20 μm.
Induction of the lytic program with sodium butyrate results in increased levels of the higher-molecular-weight form of K15P-HA and its relocalization to the cell periphery.
K15P has been suggested to play roles during both latency and lytic replication (9, 10, 17, 34, 35). We therefore next examined the K15P-HA protein during incubation of BCBL-1-HA cells with sodium butyrate, using conditions known to induce KSHV lytic reactivation (44, 46, 49). We observed that over a time course of 48 h in the presence of 1 mM sodium butyrate, the ∼23/24-kDa forms of K15P-HA gradually diminished and a protein with the predicted ∼45-kDa molecular mass of full-length K15P-HA accumulated (Fig. 4A). In contrast, identical treatment of the K15P-HA-expressing KSHV-negative B-cell lymphoma line BJAB-HA did not result in comparable accumulation of the putative full-length form of K15P-HA (Fig. 4A). For both cell lines, levels of the control cellular protein GAPDH remained approximately constant, suggesting that there were no gross effects on cellular gene expression or cell viability during the time course of the experiment. Immunocytochemical examination of BCBL-1-HA cells over a similar time course revealed that K15P-HA staining intensity substantially increased during the 48 h of incubation with sodium butyrate (Fig. 4B to D). As the ∼23/24-kDa form of K15P-HA became replaced by the higher-molecular-weight species (Fig. 4A), the protein relocalized from its compact Golgi-like staining pattern, where it colocalized with TGN46 (Fig. 4B), to a more dispersed and peripheral location (Fig. 4C and D). To specifically identify cells that had undergone KSHV lytic reactivation, the 48-h sodium butyrate-treated BCBL-1-HA cells were also immunostained for the nuclear KSHV-encoded lytic replication-associated protein K8α (RAP/K-bZIP) (50). We observed that the majority of cells exhibiting nuclear anti-K8α staining also showed elevated levels of K15P-HA immunostaining in a dispersed punctate pattern (Fig. 4D). Conversely, few of the cells lacking anti-K8α staining (and in which KSHV presumably remained latent or was at an earlier stage in its lytic reactivation program) exhibited elevated levels of K15P-HA expression (Fig. 4D). As expected, anti-K8α immunostaining was not observed in KSHV-negative BJAB cells following 48 h of sodium butyrate treatment (Fig. 4E). We also observed that in BCBL-1-HA cells, the sodium butyrate treatment resulted in relocation of TGN46 staining from its compact juxtanuclear location (Fig. 4B) to a more dispersed and dimmer pattern that in many cells became difficult to detect (Fig. 4C to E).
FIG 4.

Effects of sodium butyrate treatment on K15P-HA expression and localization. (A) Western blot for K15P-HA following sodium butyrate treatment. BCBL-1-HA, BJAB-HA, and parental BCBL-1 and BJAB cells were incubated with 1 mM sodium butyrate for 0, 24, or 48 h (as indicated above the lanes). Extracts were subjected to SDS-PAGE, Western blotted, and probed with anti-HA or anti-GAPDH antibody, as indicated at right. (B to D) Merged images of fields of BCBL-1-HA cells fixed and immunostained after incubation with sodium butyrate for 0 h (B), 30 h (C), or 48 h (D). (E) Control BJAB cells similarly incubated with sodium butyrate for 48 h. All fields were immunostained for TGN46 (red channel) and HA (green channel). Fields in panels D and E were also stained for the KSHV lytically expressed protein K8α (white channel). The anti-HA immunostaining in panel B is weaker than that shown for BCBL-1-HA cells in Fig. 3 because imaging conditions were chosen to avoid saturation of the green channel resulting from elevated expression of K15P-HA in panels C and D. Bars, 20 μm.
We further examined the dispersed pattern of K15P-HA staining in lytically reactivated BCBL-1-HA cells. In K8α-expressing cells, K15P-HA was found in a punctate distribution surrounding the nucleus (Fig. 5A to C), and in some optical sections, it also appeared to lie close to the periphery (Fig. 5D). However, in no cases did we observe colocalization of dispersed K15P-HA with the plasma membrane (Fig. 5A), mitochondria (Fig. 5C), or endoplasmic reticulum (Fig. 5D). As noted above (and see Fig. 4D and E), TGN46 immunoreactivity also dispersed from its compact Golgi-like juxtanuclear location in K8α-positive BCBL-1-HA cells (Fig. 5B). However, the staining patterns of TGN46 and K15P-HA were now distinct and no longer colocalized (Fig. 5B). We quantitated the frequencies of occurrence of the K15P-HA and TGN46 staining patterns under these conditions (and others, as described below; see Fig. 8).
FIG 5.

Effects of sodium butyrate treatment on K15P-HA localization in BCBL-1 and BJAB cells. (A to D) BCBL-1-HA cells were incubated with sodium butyrate for 48 h and then fixed and immunostained using antibodies against the KSHV lytically expressed protein K8α (white channel), HA (green channel), and the organellar markers Na+/K+ ATPase (A), TGN46 (B), SMAC (C), and calnexin (D) (red channel). For each BCBL-1-HA merged image, the individual red, white, and green channels are shown to the left. (E to H) BJAB-HA or control BJAB cells were immunostained for HA (green channel) and the plasma membrane marker Na+/K+ ATPase or the TGN marker TGN46 (red channel), as indicated. For each BJAB-HA merged image, the individual red and green channels are shown to the left. (I to L) Identical to panels E to H except that cells were incubated with sodium butyrate for 48 h before fixation and immunostaining. Bars, 20 μm.
FIG 8.

Quantitation of K15P-HA and TGN46 staining patterns. (A) Fields of BCBL-1-HA cells were immunostained for HA and K8α. The numbers of cells displaying compact (white bars), intermediate (gray bars), and dispersed (black bars) patterns of HA immunoreactivity (see the text for more details) were counted and plotted as percentages of the total. Cell conditions are indicated below each set of bars, as follows: latent, untreated cells that were K8α negative (250 cells counted from 4 fields); K8α+ (butyrate), cells that were K8α positive following incubation with 1 mM sodium butyrate for 48 h (97 cells counted from 6 fields); and K8α+ (spontaneous), untreated cells that were K8α positive (17 cells counted from 13 fields). (B) Identical to panel A except that cells were stained and their morphology scored for TGN46. Cell numbers were as follows: latent, 214 cells counted from 17 fields; K8α+ (butyrate), 91 cells counted from 9 fields; and K8α+ (spontaneous), 22 cells counted from 18 fields. The small numbers of spontaneously occurring K8α-positive cells that were counted reflect the rare nature of the phenotype.
We next tested the intracellular localization of K15P-HA in BJAB-HA cells. As in BCBL-1-HA cells, K15P-HA showed a compact juxtanuclear staining pattern that substantially overlapped TGN46 (Fig. 5E and F) but not markers of the plasma membrane (Fig. 5G and H) or mitochondria (data not shown). However, in contrast to that of BCBL-1-HA cells, incubation of BJAB-HA cells with sodium butyrate for 48 h did not result in redistribution of TGN46 or K15P-HA staining in these KSHV-negative lymphoma cells (Fig. 5I to L). We also did not observe an increase in the intensity of K15P-HA fluorescence staining in BJAB-HA cells following sodium butyrate treatment (data not shown); this is consistent with our finding that full-length K15P-HA expression was not upregulated under these conditions in BJAB-HA cells (Fig. 4A).
Consequences of initiating KSHV lytic reactivation with RTA.
Although sodium butyrate is routinely used to trigger KSHV lytic replication, it can have nonspecific and pleiotropic effects on cellular gene expression and function. As an alternative approach, we made use of the plasmid pSG5-Rta (51), which expresses the KSHV Orf50-encoded protein RTA. RTA is necessary and sufficient to trigger the full KSHV lytic program, including ordered expression of viral lytic proteins and the release of viral progeny (50–52). As shown in Fig. 6A, after electroporation of BCBL-1-HA cells to introduce pSG5-Rta, the levels of an ∼45-kDa anti-HA-reactive species gradually increased over time, though less dramatically than the results following sodium butyrate treatment. Levels of the ∼23/24-kDa form of K15P-HA were also somewhat reduced after 24 h (Fig. 6A). In a separate experiment, BCBL-1-HA and BCBL-1 cells were electroporated with no DNA, a control plasmid, or two different amounts of pSG5-Rta. After 48 h, we observed a pSG5-Rta dose-dependent increase in the ∼45-kDa form of K15P-HA in BCBL-1-HA cells (Fig. 6B).
FIG 6.

Effects of RTA expression on K15P-HA. (A) BCBL-1-HA or parental BCBL-1 cells were electroporated with 10 μg of the RTA expression plasmid pSG5-Rta. Zero, 24, 48, or 72 h after electroporation (as indicated above the lanes), cells were harvested, subjected to SDS-PAGE, and Western blotted with anti-HA or anti-GAPDH antibody, as indicated at right. Positions of molecular size markers are shown at left. (Top) Expression of the ∼45-kDa and ∼23-kDa forms of K15P-HA. (Middle) Region from top panel with contrast adjusted to show the ∼45-kDa K15P-HA band. (Bottom) GAPDH loading control. (B) BCBL-1-HA or parental BCBL-1 cells were electroporated with no DNA, 7 μg or 10 μg of pSG5-Rta, or a control plasmid, as indicated above the lanes. After 48 h, cells were collected and processed exactly as described for panel A. (C) BCBL-1-HA cells were electroporated with pSG5-Rta and after 48 h were attached to coverslips, fixed, stained with DAPI (blue channel), and immunostained for HA (green channel), K8α (white channel), and TGN46 (red channel). An individual K8α-positive cell is shown surrounded by K8α-negative cells. (D) Identical to panel C except that anti-TGN staining was omitted. For panels C and D, conditions were chosen to image the elevated levels of anti-HA staining in K8α-positive cells. Bars, 20 μm.
RTA-induced lytic reactivation also caused changes in K15P-HA and TGN46 intracellular localization that were similar to those seen upon sodium butyrate treatment. Cells exhibiting nuclear anti-K8α staining showed elevated levels of K15P-HA in a dispersed punctate pattern (Fig. 6C and D). Anti-K8α-positive cells also showed dispersed and faint TGN46 staining (Fig. 6C), unlike the compact Golgi apparatus seen in K8α-negative cells in the same field (Fig. 6C).
K15P in spontaneously reactivating cells.
We observed that a small number of BCBL-1-HA cells (∼1% to 5% of the total) exhibited anti-K8α immunoreactivity even in the absence of sodium butyrate treatment or RTA transfection. This is consistent with the known frequency of spontaneous KSHV lytic reactivation in PEL cell lines (50, 52); we never observed such K8α-positive immunostaining in non-KSHV-infected BJAB-HA cells (data not shown). We found that the dispersed punctate pattern of K15P-HA immunostaining in spontaneously K8α-positive cells resembled that seen in cells lytically reactivated by sodium butyrate or RTA transfection (Fig. 7). Similarly, in K8α-positive cells, the tight juxtanuclear Golgi-like staining of TGN46 was lost, and the antigen became dispersed and more difficult to detect (panels to the right of Fig. 7A to F show identical merged images, but with omission of the green channel, to aid visualization of TGN46).
FIG 7.

K15P-HA in spontaneously reactivating cells. Fields of BCBL-1-HA cells were fixed, stained with DAPI (blue channel), and immunostained for HA (green channel), K8α (white channel), and TGN46 (red channel). (A to F) Merged images of fields containing K8α-negative cells and a spontaneously arising K8α-positive cell. To the right of each merged image is an identical panel in which the green channel has been omitted to better reveal TGN46 staining. For all panels, conditions were chosen to image the elevated levels of anti-HA staining in K8α-positive cells. Bars, 20 μm.
To quantitate the changes in K15P-HA and TGN46 localization during latency and butyrate-induced or spontaneous lytic reactivation, we categorized the observed immunostaining patterns as “compact” (juxtanuclear, Golgi-like staining, as in Fig. 3 and 4B), “dispersed” (peripheral punctate staining around the perimeter of the cell, as in Fig. 5A to C), or “intermediate” (both peripheral and compact juxtanuclear staining, as in Fig. 5D). As summarized in Fig. 8A, more than 90% of BCBL-1-HA cells showed compact HA staining in latency, but 80% to 90% exhibited a dispersed pattern of staining when lytically reactivated by sodium butyrate treatment or when reactivating spontaneously. Similarly, TGN46 staining appeared compact, with a typical Golgi-like morphology, in ∼90% of latent cells but became dispersed in ∼80% of lytically reactivated sodium butyrate-treated cells (Fig. 8B). The TGN46 compartment was also redistributed in ∼90% of cells showing spontaneous lytic reactivation, though approximately half of these showed a fully dispersed staining pattern and approximately half were of the intermediate phenotype (Fig. 8B).
DISCUSSION
We investigated the biology of the full-length, 8-exon-encoded form of the K15P protein in the environment of KSHV-infected PEL cells. A limitation of our study is that we used an epitope-tagged version of K15P to facilitate detection of the protein, and it is important to be cautious in interpreting data obtained using a recombinant molecule. Nevertheless, K15 modified in this way has been a standard tool for study of K15P (and K15M) expression (10, 15, 20, 22) and subcellular localization (10, 15, 26) and the mapping of K15P/M interactions with cellular proteins involved in intracellular signaling (14, 15, 19, 22, 26). There are no known differences between the biological behaviors of untagged and epitope-tagged K15P; however, this will remain a caveat of our approach until the intracellular trafficking and processing properties of unmodified K15P have been investigated. It is also true that the PEL cells in our study presumably express endogenous KSHV-encoded K15P, including the various alternatively spliced forms of the protein; at the moment, the effects of those molecules upon the behavior of our reporter remain unknown.
We observed that during sodium butyrate-stimulated lytic reactivation in BCBL-1-HA cells, levels of the ∼23/24-kDa form of K15P-HA gradually diminished, and a protein with the predicted ∼45-kDa molecular mass of full-length K15P-HA accumulated over time (Fig. 4A). We also observed expression of the higher-molecular-weight form of K15P-HA and slightly reduced levels of the ∼23- to 24-kDa form within 24 h of electroporation, when the KSHV lytic program was activated by transfection with RTA (Fig. 6A and B). The fact that the effects of RTA were less dramatic than those of sodium butyrate treatment is to be expected, since electroporation of PEL cells is rather inefficient and only ∼10 to 20% of cells in the RTA-electroporated population were lytically reactivated as scored by K8α-positive immunostaining (data not shown). Our data are consistent with an earlier report that levels of the ∼23- to 24-kDa form of K15P decrease following TPA treatment of BC3 PEL cells (18), despite the well-established increase in K15P message levels upon lytic induction (9, 10, 13, 16, 17, 32–35).
Since BJAB-HA cells can express the ∼23- to 24-kDa form of K15P (Fig. 1A), the processing event that generates the protein must be independent of KSHV latent infection. Conversely, conditions that stimulate lytic reactivation in KSHV-infected PEL cells did not result in accumulation of ∼45-kDa K15P-HA in BJAB-HA cells (Fig. 4A and 6A and B). This suggests that accumulation of full-length K15P-HA in these B-cell-derived lines is due to the KSHV lytic program itself rather than the reactivation conditions (or that BJAB cells inherently differ from BCBL-1 cells in this regard). We propose that the simplest explanation for these data is that K15P-HA is initially synthesized as a full-length, ∼45-kDa protein which in latent cells undergoes cleavage by a cellular protease to release the ∼23/24-kDa carboxy-terminal portion of the molecule (to our knowledge, no studies have detected the corresponding amino-terminal portion of K15P). During KSHV lytic reactivation, the activity of this cellular protease (or delivery of K15P to it, as discussed below) is suppressed, and the full-length protein accumulates. To our knowledge, this is the first time that differential expression of these two molecular weight forms of K15P has been observed from the same K15P cDNA in a single cell type.
Lytic reactivation and accumulation of the full-length form of K15P-HA were accompanied by redistribution of K15P-HA staining from a compact Golgi-like juxtanuclear location to a more peripheral distribution (Fig. 4 to 6; quantitated in Fig. 8A). We saw a similar redistribution of K15P-HA in the rare PEL cells undergoing spontaneous lytic reactivation of KSHV (Fig. 7 and 8A), though we do not know if full-length K15P-HA accumulates in this case. The TGN-resident protein TGN46 similarly dispersed (quantitated in Fig. 8B). Neither K15P-HA nor TGN46 exhibited this behavior when non-KSHV-infected BJAB-HA cells were treated with sodium butyrate (Fig. 5E to L), consistent with it resulting from lytic reactivation (though, as mentioned above, we cannot rule out the possibility that BCBL-1 and BJAB cells respond differently to sodium butyrate treatment for other reasons). Fragmentation of the Golgi apparatus is known to accompany infection of some cell types by herpes simplex virus (HSV), where it is thought to be a consequence of very high levels of anterograde protein traffic during lytic replication (53). It is possible that lytic replication of KSHV has similar consequences for PEL cells. The loss of overlapping K15P-HA and TGN46 fluorescence staining upon Golgi fragmentation is consistent with juxtanuclear latent K15P-HA residing in an organelle spatially close to the TGN but physically distinct from it (such as the cis-, medial, or trans-Golgi cisternae), resulting in separation from the TGN upon Golgi dispersal. Alternatively, K15P-HA might actually reside in the TGN compartment during latency but depart from it upon lytic reactivation.
Whether due to organelle fragmentation or alternative routes of protein trafficking, our data are consistent with the possibility that the different molecular weight forms of K15P exhibit distinct patterns of intracellular localization. This might explain earlier findings reporting a wide variety of organellar locations for K15P (summarized in the introduction). It is also possible that processing of the full-length form of K15P to the smaller, ∼23/24-kDa species might require delivery of the ∼45-kDa K15P to an appropriate organelle, as occurs during proteolytic cleavage of certain cellular proteins (54, 55). Whatever the mechanism, it is not unreasonable to speculate that differing roles for K15P in the KSHV latent and lytic cycles might be accompanied by expression of alternative molecular weight forms of the protein in distinct subcellular locations.
MATERIALS AND METHODS
Cells and viruses.
BCBL-1 and BJAB cells and cell lines derived from them were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS) and 1% penicillin-streptomycin (PS) (Gibco Laboratories) and grown in a 5% CO2 atmosphere. The growth medium of lentivirus-infected K15P-HA-expressing cell lines was supplemented with 1 mg/ml G418 (Thermo Fisher Scientific).
DNA constructs and lentivirus infections.
Plasmid pVRK15P contains the complete 8-exon cDNA of K15P fused in-frame to two tandem C-terminal copies of the hemagglutinin (HA) epitope tag in the vector pVR1255 (9, 17). pVRK15P was digested with EcoRI and BamHI to generate a restriction fragment containing the entire K15P-HA ORF, which was then ligated between the EcoRI and BamHI sites of the lentivirus vector pLVX-IRES-Neo (Clontech) to generate the construct pLVX-K15P-HA-IRES-Neo. This positions the K15P-HA gene for transcription as the upstream ORF in a bicistronic message, followed by an internal ribosome entry site (IRES) and a neomycin resistance gene. For preparation of recombinant lentiviruses expressing K15P-HA, the plasmid pLVX-K15P-HA-IRES-Neo was combined with a Lenti-X HTX packaging mixture and transfected into Lenti-X 293T cells following the manufacturer's protocol (Clontech). After 48 h, Lenti-X 293T cell culture supernatants, containing recombinant lentiviruses, were harvested and stored at −80°C.
To achieve lentivirus infection, 1 × 106 BCBL-1 or BJAB cells were mixed with lentivirus-containing supernatants in RPMI-10% FCS at a multiplicity of infection (MOI) of 5 in the presence of 5 μg/ml 1,5-dimethyl-1,5-diazaundecamethylene polymethobromide (Polybrene; MilliporeSigma). Following incubation for 60 min in a tissue culture incubator, spinoculation was performed by centrifugation of the cell-lentivirus mixture at 1,500 × g for 90 min at room temperature. Pelleted cells were gently resuspended to a density of 2.5 × 105 cells/ml in fresh medium and incubated for a further 48 h to allow time for neomycin resistance expression, and then the growth conditions were adjusted to 1 mg/ml G418 (MilliporeSigma). Individual G418-resistant BCBL-1- and BJAB-derived clones were screened for expression of the K15P-HA protein by Western blotting and were named BCBL-1-HA and BJAB-HA.
Preparation of RIPA detergent extracts.
Cells were chilled to 4°C, pelleted by centrifugation at 2,000 × g, washed in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) at 4°C, and then solubilized in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris-HCl, pH 8.0) containing 5% (vol/vol) mammalian protease inhibitor cocktail (MilliporeSigma) on ice for 1 h. A 5,000 × g spin was then performed for 10 min to pellet insoluble material, and the supernatant was recovered for SDS-PAGE.
Preparation of PNS.
Cells were grown to a density of 0.8 × 106 to 1 × 106 cells/ml and then chilled on ice, and all subsequent steps were performed at 4°C. Cells were pelleted by centrifugation at 2,000 × g, washed in 250 mM sucrose, 2 mM MgCl2, 10 mM Tris-HCl, pH 7.2, pelleted at 2,000 × g, resuspended in 50 mM sucrose, 2 mM MgCl2, 10 mM Tris-HCl, pH 7.2, and then incubated on ice for 15 min. A mammalian protease inhibitor cocktail (MilliporeSigma) was added to a final concentration of 5% (vol/vol), cells were broken by 40 passages through a 27-gauge needle and centrifuged at 2,000 × g for 10 min to remove nuclei and unbroken cells, and the postnuclear supernatant (PNS) was collected. To obtain organelle-enriched and soluble fractions, the PNS was centrifuged in a Beckman TLA100.3 rotor at 50,000 × g for 30 min at 4°C, and the pellet (organelles) and supernatant (soluble cytoplasm) were recovered.
Density gradient fractionation of organelles.
Fractionation of BCBL-1-HA subcellular organelles was performed similarly to the method described previously (56). A PNS was first prepared as detailed above, except that the wash and cell breakage buffers had MgCl2 omitted and instead contained 1 mM EDTA. The PNS was applied to the top of a preformed linear density gradient prepared from successive layers of 30%, 25%, 20%, 17.5%, 15%, 12.5%, 10%, 7.5%, 5%, and 2.5% iodixanol (OptiPrep; MilliporeSigma). The gradient was centrifuged at 50,000 × g in an SW41 rotor for 4 h at 4°C, and 24 successive fractions were collected from top to bottom.
Western blotting and antibodies.
SDS-PAGE and Western blotting were performed as previously described (57), except that samples were heated at 37°C (rather than 95°C) in Laemmli buffer prior to electrophoresis. Following electrophoretic transfer, membranes were incubated overnight with the appropriate primary antibodies and for 1 h with peroxidase-conjugated anti-rabbit, anti-goat, or anti-mouse secondary antibodies (MilliporeSigma). Secondary antibodies were detected with an enhanced chemiluminescence substrate (PerkinElmer).
Primary antibodies were mouse anti-HA (clone 16B12; BioLegend), mouse anti-p58K (G2404; MilliporeSigma), mouse anti-Na+/K+ ATPase (ab7671; Abcam), mouse anti-GAPDH (60004-1-Ig [Proteintech] and MA5-15738 [Invitrogen]), rabbit anti-HA (H6980; MilliporeSigma), rabbit anti-PDI (ADI-SPA-890; Enzo Life Sciences), rabbit anti-SMAC (sc-22766; Santa Cruz Biotechnology), rabbit anti-TGN46 (ab50595; Abcam), and goat anti-cathepsin B (sc-6493; Santa Cruz Biotechnology).
Electroporation of PEL cells.
PEL cells were electroporated using a Lonza Nucleofector device and cell line Nucleofector solution V (Lonza) according to the manufacturer's instructions, as follows. One day prior to electroporation, cells were seeded to a density of 0.5 × 106 cells/ml and allowed to grow overnight, and then 3 × 106 cells were collected for each electroporation by centrifugation at 1,000 × g at room temperature. Following removal of the culture supernatant, each cell pellet was resuspended in 100 μl Nucleofector solution V, mixed with 8 μg plasmid DNA, and transferred to Nucleocuvette vessels (Lonza). Electroporation was performed using the Amaxa electroporation program T-001 according to the manufacturer's instructions and similar to previously described protocols (58, 59). Cells were then immediately removed from the cuvettes, transferred to Eppendorf tubes containing 0.5 ml RPMI-10% FCS (prewarmed to 37°C and preequilibrated with 5% CO2 but lacking PS antibiotics), and incubated at 37°C in a tissue culture incubator for 15 min. The cells were then gently pelleted at 100 × g at 37°C, resuspended in their normal growth medium, and returned to the incubator for 0, 24, or 48 h before harvesting.
Immunocytochemistry.
All procedures were performed at room temperature. Cells were centrifuged onto glass coverslips at 10,000 rpm in a Shandon Cytospin 2 centrifuge for 5 min, fixed with 4% paraformaldehyde in PBS for 10 min, rinsed three times with PBS, and permeabilized using 0.2% Triton X-100 in PBS for 10 min. Following PBS washes exactly as performed before, they were blocked in 10% FCS, PBS for 30 min. Samples were incubated with primary antibodies for 2 h and then with secondary antibodies for 1 h, in each case diluted in 10% FCS, PBS. Following four PBS washes over 20 min, the samples were subjected to a 5-min fixation with 4% paraformaldehyde in PBS, washed in PBS, and mounted using Diamond Prolong antifade reagent either containing DAPI (Thermo-Fisher Molecular Probes) or omitting DAPI (Thermo-Fisher Molecular Probes). Samples were left to cure overnight before sealing with nail polish. All imaging was performed in the Analytical Imaging Facility of the Albert Einstein College of Medicine, using a Leica TCS SP5 confocal microscope (Leica Microsystems, Mannheim, Germany). Images were collected as LIF files (Leica image file format; Leica Microsystems) and processed using Fiji and ImageJ (60, 61).
Primary antibodies were rabbit anti-TGN46 (ab50595; Abcam), rabbit anti-calnexin (ab22595; Abcam), rabbit anti-Na+/K+ ATPase (ab76020; Abcam), rabbit anti-SMAC (sc-22766; Santa Cruz Biotechnology), rabbit anti-HA (NB600-363; Novus Biologicals), mouse anti-KSHV K8 (SAB5300152; MilliporeSigma), mouse anti-Na+/K+ ATPase (ab7671; Abcam), mouse anti-calnexin (C7617; MilliporeSigma), mouse anti-HA (clone 16B12; BioLegend), and rat anti-HA (NBP2-50416; Novus Biologicals).
Secondary antibodies were all from Invitrogen and were as follows: cyanine3–goat anti-rabbit (A10520), Alexa Fluor 633–goat anti-rabbit (A21071), cyanine3–goat anti-mouse (A10521), Alexa Fluor 488–goat anti-mouse (A11001), cyanine3–goat anti-rat (A10522), and Alexa Fluor 488–goat anti-rat (A11006).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01 AI125244 (to D.W.W).
We thank Anne Muesch, Dawn Schranz, Margaret Kielian, and E. Richard Stanley for helpful discussions. Plasmid pVRK15P was a kind gift from Thomas Schulz, and the Rta expression plasmid pSG5-Rta was generously provided by Blossom Damania.
REFERENCES
- 1.Chang Y, Moore P. 2014. Twenty years of KSHV. Viruses 6:4258–4264. doi: 10.3390/v6114258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giffin L, Damania B. 2014. KSHV: pathways to tumorigenesis and persistent infection. Adv Virus Res 88:111–159. doi: 10.1016/B978-0-12-800098-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong EL, Damania B. 2005. Linking KSHV to human cancer. Curr Oncol Rep 7:349–356. doi: 10.1007/s11912-005-0061-6. [DOI] [PubMed] [Google Scholar]
- 4.Fukumoto H, Kanno T, Hasegawa H, Katano H. 2011. Pathology of Kaposi's sarcoma-associated herpesvirus infection. Front Microbiol 2:175. doi: 10.3389/fmicb.2011.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280. [PubMed] [Google Scholar]
- 6.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 7.Dimaio TA, Lagunoff M. 2012. KSHV induction of angiogenic and lymphangiogenic phenotypes. Front Microbiol 3:102. doi: 10.3389/fmicb.2012.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen KW, Damania B. 2010. Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett 289:140–150. doi: 10.1016/j.canlet.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi JK, Lee BS, Shim SN, Li M, Jung JU. 2000. Identification of the novel K15 gene at the rightmost end of the Kaposi's sarcoma-associated herpesvirus genome. J Virol 74:436–446. doi: 10.1128/JVI.74.1.436-446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glenn M, Rainbow L, Aurade F, Davison A, Schulz TF. 1999. Identification of a spliced gene from Kaposi's sarcoma-associated herpesvirus encoding a protein with similarities to latent membrane proteins 1 and 2A of Epstein-Barr virus. J Virol 73:6953–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damania B. 2004. Oncogenic gamma-herpesviruses: comparison of viral proteins involved in tumorigenesis. Nat Rev Microbiol 2:656–668. doi: 10.1038/nrmicro958. [DOI] [PubMed] [Google Scholar]
- 12.Dittmer DP, Damania B. 2013. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—an update. Curr Opin Virol 3:238–244. doi: 10.1016/j.coviro.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poole LJ, Zong JC, Ciufo DM, Alcendor DJ, Cannon JS, Ambinder R, Orenstein JM, Reitz MS, Hayward GS. 1999. Comparison of genetic variability at multiple loci across the genomes of the major subtypes of Kaposi's sarcoma-associated herpesvirus reveals evidence for recombination and for two distinct types of open reading frame K15 alleles at the right-hand end. J Virol 73:6646–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pietrek M, Brinkmann MM, Glowacka I, Enlund A, Havemeier A, Dittrich-Breiholz O, Kracht M, Lewitzky M, Saksela K, Feller SM, Schulz TF. 2010. Role of the Kaposi's sarcoma-associated herpesvirus K15 SH3 binding site in inflammatory signaling and B-cell activation. J Virol 84:8231–8240. doi: 10.1128/JVI.01696-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, Brinkmann MM, Pietrek M, Ottinger M, Dittrich-Breiholz O, Kracht M, Schulz TF. 2007. Functional characterization of the M-type K15-encoded membrane protein of Kaposi's sarcoma-associated herpesvirus. J Gen Virol 88:1698–1707. doi: 10.1099/vir.0.82807-0. [DOI] [PubMed] [Google Scholar]
- 16.Tsai YH, Wu MF, Wu YH, Chang SJ, Lin SF, Sharp TV, Wang HW. 2009. The M type K15 protein of Kaposi's sarcoma-associated herpesvirus regulates microRNA expression via its SH2-binding motif to induce cell migration and invasion. J Virol 83:622–632. doi: 10.1128/JVI.00869-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brinkmann MM, Pietrek M, Dittrich-Breiholz O, Kracht M, Schulz TF. 2007. Modulation of host gene expression by the K15 protein of Kaposi's sarcoma-associated herpesvirus. J Virol 81:42–58. doi: 10.1128/JVI.00648-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, Du MQ, Boshoff C. 2002. K15 protein of Kaposi's sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol 76:802–816. doi: 10.1128/JVI.76.2.802-816.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Havemeier A, Gramolelli S, Pietrek M, Jochmann R, Sturzl M, Schulz TF. 2014. Activation of NF-κB by the Kaposi's sarcoma-associated herpesvirus K15 protein involves recruitment of the NF-κB-inducing kinase, IκB kinases, and phosphorylation of p65. J Virol 88:13161–13172. doi: 10.1128/JVI.01766-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, Schulz TF. 2003. Activation of mitogen-activated protein kinase and NF-κB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J Virol 77:9346–9358. doi: 10.1128/JVI.77.17.9346-9358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho NH, Choi YK, Choi JK. 2008. Multi-transmembrane protein K15 of Kaposi's sarcoma-associated herpesvirus targets Lyn kinase in the membrane raft and induces NFAT/AP1 activities. Exp Mol Med 40:565–573. doi: 10.3858/emm.2008.40.5.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bala K, Bosco R, Gramolelli S, Haas DA, Kati S, Pietrek M, Havemeier A, Yakushko Y, Singh VV, Dittrich-Breiholz O, Kracht M, Schulz TF. 2012. Kaposi's sarcoma herpesvirus K15 protein contributes to virus-induced angiogenesis by recruiting PLCγ1 and activating NFAT1-dependent RCAN1 expression. PLoS Pathog 8:e1002927. doi: 10.1371/journal.ppat.1002927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gramolelli S, Weidner-Glunde M, Abere B, Viejo-Borbolla A, Bala K, Ruckert J, Kremmer E, Schulz TF. 2015. Inhibiting the recruitment of PLCγ1 to Kaposi's sarcoma herpesvirus K15 protein reduces the invasiveness and angiogenesis of infected endothelial cells. PLoS Pathog 11:e1005105. doi: 10.1371/journal.ppat.1005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kati S, Tsao EH, Gunther T, Weidner-Glunde M, Rothamel T, Grundhoff A, Kellam P, Schulz TF. 2013. Activation of the B cell antigen receptor triggers reactivation of latent Kaposi's sarcoma-associated herpesvirus in B cells. J Virol 87:8004–8016. doi: 10.1128/JVI.00506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120:939–949. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim CS, Seet BT, Ingham RJ, Gish G, Matskova L, Winberg G, Ernberg I, Pawson T. 2007. The K15 protein of Kaposi's sarcoma-associated herpesvirus recruits the endocytic regulator intersectin 2 through a selective SH3 domain interaction. Biochemistry 46:9874–9885. doi: 10.1021/bi700357s. [DOI] [PubMed] [Google Scholar]
- 27.Matolcsy A, Nador RG, Cesarman E, Knowles DM. 1998. Immunoglobulin VH gene mutational analysis suggests that primary effusion lymphomas derive from different stages of B cell maturation. Am J Pathol 153:1609–1614. doi: 10.1016/S0002-9440(10)65749-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arvanitakis L, Mesri EA, Nador RG, Said JW, Asch AS, Knowles DM, Cesarman E. 1996. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 88:2648–2654. [PubMed] [Google Scholar]
- 29.Arguello M, Sgarbanti M, Hernandez E, Mamane Y, Sharma S, Servant M, Lin R, Hiscott J. 2003. Disruption of the B-cell specific transcriptional program in HHV-8 associated primary effusion lymphoma cell lines. Oncogene 22:964–973. doi: 10.1038/sj.onc.1206270. [DOI] [PubMed] [Google Scholar]
- 30.Steinbruck L, Gustems M, Medele S, Schulz TF, Lutter D, Hammerschmidt W. 2015. K1 and K15 of Kaposi's sarcoma-associated herpesvirus are partial functional homologues of latent membrane protein 2A of Epstein-Barr virus. J Virol 89:7248–7261. doi: 10.1128/JVI.00839-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinkmann MM, Schulz TF. 2006. Regulation of intracellular signalling by the terminal membrane proteins of members of the Gammaherpesvirinae. J Gen Virol 87:1047–1074. doi: 10.1099/vir.0.81598-0. [DOI] [PubMed] [Google Scholar]
- 32.Jenner RG, Alba MM, Boshoff C, Kellam P. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J Virol 75:891–902. doi: 10.1128/JVI.75.2.891-902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulose-Murphy M, Ha NK, Xiang C, Chen Y, Gillim L, Yarchoan R, Meltzer P, Bittner M, Trent J, Zeichner S. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J Virol 75:4843–4853. doi: 10.1128/JVI.75.10.4843-4853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong EL, Damania B. 2006. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus K15 gene. J Virol 80:1385–1392. doi: 10.1128/JVI.80.3.1385-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guito J, Lukac DM. 2012. KSHV Rta promoter specification and viral reactivation. Front Microbiol 3:30. doi: 10.3389/fmicb.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Damania B. 2008. Kaposi's sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res 68:4640–4648. doi: 10.1158/0008-5472.CAN-07-5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma-Walia N, Paul AG, Bottero V, Sadagopan S, Veettil MV, Kerur N, Chandran B. 2010. Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog 6:e1000777. doi: 10.1371/journal.ppat.1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiller K, Grote A, Scheer M, Munch R, Jahn D. 2004. PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res 32:W375–W379. doi: 10.1093/nar/gkh378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 41.Sander G, Konrad A, Thurau M, Wies E, Leubert R, Kremmer E, Dinkel H, Schulz T, Neipel F, Sturzl M. 2008. Intracellular localization map of human herpesvirus 8 proteins. J Virol 82:1908–1922. doi: 10.1128/JVI.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Komar AA, Hatzoglou M. 2011. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 10:229–240. doi: 10.4161/cc.10.2.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Komar AA, Mazumder B, Merrick WC. 2012. A new framework for understanding IRES-mediated translation. Gene 502:75–86. doi: 10.1016/j.gene.2012.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu Y, Black JB, Goldsmith CS, Browning PJ, Bhalla K, Offermann MK. 1999. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J Gen Virol 80:83–90. doi: 10.1099/0022-1317-80-1-83. [DOI] [PubMed] [Google Scholar]
- 45.Menezes J, Leibold W, Klein G, Clements G. 1975. Establishment and characterization of an Epstein-Barr virus (EBC)-negative lymphoblastoid B cell line (BJA-B) from an exceptional, EBV-genome-negative African Burkitt's lymphoma. Biomedicine 22:276–284. [PubMed] [Google Scholar]
- 46.Cannon JS, Ciufo D, Hawkins AL, Griffin CA, Borowitz MJ, Hayward GS, Ambinder RF. 2000. A new primary effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma herpesvirus-containing supernatant. J Virol 74:10187–10193. doi: 10.1128/JVI.74.21.10187-10193.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horenstein MG, Nador RG, Chadburn A, Hyjek EM, Inghirami G, Knowles DM, Cesarman E. 1997. Epstein-Barr virus latent gene expression in primary effusion lymphomas containing Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8. Blood 90:1186–1191. [PubMed] [Google Scholar]
- 48.Gao YS, Vrielink A, MacKenzie R, Sztul E. 2002. A novel type of regulation of the vimentin intermediate filament cytoskeleton by a Golgi protein. Eur J Cell Biol 81:391–401. doi: 10.1078/0171-9335-00260. [DOI] [PubMed] [Google Scholar]
- 49.Miller G, Heston L, Grogan E, Gradoville L, Rigsby M, Sun R, Shedd D, Kushnaryov VM, Grossberg S, Chang Y. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J Virol 71:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Purushothaman P, Uppal T, Verma SC. 2015. Molecular biology of KSHV lytic reactivation. Viruses 7:116–153. doi: 10.3390/v7010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez CM, Wong EL, Bowser BS, Hong GK, Kenney S, Damania B. 2006. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J Virol 80:3062–3070. doi: 10.1128/JVI.80.6.3062-3070.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lukac DM, Yuan Y. 2007. Reactivation and lytic replication of KSHV, p 434–460. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 53.Campadelli G, Brandimarti R, Di Lazzaro C, Ward PL, Roizman B, Torrisi MR. 1993. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc Natl Acad Sci U S A 90:2798–2802. doi: 10.1073/pnas.90.7.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown MS, Ye J, Rawson RB, Goldstein JL. 2000. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100:391–398. doi: 10.1016/S0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 55.Schwake M, Schroder B, Saftig P. 2013. Lysosomal membrane proteins and their central role in physiology. Traffic 14:739–748. doi: 10.1111/tra.12056. [DOI] [PubMed] [Google Scholar]
- 56.Chen D, Sandford G, Nicholas J. 2009. Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J Virol 83:722–733. doi: 10.1128/JVI.01517-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J Virol 82:7388–7394. doi: 10.1128/JVI.00225-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prasad A, Lu M, Lukac DM, Zeichner SL. 2012. An alternative Kaposi's sarcoma-associated herpesvirus replication program triggered by host cell apoptosis. J Virol 86:4404–4419. doi: 10.1128/JVI.06617-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rainy N, Zayoud M, Kloog Y, Rechavi O, Goldstein I. 2016. Viral oncomiR spreading between B and T cells is employed by Kaposi's sarcoma herpesvirus to induce non-cell-autonomous target gene regulation. Oncotarget 7:41870–41884. doi: 10.18632/oncotarget.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]