

Abstract
Background
Cancers are caused by the acquisition of somatic mutations. Numerous efforts have been made to characterize the key driver genes and pathways in glioma, however, the etiology of glioma is still not completely known. This study was implemented to characterize driver genes in glioma independently of somatic mutation frequencies.
Methods
Driver genes and pathways were predicted by OncodriveCLUST, OncodriveFM, Icages, Drgap and Dendrix in glioma using 31,958 somatic mutations from TCGA, followed by an integrative characterization of driver genes.
Results
Overall, 685 driver genes and 215 driver pathways were determined by the five tools. FSTL5, HCN1, TMEM132D, TRHDE and KRT222 showed the strongest expression correlation with other genes in the co-expression network of glioma tissues. ST6GAL2, PIK3CA, PIK3R1, TP53 and EGFR are at the core of the protein–protein interaction network. 133 driver genes were up-regulated and associated to poor prognosis, 43 driver genes were down-regulated and related to favorable clinical outcome in glioma patients. The driver genes such as MSH6 and RUNX1T1 might serve as candidate prognostic biomarkers and therapeutic targets in glioma.
Conclusions
The set of new cancer genes and pathways sheds insights into the tumorigenesis of glioma and paves the way for developing driver gene-targeted therapy and prognostic biomarkers in glioma.
Electronic supplementary material
The online version of this article (doi:10.1186/s12935-017-0458-y) contains supplementary material, which is available to authorized users.
Keywords: Glioma, Driver gene, Driver pathway
Introduction
Gliomas are tumors that arise from glial cells and common primary brain tumors in adults, with an incidence rate of 6.03 per 100,000 in USA [1]. Gliomas are classified into a variety of subtypes, including astrocytoma, glioblastoma, oligodendroglioma, ependymoma, mixed glioma, malignant glioma, and a few more rare histologies. Of them, astrocytoma accounts for about 70% of glioma cases [1]. Low grade glioma is a lethal disease in young adults, with an average survival time of 7 years, only 20% of low grade glioma patients survived for more than two decades [2].
Recently, large-scale genomics studies have been conducted to determine the core genes and pathways underlying gliomagenesis and to define molecular subtypes in glioblastoma and lower-grade gliomas [3–5]. For instance, 87% of glioblastomas have genetic alterations in the TP53/MDM2/MDM4/p14ARF pathway, including TP53 mutations or homozygous deletion (35%), MDM2 amplification (14%), MDM4 amplification (7%), or p14ARF homozygous deletion or mutation (49%) [3]. IDH1 mutation combined with either TP53 mutation or total 1p/19q loss is a frequent and early change in the majority of common adult gliomas but not in primary glioblastomas [6]. Lower-grade glioma patients with an IDH mutation and 1p/19q co-deletion showed favorable clinical outcomes in lower-grade gliomas [5]. The DNA repair enzyme MGMT is frequently methylated in glioma, methylation of the CpG islands of the MGMT gene prevents transcription, which may increase the sensitivity of glioma to alkylating agents [7–9].
Though considerable progresses have been achieved, the etiology of glioma is still not completely understood. In this study, we performed genome-wide analyses of 576 gliomas, incorporating exome sequence, mRNA expression, protein–protein interaction, DNA copy number variations and clinical outcome from The Cancer Genome Atlas (TCGA) database. We revealed a list of cancer-driving genes and pathways and many driver genes were aberrantly expressed, co-expressed with other driver genes, involved in copy number variations and correlated with prognosis of glioma patients. Our study is of importance to characterize cancer biology and identify potential therapeutic targets and prognostic biomarkers in glioma.
Methods and materials
Classification of cancer mutations
31,958 somatic mutations generated by whole-exome sequencing of 576 pairs of glioma tumor/normal samples were downloaded from TCGA database at Broad Institute (http://firebrowse.org/?cohort=GBMLGG&download_dialog=true, download on April 15, 2017) [10]. Functional impact of somatic mutations was evaluated by Ensembl Variant Effect Predictor (VEP) [11] and then all mutations were classified into 11 categories according to their functional impact.
Prediction of driver genes and pathways
Driver gene candidates were predicted by five distinct tools, including OncodriveCLUST [12], OncodriveFM [13] (https://www.intogen.org), drgap [14] (https://code.google.com/archive/p/drgap), icages [15] (http://icages.wglab.org/) and Dendrix [16] (http://compbio.cs.brown.edu/projects/dendrix/) with default parameters. The following criteria were applied to determine driver genes or pathways: (1) genes have q values smaller than 0.05 (OncodriveCLUST and OncodriveFM), (2) genes or pathways have adjusted P values less than 0.05 (drgap), (3) genes were classified as drivers by icages and showed icagesGeneScores above 0.5 (icages), (4) the selected genes were sampled at least 10% (100/1000) of the times in the mutually exclusive analysis (Dendrix). Then, 576 glioma patients were stratified into IDH1-mutated (236) and non-IDH1-mutated (340) groups. Driver gene and pathway analyses were performed in the two groups using the same methods as described above.
GO, KEGG pathway enrichment analyses
In order to characterize the functional enrichment of driver genes, GO term and KEGG pathway enrichment analyses were implemented for all the driver genes on the home page of STRING [17] (http://string.embl.de/). GO terms and KEGG pathways were considered to be significantly enriched for driver genes with the cutoff of false discovery rate (FDR) < 0.05.
Expression and co-expression network analyses
RNA-seq data of 75 glioma and 17 normal brain tissues were obtained from the study of Gill et al. (GSE59612) [18]. Gene expression values expressed as Fragments per kilobase of exon per million fragments mapped (FPKM) were compared between glioma and normal brain tissues for all driver genes with t test, P values were adjusted using False Discovery Rate (FDR) in R. Genes were regarded as significantly differentially expressed with the cutoff of adjusted P value < 0.05. Next, Co-expression network was constructed by weighted correlation network analysis (WGCNA) using gene expression log2(FPKM + 1) [19]. All parameters were used with the default values except for the softpower (12) and threshold (0.004). Degree centrality is defined as the number of connections one node has to another. Hub genes which have the highest degrees connect most adjacent genes and build the structure of the network.
Protein–protein interaction network analysis
Protein–protein interaction (PPI) network was constructed using STRING to prioritize the core driver genes in glioma. As for each driver gene, combined STRING scores of all protein–protein interactions were summed as total STRING score which represents the number of interactions the driver gene has with other genes.
Sources of copy number variation and survival analyses
Focal copy number variations (CNVs) were acquired from 52 glioma samples at broad institute [10] (http://firebrowse.org/?cohort=GBMLGG&download_dialog=true#). TCGA RNAseq and clinical outcome data were retrieved to assess whether the expression of driver genes could be associated to patients’ survival in glioma. Multivariate Cox regressions were performed with the coxph function from the R survival library using gene expression, sex, age, and grade or histology as multivariates [20]. Kaplan–Meier survival curves were plotted on the website of oncolnc [21] (http://www.oncolnc.org/), log rank test was used to compare the survival rates between high and low expression groups which refer to 25% (127/508) of glioma patients that have the highest or lowest RNA expression levels respectively.
Results
The catalogue of somatic mutations
31,958 somatic mutations comprise 30,216 single-nucleotide variants (SNVs) and 1742 small insertions or deletions (indels). The SNVs included 7818 silent, 20,311 missense, 1248 nonsense, 713 splice-site, 119 RNA, 71 translation start site and 24 nonstop mutations, 1259 indels caused reading frame shifts, 404 indels were in frame mutations. Over 67.46% (21,559/31,958) of variants were non-synonymous mutations (Fig. 1a). C>T/G>A, T>C/A>G and T>A/A>T accounted for 54.18, 17.38 and 13.05% of the variant types in the non-CpG sites, 6.90, 1.06 and 0.98% of variant types in the CpG islands respectively. Therefore, C>T/G>A, T>C/A>G and T>A/A>T were the three predominant transitions in glioma (Fig. 1b).
Fig. 1.
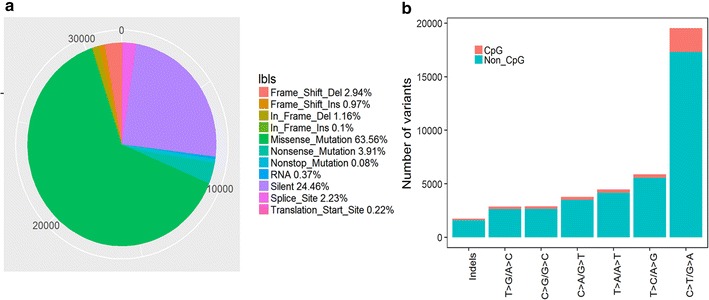
Characterization of somatic mutations in glioma. a The number and proportion of mutation classes with different functional impact in glioma. b The somatic mutation signatures in glioma
Cancer driver genes and pathways in glioma
Overall, there were 15, 68, 221 and 445 driver genes determined by OncodriveCLUST, OncodriveFM, icages and drgap respectively (Additional file 1: Table S1). Dendrix reported 11,814 genes mutated in at least one patient. We performed Dendrix analyses for sets of size ranging from 2 to 4. When k = 2, the pair IDH1 and PTEN was sampled 95.3% (953/1000) of the times. When k ≥ 3, the triple IDH1, Unknown and PTEN, IDH1, EGFR and PTEN were sampled 69.2% (692/1000) and 30.7% (307/1000) of the times respectively. Therefore, IDH1, PTEN and EGFR showed mutational exclusivity in glioma (Additional file 1: Table S1). EGFR is the only common gene detected by all five tools (Fig. 2a), suggesting that EGFR plays a pivotal role in the tumorigenesis of glioma. Of 685 driver genes, the majority of driver genes are low-frequency mutated genes in glioma, with an average mutation rate of 1.39% (Fig. 2b). IDH1, TP53, ATRX, TTN, PTEN, EGFR and MUC16 were known recurrently mutated genes and showed mutation rates of 40.97, 39.93, 23.78, 22.92, 17.88,16.32 and 15.10% across all samples (Fig. 2c). Besides the list of driver genes, Drgap also reported 215 driver pathways in glioma, such as Hedgehog signaling pathway, mTOR signaling pathway, glioma and acute myeloid leukemia (Additional file 1: Table S2).
Fig. 2.

Driver genes in glioma. a The overlap of driver genes predicted by five distinct methods. b The distribution of mutation frequencies of driver genes. c The mutation patterns of top 20 most frequently mutated driver genes across 576 glioma samples
The IDH1 gene encoding isocitrate dehydrogenase 1 (IDH1) catalyzes the oxidative carboxylation of isocitrate to a-ketoglutarate, causing production of NADPH in the citric acid (Krebs) cycle [22]. IDH1 is frequently mutated in the majority of low grade gliomas and secondary high grade gliomas. IDH1 mutations change the function of the enzymes, increase DNA methylation and correlate with improved prognosis in glioma [23]. 576 glioma patients were classified into IDH1-mutated (236 patients) and non-IDH1-mutated groups (340 patients) based on the occurrence of IDH1 mutation. Then we applied OncodriveCLUST, OncodriveFM, icages, drgap and Dendrix to detect driver genes and pathways in the two distinct groups. The number of driver genes detected by the five tools was largely different between IDH1-mutated and non-IDH1-mutated groups. There were only 2, 5, 46, 8 and 1 overlapping driver genes predicted by OncodriveCLUST, OncodriveFM, icages, drgap and Dendrix respectively (Additional file 1: Figure S1, Tables S3, S4). Moreover, a set of driver genes were predicted to be IDH1-associated, such as IDH1, NOTCH1, FUBP1, ARID1A, KAT6B and ARID2, while others were IDH1-independent, such as EGFR, PIK3CA, BRAF, RB1 and PTGS2 (Additional file 1: Tables S3, S4, Figure S1). However, the majority of driver pathways (196/209, 93.78%) unraveled by drgap in glioma patients with IDH1 mutations overlapped with those (196/214, 91.59%) in glioma patients without IDH1 mutations. All the results suggest that IDH1 and non-IDH1 mutated glioma subtypes are caused by mutational disruption of different genes but similar pathways.
GO term and KEGG pathway enrichment analyses
The enrichment of GO terms and KEGG pathways was analyzed for 685 driver genes on the home page of STRING. GO enrichment analysis indicated driver genes were significantly enriched in 1750 biological processes (FDR < 0.05). The main GO biological process terms showed a wide spectrum of functional processes ranging from cellular developmental process, cell differentiation, programmed cell death, apoptotic process to regulation of metabolic processes (Additional file 1: Table S5). STRING also revealed 145 KEGG pathways significantly enriched for driver genes, such as glioma, melanoma, thyroid cancer, pancreatic cancer, renal cell carcinoma, bladder cancer, colorectal cancer, non-small cell lung cancer, endometrial cancer, prostate cancer, acute myeloid leukemia, MAPK signaling pathway, mTOR signaling pathway and cell cycle (FDR < 0.05, Additional file 1: Table S6).
Expression profiling in glioma
In order to analyze the driver gene expression profile in glioma, RNA-seq data of 75 glioma and 17 normal brain tissues were obtained from Gill’s study. Overall, we found 428 differentially expressed driver genes, including 330 up-regulated and 98 down-regulated genes (Additional file 1: Figure S2). Next, we built co-expression networks based on the expression correlation of differentially expressed driver genes in cancer tissues and normal brain tissues respectively. The genes which have high degrees possess intensive interactions with other genes and may act as key genes in the co-expression network. As shown Fig. 3, FSTL5, HCN1, TMEM132D, TRHDE, KRT222, CACNA1B, CDH9, SLC22A9, GABRA1 and GABRB2 showed the strongest expression correlation with other genes in glioma tissues (Additional file 1: Table S7), while, PRKAR2B, CCND2, C1orf173, WBSCR17, STXBP5L, PRKCE, KIF3A, GRAMD1B, SLC1A6 and ADCY1 were the hub genes in the co-expression network of normal brain tissues (Fig. 4; Additional file 1: Table S8).
Fig. 3.

Co-expressed differentially expressed driver genes and their network in glioma tissues; Of note, the greater the sizes of nodes are, the stronger the differentially expressed driver genes are co-expressed with surrounding genes. The nodes that have the same color belong to the same module detected by WGCNA
Fig. 4.

Co-expression networks of differentially expressed driver genes in normal brain tissue
Protein–protein interaction network analysis
In addition to co-expression analysis on driver genes at the mRNA level, we also wanted to know the interactions of driver genes in glioma at the protein level. To this aim, we applied STRING to construct a protein–protein interaction network using 685 driver genes. A high degree protein regulates or is regulated by many other proteins, suggesting an important role in the network of interactions. SRC, ST6GAL2, PIK3CA, PIK3R1, CREBBP, TP53, SOS1, EGFR, EGR1 and EIF1AX are at the core of the protein–protein interaction network (Total STRING score > 29.20, Additional file 1: Table S9, Figure S3). They are responsible for regulation of programmed cell death, protein metabolic process, apoptotic process, EGFR signaling pathway and signaling transduction, suggesting they may play key roles in glioma [24].
Copy number variations in glioma
We also obtained focal CNVs of 52 glioma samples at broad institute. Significant focal gains and deletions (q < 0.25) were found in 29 samples (29/52, 55.77%) at 5 loci (3 amplifications and 2 deletions). Among them, deletions at 9p21.3, 7p11.2 and amplification at 1q32.1 were the most frequent CNVs in glioma, with occurrence rates of 32.69% (17/52), 26.92% (14/52) and 13.46% (7/52) respectively (Additional file 1: Figure S4). 10 driver genes were involved in CNVs, including known tumor suppressors and oncogenes, such as EGFR (amplification, 7p11.2) and MET (amplification, 12q14.1). Many driver genes were also found to be implicated in the CNVs, including PIP4K2C (amplification, 12q14.1), REN (amplification, 1q32.1), PIK3C2B (amplification, 1q32.1), CDKN2B and CDKN2A (deletion, 9p21.3), COL6A3 and NEU2 (deletion, 2q37.3) and HDAC4 (deletion, 2q37.3).
Survival analyses in glioma
We acquired RNAseq and clinical outcome data from TCGA to evaluate whether the expression of driver genes is associated to survival of glioma patients. Overall, Kaplan–Meier survival analyses showed that the expression of 268 driver genes was significantly related to clinical outcomes of glioma patients. The high expression of 162 driver genes was negatively correlated with survival rates of glioma patients, such as SAMD9L, SAMD9, VAV3, FLNA, KDELC2, BRCA2, MAP3K1, BRCA1, LAMA2 and PDGFD (Additional file 1: Table S10). By contrast, 106 driver genes showed positive correlation with patients’ prognosis, such as BMP2, CSMD3, SMOC1, FAM110B, SLC1A6, GABRB3, BAG1, SNAP91, CALN1 and MAPK9 (Additional file 1: Table S10). 133 driver genes were up-regulated and associated to poor prognoses in glioma patients, such as NOTCH2, STAT3, IDH1, ARID1A and MSH6 (Fig. 5a). On the contrary, 43 driver genes were down-regulated expression and related to favorable clinical outcomes in glioma patients, such as PIK3R1, FLT3, PIK3R1 and RUNX1T1 (Fig. 5b). These driver genes might be potential prognostic biomarkers for glioma patients in the future.
Fig. 5.
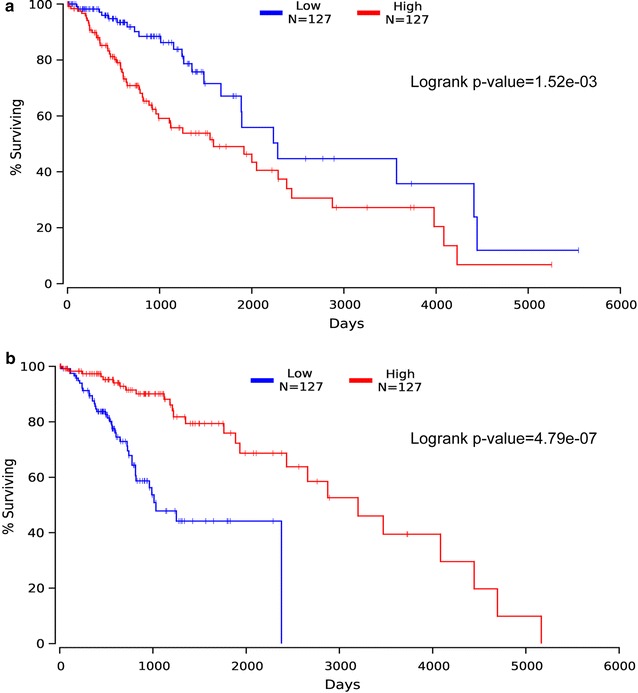
Correlation of MSH6 and RUNX1T1 expression with patients’ prognosis in glioma. a Patients with high expression of MSH6 (log2 normalized count was above 146.48, red) showed a poor survival rate as compared to glioma patients with low expression of MSH6 (log2 normalized count was smaller than 47.69, blue). b Glioma patients with high expression of RUNX1T1 (log2 normalized count was above 1035.7, red) showed a better prognosis than patients with low expression of RUNX1TX (log2 normalized count was less than 759.39, blue)
Discussion
In this study, we applied OncodriveCLUST, OncodriveFM, Icages, Drgap and Dendrixto 576 pairs of glioma tumor/normal samples and identified 685 driver genes and 215 driver pathways in glioma. Only a small number of driver genes are recurrently mutated in glioma samples, such as IDH1, TP53, ATRX, TTN, PTEN, EGFR and MUC16. By comparing the list of driver genes to annotated oncogene [25] and tumor suppressor gene [26] databases, we found 76 known oncogenes, such as BRAF, FOXO1, KRAS,MET and MTOR as well as 61 tumor suppressor genes, such as ATM, BRCA1,CHEK1, FOXO1 and NOTCH1. The majority of driver genes showed low or middle mutation frequencies and were first determined as driver genes in glioma, such as BCOR, FRG1B, GABRA6 and LRP1B. In addition, multiple IDH1-dependent driver genes were also detected by Oncodrive-FM and drgap, for instance NOTCH1 and ARID1A, suggesting these drivers might be therapeutic targets for IDH1-mutated gliomas.
In addition, we also uncovered 428 differentially expressed driver genes, including 330 up-regulated and 98 down-regulated genes, as well as 10 driver genes involved in CNVs, suggesting these genes might contribute to gliomagenesis in a variety of fashions. On the basis of the 428 differentially expressed driver genes, we built gene co-expression networks in glioma tissues and normal brain tissues. No significant loss of connections between genes were observed in glioma, which contrasts with Hao Li’s study [27], in which most of the gene–gene interactions and linkages in normal tissues had been broken or lost in the gastric cancer tissues. The inconsistent findings might be attributed to two factors. First of all, cancer types are greatly different in the two studies. Secondly, the co-expression networks were built using two distinct subset of genes, including differentially expressed driver genes in our study and differentially expressed genes in Hao’s study. The PPI network analysis indicated the top ranking genes are responsible for regulation of programmed cell death, protein metabolic process, apoptotic process, EGFR signaling pathway and signaling transduction, suggesting they may play pivotal roles in glioma [24].
Lastly, 268 driver genes were significantly correlated to clinical outcomes of glioma patients. Of them, two driver genes, MSH6 and RUNX1T1, drew our attention, as they have been repeatedly reported to be involved in multiple cancer types [28–39]. The MSH6 is a member of mismatch repair (MMR) genes, germline mutations in MMR genes, predominantly in MLH1, MSH2 and MSH6, are responsible for hereditary nonpolyposis colorectal cancer [28, 29], prostate cancer [30] and endometrial cancer [31]. In line with our study, high expression of MSH6 was significantly associated with poor survival rates in melanoma [32] and osteosarcoma [33]. The second gene, RUNX1T1, encodes a member of myeloid translocation genes. The chromosomal translocation t(8;21)(q22;q22) generates the RUNX1/RUNX1T1 fusion gene, which supports human haematopoietic stem cell self-renewal as well as leukaemic proliferation and clonogenicity in vivo [34–37]. The expression of RUNX1T1 was severely down-regulated in colorectal cancer (CRC), increased expression of RUNX1T1 suppressed cellular proliferation and sensitized CRC cells to 5-fluorouracil [38]. RUNX1T1 was frequently hypermethylated in ovarian tumors with high clinical stages and primary ovarian cancer-initiating cells. Enhanced RUNX1T1 expression inhibited ovarian cancer cell growth [39]. The results obtained in our study in combination with published reports support that MSH6 and RUNX1T1 have oncogenic and tumor suppressive functions respectively in cancers.
Conclusions
In conclusion, we performed an integrative study on the set of driver genes detected by five distinct computational tools, which enhanced our understanding of tumorigenesis and progression of glioma. The driver genes and pathways identified herein such as MSH6 and RUNX1T1 might be candidate prognostic biomarkers and therapeutic targets in glioma.
Authors’ contributions
BZ downloaded cancer mutations, copy number variations and patient clinical outcomes from TCGA, RNA-seq data from GEO. AL applied computational tools to predict cancer genes and pathways, conducted differentially expressed gene expression, Co-expression and PPI network and survival analyses. AL and WS wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
None.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- DEGs
differentially expressed genes
- GO
gene ontology
- PPI
protein–protein interaction
- CNV
copy number variation
- FPKM
fragments per kilobase of exon per million fragments mapped
- FDR
false discovery rate
- CRC
colorectal cancer
Additional file
Additional file 1: Figure S1. Driver genes in IDH1-mutated, non-IDH1-mutated and total glioma groups. A. The overlap of driver genes predicted by OncodriveCLUST in the three groups; B. The overlap of driver genes predicted by OncodriveFM in the three groups; C. The overlap of driver genes predicted by Icages in the three groups; D. The overlap of driver genes predicted by Drgap in the three groups; E. The overlap of driver genes predicted by Dendrix in the three groups; F. The overlap of driver pathways predicted by Dendrix in the three groups. Figure S2. Expression profiling of 428 driver genes between glioma and normal brain samples, expression values of each gene was presented as log(FPKM + 0.001). Figure S3. The protein–protein interaction network of driver genes. Network nodes represent proteins, network edges represent protein–protein associations. Notably, disconnected nodes were disabled in the network. Colored nodes refer to query proteins and first shell of interactors, white nodes are second shell of interactors. Blue lines represent known interactions from curated Kyoto Encyclopedia of Genes and Genomes database. Pink lines represent experimentally determined protein–protein interactions. Green lines denote genes that are frequently observed in each other’s genomic neighborhood. Black lines stand for genes whose expression is correlated across a large number of experiments. Dark blue gene families show similar occurrence patterns across genomes, light blue proteins show amino acid sequence similarity. Figure S4. The frequencies of copy number variations in 52 glioma samples.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12935-017-0458-y) contains supplementary material, which is available to authorized users.
Contributor Information
Aijun Liang, Email: 22251881@qq.com.
Bin Zhou, Email: 275248220@qq.com.
Wei Sun, Email: doctorsunwei000@163.com.
References
- 1.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, et al. NEURO-ONCOLOGY CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013;12:28–36. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claus EB, Walsh KM, Wiencke JK, Molinaro AM, Wiemels JL, Schildkraut JM, et al. Survival and low-grade glioma: the emergence of genetic information. Neurosurg Focus. 2015;38:1–10. doi: 10.3171/2014.10.FOCUS12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis GM, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ichimura K, Pearson DM, Kocialkowski S, Bäcklund LM, Chan R, Jones DTW, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009;11:341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silber JR, Blank A, Bobola MS, Ghatan S, Kolstoe DD, Berger MS. O6-methylguanine-DNA methyltransferase-deficient phenotype in human gliomas: frequency and time to tumor progression after alkylating agent-based chemotherapy. Clin Cancer Res. 1999;5:807–814. [PubMed] [Google Scholar]
- 8.Jaeckle KA, Eyre HJ, Townsend JJ, Schulman S, Knudson HM, Belanich M, et al. Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: a Southwest Oncology Group study. J Clin Oncol. 1998;16:3310–3315. doi: 10.1200/JCO.1998.16.10.3310. [DOI] [PubMed] [Google Scholar]
- 9.Hegi ME, Diserens A, Godard S, Dietrich P, Regli L, Ostermann S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–1874. doi: 10.1158/1078-0432.CCR-03-0384. [DOI] [PubMed] [Google Scholar]
- 10.Chang K, Creighton CJ, Davis C, Donehower L, Drummond J, Wheeler D, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Cunningham F, Rios D, McLaren WM, Smith J, Pritchard B, et al. Ensembl variation resources. BMC Genom. 2010;11:293. doi: 10.1186/1471-2164-11-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamborero D, Gonzalez-perez A, Lopez-bigas N. Genome analysis OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics. 2013;29:2238–2244. doi: 10.1093/bioinformatics/btt395. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012;40:1–10. doi: 10.1093/nar/gks743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hua X, Xu H, Yang Y, Zhu J, Liu P, Lu Y. DrGaP: a powerful tool for identifying driver genes and pathways in cancer sequencing studies. Am J Hum Genet. 2013;93:439–451. doi: 10.1016/j.ajhg.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong C, Yang H, He Z, Liu X, Wang K. iCAGES: integrated cancer genome score for comprehensively prioritizing cancer driver genes in personal genomes. Genome Med. 2016;8:135. doi: 10.1186/s13073-016-0390-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vandin F, Upfal E, Raphael BJ, Hormozdiari F, Hajirasouliha I, Mcpherson A. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012;22:375–385. doi: 10.1101/gr.120477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gill BJ, Pisapia DJ, Malone HR, Goldstein H, Lei L, Sonabend A, et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc Natl Acad Sci USA. 2014;111:12550–12555. doi: 10.1073/pnas.1405839111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Therneau T. Survival analysis. Cran. 2017. https://cran.r-project.org/web/packages/survival/survival.pdf.
- 21.Anaya J. OncoLnc : linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. 2016.
- 22.Geisbrecht BV, Gould SJ. The human PICD gene encodes a cytoplasmic and peroxisomal NADP+-dependent isocitrate dehydrogenase. J Biol Chem. 1999;274:30527–30533. doi: 10.1074/jbc.274.43.30527. [DOI] [PubMed] [Google Scholar]
- 23.Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13:345. doi: 10.1007/s11910-013-0345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Sun J, Zhao M. ONGene: a literature-based database for human oncogenes. J Genet Genom. 2016;44:2016–2018. doi: 10.1016/j.jgg.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Zhao M, Sun J, Zhao Z. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res. 2013;41:970–976. doi: 10.1093/nar/gks937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Yu B, Li J, Su L, Yan M, Zhang J, et al. Characterization of differentially expressed genes involved in pathways associated with gastric cancer. PLoS ONE. 2015;10:1–17. doi: 10.1371/journal.pone.0125013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plaschke J, Krüger S, Dietmaier W, Gebert J, Sutter C, Mangold E, et al. Eight novel MSH6 germline mutations in patients with familial and nonfamilial colorectal cancer selected by loss of protein expression in tumor tissue. Hum Mutat. 2004;23:285. doi: 10.1002/humu.9217. [DOI] [PubMed] [Google Scholar]
- 29.Kolodner R, Tytell J, Schmeits J, Kane MF, Gupta RD, Wahlber S, et al. Germline msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–5074. [PubMed] [Google Scholar]
- 30.Grindedal EM, Møller P, Eeles R, Stormorken AT, Bowitz-Lothe IM, Landrø SM, et al. Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol Biomark Prev. 2009;18:2460–2467. doi: 10.1158/1055-9965.EPI-09-0058. [DOI] [PubMed] [Google Scholar]
- 31.Devlin LA, Graham CA, Price JH, Morrison PJ. Germline MSH6 mutations are more prevalent in endometrial cancer patient cohorts than hereditary non polyposis colorectal cancer cohorts. Ulster Med J. 2008;77:25–30. [PMC free article] [PubMed] [Google Scholar]
- 32.Alvino E, Passarelli F, Cannavò E, Fortes C, Mastroeni S, Caporali S, et al. High expression of the mismatch repair protein MSH6 is associated with poor patient survival in melanoma. Am J Clin Pathol. 2014;142:121–132. doi: 10.1309/AJCPCX2D9YULBBLG. [DOI] [PubMed] [Google Scholar]
- 33.Jentzsch T, Robl B, Husmann M, Bode-Lesniewska B, Fuchs B. Expression of MSH2 and MSH6 on a tissue microarray in patients with osteosarcoma. Anticancer Res. 2014;34:6961–6972. [PubMed] [Google Scholar]
- 34.Mulloy JC, Cammenga J, Berguido FJ, Wu K, Zhou P, Comenzo RL, et al. Maintaining the self-renewal and differentiation potential of human CD34+ hematopoietic cells using a single genetic element. Blood. 2003;102:4369. doi: 10.1182/blood-2003-05-1762. [DOI] [PubMed] [Google Scholar]
- 35.Heidenreich O, Riehle H, Hadwiger P, John M, Heil G, Vornlocher H, et al. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t (8;21)-positive leukemic cells. Gene Expr. 2003;101:3157–3163. doi: 10.1182/blood-2002-05-1589. [DOI] [PubMed] [Google Scholar]
- 36.Martinez N, Drescher B, Riehle H, Cullmann C, Vornlocher H-P, Ganser A, et al. The oncogenic fusion protein RUNX1-CBFA2T1 supports proliferation and inhibits senescence in t(8;21)-positive leukaemic cells. BMC Cancer. 2004;4:44. doi: 10.1186/1471-2407-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez Soria N, Tussiwand R, Ziegler P, Manz MG, Heidenreich O. Transient depletion of RUNX1/RUNX1T1 by RNA interference delays tumour formation in vivo. Leukemia. 2009;23:188–190. doi: 10.1038/leu.2008.157. [DOI] [PubMed] [Google Scholar]
- 38.Alfayez M, Vishnubalaji R, Alajez NM. Runt-related transcription factor 1 (runx1t1) suppresses colorectal cancer cells through regulation of cell proliferation and chemotherapeutic drug resistance. Anticancer Res. 2016;36:5257–5263. doi: 10.21873/anticanres.11096. [DOI] [PubMed] [Google Scholar]
- 39.Yeh KT, Chen TH, Yang HW, Chou JL, Chen LY, Yeh CM, et al. Aberrant TGFβ/SMAD4 signaling contributes to epigenetic silencing of a putative tumor suppressor, RunX1T1 in ovarian cancer. Epigenetics. 2011;6:727–739. doi: 10.4161/epi.6.6.15856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.