

Abstract
The morphology of a population of mitochondria is the result of several interacting dynamical phenomena, including fission, fusion, movement, elimination and biogenesis. Each of these phenomena is controlled by underlying molecular machinery, and when defective can cause disease. New understanding of the relationships between form and function of mitochondria in health and disease is beginning to be unraveled on several fronts. Studies in mammals and model organisms have revealed that mitochondrial morphology, dynamics and function appear to be subject to regulation by the same proteins that regulate apoptotic cell death. One protein family that influences mitochondrial dynamics in both healthy and dying cells is the Bcl-2 protein family. Connecting mitochondrial dynamics with life-death pathway forks may arise from the intersection of Bcl-2 family proteins with the proteins and lipids that determine mitochondrial shape and function. Bcl-2 family proteins also have multifaceted influences on cells and mitochondria, including calcium handling, autophagy and energetics, as well as the subcellular localization of mitochondrial organelles to neuronal synapses. The remarkable range of physical or functional interactions by Bcl-2 family proteins is challenging to assimilate into a cohesive understanding. Most of their effects may be distinct from their direct roles in apoptotic cell death and are particularly apparent in the nervous system. Dual roles in mitochondrial dynamics and cell death extend beyond BCL-2 family proteins. In this review, we discuss many processes that govern mitochondrial structure and function in health and disease, and how Bcl-2 family proteins integrate into some of these processes.
Keywords: Mitochondria, Apoptosis, Mitochondrial dynamics, Neurons, Bcl-xL, Drp1
1. Introduction
Mitochondria are considered to have originated from the endosymbiosis of an α-proteobacterium. Vestiges of their bacterial ancestry are apparent by their micrometric size (similar to bacteria), double membrane, circular DNA, unique genetic code and ribosomes, and their semblance to bacterial cell division and movement. However, these microbial resemblances have been dramatically altered by 1–2 billion years of evolution resulting in the progressive loss of independence and further conversion from mitochondrial ancestor to bona fide eukaryotic organelle. This intracellular domestication was accompanied by transfer of most protomitochondrial genes to the nuclear genome and many other changes related to the intracellular environment of eukaryotic cells. The small number of mitochondria-encoded proteins (13 in humans) is counterbalanced by a rich mitochondrial proteome of nuclear-encoded proteins targeted to mitochondria by a diverse complement of signal sequences (Cotter et al., 2004).
Conversely, important eukaryotic traits apparently emerged from microbial acquisition, including a more advanced energy metabolism. Mitochondrial structure appears to have been exploited to take over not only energy production, but additional tasks to meet the needs of cells and organisms. Mitochondria of modern-day eukaryotic cells fulfill multiple cellular functions such as maintenance of ion homeostasis (e.g. calcium), steroid production, biosynthesis of specialized iron-containing complexes, heat production, cellular redox state regulation, generation of reactive oxygen species (ROS) (e.g. as second messengers) and initiation of apoptotic cell death by liberating key components into the cytosol, a process regulated by Bcl-2 family proteins (McBride et al., 2006; Nunnari and Suomalainen, 2012).
Like “soft robots” (Kim et al., 2013), mitochondria are highly dynamic organelles that emulate diverse outputs by adapting their number, shape, position, connectivity and motion in response to intracellular inputs and extracellular environment. Thus, mitochondrial morphology is intertwined with mitochondrial function, but most of the details are not yet known (Benard and Rossignol, 2008; Bindoff et al., 1991; Guillery et al., 2008; Wai and Langer, 2016). However, interest in this direction is spawned by the alterations in mitochondrial structure and function associated with aging, cancer, heart disease and a growing number of neurological disorders including epilepsy, stroke, Wolfram syndrome, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease (Archer, 2013; Cagalinec et al., 2016; Corrado et al., 2012; Knott et al., 2008; Lee et al., 2016b; Rugarli and Langer, 2012; Schon and Przedborski, 2011; Vafai and Mootha, 2012). Much less is known about ultrastructural morphology and dynamics of mitochondrial inner membrane cristae despite being disrupted in neurodegenerative and other disease states. Research over the last two decades has begun to reveal the nature and complexity of cellular signaling pathways that sculpt the mitochondrial network (Burte et al., 2015; Nasrallah and Horvath, 2014).
One key protein family that influences both mitochondrial structure and function in both healthy and dying cells is the extended Bcl-2 family that apparently arose with metazoans (Aouacheria et al., 2013; Bhola and Letai, 2016). Members of this extended Bcl-2 protein conglomerate are thought to exert many of their functions on the cytosolic side of the mitochondrial membrane, where most research has focused on the events leading to apoptosis. A key event during apoptosis is pore-formation in the mitochondrial outer membrane induced by oligomerization of the pro-apoptotic Bax protein, resulting in permeabilization of mitochondria and subsequent caspase activation in the cytosol to orchestrate apoptosis (Basanez et al., 1999; Kuwana et al., 2002).
In addition to apoptotic cell death, the extended Bcl-2 family of proteins (Aouacheria et al., 2015), interact to exert many effects in healthy cells that are distinct from apoptotic processes (Chau et al., 2000; Chen et al., 2011; Cheng et al., 1996; Chipuk et al., 2005; Fannjiang et al., 2003; Gimenez-Cassina et al., 2014; Hosoi et al., 2017; White et al., 2005; Yi et al., 2011). In this capacity, Bcl-2 family proteins interact physically or functionally with a remarkably broad range of other proteins and lipids, the full extent of which is difficult to assimilate and comprehend. However, some of the alternative “day-job” functions identified for Bcl-2 family proteins have been confirmed, suggesting that the tie that binds them together (e.g. an underlying biochemical function of Bcl-2) remains elusive. Here we discuss the dynamical phenomena and underlying mechanisms contributing to mitochondrial morphology, focusing on the nervous system. We also explore the integration of Bcl-2 family proteins into many of processes influencing mitochondrial structure and function in both healthy and dying cells, while covering conceptual and methodological topics related to mitochondrial content, morphology and dynamics.
2. Mitochondrial dynamics defined by five phenomena
2.1. Deciphering the core mitochondrial dynamics that underlie morphology
Mitochondrial organelle shapes and patterns (e.g. fragmented, elongated, branched, reticulated and number of organelles per area) change rapidly with conditions and can differ greatly between cell types and disease states (see section 10). Objective parameterization of mitochondrial shapes requires sophisticated image analysis methods and software (Giedt et al., 2016; McClatchey et al., 2016; Vinegoni et al., 2016). These morphological quantifications are useful experimental readouts, but it is important to appreciate that morphology is not a unique determinant of the underlying dynamical phenomena (Bordt et al., 2017). For example, elongated mitochondrial shape could arise from increased fusion, decreased fission, increased growth or any combination of these phenomena. Thus, it is important to distinguish mitochondrial morphology from mitochondrial dynamics. The former is a physical pattern that characterizes a mitochondrial population, while the latter refers to a set of dynamical phenomena whose interaction determines the observed morphological patterns.
The appellation of ‘mitochondrial dynamics’ is used commonly to refer to only two dynamic phenomena, fission and fusion. Instead, at least five dynamical phenomena determine mitochondrial morphology: fission, fusion, movement, degradation/removal and growth/biogenesis (Fig. 1). Each of these five phenomena are in turn determined by underlying biochemical mechanisms that act on mitochondria both directly (e.g. the GTPase activity of fission and fusion proteins; sections 4 and 5) and indirectly (e.g. cytoskeleton-driven architectural changes; see sections 6 and 10) (Westrate et al., 2014). Is there a strategy to tease apart the dynamical behavior and underlying mechanisms based only on the quantified morphological features?
Figure 1. Causal relationship between molecular machinery, mitochondrial dynamics and mitochondrial morphology.
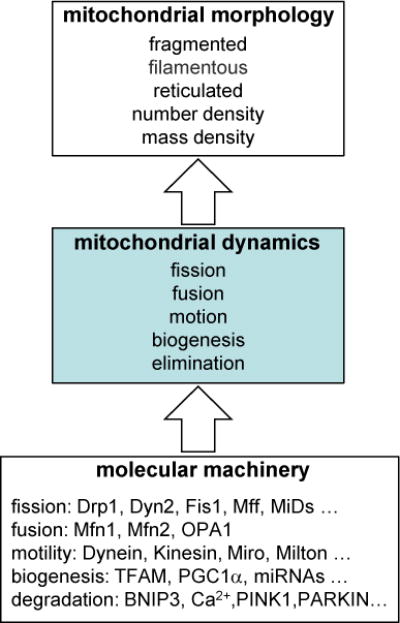
Schema summarizes the relationship between the underlying biochemical mechanisms that drive morphology through their effects on mitochondrial dynamics. Note that individual molecular species may in fact have multiple direct influences on dynamical phenomena.
A valid strategy for understanding mitochondrial dynamics and morphology is to apply the quantitative reasoning of “dynamical systems theory” to the task of dissecting a relatively simple biological model system. The complexity of mitochondrial morphology in a simple system should be limited and therefore relatively easier to measure and parameterize. The axons and dendrites of neurons provide such a system. The confined and constrained environment of linear neuronal processes limits allowed morphology to essentially one-dimensional structures.
Moreover, dynamical phenomena are also relatively easier to observe via methods developed for direct and simultaneous observation of fission, fusion and movement of fluorescently-tagged mitochondria (Berman et al., 2009; Berman et al., 2008). This approach allows the biological system to be understood in terms of the dynamic properties of an equilibrium state, wherein biochemical mechanisms determine mitochondrial dynamics, which in turn induce a dynamical system that determines morphology (Fig. 1). Thus, there is an emerging understanding of how fission, fusion, degradation, growth and transport, interact to determine mitochondrial morphology and content.
2.2. Neurons have unbalanced ratios of fission and fusion
Consider, for example, a study of the anti-apoptotic Bcl-2 family protein Bcl-xL in neuronal processes of healthy cultured rat cortical neurons using live video microscopy (Berman et al., 2009; Berman et al., 2008). Bcl-xL is essential for normal brain development and is abundantly expressed on mitochondria in adult neurons (Chen et al., 2011) (see sections 8 and 9). Expressed and endogenous Bcl-xL increased the length/size of mitochondrial and the localization of mitochondria to synapses (Berman et al., 2008; Li et al., 2008). To investigate how Bcl-xL alters mitochondrial dynamics, mitochondria in neuronal processes of healthy cultured rat cortical neurons were observed by live video microscopy. This study measured several morphological parameters (mitochondrial number density, mass density and distribution of lengths) (see sections 10.1 and 10.2), as well as dynamical parameters (mitochondrial fission rates per unit length, fusion rates per encounter and velocities). To accurately quantify these parameters, mitochondria were tagged with RFP to mark all mitochondria and with photoactivatable-GFP to detect fusion events leading to rapid spread of GFP throughout the red-only mitochondria (Berman et al., 2009; Berman et al., 2008).
Contrary to original expectation of a balance between fission and fusion rates at steady state, the data clearly revealed that fission rates could be several-fold higher than fusion rates (Berman et al., 2009). Therefore to maintain constant mitochondrial numbers and mass, mitochondrial biogenesis (growth in length) and degradation/elimination must compensate for the observed unbalanced ratios of fission and fusion. In particular, simple counting arguments reveal that if the number of mitochondrial organelles is in equilibrium, then the average rates of binary fission Rfission (one organelle divides into two organelles), binary fusion Rfusion (two organelles fuse into one organelle) and degradation Rdegradation (removal of one organelle from the system), must be related by the equation:
Thus, at equilibrium, a measured ratio of 4:1 in fission:fusion rates requires a ratio of 4:3 in fission:degradation rates. This implies that mitochondrial degradation/elimination rates could be nearly as high as fission rates in some scenarios. Another important consequence of this counting argument is that at equilibrium, growth/biogenesis of mitochondrial organelles is required to make up for loss of mitochondrial biomass due to degradation/elimination (Berman et al., 2009; Berman et al., 2008). Thus, this analysis revealed that degradation and consequent biogenesis of mitochondria are more important contributors to mitochondrial dynamics than previously anticipated, and that mitochondrial morphology is not determined simply by the ratio of fission to fusion.
This quantitative model of an essentially one-dimensional environment of neuronal processes proved to be a powerful model system for teasing apart how the interactions between the five dynamical processes produce morphology. For example, this approach revealed a role for fission factor Drp1 in the mitochondrial morphology changes observed with the anti-apoptotic protein Bcl-xL (Berman et al., 2009). The results further revealed that Bcl-xL has additional Drp1-independent functions distinct from its known role as a direct inhibitor of Bax/Bak-dependent apoptosis. Using both overexpression and neuron-specific Bcl-xL knockout mice, measurements of mitochondrial dynamic properties indicate that Bcl-xL increases mitochondrial length, not by inhibiting fission, but by increasing the rates of both mitochondrial fission and fusion, implying that the increased mitochondrial length observed requires an increase in mitochondrial biomass/growth at steady state, which was only partly dependent on the fission factor Drp1 (Berman et al., 2009) (see section 8). More generally, it is possible that the principles of mitochondrial dynamics that were identified in this geometrically constrained neural system could be extended more broadly to understand the formation of three-dimensional morphology (e.g. reticulated structures) found in other cell types.
2.3. Balancing the mitochondrial dynamics equation versus neurodegeneration
A difficult question that arose but was not answered in the study by Berman et al. (Berman et al., 2009) was how and where does elimination of mitochondrial organelles occur? Because no degradation events were directly observed within the neuronal segments being monitored, the simple counting arguments strongly imply that mitochondria are being transported out of the view fields for degradation elsewhere (see section 7.4). There is compelling evidence from in vivo studies that damaged mitochondria are transported from neuronal processes towards the cell body for degradation (Dukes et al., 2016). Alternative mechanisms may also exist to eliminate mitochondria from neuronal processes. This possibility is appealing to avoid potential disadvantages associated with long-distance transport to the cell body for degradation, which on average would require transport over half the length of the dendrite or axon. This would imply a large energetic cost associated with degradation as well as a long lifetime for damaged mitochondria. It also begs the question: where would the energy come from to transport mitochondria if not from these damaged mitochondria? Furthermore, given that the volume of axonal/dendritic processes exceeds that of the soma, reliance solely on long-distance transport back to the cell body for degradation would imply the possibility of traffic jams of damaged mitochondria at a junction near the cell body. Moreover, damaged mitochondria are suggested to over-produce ROS, so their trip over long distances could potentially impair cellular constituents (Harman, 1972; Tal et al., 2009). All this presents a conundrum.
A possible solution to the conundrum comes from the observation of mitochondria in vivo, rather than in cultures of isolated neurons as recently reported for C. elegans (Melentijevic et al., 2017). For some years, the researchers noted that fluorescently labeled components of the six gentle touch neurons of C. elegans were found outside the cell particularly during some stages of life. Using microscopy and genetics, they found that whole mitochondrial organelles as well as protein aggregates, including neurotoxic aggregates of Huntingtin polyQ protein (Htt-Q128), were released from neurons in large membrane-bound structures approximately the size of the cell soma, which they refer to as exophers (Melentijevic et al., 2017). By unknown mechanisms, exophers appear to preferentially package damaged over healthy mitochondria and aggregating fluorescent proteins over non-aggregating versions (Melentijevic et al., 2017) (see section 7.4). A potentially analogous process was identified in retinal ganglion cells in mice expressing fluorescently tagged mitochondria, revealing that labeled mitochondria derived from neurons were present in neighboring astrocytes where they were subsequently degraded (Davis et al., 2014). Treatment with inhibitors of the mitochondrial respiratory chain enhanced this process implying that the process is selective for damaged mitochondria. Similar phenomena have been described in past decades, but these elegant studies provide compelling new evidence (see section 7.4).
3. Many events and conditions influence mitochondrial dynamics
3.1. Generalizations regarding energetic stress
Mitochondrial dynamics can be described by the five dynamic but fundamental processes: fission, fusion, growth/biogenesis, degradation and the movement/transport that allows organelles to come into contact with one another (section 2.1.–2.3. and 10.3–4.). Within minutes, the interaction of these processes can cause dramatic shape changes in mitochondria, the most rapid of which may be fission (Berman et al., 2009; Liesa and Shirihai, 2013). These dynamical processes respond to cellular conditions to affect morphology and function. There is no formula known that connects form and function of mitochondria, yet distortion of mitochondrial morphology as well as ultrastructure is associated with many neurodegenerative disorders (Bindoff et al., 1991; Cagalinec et al., 2016; Corrado et al., 2012; Knott et al., 2008; Rugarli and Langer, 2012; Schon and Przedborski, 2011). At the level of whole organelles, the predominate theory is that when bioenergetic needs are high, for example during starvation, mitochondria are more tubular in connected networks, thereby preventing the degradation of small mitochondria by mitophagy (Gomes et al., 2011a; Rambold et al., 2011). One exception is during glucose deprivation, mitochondrial elongation is inhibited if amino acid availability is sustained (Gomes et al., 2011b). Conversely, during times of catabolism or cell death, mitochondria undergo fission/fragment into a discontinuous network, a process promoted by the GTPase Drp1 and pro-apoptotic Bcl-2 family proteins Bax and Bak (see section 4). In contrast to dying cells, deletion of both Bax and Bak in fibroblasts results in fragmented mitochondrial morphology suggesting alternative functions of Bax and Bak in healthy cells (Karbowski et al., 2006; Zhang et al., 2013) (see section 8.3).
3.2. Biogenesis or import of mitochondria
Because cells lack the capability to synthesize mitochondria de novo, there are only two known mechanisms for increasing mitochondrial number, by mitochondrial fission and by direct transfer from another cell. Although the export of mitochondria from neurons is considered a protective mechanism to eliminate damaged organelles (see section 7.4), evidence suggests that useful mitochondria can be acquired, such as the donation of astrocyte mitochondria to neurons following stroke (Hayakawa et al., 2016).
3.3. Benefits and restrictions on fusion and fission
The key proteins responsible for mediating mitochondrial fission and fusion belong to a family of large GTPases, and GTP hydrolysis provides energy and force required to split or fuse mitochondrial membranes. Seminal studies in model organisms originally identified these key mediators, which are conserved in mammals. In mammals, Drp1 is responsible for fission, while Mitofusin 1 and 2 (Mfn1/2) and Opa1 carry out fusion of the mitochondrial outer and inner membranes, respectively (Chan, 2012; van der Bliek et al., 2013). All appear to be critical for neuronal health as pathogenic mutations in each have been shown to manifest in neurological diseases ranging from severe, early onset to isolated optic atrophy (see sections 4 and 5).
Fission and fusion together with three other parameters (growth, degradation and movement/transport) control the dynamic shape changes of mitochondrial organelles (see section 2). Fusion events enable mixing of mitochondrial contents and can prevent mitochondrial degradation (Chen and Chan, 2010). Thus, fusion of a damaged mitochondrion to a healthy mitochondrion is suggested to restore loss of function. Interestingly, fusion appears to be dependent on mitochondrial membrane potential Δψm, as mitochondrial membrane depolarization inhibits fusion (Legros et al., 2002; Meeusen et al., 2004; Twig et al., 2008). Severely damaged mitochondria with mtDNA mutations or oxidized proteins may also be less likely to fuse to the network, providing a first layer of quality control. On the other hand, fission generates small mitochondrial fragments that can subsequently be targeted for degradation by mitophagy (Twig et al., 2008; van der Bliek et al., 2013; Westermann, 2012), providing a second layer of quality control. However, mitochondrial fission may not be a prerequisite for the elimination of mitochondria that are exported to neighboring cells via large vesicles such as neuronal exophers (~4 μm) (Melentijevic et al., 2017) or large-diameter tunneling nanotubes that connect macrophages (>0.7 μm) (Onfelt et al., 2006) (see section 7.4).
Mitochondrial fission and fusion are essential for mitochondrial remodeling during several important homeostatic cell processes such as cell cycle (fission being a requisite step for mitosis), cellular differentiation, and release of pro-apoptotic proteins during apoptotic cell death (Bender and Martinou, 2013; Lackner, 2014; van der Bliek et al., 2013). The importance of mitochondrial dynamics in disease extends beyond the host cell to include human pathogens, as tubulation of mitochondria in the fungal pathogen Cryptococcus gattii has gained attention as a marker of virulence (Engelthaler et al., 2014; Voelz et al., 2014).
3.4. The extended Bcl-2 family enters the fray
Bcl-2 family proteins have a seemingly unfathomable array of functions reported to affect mitochondria, cells and organisms, but one emerging theme is their role in mitochondrial dynamics. Their diverse functions stem in part from the unconventional grouping of diverse proteins into the extended Bcl-2 protein “family”. In addition to the homologous anti-apoptotic (e.g. Bcl-2 and Bcl-xL) and pro-apoptotic (Bax and Bak) proteins that share overall sequence similarity, the Bcl-2 “family” typically includes an honorary subfamily of eight non-homologous “BH3-only” proteins. These eight canonical BH3-only proteins are also unrelated to each other but share one of four Bcl-2 homology (BH) motifs present in the anti-/pro-death Bcl-2 homologs (Aouacheria et al., 2015) (see sections 8 and 9).
3.4.1. The conglomerate of BH3-containing proteins
BH3-only proteins have garnered considerable attention because their BH3 motifs bind and regulate both anti- and pro-death Bcl-2 homologs in response to different stress-induced cues (Bhola and Letai, 2016; Youle and Strasser, 2008). These features of BH3-only proteins have driven the development of chemical and peptide BH3 mimetics to inhibit both cellular and viral Bcl-2 homologs for clinical applications (Oltersdorf et al., 2005; Petros et al., 2004). The anti-cancer therapeutic Venetoclax, which binds the BH3-binding groove of Bcl-2 to promote cell death, is the first of these to be FDA approved (Green, 2016). BH3 mimetics may have other potential uses. Approximately 40 additional proteins from an array of different protein families have been reported to contain a functional BH3 motif that connects Bcl-2 family proteins to many other cellular functions, including mitophagy regulator Bnip3 (Aouacheria et al., 2015; Hamacher-Brady and Brady, 2016), although many BH3-containing candidates remain unconfirmed. Additional factors, such as MTCH2, a mitochondrial receptor for BH3-only protein BID, are also suggested to modulate mitochondrial morphology (Gross and Katz, 2017).
3.4.2. Bcl-2 homologs meet mitochondrial fission factor Drp1
The intersection of mitochondrial dynamics and life-death decisions may arise from Bcl-2 family protein interactions with the large GTPase that mediates mitochondrial fission, Drp1, which itself can contribute to cell death. The death-promoting effects of Drp1 in stressed cells may extend beyond classical apoptosis as this function is apparently conserved in yeast (which do not undergo classical apoptosis) as well as worms and mammals based on enhanced survival of Drp1/Dnm1-deficient cells (Cheng et al., 2008a; Cheng et al., 2008b; Delivani et al., 2006; Fannjiang et al., 2004; Frank et al., 2001; Jagasia et al., 2005; Lee et al., 2016a; Lu et al., 2011). Although not definitively demonstrated, it is possible that a direct role of Bcl-2 family proteins in regulating mitochondrial dynamics is an important underlying mechanism for their diverse effects on cells (e.g. localization of mitochondria at neuronal synapses, calcium handling at mitochondria-associated ER membranes or MAM, mitochondrial energetics, etc.).
3.4.3. The emerging non-death functions of Bcl-2 homologs
The best characterized function of the Bcl-2 family is their control of mitochondrial outer membrane permeability to release selected apoptosis-inducing factors into the cytosol during apoptosis. However, mounting evidence has revealed alternative non-death roles for many BCL-2-related proteins in regulating mitochondrial dynamics (sections 2.2), autophagy (section 7.2), mitochondrial energetics and neuronal activity (section 8), and membrane curvature and channel activity (section 9). Delineation of these detailed mechanisms remains an emerging field but is rapidly gaining new ground. For example, the pro-apoptotic Bax-like protein Bak is inhibited when bound to VDAC2 on the outer mitochondrial membrane (Cheng et al., 2003). While disruption of this interaction can liberate Bak to promote apoptosis, recent work revealed an alternative function of Bak important for peroxisome biogenesis that may help explain the disease mechanisms of some patients (Fujiki et al., 2017; Hosoi et al., 2017). While excess Bak on peroxisomal membranes can indeed cause trouble, presumably analogous to its effects on mitochondria, additional intriguing evidence suggests that a normal function of Bak on peroxisomes is to facilitate the controlled release of catalase, which has been observed previously in the cytosol (Fujiki et al., 2017; Hosoi et al., 2017). Thus Bak is yet another factor shared between peroxisomes and mitochondria (Sugiura et al., 2017).
Anti-apoptotic Bcl-2 family proteins also have non-death roles involving membrane permeability. Rather than increasing membrane permeability, neurons lacking the anti-apoptotic Bcl-2 homolog Bcl-xL have increased ion flux and excessive swings in membrane potential across the inner mitochondrial membrane where a significant portion of endogenous Bcl-xL protein resides in the brain, potentially for the purpose of regulating synaptic activity (Alavian et al., 2011; Chen et al., 2011). Both anti- and pro-apoptotic Bcl-2 family proteins as well as pro-apoptotic caspases can also modulate synaptic activity in neurons, and the mechanisms are under investigation (see sections 8.2 and 8.3).
4. Cellular machinery of mitochondrial fission
Mitochondrial fission appears to be mediated by two large GTPases, the dynamin-related protein 1 (Drp1) and more recently, dynamin-2 (Dyn2) (Lee et al., 2016a). Human mutations in DRP1 or DYN2 have been associated with neurological impairments (Gonzalez-Jamett et al., 2014; Vanstone et al., 2016; Waterham et al., 2007). Drp1, despite lacking a transmembrane domain, cycles from the cytosol to spots on the mitochondrial outer membrane (Smirnova et al., 1998) by interacting with several proteins, including Fis1, Mff, MiD49, MiD51 and a BAR-domain protein endophilin B1 (Cerveny et al., 2001; Cherok et al., 2017; Fekkes et al., 2000; Gandre-Babbe and van der Bliek, 2008; Otera et al., 2010; Palmer et al., 2011; Tieu and Nunnari, 2000; Zhao et al., 2011). The mitochondrial receptors are suggested to regulate Drp1-mediated fission in response to other signals (Koirala et al., 2013; Lackner et al., 2009; Loson et al., 2014; Loson et al., 2013). At the mitochondria, Drp1 drives fission by wrapping around the outer surface of organelles at sites of ER-mitochondria contacts (Friedman et al., 2011; Shim et al., 2012; Stavru et al., 2013), where actin is in close apposition (Li et al., 2015; Moore et al., 2016), and where pro-apoptotic Bcl-2 family protein Bax is also concentrated (Karbowski and Youle, 2003).
Many labs have reported that Drp1 contributes to apoptotic cell death by inducing mitochondrial fission/fragmentation. The details of how Bax and Drp1 contribute individually or cooperate to damage mitochondrial membranes are currently debated. Multiple cell stress signals can trigger the recruitment of Bax monomers to the outer mitochondrial membrane where Bax oligomerizes, resulting in MOMP (mitochondrial outer membrane permeabilization), a hallmark of apoptotic cell death (Bhola and Letai, 2016). More recently, Drp1 and its mitochondrial receptors have been linked to Bcl-2 family proteins in a different manner. Deletion of either Drp1 or Mff results in increased ubiquitination and degradation of MiD49 and the anti-apoptotic Bcl-2 family protein Mcl-1, which is mediated by E3 ubiquitin ligase MARCH5 located on the the outer mitochondrial membrane (Cherok et al., 2017). Drp1 is itself posttranslationally regulated by activating phosphorylation at Ser616 and inhibitory phosphorylation at Ser637. Oncogenic Ras may promote tumorigenesis in part by stimulating mitochondrial fission dependent on ERK phosphorylation on Drp1 Ser616, consistent with high levels of phospho-S616 in pancreatic cancer (Kashatus et al., 2015), and possibly also glioblastoma (Xie et al., 2015). However, Drp1-dependent fission induced by some death stimuli can induce MOMP independently of Bax, Bak or other apoptosis factors, implying the involvement of other factors (Oettinghaus et al., 2016). Drp1 inhibitors are being developed as potential therapeutics to suppress neurodegeneration. However, the fission inhibitor mdivi-1, originally identified as a direct inhibitor of Drp1, instead appears to be an inhibitor of Complex I of the mitochondrial respiratory chain (Bordt et al., 2017).
At least one other GTPase involved in mitochondrial fission is the ubiquitously expressed dynamin-2 protein (Lee et al., 2016a). Dyn2 was recently reported to function in concert with Drp1 by taking charge of the final step of mitochondrial division (Lee et al., 2016a). The ER-associated protein Inf2 (Korobova et al., 2014; Korobova et al., 2013), in conjunction with the actin nucleation factor Spire, was shown to promote actin polymerization at mitochondrial fission sites (Manor et al., 2015). Although not precisely mapped, these data suggest that the ER and local actinomyosin cytoskeleton delineate mitochondrial division sites where Drp1 can be recruited (De Vos et al., 2005). As the diameter of mitochondria is larger than oligomerized Drp1, initial constriction of mitochondria via the ER and actin cytoskeleton may facilitate Drp1 recruitment allowing its oligomerization. Interestingly, Dyn2 is recruited to Drp1 spots on mitochondria, which further constricts mitochondrial organelles to mediate fission and organelle division (Lee et al., 2016a). Dyn-2- and Drp1-double-deficient cells have delayed mitochondrial fragmentation and recruitment of pro-apoptotic Bax to mitochondria following staurosporine treatment (Lee et al., 2016a). Although the relationship between Bax and the fission machinery is being debated, several groups have observed Bcl-2 family proteins to interact with and modulate mitochondrial fission and fusion machinery (see sections 3.3 and 3.4).
Mitochondrial fission is also highly dependent on interactions between mitochondria and both the actin cytoskeleton and microtubule networks. Disruption of actin filaments with Latrunculin A induces Fis-1/Dnm1-dependent fission in yeast (Fannjiang et al., 2004; Jensen et al., 2000). Drp1 itself was shown to interact with actin filaments, which have been proposed to act as Drp1 reservoirs that can be mobilized for fission (Hatch et al., 2016). Recently, Moore et al. (Moore et al., 2016) have conducted elegant experiments demonstrating that actin filaments transiently associate with mitochondria at ER-mitochondrial contact sites to promote mitochondrial fission. Actin was found to cycle through all cellular mitochondria (irrespective of their membrane potential) in about 15 min, with actin-dependent fission events undergoing sometimes spectacular clockwise or anti-clockwise rotation around the cytoplasm (Moore et al., 2016).
5. Cellular machinery of mitochondrial fusion
5.1. Outer mitochondrial membrane fusion
In mammalian cells, fusion of the outer mitochondrial membranes of neighboring mitochondrial organelles is governed by the dynamin-like GTPases mitofusin 1 and 2 (Mfn1, Mfn2) (Koshiba et al., 2004; Santel et al., 2003), whereas a third GTPase, Opa1, is localized to the inner mitochondrial membrane and mediates fusion of the inner mitochondrial membranes (Olichon et al., 2003). Mutations in human MFN2 cause a neuromuscular disorder, Charcot-Marie-Tooth disease Type 2A, and OPA1 mutations account for 40–60% of cases of Type 1 dominant optic atrophy, a neurological disorder resulting in the progressive degeneration of retinal ganglion cells leading to blindness (Burte et al., 2015). Although cultured mammalian cells lacking either Mfn1 or Mfn2 lack obvious mitochondrial morphology defects, deletion of both genes causes severe mitochondrial fragmentation, allowing unimpeded Drp1-dependent fission (Roy et al., 2015). Double Mfn1/2 knockout cells also have less mitochondrial DNA and compromised respiration and mitochondrial trafficking. Consistent with high levels of Mfn2 expression in the brain, mutations in Mfn2 lead to neurological defects, corroborated by genetic studies in mice, where Mfn2-deficiency results in cerebellar degeneration, severe ataxia and death around 2-weeks of age (Chen et al., 2003; Chen et al., 2007; Eura et al., 2003). Purkinje cells in Mfn2 knockout mice have severely compromised arborization of dendrites and accumulation of mtDNA mutations (Chen et al., 2007; Chen et al., 2010).
The mechanisms of fusion by Mfn1 and Mfn2 have recently been advanced though a general consensus has not yet emerged. In contrast to the fission protein Drp1, Mfn1/2 are integral membrane proteins and structure-function studies indicate that most of the protein faces the cytoplasm, consistent with the requirement for Mfn proteins on opposing membranes to mediate fusion (Koshiba et al., 2004). Mitofusins on adjacent mitochondria can form hetero- or homotypic oligomers with each other, tethering the two mitochondria in close proximity (Chen et al., 2003; Shutt et al., 2012). This homo/hetero-oligomeric feature may help to explain why single knockout cells have relatively normal mitochondrial network morphology. Structure determinations for the bacterial dynamin-like fusion protein, BDLP, and human Mfn1 help provide new insights into the role of GTPase activity in the fusion reaction (Cao et al., 2017; Low et al., 2009). Upon GTP hydrolysis, Mfn1 folds back into the closed state to bring the opposing membranes in close proximity, perhaps facilitating fusion. Recently, cryo-electron tomography experiments revealed that macromolecular complexes containing yeast Fzo1, homolog of Mfn1/2, assemble in the form of a docking ring at the junction sites between mitochondrial outer membranes from two adjacent mitochondria, prior to their fusion (Brandt et al., 2016).
The possibility of restoring fusion activity to patient mutants of Mfn2 is suggested by the development of peptide mimics of the heptad-repeat (HR)-1 region of Mfn2 (Franco et al., 2016). This HR-1 mimetic strategically interacts with HR-1 to disrupt intramolecular interactions between HR-1 the HR-2 of Mfn2 and induce conformational changes. Addition of these cell-permeable peptides to neurons from mice with mutant Mfn2 restored mitochondrial network morphology from fragmented to reticular, although the function of restored mitochondria was not yet tested (Franco et al., 2016). In this rapidly evolving arena, these mechanistic details still need to be reconciled with other models, including a recent structure of Mfn1 (Cao et al., 2017).
Another role for mitochondrial fusion is to distribute outer membrane proteins, based on the uneven distribution of various outer membrane proteins on mitochondria in Mfn1/2-deficient cells (Weaver et al., 2014). This uneven distribution may protect from the transfer of pro-apoptotic Bak, resulting in apoptosis (Weaver et al., 2014). On another front, geometrical parameters capable of predicting whether a given mitochondria will fuse or fragment were recently reported, suggesting that mitochondria undergo (presumably cytoskeleton-driven) architectural changes before engaging into fission or fusion (Westrate et al., 2014).
5.2. Inner mitochondrial membrane fusion
Eight variant OPA1 transcripts have been identified, all of which encode a mitochondrial import sequence (MIS). This import signal directs Opa1 protein to mitochondria, where it is removed by proteolytic cleavage, which generates the inner membrane-anchored long isoform of Opa1, L-Opa1 (Bertholet et al., 2016; Song et al., 2007). Under steady-state conditions, a fraction of the L-Opa1 pool is further proteolytically cleaved at two sites S1 and S2 to generate a short isoform lacking the transmembrane domain, S-Opa1 (Bertholet et al., 2016; Ishihara et al., 2006). In contrast to initial reports suggesting that the rhomboid protease PARL and the mAAA+ protease paraplegin were responsible, genetic deletion of both did not alter the ratio of long and short Opa1 (Anand et al., 2014; Ishihara et al., 2006). Subsequent studies indicate that the iAAA+ protease Yme1L cleaves at S2, while the metallopeptidase Oma1 cleaves at S1, and these two proteases predominantly maintain the homeostatic ratio of L-/S-Opa1 (Anand et al., 2014; Bertholet et al., 2016; Ehses et al., 2009; Griparic et al., 2007; Head et al., 2009). Cell stress and reduced mitochondrial membrane potential can result in nearly complete conversion of L-Opa1 to S-Opa and increases sensitivity to apoptosis (Duvezin-Caubet et al., 2006; Griparic et al., 2004). Cleavage by Yme1L is thought to link fusion to normal cellular bioenergetics (Mishra et al., 2014). The ratio of L/S-Opa1 appears to influence fusion events, however a consensus on the molecular details has not been reached.
Of note, depending on its levels, Opa1 was suggested to induce two types of fusion. The first, termed “transient fusion”, results in rapid exchange of soluble intermembrane and matrix components without affecting mitochondrial morphology. The second type of fusion, “complete fusion”, exchanges all mitochondrial components and changes morphology (Liu et al., 2009). Complete fusions are believed to result from the merge of two mitochondria moving towards each other along the same microtubule, and transient fusion events occur between mitochondria traveling along separate microtubules (Liu et al., 2009). In this latter case, merging mitochondria first appear to quickly engage oblique interactions before undergoing Drp1-induced fission due to the pulling forces exerted by the microtubule motor proteins, in a transient or kiss-and-run manner, and the mechanisms are currently under investigation (Jiang et al., 2017; Lavorato et al., 2017; Picard et al., 2015).
6. Nanotubulation-reticulation
In cytoplasmic volumes unconfined to neuronal processes, fused mitochondria can further connect to form a large three-dimensional lattice referred to as ‘reticulation’. Lattices can exist as sub-networks of interconnected mitochondria or a single organelle such as the giant mitochondrion in trypanosomes (Stuart, 1983), differentiating sperm cells in the fly (Noguchi et al., 2011), and the single reticulum of interconnected mitochondrial nanotubules in mammalian cardiomyocytes (Huang et al., 2013). The mechanism of mitochondrial reticulation is not well understood and could result from increased fusion events with or without growth/biogenesis. Alternatively, it may be possible that mitochondrial networks can reticulate by sliding junctions without fission or fusion, analogous to that observed in ER networks (Nixon-Abell et al., 2016), or other mechanisms.
6.1. Reticulation by hyperfusion
Elongated and networked mitochondria have been observed under specific conditions such as nutrient starvation (Gomes et al., 2011a; Rambold et al., 2011) and cellular senescence (Lee et al., 2007). In the case of starvation, the elongation of mitochondria appears to be conserved across species and occurs in vivo, for example the “grooved carpet shell” clam Ruditapes decussatus (Fig. 2). In the fed state, the clam’s digestive gland cells have small mitochondria that can be observed within a sea of ER, while in severely starved animals, mitochondria are highly tubular and surrounded as observed here among secretory vesicles (Fig. 2). Reticulation is suggested to confer some advantages compared to individual organelles (Glancy et al., 2015; Hoitzing et al., 2015). One thought is that the formation of fused mitochondrial networks might increase ATP synthesis by increasing the density of electron transport chain components (Gomes et al., 2011a; Liesa and Shirihai, 2013; Mitra et al., 2009; Rambold et al., 2011). However, it is unclear whether increased ATP production is a direct consequence of mitochondrial hyperfusion as some models observe increased ATP prior to fusion (Mishra et al., 2014). Another possibility is that fusion could confer robustness to the mitochondrial pool by dampening biochemical fluctuations (Mai et al., 2010) and by allowing functional complementation through the exchange of mitochondrial contents (Nakada et al., 2001; Ono et al., 2001). Fusion may also protect healthy mitochondria from being targeted for degradation (Twig et al., 2008) or enable efficient energy transmission along intracellular power cables constituted by filamentous mitochondria (Amchenkova et al., 1988; Glancy et al., 2015; Scholkmann, 2016; Skulachev, 2001) (see section 5).
Figure 2. Mitochondrial elongation is a conserved process across metazoans.
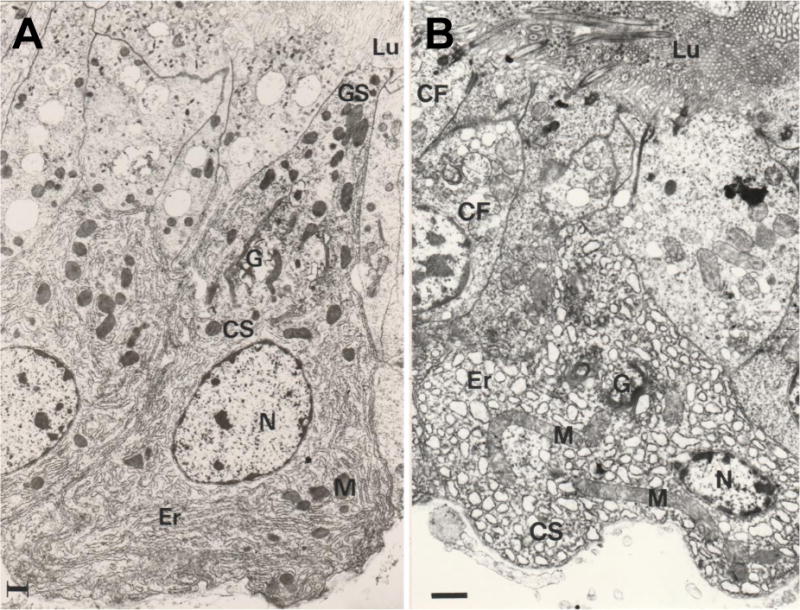
Electron microscopy images of digestive gland cells from the bivalve molusc Ruditapes decussatus that was normally fed (A), or subjected to strict experimental starvation for 45 days (B). M: mitochondrion; Lu: lumen; Er: endoplasmic reticulum; G: Golgi apparatus; N: nucleus; GS: secretary granule; CF: flagellate cell. Scale bars: 1 μm (A); 2 μm (B).
Analysis of the shape of reticulated mitochondrial networks at the scale of the entire cell necessitates additional methods such as those used to model neuronal networks, with quantification of branch properties, branching points and number of branch endpoints (Leonard et al., 2015; Lihavainen et al., 2012; Nikolaisen et al., 2014). Although concepts and tools derived from graph theory can be useful in studying static mitochondrial networks as captured in conventional fluorescence images, time-lapse imaging revealed that the interactions between individual mitochondria and segregated mitochondrial subnetworks change over time. Such ‘temporal networks’ have to be modeled using specific methods such as agent-based modeling (Dalmasso et al., 2017) or metrics developed for time-varying graphs, taking into account movement and connectivity of mitochondrial objects (Holme, 2016).
6.2. Fusion-independent reticulation
The molecular mechanisms involved in mitochondrial reticulation are far less well understood. Yeast cells that are genetically deficient in the two major components of the fission-fusion machinery (through mutations in the Drp1 homolog Dnm1 and in the mitofusin homolog Fzo1) seemingly have a normal interconnected network similar to that of wild-type cells (Sesaki and Jensen, 1999). Moreover, prompting the fusion of individual mitochondria did not prove sufficient to form networks in vitro (Meeusen et al., 2004), raising the possibility that distinct or additional mechanisms are necessary for mitochondrial reticulation with respect to small-scale fusion events. One molecular actor that was recently implicated in the ‘dynamic tubulation’ of mitochondria is the motor protein KIF5B (Wang et al., 2015), which was known from earlier findings to drive mitochondrial transport along microtubules (Tanaka et al., 1998). KIF5B was reported to trigger outward expansion of the mitochondrial network from the perinuclear area to the cell periphery, by pulling thin tubules out of the mitochondria and fusing them regionally to create subnetworks (Huang et al., 2013). Genetic deletion of Mfn1 and Mfn2 did not impair the process of dynamic tubulation in mouse embryonic fibroblasts, but these tubules failed to fuse into high-order networks, indicating that other actors are probably needed for the fusion of mitochondrial lattices into a full network. Interestingly, KIF5B was found to be dispensable for mitochondrial network formation in the perinuclear area, suggesting that the mitochondrial reticulum is actually composed of different mitochondrial regions. In support of this, at least three mitochondrial pools might coexist within the cytoplasm (Chevrollier et al., 2012; Collins et al., 2002): perinuclear, periplasmic and around the MTOC (microtubule organizing center), each of which exhibit different susceptibilities to defects in mitochondrial fusion genes (Chevrollier et al., 2012). These findings raise the intriguing possibility that distinct mechanisms of mitochondrial reticulation operate in each of these cytoplasmic regions.
7. Mitochondrial turnover
7.1 Piecemeal turnover of mitochondrial components
There appear to be three general categories of mitochondrial turnover mechanisms, two involving whole organelle turnover and a third microscale catchall category we refer to as piecemeal disposal about which little is known. The best characterized of these is whole organelle turnover by lysosome-dependent degradation of mitochondrial organelles in autophagosomes, known as mitophagy. A second, less well characterized process of whole mitochondrial turnover is organelle export (e.g. from neurons) to neighboring cells (e.g. astrocytes) for degradation (see section 3.3.). The third process refers to a conglomerate of microscale processes. Proteosome-dependent molecular disposal/replacement has been predicted to have significant impact on mitochondrial maintenance, but could be considered more of a repair process. Molecular replacement strategies maintain the whole organelle by replacement of parts, perhaps analogous to the restoration of historic buildings. This third catchall category may involve the degradation and replacement of individual proteins, protein complexes or possibly the export of material destined for degradation through mitochondria-derived transport vesicles that pinch off from mitochondria independently of fission machinery or canonical autophagy machinery (McLelland et al., 2016; Soubannier et al., 2012; Sugiura et al., 2014), or by extraction directly from the tubular sides of mitochondria by attacking autophagosomes (Yamashita et al., 2016). Although several have estimated that at basal rates, cultured cells produce 50 autophagosomes per hour, it is currently not known what volume of mitochondrial biomass is turned over by any of these processes in neurons or other cell types, nor whether all of the different processes work in concert.
7.2. Mitochondrial turnover via autophagy
Over the past 15 years, novel pathways that specifically target the engulfing autophagosome to mitochondria have begun to be delineated. Receptors on the mitochondrial outer membrane serve to physically recruit mitochondria to LC3-II-decorated preautophagosomal membranes by their LIR (LC3 interacting region). A major focus of attention in the mitophagy field is the interaction of ubiquitin ligase Parkin and the accumulation of ubiquitinated proteins such as PINK1 kinase, and more recently prohibitin, that recruit autophagy receptors to the outer mitochondrial membrane (Lazarou et al., 2015; Wei et al., 2017). This process is triggered by failed mitochondrial protein import upon collapse of the mitochondrial membrane potential, at least in non-neuronal cells (see section 7.3). However, additional roles for PINK1 and Parkin are being uncovered. In contrast to depolarization-induced mitophagy, PINK1 and Parkin are required during oxidative stress for the degradation of oxidized mitochondrial matrix components via mitochondria-derived vesicles, consistent with evidence from Drosophila (Soubannier et al., 2012; Sugiura et al., 2014; Vincow et al., 2013). In addition, the fusion protein, Mfn2, when ubiquitinated by the E3-ligase Parkin, was reported to serve as a platform for recruitment and binding of the PINK1 kinase (Chen and Dorn, 2013). Both Bcl-2 homologs and BH3-only proteins have been assigned roles in regulating mitophagy, including functioning as regulatory molecules (e.g. Bcl-2-Beclin1 inhibitory interaction) (Pattingre et al., 2005) and as mitophagy-promoting mitochondrial receptors (e.g. Bnip3, Bnip3L/Nix, and Bcl2L13/Bcl-Rambo) (Hanna et al., 2012; Murakawa et al., 2015; Novak et al., 2010; Zhu et al., 2013). By coupling autophagy machinery with apoptosis regulators, the cell may attempt to resolve cellular stresses (e.g. loss of membrane potential, or mtDNA damage) while preventing death via apoptosis pathways. Further details of mitophagy were recently reviewed by others (Hamacher-Brady and Brady, 2016; Martinez-Vicente, 2017).
Regarding the role of mitochondrial fission in mitophagy, the general consensus is that Drp1-mediated mitochondrial fission divides tubular mitochondria into segments of sufficiently small size to be engulfed by autophagosomes (Twig et al., 2008). However, more recent work identified a Drp1/Dnm1-independent mitophagy process conserved in yeast and mammals (Yamashita et al., 2016). In this case, distinct from organelle fission, small fragments of mitochondria are generated in the absence of Drp1/Dnm1 because they are apparently extracted from the sides of mitochondrial tubules by attacking autophagosomes (Yamashita et al., 2016). In this case, the autophagosome itself appears to form at spots along mitochondria with or without Drp1/Dnm1 to consume a bulge along the mitochondrial tubule (Yamashita et al., 2016) (see section 7.4).
7.3. What are the neuron-specific triggers of mitophagy?
The topic of mitophagy has been a major focus in recent years following the discovery that depolarization of mitochondria (e.g. with CCCP) rapidly triggers relocalization of Parkin, an E3 ligase associated with familial Parkinson’s disease, from the cytosol to mitochondria within minutes and subsequent degradation by mitophagy (Narendra et al., 2008; Youle and Narendra, 2011). Intriguingly, despite the association between Parkin mutations in neuronal loss in Parkinson’s disease, this robust model system to study mitophagy in HeLa cells may not be as useful for neurons, as mitochondrial depolarization in cultured neurons does not trigger relocalization of Parkin to mitochondria, for example when HeLa cells and neurons were compared side-by-side (Van Laar et al., 2011). However, Parkin can be coaxed to a fraction of neuronal mitochondria upon brief exposure to glutamate to trigger excitotoxicity (Van Laar et al., 2015). The recruitment is surprisingly enhanced (not suppressed) if basal ROS is scavenged, possibly suggesting that non-ROS-producing mitochondria may be recognized as non-functional (Berman and Hardwick, 2015; Van Laar et al., 2015). Perhaps there a many distinct signals that trigger neuronal mitophagy and that differ between different cell types.
7.4. Transfer of neuronal mitochondria to neighboring cells for degradation
In the optic nerve head of mice, it was observed that neuronal mitochondria tagged with a tandem acid-resistant/sensitive red/green fluorescent reporter collected in protrusions and evulsions along the lengths of axons (Davis et al., 2014). Remarkably, the content of these protrusions and evulsions appear to be degraded subsequently by lysosomes in adjacent astrocytes. Furthermore, impaired mitochondria appear to be preferentially targeted as this process was enhanced following treatment with the Complex I inhibitor rotenone. Additional clues from this study suggest that external degradation of neuronal mitochondria may be a widespread phenomenon in the brain (Davis et al., 2014). Moreover, a similar and possibly conserved mechanism was recently reported in C. elegans wherein adult neurons extrude large membrane-surrounded vesicles, called exophers (Melentijevic et al., 2017). In worms expressing fluorescent reporters, exophers preferentially sequestered compromised mitochondria and neurotoxic protein aggregates including those associated with human neurodegenerative disorders (Melentijevic et al., 2017). This process involves the worm homologs of Parkinson’s disease genes PINK1, PARK2 and the BNIP3 protein (C. elegans DCT-1), which is a non-canonical BH3-only protein akin to Bcl-2 family proteins (Melentijevic et al., 2017). Furthermore, protein aggregates and mitochondria can be degraded upon engulfment of exophers by other cells using some of the same machinery responsible for engulfing apoptotic cells. However, this process appears to be distinct from classical apoptosis as exophers lack the apoptotic “eat-me” signal phosphatidylserine that characterizes apoptotic bodies, and exopher release does not kill neurons, but rather improves neuronal activity (Melentijevic et al., 2017). Curiously, exophers often have long thin tethers to the neuron, which resemble tunneling nanotubes reported to transfer huntingtin protein aggregates between cells in mammals and Drosophila (Costanzo et al., 2013; Pearce et al., 2015) (see 3.3. and 5.3).
8. Bcl-2 family proteins alter mitochondrial dynamics and neuronal activity
Although initially recognized for their importance in regulating apoptosis, it is now widely appreciated that both anti- and pro-apoptotic Bcl-2 family members have other roles in cellular function (Gross and Katz, 2017; Hardwick et al., 2012). These non-apoptotic “day-jobs” are not as well characterized but are also important determinants of cell death versus survival. For example, the effects of anti-apoptotic Bcl-2 and Bcl-xL on the energetic state of neurons and other cell types prior to apoptosis induction appears to suppress death independently of their canonical anti-apoptotic function (Alavian et al., 2011; Chen et al., 2011; Yi et al., 2011).
8.1. Non-apoptotic functions for Bcl-2 family proteins in neurons
Bcl-xL is expressed in many tissues but is essential for hematopoietic functions during early embryogenesis in mice as its genetic deletion is lethal at around embryonic day 12.5 (Motoyama et al., 1995). However, Bcl-xL is also critical for normal brain development and conditional deletion of Bcl-xL in a subset of cortical neurons severely compromises neuron survival and brain development (Zhang et al., 2005). While other Bcl-2 family members substantially decline during development (Krajewska et al., 2002), Bcl-xL expression is maintained throughout the adult brain (Gonzalez-Garcia et al., 1995) and Bcl-w is abundant in the adult cerebellum (Hamner et al., 1999). Interestingly, Bcl-xL may not be essential for neuronal survival until receiving specific development cues or stress conditions (Berman et al., 2009; Chen et al., 2011; Gonzalez-Garcia et al., 1995; Zheng et al., 2006). This is consistent with Bcl-xL exerting a critical function in mature, long-lived neurons. Although the observed gradual neuronal loss, motor-learning difficulties, increased risk-taking and self-injurious behaviors of Bcl-xL-deficient mice (Nakamura et al., 2016) are generally attributed to increased apoptosis, the underlying mechanisms may also involve non-apoptotic functions.
More complex roles for Bcl-xL are consistent with earlier studies of Bcl-x knockout mice with simultaneously deletion of either caspase-9 or caspase-3 (which are activated post mitochondrial permeabilization by Bax downstream of Bcl-xL in apoptosis). Double Bcl-x/caspase-3 or -9 knockouts survived beyond embryonic day 12, but died thereafter as no double knockouts were born (Roth et al., 2000; Shindler et al., 1998; Zaidi et al., 2001). However, deletion of caspase-3 or caspase-9 markedly decreased the death of some embryonic neurons in dorsal root ganglia and of Bcl-x-deficient telencephalic cells in culture (Roth et al., 2000; Shindler et al., 1998; Zaidi et al., 2001). However, simultaneous deletion of both Bcl-xL and Bax, which prevented caspase-3 activation, failed to rescue embryonic lethality by day 13 in mice (Roth et al., 2000; Shindler et al., 1998; Zaidi et al., 2001). Bcl-x-deficiency also did not rescue the highly abnormal developmental brain pathology and extra neurons observed in the caspase-9 knockout mice (Zaidi et al., 2001). While gene compensation, developmental mismatches and non-death functions of caspases in neurons may confound the interpretations of these approaches (Li et al., 2010; Ofengeim et al., 2012; Tang et al., 2015), the overall conclusion is that Bcl-xL has functions distinct from classical apoptosis. Consistent with this conclusion, Bcl-xL, Bax and Bak also regulate neuronal activity in healthy neurons, presumably independent of classical apoptosis processes (sections 8.2 and 8.3). Nevertheless, a recent study found that neuron-targeted triple knockout of anti-apoptotic Bcl-xL plus pro-apoptotic Bax and Bak rescued neuronal survival, unlike double knockouts of Bcl-xL with either Bax or Bak (Nakamura et al., 2016).
8.2. Bcl-xL alters mitochondrial dynamics and synaptic transmission
How does Bcl-xL function in neurons to protect from death independently of its canonical role of inhibiting Bax and Bak? Although the biochemical mechanisms are still being investigated, several studies have provided hints. Cortical neurons lacking Bcl-xL display fragmented mitochondria and restoring Bcl-xL leads to elongated mitochondria (Berman et al., 2009). This could suggest that Bcl-xL enhances mitochondrial fusion rates or inhibits mitochondrial fission rates (see section 2.1. – 2.3.). However, while Bcl-xL indeed increases the rate of mitochondrial fusion, surprisingly Bcl-xL induced an even greater increase in the rate of fission (Berman et al., 2009; Berman et al., 2008). In addition, Bcl-xL also increased total mitochondrial biomass (length) in cultured hippocampal neurons and cortical neurons (Alavian et al., 2011; Berman et al., 2008). These findings are consistent with the observation that Bcl-xL, as well as the C. elegans homolog CED-9 can interact with the mitochondrial fission-fusion machinery (Jagasia et al., 2005; Li et al., 2008). Thus, it is conceivable that Bcl-xL could maintain neuronal survival by regulating mitochondrial dynamics. Moreover, recent work has also demonstrated that Bcl-xL can coopt the fission protein Drp1 to perform functions other than mitochondrial division. In hippocampal neurons, Bcl-xL increases the numbers of synapses and synaptic vesicles, as well as mitochondrial localization to vesicle clusters and synapses in a Drp1-dependent manner (Li et al., 2013). These findings could potentially explain previous results documenting the ability of Bcl-xL to increase synaptic transmission within minutes when acutely injected into the presynaptic terminus, therefore acting independently of longer-term cell remodeling effects (Hickman et al., 2008; Jonas et al., 2005b; Jonas et al., 2003). Follow-up studies found that Bcl-xL in a complex with Drp1, Mff and clathrin regulates synaptic vesicle dynamics during endocytosis (Li et al., 2013). Following exocytosis, recovery of neurotransmitters is mediated via vesicle retrieval from the plasma membrane as well as from reserve pools within the synapse, both of which are dependent on mitochondrial ATP. Thus, the underlying molecular details of how Bcl-xL alters neuronal synapses may stem from its direct effects on mitochondrial protein complexes and lipids of the inner mitochondrial membrane (Alavian et al., 2011; Chen et al., 2011).
8.3. Pro-apoptotic Bax and Bak regulate neuronal activity to protect neurons
Pro-apoptotic Bcl-2 family proteins Bax and Bak also regulate neuronal activity (Fannjiang et al., 2003; Jonas et al., 2005a). Although Bak knockout mice are generally not found to have obvious phenotypes (Lindsten et al., 2003), in fact they have several neuronal phenotypes. Acute hippocampal slices from Bak knockout mice have evidence of altered spontaneous and evoked neurotransmitter release (Fannjiang et al., 2003). The observed increase in excitatory inputs and decrease in inhibitory nerve terminals may contribute to their increased seizure susceptibility of Bak knockout mice. This function may contribute to the protective effects of Bak in mice infected with the neuronotropic encephalitis virus, Sindbis virus, an alphavirus transmitted by mosquitoes in nature (Cheng et al., 1996; Fannjiang et al., 2003; Levine et al., 1993; Lewis et al., 1999). Both Bax and Bak knockout mice have 60–80% higher mortality rates and increased neuronal death several days following infection with Sindbis virus relative to wild type and heterozygous littermates. Strikingly, reexpression of Bax or Bak proteins by using Sindbis virus itself as a delivery vector to restore Bax or Bak to their respective knockout mice, rescues neuron survival and reduces mouse mortality to 25–30%, while control viruses had no ability to rescue (Fannjiang et al., 2003; Lewis et al., 1999). This finding argues against non-neuronal roles in fighting infection as an alternative explanation for these unexpected protective effects of Bax and Bak in the brain.
9. Impact of Bcl-2 family proteins on mitochondria-like membranes
Although the detailed mechanisms explaining the effects of Bcl-2 family proteins on mitochondrial dynamics are not known, emerging insights into how Bcl-2 homologs engage membranes has recently emerged from biophysical studies. Most anti- and pro-apoptotic Bcl-2 homologs are tail-anchored proteins. This transmembrane tail-anchor is the hydrophobic C-terminal helix and flanked by positive charges that also serves to target different family members to the mitochondrial and ER membranes, but also to other cytoplasmic membranes. In solution, Bcl-2 homologs form a single structural domain known as the Bcl-2-fold, which is composed of approximately 9 alpha-helices and intervening loops. Each protein has a deep binding cleft on one side to receive a helical BH3 motif donated by another Bcl-2 family protein, particularly a pro-death family member, although the BH3 is present in almost all family members (Aouacheria et al., 2015; Aouacheria et al., 2013). However, Bcl-2 proteins localize to membranes, which is required for some of their interactions, but structures of membrane-inserted Bcl-2 proteins have only recently been advanced.
9.1. Evolving role for the helical hairpin of Bcl-2 homologs in membrane insertion
In addition to the C-terminal transmembrane tail-anchor, Bcl-2 proteins were originally proposed to fully insert their central helical hairpin (alpha helices 5–6) characteristic of the Bcl-2-fold into membranes perpendicular to the membrane bilayer (reviewed in (Petros et al., 2004). Upon membrane insertion of this hairpin, the surrounding helices were suggested to open in an umbrella-like manner, based in part on their ability of a peptide fragment containing alpha 5–6 to induce pore formation in liposomes (Garcia-Saez et al., 2006) and in cells (Annis et al., 2005; Valero et al., 2011). These conclusions were later challenged by the finding that residues throughout helices α5 and α6 of Bax or Bak remained accessible to IASD-labeling before, during, and after tBid-activation of Bax/Bak, leading to the conclusion that α5–α6 helices lie on the surface of the membrane, rather than being deeply embedded or transmembrane (Westphal et al., 2014). In contrast to the earlier model, new structure determinations and biophysical approaches using DEER (double electron-electron resonance) and cell-based reporter assays have provided strong evidence that the central α5–α6 hairpin, instead of inserting like a spear into lipid bilayers, opens at its hinge thereby separating the two halves of the Bcl-2 family molecule (Czabotar et al., 2013). In this state, the protein either lays partially embedded on one side of the membrane, or traverses a lipidic pore possibly equivalent to the Bax apoptotic pore. This “unhinged” hypothesis has subsequently been supported by evidence from several groups and confirmed for Bak (Bleicken et al., 2014; Brouwer et al., 2014; Sung et al., 2015). Furthermore, structures of unhinged monomers within domain-swap dimers have been described for other Bcl-2 family members (Lee et al., 2011; O’Neill et al., 2006). However the unhinged dimers are not believed to function in a death pathway (Czabotar et al., 2013), leaving open the possibility that domain-swapped dimers of Bax and others may engage exclusively in novel “day jobs”. Despite observing a similar unlatching of the α5–α6 hinge, the Bordignon group (Bleicken et al., 2014) proposed an intriguing “clamp” model of Bax membrane engagement, in which the opened α5–α6 helices extend the N-terminus to facilitate dimerization with another Bax molecule extended from the other side of the bilayer. This lack of consensus regarding the role of α5–α6 in membrane engagement, testifies that Bcl-2 family interactions with membranes are complex and have yet to be fully delineated.
9.2. The Bcl-2 family transmembrane tail-anchors may intertwine away from the pore
Similar experimental approaches were used to confirm the membrane targeting function of the C-terminus, but also suggest that the tail-anchor regions of Bcl-2 family proteins undergo a novel intramembrane dimerization (Andreu-Fernandez et al., 2017; Bleicken et al., 2014) where the C-terminal transmembrane domain, helix α9, of two Bax molecules interact either in parallel or anti-parallel angles within the membrane and away from the apoptotic pore (Bleicken et al., 2014; Iyer et al., 2015; Liao et al., 2016). C-terminal dimer interactions within the mitochondrial membrane were not required for release of small molecules, such as cytochrome c, but were needed to form larger pores, suggesting that these tail-tail interactions are required for pore expansion (Zhang et al., 2016). The analogous C-terminus of BAK was also demonstrated to interact within membranes, and importantly, the tail-anchor regions were protected from biochemical labeling, suggesting that these proteins do not themselves constitute a proteinaceous pore (Iyer et al., 2015). However, it remains entirely unclear how such structures might explain either the ability of Bcl-xL to retrotranslocate Bax from mitochondrial membranes (to prevent apoptosis), or the emerging non-canonical functions of Bcl-2 proteins at membranes, particularly in neurons, including regulating channel activity, synaptic transmission, calcium homeostasis, and autophagy (D’Orsi et al., 2015; Hardwick and Soane, 2013; Hickman et al., 2008; Jiao and Li, 2011; Jonas et al., 2005a; Jonas et al., 2003).
9.3. Membrane interactions by anti-apoptotic Bcl-2 homologs
Much effort has focused on the membrane integration and conformational changes associated with the pro-apoptotic BCL-2 proteins, but less emphasis has been devoted to the anti-death counterparts. Despite possessing opposite activities, pro- and anti-death BCL-2 proteins share structural similarity and, thus far, the exact features of these proteins that govern their respective activity are not fully grasped (Hardwick et al., 2012; Youle and Strasser, 2008). To identify distinguishing features between anti- and pro-death protein, one recent study suggested that apoptotic activity correlates with membrane binding potential of the central helices α5–α6, charge distribution at the termini of α5–α6, and the presence of a strong inhibitory N-terminal domain (Xiao et al., 2016), but this requires additional confirmation. Since cleavage of the N-terminus of anti-death family members (Bcl-2, Bcl-xL, Bfl-1 and Mcl-1) can potently convert them to potent pro-death factors (Cheng et al., 1997; Clem et al., 1998; Xiao et al., 2016), it is reasonable to infer that anti-death Bcl-2 family members have the potential to engage membranes similar to Bax and Bak. A related model argues that anti-apoptotic Bcl-2/Bcl-xL are dominant negative versions of Bax/Bak (Leber et al., 2010; Reed, 2006; Westphal et al., 2014).
Both Bcl-xL and Bcl-2 preferentially localize to mitochondrial and ER membranes, respectively via their C-terminal tail anchor. However, a range of techniques has made clear that Bcl-xL is enriched in the inner mitochondrial membrane, in contrast to central dogma restricting Bcl-2 proteins to the outer mitochondrial membrane (Hardwick et al., 2012). Although the import mechanisms are not yet known, Bcl-xL may be targeted to the mitochondrial inner membrane via its C-terminal transmembrane domain, with help from the N-terminus of Bcl-xL, which contains a moderately strong mitochondrial presequence that is sufficient to efficiently target GFP to mitochondria (Chen et al., 2011; McNally et al., 2013). The N-terminus of Mcl-1 also contains a mitochondrial import sequence (Perciavalle et al., 2012). Bcl-xL is specifically enriched in sub-mitochondrial fractions containing the F1Fo ATP synthase, an observation made by many labs (Chen et al., 2011; Hardwick et al., 2012).
9.4. Bcl-2 proteins on the inner mitochondrial membrane
Experimental evidence using inhibitors directed at either the F1 and Fo subunits of the ATP synthase suggests that in Bcl-xL knockout neurons, F1 and Fo are not properly coupled, leading to unstable excess ion flux across the mitochondrial inner membrane and compromised reserve capacity (Chen et al., 2011). By measuring oxygen consumption with an oscillating probe, it was found that Bcl-xL-expressing neurons consume less oxygen but produce equivalent amounts of ATP (Alavian et al., 2011). By decreasing excess ion flux across the inner membrane, Bcl-xL could potentially increase the energetic efficiency of neurons, thereby increasing tolerance to cell stress independently of its classical anti-apoptotic, Bax-interacting functions.
Experimental evidence has shown many strategies in which Bcl-xL can protect neurons. However, Bcl-xL can also be a detriment to neurons if cleaved by caspases. Following an insult, Bcl-xL can be cleaved by caspases near its N-terminus and the resultant C-terminal fragment carries an intrinsic pro-death function, analogous to the cleavage and activation of Bid. When added to cells or artificial liposomes, ΔN-Bcl-xL can induce the release of cytochrome c into the cytosol, facilitating apoptosis (Basanez et al., 2002; Basanez et al., 2001). The ΔN-Bcl-xL fragment has also been shown to be neurotoxic and to promote neuronal injury in an ischemia-reperfusion model (Ofengeim et al., 2012). By mutating the two caspase-cleavage sites within Bcl-xL, mice expressing caspase-resistant Bcl-xL were protected from neuronal death following the ischemic injury (Ofengeim et al., 2012). While recombinant, full-length Bcl-xL can increase synaptic plasticity within minutes after being injected into the presynaptic terminus in preparations of the squid giant axon, ΔN-Bcl-xL induces synaptic depression that is similar to the synaptic rundown observed under hypoxic conditions (Hickman et al., 2008; Jonas et al., 2004; Jonas et al., 2005b; Jonas et al., 2003). All of these diverse experimental approaches point to an ability of Bcl-xL to alter channel conductance. However, the context is critical. While full length Bcl-xL can enable ADP/ATP exchange and closedown the wasteful leak of protons in mitochondria (Chen et al., 2011), ΔN-Bcl-xL and ΔN-Bcl-2 can facilitate release of pro-apoptotic factors such as cytochrome c (Cheng et al., 1997; Kirsch et al., 1999). Therefore, a delicate balance between pro- and anti-apoptotic roles, as well as non-apoptotic pro-survival roles of Bcl-xL may impact mitochondrial metabolism during stress to control neuronal health and survival.
10. Descriptions of mitochondrial morphology
Mitochondrial morphology was shown to vary depending upon cell types (Lennon et al., 2016; Palmer et al., 2011; Vafai and Mootha, 2012) and conditions, including metabolic needs, stress and redox state (Ahmad et al., 2013; Gomes et al., 2011b; van der Bliek, 2009). Mitochondria can shift from small rounded structures to a more elongated state, and can even develop into filamentous networks (for instance upon nutrient starvation (Gomes et al., 2011a; Rambold et al., 2011) resembling interconnected neurons of neural networks (Nikolaisen et al., 2014). They can also become swollen (Bonora et al., 2016; Yasuda et al., 2006) or highly fragmented during apoptosis induction (Bhola and Letai, 2016), neurodegeneration (Knott et al., 2008) and cell division (Lopez-Mejia and Fajas, 2015; Westrate et al., 2014). The “mitochondriome” is therefore a particularly polymorph entity – it exists in several distinct shapes both at the nanometer scale, at the level of individual mitochondria, and as the micrometer scale, at the level of the mitochondrial network. Mitochondrial morphology and mitochondrial network topology can be described by a number of features, some of which are geometrical such as size, shape, position, whereas others are mechanical, including dynamics (mitochondrial fission and fusion), movement and communication. These features are often interdependent. For example, mitochondrial size will increase with mitochondrial perimeter (the number of pixels forming the boundary of a mitochondrial object) (Wiemerslage and Lee, 2016) and mitochondrial movement is a decreasing function of mitochondrial size (with small mitochondria moving more rapidly than large mitochondrial sub-networks) (Caino et al., 2016; Narayanareddy et al., 2014). Mitochondrial movement also varies in relation to the position inside the cell (with peripheral mitochondria moving faster than mitochondria located in the more densely populated perinuclear region) (Kiryu-Seo et al., 2010; Watanabe et al., 2007).
10.1. Mitochondrial size and mass density
Based on their bacterial ancestry, it is reasonable to assume that a ‘single’ mitochondrial segment contains no more than one mitochondrial DNA cluster, defining a basic mitochondrial ‘unit’. These units may be of various size, generally having a diameter of 0.5–1 μm (Finley et al., 2012; Holloway et al., 2010; Karam et al., 2003; McClatchey et al., 2016; Moran et al., 2010) and a length on average 1–15 μm (Bereiter-Hahn, 1990; Cagalinec et al., 2013; Choi et al., 2015; Korobova et al., 2014; Korobova et al., 2013; McClatchey et al., 2016; Nozaki et al., 2001; Rafelski and Marshall, 2008; Rintoul et al., 2003; Wiemerslage and Lee, 2016). In addition to the size of the organelles bounded by their outer membrane, the area of all mitochondrial units forming the mitochondrial network should also be considered. This spatial density (mass density) of mitochondria within a cell can be estimated by calculating the ratio of mitochondrial size to cell size (Koopman et al., 2006; Posakony et al., 1977; Rafelski et al., 2012). By applying these metrics, Koopman et al. (Koopman et al., 2006) determined that about 17% of the cell area is occupied by mitochondria in skin fibroblasts (a value falling in the expected range of 10–40% (Posakony et al., 1977). Interestingly, by culturing fibroblasts on micropatterned coverslips of various sizes, it was shown that mitochondrial network volume primarily depends on the size and shape of the cell (Chevrollier et al., 2012). Shape complexity of individual mitochondria can be described by several parameters, such as ‘solidity’ (compactness) (Westrate et al., 2014), ‘tortuosity’ (for twisted mitochondria) (Lihavainen et al., 2012) ‘Feret ratio’ (ratio between length and width, a descriptor of filamentous character) (Hodneland Nilsson et al., 2015), or Form Factor (a combined measure of mitochondrial length and degree of branching) (Koopman et al., 2008). Other morphological parameters, like the number of branches or branch intersections, give an estimation of shape complexity at the level of the mitochondrial network (Lihavainen et al., 2012; Nikolaisen et al., 2014; Westrate et al., 2014). In multi-scale representation systems, mitochondrial morphology is depicted on a linear scale ranging from “fragmented” to “filamentous”, before adding two or more categories (e.g. “compact” versus “branched”, “non-tubular” versus “tubular”) (Vowinckel et al., 2015). When consensus clustering was applied to automatically define morphological subtypes in mitochondrial images, a total of 19 distinct morphological clusters were identified, suggesting that mitochondrial shape complexity is under-appreciated even when a single cell type is considered (Peng et al., 2011). The diversity in mitochondrial morphology may be far greater considering the extent to which mitochondria are specialized between the various cell types (Vafai and Mootha, 2012). Additional descriptors and the application of deep learning methods may thus be required to fully decipher this morphological ‘mitodiversity’ (Popkov et al., 2015).
Of note, the structure of the mitochondrial cristae also shows a high degree of pleiomorphism (e.g. lamellar, tubular, disc-like, helical) within a given cell, between different cells and between cell types (Mannella, 2006). Unfortunately, in live cells, structure of the cristae appears essentially blurred even when live-cell super-resolution microscopy techniques are applied (Hayashi and Okada, 2015; Shao et al., 2011; Shim et al., 2012). The nature of the design principles governing the shape of the inner mitochondrial membrane, and which quantitative parameters are the most relevant for evaluating mitochondrial cristae structure in live cells, represent fundamental unanswered questions.
10.2. Mitochondrial position and number density
Positional parameters (such as the least distance to the nearest mitochondrial neighbor) estimate the relative density of mitochondria in the neighborhood of a given mitochondrion, which can be measured by the proximity between two mitochondria and is directly correlated to the likelihood of fusion (Westrate et al., 2014). Mitochondrial movement also increases the likelihood of fusion events by favoring interactions between proximal but also distal mitochondria (Twig et al., 2008). Position and movement of mitochondria are largely conditioned by the cytoskeleton, and more particularly by microtubules (Anesti and Scorrano, 2006; Detmer and Chan, 2007; Heggeness et al., 1978; Karbowski et al., 2001; Rafelski, 2013). In a seminal study devoted to cellular ‘tensegrity’, the study of structures that stabilize their shape by continuous tension rather than by continuous compression (Ingber, 2003), micropipettes were used to induce a local stress to the plasma membrane of living cells by pulling on RGD-coated microbeads bound to cell surface integrins (Wang et al., 2001). Mechanical stress resulted in displacement of mitochondria up to 20 μ m away in the depth of the cytoplasm, followed by immediate reset to the initial mitochondrial position upon force removal. As mechanical stress application exerts its effects by inducing local changes in the position of microtubule tracks, this experiment substantiated that subcellular localization of mitochondria is critically linked to the microtubule cytoskeleton through dynamic self-organizing principles.
Mitochondria tend to aggregate around the nucleus in close proximity to the microtubule-organizing center (MTOC) in stressed cells (Westrate et al., 2014). This phenomenon of perinuclear clustering appears to be cell type- and condition-dependent. Perinuclear mitochondria were shown to display a lower Δψm than peripheral mitochondria (Collins et al., 2002; Pletjushkina et al., 2006), highlighting the relationships existing between functional characteristics and positional features. Potentially related phenomena may explain the curious of mitochondria around the nucleus in cells overexpressing anti-apoptotic Bcl-xL, which are apoptosis-resistant. Alternative possibilities include the effects of Bcl-2 family proteins on mitochondria dynamics (see section 8), on membrane curvature, supported by in vitro studies liposomes (Basanez et al., 2002; Basanez et al., 2001), or other phenomenon.
There are also important structural and functional links between mitochondria and the ER (Armstrong et al., 1973; Friedman et al., 2011; Giorgi et al., 2010; Stoica et al., 2014), and mitochondrial translocation to perinuclear ER has been observed during early phases of ER stress (Bravo et al., 2011). Beyond physical principles of cellular mechanics, mitochondrial distribution must be actively controlled to meet the needs of the cell. In that respect, mitochondrial enrichment has been described at subcellular areas with high ATP consumption, including axonal growth cones (Morris and Hollenbeck, 1993), presynaptic terminals, astrocytic processes near synapses (Stephen et al., 2015), viral assembly sites in ASFV (African swine fever virus)-infected cells (Rojo et al., 1998), and in cortical cytoskeleton of tumor cells (Desai et al., 2013) and oocytes (Guillemin et al., 2009; Van Blerkom and Davis, 2006).
A number of topological descriptors have been computed to describe the distribution of mitochondria relative to the nucleus (or to the plasma membrane) in quantitative terms. For instance, McClatchey et al. (McClatchey et al., 2016) calculated a “perinuclear preference” index based on the ratio between mitochondrial abundance measured in the third of the cytoplasm closest and furthest from the nearest nucleus. Kanfer et al. (Kanfer et al., 2015) computed the moment of inertia of the mitochondrial network around the cell center of mass to give an estimate of the spreading of the mitochondrial network. Using this metrics, they showed that the mitochondrial network spreading was diminished in cells deficient for Miro, a mitochondrial GTPase involved in mitochondrial trafficking (Fransson et al., 2006).
10.3. Mitochondrial mobility
Mitochondria are mobile organelles, able to move long distances or travel back and forth along microtubules with numerous starts and stops (Lin and Sheng, 2015; Morris and Hollenbeck, 1995). Motile mitochondria are present not only within cells but may also be present between cells. Recent studies have observed mitochondria inside microtubule-containing ‘membrane nanotubes’ connecting neighboring cells (Antanaviciute et al., 2014; Liu and Hajnoczky, 2011; Lu et al., 2017; Scholkmann, 2016). Endogenous and expressed Bcl-xL in neurons increases the number of synapses that contain a mitochondrion immediately adjacent to the synaptic density (Li et al., 2013; Li et al., 2008). Although the mechanisms are not known, we consider some of the potentially important players.
Although mitochondrial movement is likely important in all cell types, most studies investigate mitochondrial transport in neuronal axons and dendrites for three main reasons. First, mitochondria of cultured neurons are well-resolved by fluorescence imaging, and they are transported bidirectionally on microtubules towards the growth cone (Cai et al., 2011; Cameron et al., 1991; Morris and Hollenbeck, 1993; Pilling et al., 2006; Popov et al., 2005; Zinsmaier et al., 2009), rendering this system particularly suitable for in vitro particle tracking experiments. Since neuronal processes and axons can be very long (up to one meter in humans), this system ensures that mitochondria are transported to distant regions to support the local ATP demand. Second, in addition to their bioenergetics function, mitochondria play additional crucial roles in neurons including calcium homeostasis, plasma membrane potential maintenance, synaptic formation, and neurotransmission (Flippo and Strack, 2017; Li et al., 2004).
Lastly, abnormal mitochondrial distribution and trafficking have been described in many neurodegenerative disease models such as Alzheimer’s disease (Pigino et al., 2003; Rui et al., 2006; Thies and Mandelkow, 2007), Huntington’s disease (Trushina et al., 2004), Parkinson’s disease (Blesa et al., 2015), Hereditary Spastic Paraplegia (Ferreirinha et al., 2004; McDermott et al., 2003), Charcot-Marie-Tooth (Baloh et al., 2007) and Amyotrophic Lateral Sclerosis (De Vos et al., 2007). Using cultured neurons as a model system, mitochondria were found to move at speeds ranging from 0.05 μm/s (representing a possible threshold to define motile mitochondria (Chen et al., 2016) to 1–1.5 μm/s (Stepkowski et al., 2016), consistent with previous studies (Ashrafi et al., 2014; Wang and Schwarz, 2009). Therefore, observed average velocities vary across a wide range. For example, astrocyte mitochondria move slower than neuronal ones, perhaps pertaining to cell type-specific differences in the trafficking machinery (Fiacco and McCarthy, 2004). However astrocyte mitochondria are still capable of covering the same distances as neuronal mitochondria (Stephen et al., 2015). Hence, in addition to mitochondrial velocity, assessment of the distance covered by moving mitochondria may be of interest. Run times (Miller et al., 2015) and percent of time spent in motion (‘processivity’) (Caino et al., 2016) represents other numerical parameters that could be used to classify mitochondria on the basis of their movement. Other factors that may influence measured mitochondrial speeds are: (i) fission-fusion (Liu et al., 2009); (ii) experimental conditions, (iii) local cytoskeletal environment (Moore et al., 2016); (iv) mitochondrial morphology and network complexity (Caino et al., 2016; Rafelski, 2013).
10.4. Molecular control of mitochondrial mobility and density
At the molecular level, mitochondrial transport is mediated by ATP-dependent kinesin and dynein families of motor proteins (Barnhart, 2016; Hirokawa and Tanaka, 2015; MacAskill and Kittler, 2010; Stepkowski et al., 2016; Wilson et al., 2013). In neurons, anterograde mitochondrial transport is mainly driven by KIF5, whereas retrograde transport is driven by the dynein complex. These motor proteins are complexed with mitochondria by protein adaptors, like syntabulin or Milton. In flies, Milton recruits KIF5 to mitochondria by binding to MIRO, a Rho GTPase located at the mitochondrial outer membrane. Similarly, TRAK1 and TRAK2, the mammalian homologs of Milton, were reported to bind to the MIRO homologues MIRO1 and MIRO2 in vertebrate cells (Beck et al., 2002; Cai et al., 2011; Iyer et al., 2003; Macaskill et al., 2009; Stowers et al., 2002). The Milton-MIRO complex is sensitive to cytosolic calcium levels, with elevation of Ca2+ producing stationary mitochondria and diminution of Ca2+ restoring mitochondrial motility (Macaskill et al., 2009; Saotome et al., 2008; Wang and Schwarz, 2009). This calcium-responsive switch is not the unique element governing mitochondrial motion: syntaphilin (SNPH) was reported to constitute a ‘stop’ signaling molecule by behaving as a negative regulator of KIF5 activity in axonal mitochondria of active neurons (Chen and Sheng, 2013). Interestingly, non-neuronal tumor cells might exploit this molecular ‘brake’ system by silencing SNPH expression, which translates into enhanced mitochondrial movements and targeting of ATP-producing mitochondria to the peripheral cytoskeleton of the tumor cells to fuel cell motility and invasion (Caino et al., 2016). Still, regulation of SNPH recruitment and activity remains elusive, as do the molecular pathways controlling the duration of mitochondrial retention at specific sites and the frequent reversal of motion direction following the pausing events (Bertholet et al., 2016; Chen et al., 2016; Ligon and Steward, 2000).
It is likely that selective transport and retention pathways exist to coordinate subcellular mitochondrial distribution in relation to specific changes in the cellular state, including cell cycle phases. For instance, recent evidence shows that during mitosis, MIRO directly recruits the centromere protein F (Cenp-F) to mitochondria, thereby ensuring anterograde mitochondrial movement for proper segregation of mitochondria in daughter cells (Kanfer et al., 2015). Interestingly, the MIRO-Cenp-F interaction was suggested to permit mitochondrial attachment to the tips of polymerizing microtubules, similar to ER ‘tip-tracking’ movements in which the tip of the ER tubule attaches to the tip of microtubule (+) ends (Grigoriev et al., 2008; Pendin et al., 2011).
The mechanisms of interaction between mitochondria and the cytoskeleton and their effects on mitochondrial motility remain matters of intense debate. Consistent with the ‘tip-tracking’ model described above, a number of research studies showed that mitochondrial motility was impaired upon microtubule depolymerization (Kandel et al., 2015; Ligon and Steward, 2000; Morris and Hollenbeck, 1995; Muller et al., 2005; Yi et al., 2004). While a consensus has emerged regarding the role of the microtubular cytoskeleton, conflicting results have been published on the relationships between the actin cytoskeleton and mitochondrial movements. Actin filament depolarization was shown to influence mitochondrial motility either negatively (Muller et al., 2005; Quintero et al., 2006) or positively (Ligon and Steward, 2000; Morris and Hollenbeck, 1995), partly depending on the cell type analyzed (Kandel et al., 2015). Interestingly, depolymerizing the actin cytoskeleton was recently suggested to blunt mitochondrial motility only in neuronal processes, and not in the cell body (Narayanareddy et al., 2014), providing further support to the idea that distinct control mechanisms may operate in different spatially-restricted cytoplasmic regions.
11. Future challenges
The next generation of microscope instruments and future increase in computational resources will help provide a global knowledge of mitochondrial morphology, trajectories and dynamics, and information about how cellular mitochondrial networks can be affected by treatment, disease and aging. Additional numerical descriptors will need to be incorporated in quantitative analysis of mitochondrial morphology and have their level of redundancy clearly assessed, in order to produce phenotypic ‘fingerprints’ or ‘mitograms’ at the level of individual mitochondria and mitochondrial networks, as recently anticipated (Blanchet et al., 2015; Iannetti et al., 2016). Development of a unified nomenclature of mitochondrial parameters and novel graphical representation systems might be useful in order to define how to best describe the morphology and dynamics of these organelles. This effort will also benefit from simultaneous measurements of functional state indicators for calcium, ATP and ROS levels, collectively know as the ‘mitochondrial love-hate triangle’ (Brookes et al., 2004), and from co-staining experiments for other cell components (cytoskeletal proteins, signaling molecules, autophagosomes and other organelles). Implementation of more ‘positive’ markers of mitochondrial damage (currently assessed by Δψm loss) is also awaited.
Research will have to explore new frontiers such as defining the molecular mechanisms that give rise to the tremendous heterogeneity in mitochondrial morphology between cell types and across species. Application of novel microscopy methods should allow resolving the fine structure and dynamics of mitochondrial membranes and mitochondrion-derived vesicles (Hayashi and Okada, 2015) in living cells, and develop a more detailed understanding of local interactions among mitochondria, formation of clusters and the morphological responses to cellular stresses (i.e. criticality and percolation) (Aon et al., 2004).
Conclusions
It remains a mystery how Bcl-2 proteins, which are metazoan-specific (Aouacheria et al., 2005), evolved to exert tight control on various mitochondrial features, such as integrity of the mitochondrial surface, dynamic processes like fission-fusion events, and many other effects potentially resulting from their unique interactions with membranes. Because most of these features and processes of Bcl-2 family proteins were already present in the unicellular ancestors of metazoans, it is likely that Bcl-2 proteins were co-opted to ensure their regulation in more complex organisms, interfacing with many preexisting molecular actors at the mitochondria and within other cell compartments. In addition to biochemical and structural works, future studies will have to carefully assess the roles of pro- and anti-apoptotic members of the Bcl-2 group by using quantitative measurements of mitochondrial parameters, including structural, organizational and functional readouts as those described in this review. Similar analysis could then be extended to other protein groups (e.g. BH3-only proteins, dynamins and KIF-family proteins) and other endomembrane systems with shared functions and components with mitochondria, such as the ER and peroxisomes.
Highlights.
Multiple features of mitochondrial morphology
Strategies for deducing mitochondrial dynamics from morphological features
Eliminating neuronal mitochondria by diverse strategies
Bcl-2 family proteins influence mitochondrial dynamics in neurons
Technologies for quantifying mitochondrial dynamics
Acknowledgments
This work was supported by grants from National Institutes of Health grants to J.M.H. (NS37402, GM077875, NS096677), and from the French National Center for Scientific Research (CNRS) Institute of Mechanobiology (MECANOBIO) and the SILAB to A.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, Tiwari BK, Kumar M, Micheal A, Mabalirajan U, Ghosh B, Sinha Roy S, Agrawal A. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013;4:e461. doi: 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP, Zorov DB. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J Cell Biol. 1988;107:481–495. doi: 10.1083/jcb.107.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu-Fernandez V, Sancho M, Genoves A, Lucendo E, Todt F, Lauterwasser J, Funk K, Jahreis G, Perez-Paya E, Mingarro I, Edlich F, Orzaez M. Bax transmembrane domain interacts with prosurvival Bcl-2 proteins in biological membranes. Proc Natl Acad Sci U S A. 2017;114:310–315. doi: 10.1073/pnas.1612322114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta. 2006;1757:692–699. doi: 10.1016/j.bbabio.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews DW. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005;24:2096–2103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antanaviciute I, Rysevaite K, Liutkevicius V, Marandykina A, Rimkute L, Sveikatiene R, Uloza V, Skeberdis VA. Long-distance communication between laryngeal carcinoma cells. PLoS One. 2014;9:e99196. doi: 10.1371/journal.pone.0099196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Cortassa S, O’Rourke B. Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci U S A. 2004;101:4447–4452. doi: 10.1073/pnas.0307156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouacheria A, Brunet F, Gouy M. Phylogenomics of life-or-death switches in multicellular animals: Bcl-2, BH3-Only, and BNip families of apoptotic regulators. Mol Biol Evol. 2005;22:2395–2416. doi: 10.1093/molbev/msi234. [DOI] [PubMed] [Google Scholar]
- Aouacheria A, Combet C, Tompa P, Hardwick JM. Redefining the BH3 Death Domain as a ‘Short Linear Motif’. Trends Biochem Sci. 2015;40:736–748. doi: 10.1016/j.tibs.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouacheria A, Rech de Laval V, Combet C, Hardwick JM. Evolution of Bcl-2 homology motifs: homology versus homoplasy. Trends Cell Biol. 2013;23:103–111. doi: 10.1016/j.tcb.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- Armstrong EM, More IA, McSeveney D, Carty M. The giant mitochondrion-endoplasmic reticulum unit of the human endometrial glandular cell. J Anat. 1973;116:375–383. [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206:655–670. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27:422–430. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart EL. Mechanics of mitochondrial motility in neurons. Curr Opin Cell Biol. 2016;38:90–99. doi: 10.1016/j.ceb.2016.02.022. [DOI] [PubMed] [Google Scholar]
- Basanez G, Nechushtan A, Drozhinin O, Chanturiya A, Choe E, Tutt S, Wood KA, Hsu Y, Zimmerberg J, Youle RJ. Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc Natl Acad Sci U S A. 1999;96:5492–5497. doi: 10.1073/pnas.96.10.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanez G, Sharpe JC, Galanis J, Brandt TB, Hardwick JM, Zimmerberg J. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem. 2002;277:49360–49365. doi: 10.1074/jbc.M206069200. [DOI] [PubMed] [Google Scholar]
- Basanez G, Zhang J, Chau BN, Maksaev GI, Frolov VA, Brandt TA, Burch J, Hardwick JM, Zimmerberg J. Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. J Biol Chem. 2001;276:31083–31091. doi: 10.1074/jbc.M103879200. [DOI] [PubMed] [Google Scholar]
- Beck M, Brickley K, Wilkinson HL, Sharma S, Smith M, Chazot PL, Pollard S, Stephenson FA. Identification, molecular cloning, and characterization of a novel GABAA receptor-associated protein, GRIF-1. J Biol Chem. 2002;277:30079–30090. doi: 10.1074/jbc.M200438200. [DOI] [PubMed] [Google Scholar]
- Benard G, Rossignol R. Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid Redox Signal. 2008;10:1313–1342. doi: 10.1089/ars.2007.2000. [DOI] [PubMed] [Google Scholar]
- Bender T, Martinou JC. Where killers meet–permeabilization of the outer mitochondrial membrane during apoptosis. Cold Spring Harb Perspect Biol. 2013;5:a011106. doi: 10.1101/cshperspect.a011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn J. Behavior of mitochondria in the living cell. Int Rev Cytol. 1990;122:1–63. doi: 10.1016/s0074-7696(08)61205-x. [DOI] [PubMed] [Google Scholar]
- Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman SB, Hardwick JM. Neuronal mitochondria are different: relevance to neurodegenerative diseas. In: Gribkoff VK, Jonas EA, Hardwick JM, editors. The Functions, Disease-Related Dysfunctions and Therapeutic Targeting of Neuronal Mitochondria. John Wiley & Sons, Inc; New York: 2015. pp. 221–239. [Google Scholar]
- Berman SB, Pineda FJ, Hardwick JM. Mitochondrial fission and fusion dynamics: the long and short of it. Cell Death Differ. 2008;15:1147–1152. doi: 10.1038/cdd.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnaune-Pelloquin L, Davezac N, Mils V, Miquel MC, Rojo M, Belenguer P. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis. 2016;90:3–19. doi: 10.1016/j.nbd.2015.10.011. [DOI] [PubMed] [Google Scholar]
- Bhola PD, Letai A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol Cell. 2016;61:695–704. doi: 10.1016/j.molcel.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindoff LA, Birch-Machin MA, Cartlidge NE, Parker WD, Jr, Turnbull DM. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J Neurol Sci. 1991;104:203–208. doi: 10.1016/0022-510x(91)90311-t. [DOI] [PubMed] [Google Scholar]
- Blanchet L, Smeitink JA, van Emst-de Vries SE, Vogels C, Pellegrini M, Jonckheere AI, Rodenburg RJ, Buydens LM, Beyrath J, Willems PH, Koopman WJ. Quantifying small molecule phenotypic effects using mitochondrial morpho-functional fingerprinting and machine learning. Sci Rep. 2015;5:8035. doi: 10.1038/srep08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, Garcia-Saez AJ, Bordignon E. Structural model of active Bax at the membrane. Mol Cell. 2014;56:496–505. doi: 10.1016/j.molcel.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR. Oxidative stress and Parkinson’s disease. Front Neuroanat. 2015;9:91. doi: 10.3389/fnana.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Morganti C, Morciano G, Giorgi C, Wieckowski MR, Pinton P. Comprehensive analysis of mitochondrial permeability transition pore activity in living cells using fluorescence-imaging-based techniques. Nat Protoc. 2016;11:1067–1080. doi: 10.1038/nprot.2016.064. [DOI] [PubMed] [Google Scholar]
- Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell. 2017;40:583–594. e586. doi: 10.1016/j.devcel.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt T, Cavellini L, Kuhlbrandt W, Cohen MM. A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. Elife. 2016;5 doi: 10.7554/eLife.14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, Iglewski M, Chiong M, Simmen T, Zorzano A, Hill JA, Rothermel BA, Szabadkai G, Lavandero S. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011;124:2143–2152. doi: 10.1242/jcs.080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55:938–946. doi: 10.1016/j.molcel.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- Cagalinec M, Liiv M, Hodurova Z, Hickey MA, Vaarmann A, Mandel M, Zeb A, Choubey V, Kuum M, Safiulina D, Vasar E, Veksler V, Kaasik A. Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLoS Biol. 2016;14:e1002511. doi: 10.1371/journal.pbio.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagalinec M, Safiulina D, Liiv M, Liiv J, Choubey V, Wareski P, Veksler V, Kaasik A. Principles of the mitochondrial fusion and fission cycle in neurons. J Cell Sci. 2013;126:2187–2197. doi: 10.1242/jcs.118844. [DOI] [PubMed] [Google Scholar]
- Cai Q, Davis ML, Sheng ZH. Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci Res. 2011;70:9–15. doi: 10.1016/j.neures.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caino MC, Seo JH, Aguinaldo A, Wait E, Bryant KG, Kossenkov AV, Hayden JE, Vaira V, Morotti A, Ferrero S, Bosari S, Gabrilovich DI, Languino LR, Cohen AR, Altieri DC. A neuronal network of mitochondrial dynamics regulates metastasis. Nat Commun. 2016;7:13730. doi: 10.1038/ncomms13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, Kaliszewski CK, Greer CA. Organization of mitochondria in olfactory bulb granule cell dendritic spines. Synapse. 1991;8:107–118. doi: 10.1002/syn.890080205. [DOI] [PubMed] [Google Scholar]
- Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, Li YJ, Yang JY, Liao S, Chan DC, Gao S. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature. 2017;542:372–376. doi: 10.1038/nature21077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny KL, McCaffery JM, Jensen RE. Division of mitochondria requires a novel DMN1-interacting protein, Net2p. Mol Biol Cell. 2001;12:309–321. doi: 10.1091/mbc.12.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- Chau BN, Cheng EH, Kerr DA, Hardwick JM. Aven, a novel inhibitor of caspase activation, binds Bcl-xL and Apaf-1. Mol Cell. 2000;6:31–40. [PubMed] [Google Scholar]
- Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010;1201:21–25. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Li Y, Yang M, Chen X, Chen Y, Yang F, Lu S, Yao S, Zhou T, Liu J, Zhu L, Du S, Wu JY. A new method for quantifying mitochondrial axonal transport. Protein Cell. 2016;7:804–819. doi: 10.1007/s13238-016-0268-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sheng ZH. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol. 2013;202:351–364. doi: 10.1083/jcb.201302040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, Hardwick JM. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Levine B, Boise LH, Thompson CB, Hardwick JM. Bax-independent inhibition of apoptosis by Bcl-XL. Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- Cheng WC, Leach KM, Hardwick JM. Mitochondrial death pathways in yeast and mammalian cells. Biochim Biophys Acta. 2008a;1783:1272–1279. doi: 10.1016/j.bbamcr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WC, Teng X, Park HK, Tucker CM, Dunham MJ, Hardwick JM. Fis1 deficiency selects for compensatory mutations responsible for cell death and growth control defects. Cell Death Differ. 2008b;15:1838–1846. doi: 10.1038/cdd.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherok E, Xu S, Li S, Das S, Meltzer WA, Zalzman M, Wang C, Karbowski M. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol Biol Cell. 2017;28:396–410. doi: 10.1091/mbc.E16-04-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrollier A, Cassereau J, Ferre M, Alban J, Desquiret-Dumas V, Gueguen N, Amati-Bonneau P, Procaccio V, Bonneau D, Reynier P. Standardized mitochondrial analysis gives new insights into mitochondrial dynamics and OPA1 function. Int J Biochem Cell Biol. 2012;44:980–988. doi: 10.1016/j.biocel.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- Choi HW, Kim JH, Chung MK, Hong YJ, Jang HS, Seo BJ, Jung TH, Kim JS, Chung HM, Byun SJ, Han SG, Seo HG, Do JT. Mitochondrial and metabolic remodeling during reprogramming and differentiation of the reprogrammed cells. Stem Cells Dev. 2015;24:1366–1373. doi: 10.1089/scd.2014.0561. [DOI] [PubMed] [Google Scholar]
- Clem RJ, Cheng EH, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, Hardwick JM. Modulation of cell death by Bcl-XL through caspase interaction. Proc Natl Acad Sci U S A. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado M, Scorrano L, Campello S. Mitochondrial dynamics in cancer and neurodegenerative and neuroinflammatory diseases. Int J Cell Biol. 2012;2012:729290. doi: 10.1155/2012/729290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P, Zurzolo C. Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci. 2013;126:3678–3685. doi: 10.1242/jcs.126086. [DOI] [PubMed] [Google Scholar]
- Cotter D, Guda P, Fahy E, Subramaniam S. MitoProteome: mitochondrial protein sequence database and annotation system. Nucleic Acids Res. 2004;32:D463–467. doi: 10.1093/nar/gkh048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- D’Orsi B, Kilbride SM, Chen G, Perez Alvarez S, Bonner HP, Pfeiffer S, Plesnila N, Engel T, Henshall DC, Dussmann H, Prehn JH. Bax regulates neuronal Ca2+ homeostasis. J Neurosci. 2015;35:1706–1722. doi: 10.1523/JNEUROSCI.2453-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmasso G, Marin Zapata PA, Brady NR, Hamacher-Brady A. Agent-Based Modeling of Mitochondria Links Sub-Cellular Dynamics to Cellular Homeostasis and Heterogeneity. PLoS One. 2017;12:e0168198. doi: 10.1371/journal.pone.0168198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111:9633–9638. doi: 10.1073/pnas.1404651111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Allan VJ, Grierson AJ, Sheetz MP. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr Biol. 2005;15:678–683. doi: 10.1016/j.cub.2005.02.064. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–773. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J. 2013;104:2077–2088. doi: 10.1016/j.bpj.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Dukes AA, Bai Q, Van Laar VS, Zhou Y, Ilin V, David CN, Agim ZS, Bonkowsky JL, Cannon JR, Watkins SC, Croix CM, Burton EA, Berman SB. Live imaging of mitochondrial dynamics in CNS dopaminergic neurons in vivo demonstrates early reversal of mitochondrial transport following MPP(+) exposure. Neurobiol Dis. 2016;95:238–249. doi: 10.1016/j.nbd.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelthaler DM, Hicks ND, Gillece JD, Roe CC, Schupp JM, Driebe EM, Gilgado F, Carriconde F, Trilles L, Firacative C, Ngamskulrungroj P, Castaneda E, Lazera Mdos S, Melhem MS, Perez-Bercoff A, Huttley G, Sorrell TC, Voelz K, May RC, Fisher MC, Thompson GR, 3rd, Lockhart SR, Keim P, Meyer W. Cryptococcus gattii in North American Pacific Northwest: whole-population genome analysis provides insights into species evolution and dispersal. MBio. 2014;5:e01464–01414. doi: 10.1128/mBio.01464-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eura Y, Ishihara N, Yokota S, Mihara K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem. 2003;134:333–344. doi: 10.1093/jb/mvg150. [DOI] [PubMed] [Google Scholar]
- Fannjiang Y, Cheng WC, Lee SJ, Qi B, Pevsner J, McCaffery JM, Hill RB, Basanez G, Hardwick JM. Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev. 2004;18:2785–2797. doi: 10.1101/gad.1247904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannjiang Y, Kim CH, Huganir RL, Zou S, Lindsten T, Thompson CB, Mito T, Traystman RJ, Larsen T, Griffin DE, Mandir AS, Dawson TM, Dike S, Sappington AL, Kerr DA, Jonas EA, Kaczmarek LK, Hardwick JM. BAK alters neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev Cell. 2003;4:575–585. doi: 10.1016/s1534-5807(03)00091-1. [DOI] [PubMed] [Google Scholar]
- Fekkes P, Shepard KA, Yaffe MP. Gag3p, an outer membrane protein required for fission of mitochondrial tubules. J Cell Biol. 2000;151:333–340. doi: 10.1083/jcb.151.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V, Auricchio A, Piemonte F, Tozzi G, Gaeta L, Casari G, Ballabio A, Rugarli EI. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest. 2004;113:231–242. doi: 10.1172/JCI20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LW, Lee J, Souza A, Desquiret-Dumas V, Bullock K, Rowe GC, Procaccio V, Clish CB, Arany Z, Haigis MC. Skeletal muscle transcriptional coactivator PGC-1alpha mediates mitochondrial, but not metabolic, changes during calorie restriction. Proc Natl Acad Sci U S A. 2012;109:2931–2936. doi: 10.1073/pnas.1115813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flippo KH, Strack S. Mitochondrial dynamics in neuronal injury, development and plasticity. J Cell Sci. 2017;130:671–681. doi: 10.1242/jcs.171017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW., II Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature. 2016;540:74–79. doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–510. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki Y, Miyata N, Mukai S, Okumoto K, Cheng EH. BAK regulates catalase release from peroxisomes. Molecular and Cellular Oncology. 2017 doi: 10.1080/23723556.2017.1306610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Saez AJ, Coraiola M, Serra MD, Mingarro I, Muller P, Salgado J. Peptides corresponding to helices 5 and 6 of Bax can independently form large lipid pores. FEBS J. 2006;273:971–981. doi: 10.1111/j.1742-4658.2006.05123.x. [DOI] [PubMed] [Google Scholar]
- Giedt RJ, Fumene Feruglio P, Pathania D, Yang KS, Kilcoyne A, Vinegoni C, Mitchison TJ, Weissleder R. Computational imaging reveals mitochondrial morphology as a biomarker of cancer phenotype and drug response. Sci Rep. 2016;6:32985. doi: 10.1038/srep32985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Cassina A, Garcia-Haro L, Choi CS, Osundiji MA, Lane EA, Huang H, Yildirim MA, Szlyk B, Fisher JK, Polak K, Patton E, Wiwczar J, Godes M, Lee DH, Robertson K, Kim S, Kulkarni A, Distefano A, Samuel V, Cline G, Kim YB, Shulman GI, Danial NN. Regulation of hepatic energy metabolism and gluconeogenesis by BAD. Cell Metab. 2014;19:272–284. doi: 10.1016/j.cmet.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, Rizzuto R, Tacchetti C, Pinton P, Pandolfi PP. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330:1247–1251. doi: 10.1126/science.1189157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, Subramaniam S, Balaban RS. Mitochondrial reticulum for cellular energy distribution in muscle. Nature. 2015;523:617–620. doi: 10.1038/nature14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011a;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. Essential amino acids and glutamine regulate induction of mitochondrial elongation during autophagy. Cell Cycle. 2011b;10:2635–2639. doi: 10.4161/cc.10.16.17002. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia M, Garcia I, Ding L, O’Shea S, Boise LH, Thompson CB, Nunez G. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci U S A. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Jamett AM, Haro-Acuna V, Momboisse F, Caviedes P, Bevilacqua JA, Cardenas AM. Dynamin-2 in nervous system disorders. J Neurochem. 2014;128:210–223. doi: 10.1111/jnc.12455. [DOI] [PubMed] [Google Scholar]
- Green DR. A BH3 Mimetic for Killing Cancer Cells. Cell. 2016;165:1560. doi: 10.1016/j.cell.2016.05.080. [DOI] [PubMed] [Google Scholar]
- Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, Splinter D, Steinmetz MO, Putney JW, Jr, Hoogenraad CC, Akhmanova A. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18:177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. 2004;279:18792–18798. doi: 10.1074/jbc.M400920200. [DOI] [PubMed] [Google Scholar]
- Gross A, Katz SG. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017 doi: 10.1038/cdd.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin Y, Lalle P, Gillet G, Guerin JF, Hamamah S, Aouacheria A. Oocytes and early embryos selectively express the survival factor BCL2L10. J Mol Med (Berl) 2009;87:923–940. doi: 10.1007/s00109-009-0495-7. [DOI] [PubMed] [Google Scholar]
- Guillery O, Malka F, Frachon P, Milea D, Rojo M, Lombes A. Modulation of mitochondrial morphology by bioenergetics defects in primary human fibroblasts. Neuromuscul Disord. 2008;18:319–330. doi: 10.1016/j.nmd.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Hamacher-Brady A, Brady NR. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci. 2016;73:775–795. doi: 10.1007/s00018-015-2087-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamner S, Skoglosa Y, Lindholm D. Differential expression of bcl-w and bcl-x messenger RNA in the developing and adult rat nervous system. Neuroscience. 1999;91:673–684. doi: 10.1016/s0306-4522(98)00642-3. [DOI] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Chen YB, Jonas EA. Multipolar functions of BCL-2 proteins link energetics to apoptosis. Trends Cell Biol. 2012;22:318–328. doi: 10.1016/j.tcb.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Hatch AL, Ji WK, Merrill RA, Strack S, Higgs HN. Actin filaments as dynamic reservoirs for Drp1 recruitment. Mol Biol Cell. 2016;27:3109–3121. doi: 10.1091/mbc.E16-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–555. doi: 10.1038/nature18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Okada Y. Ultrafast superresolution fluorescence imaging with spinning disk confocal microscope optics. Mol Biol Cell. 2015;26:1743–1751. doi: 10.1091/mbc.E14-08-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggeness MH, Simon M, Singer SJ. Association of mitochondria with microtubules in cultured cells. Proc Natl Acad Sci U S A. 1978;75:3863–3866. doi: 10.1073/pnas.75.8.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman JA, Hardwick JM, Kaczmarek LK, Jonas EA. Bcl-xL inhibitor ABT-737 reveals a dual role for Bcl-xL in synaptic transmission. J Neurophysiol. 2008;99:1515–1522. doi: 10.1152/jn.00598.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Tanaka Y. Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Exp Cell Res. 2015;334:16–25. doi: 10.1016/j.yexcr.2015.02.016. [DOI] [PubMed] [Google Scholar]
- Hodneland Nilsson LI, Nitschke Pettersen IK, Nikolaisen J, Micklem D, Avsnes Dale H, Vatne Rosland G, Lorens J, Tronstad KJ. A new live-cell reporter strategy to simultaneously monitor mitochondrial biogenesis and morphology. Sci Rep. 2015;5:17217. doi: 10.1038/srep17217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoitzing H, Johnston IG, Jones NS. What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research. Bioessays. 2015;37:687–700. doi: 10.1002/bies.201400188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway GP, Gurd BJ, Snook LA, Lally J, Bonen A. Compensatory increases in nuclear PGC1alpha protein are primarily associated with subsarcolemmal mitochondrial adaptations in ZDF rats. Diabetes. 2010;59:819–828. doi: 10.2337/db09-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holme P. Temporal network structures controlling disease spreading. Phys Rev E. 2016;94:022305. doi: 10.1103/PhysRevE.94.022305. [DOI] [PubMed] [Google Scholar]
- Hosoi KI, Miyata N, Mukai S, Furuki S, Okumoto K, Cheng EH, Fujiki Y. The VDAC2-BAK axis regulates peroxisomal membrane permeability. J Cell Biol. 2017;216:709–722. doi: 10.1083/jcb.201605002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Sun L, Ji S, Zhao T, Zhang W, Xu J, Zhang J, Wang Y, Wang X, Franzini-Armstrong C, Zheng M, Cheng H. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc Natl Acad Sci U S A. 2013;110:2846–2851. doi: 10.1073/pnas.1300741110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannetti EF, Smeitink JA, Beyrath J, Willems PH, Koopman WJ. Multiplexed high-content analysis of mitochondrial morphofunction using live-cell microscopy. Nat Protoc. 2016;11:1693–1710. doi: 10.1038/nprot.2016.094. [DOI] [PubMed] [Google Scholar]
- Ingber DE. Tensegrity II. How structural networks influence cellular information processing networks. J Cell Sci. 2003;116:1397–1408. doi: 10.1242/jcs.00360. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S, Bell F, Westphal D, Anwari K, Gulbis J, Smith BJ, Dewson G, Kluck RM. Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death Differ. 2015;22:1665–1675. doi: 10.1038/cdd.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SP, Akimoto Y, Hart GW. Identification and cloning of a novel family of coiled-coil domain proteins that interact with O-GlcNAc transferase. J Biol Chem. 2003;278:5399–5409. doi: 10.1074/jbc.M209384200. [DOI] [PubMed] [Google Scholar]
- Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- Jensen RE, Hobbs AE, Cerveny KL, Sesaki H. Yeast mitochondrial dynamics: fusion, division, segregation, and shape. Microsc Res Tech. 2000;51:573–583. doi: 10.1002/1097-0029(20001215)51:6<573::AID-JEMT7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Jiang YF, Lin SS, Chen JM, Tsai HZ, Hsieh TS, Fu CY. Electron tomographic analysis reveals ultrastructural features of mitochondrial cristae architecture which reflect energetic state and aging. Sci Rep. 2017;7:45474. doi: 10.1038/srep45474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Li Z. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron. 2011;70:758–772. doi: 10.1016/j.neuron.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hardwick JM, Kaczmarek LK. Actions of BAX on mitochondrial channel activity and on synaptic transmission. Antioxid Redox Signal. 2005a;7:1092–1100. doi: 10.1089/ars.2005.7.1092. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Chachar M, Polster BM, Brandt TA, Fannjiang Y, Ivanovska I, Basanez G, Kinnally KW, Zimmerberg J, Hardwick JM, Kaczmarek LK. Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc Natl Acad Sci U S A. 2004;101:13590–13595. doi: 10.1073/pnas.0401372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Hardwick JM, Kaczmarek LK. Exposure to hypoxia rapidly induces mitochondrial channel activity within a living synapse. J Biol Chem. 2005b;280:4491–4497. doi: 10.1074/jbc.M410661200. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Hoit D, Hickman JA, Brandt TA, Polster BM, Fannjiang Y, McCarthy E, Montanez MK, Hardwick JM, Kaczmarek LK. Modulation of synaptic transmission by the BCL-2 family protein BCL-xL. J Neurosci. 2003;23:8423–8431. doi: 10.1523/JNEUROSCI.23-23-08423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel J, Chou P, Eckmann DM. Automated detection of whole-cell mitochondrial motility and its dependence on cytoarchitectural integrity. Biotechnol Bioeng. 2015;112:1395–1405. doi: 10.1002/bit.25563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanfer G, Courtheoux T, Peterka M, Meier S, Soste M, Melnik A, Reis K, Aspenstrom P, Peter M, Picotti P, Kornmann B. Mitotic redistribution of the mitochondrial network by Miro and Cenp-F. Nat Commun. 2015;6:8015. doi: 10.1038/ncomms9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam SM, Straiton T, Hassan WM, Leblond CP. Defining epithelial cell progenitors in the human oxyntic mucosa. Stem Cells. 2003;21:322–336. doi: 10.1634/stemcells.21-3-322. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Spodnik JH, Teranishi M, Wozniak M, Nishizawa Y, Usukura J, Wakabayashi T. Opposite effects of microtubule-stabilizing and microtubule-destabilizing drugs on biogenesis of mitochondria in mammalian cells. J Cell Sci. 2001;114:281–291. doi: 10.1242/jcs.114.2.281. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57:537–551. doi: 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Laschi C, Trimmer B. Soft robotics: a bioinspired evolution in robotics. Trends Biotechnol. 2013;31:287–294. doi: 10.1016/j.tibtech.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274:21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- Kiryu-Seo S, Ohno N, Kidd GJ, Komuro H, Trapp BD. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci. 2010;30:6658–6666. doi: 10.1523/JNEUROSCI.5265-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, Shaw JM. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci U S A. 2013;110:E1342–1351. doi: 10.1073/pnas.1300855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman WJ, Distelmaier F, Esseling JJ, Smeitink JA, Willems PH. Computer-assisted live cell analysis of mitochondrial membrane potential, morphology and calcium handling. Methods. 2008;46:304–311. doi: 10.1016/j.ymeth.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Visch HJ, Smeitink JA, Willems PH. Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytometry A. 2006;69:1–12. doi: 10.1002/cyto.a.20198. [DOI] [PubMed] [Google Scholar]
- Korobova F, Gauvin TJ, Higgs HN. A role for myosin II in mammalian mitochondrial fission. Curr Biol. 2014;24:409–414. doi: 10.1016/j.cub.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–467. doi: 10.1126/science.1228360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Mai JK, Zapata JM, Ashwell KW, Schendel SL, Reed JC, Krajewski S. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ. 2002;9:145–157. doi: 10.1038/sj.cdd.4400934. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Lackner LL. Shaping the dynamic mitochondrial network. BMC Biol. 2014;12:35. doi: 10.1186/1741-7007-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner LL, Horner JS, Nunnari J. Mechanistic analysis of a dynamin effector. Science. 2009;325:874–877. doi: 10.1126/science.1176921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavorato M, Iyer VR, Dewight W, Cupo RR, Debattisti V, Gomez L, De la Fuente S, Zhao YT, Valdivia HH, Hajnoczky G, Franzini-Armstrong C. Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges. Proc Natl Acad Sci U S A. 2017;114:E849–E858. doi: 10.1073/pnas.1617788113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber B, Lin J, Andrews DW. Still embedded together binding to membranes regulates Bcl-2 protein interactions. Oncogene. 2010;29:5221–5230. doi: 10.1038/onc.2010.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EF, Dewson G, Smith BJ, Evangelista M, Pettikiriarachchi A, Dogovski C, Perugini MA, Colman PM, Fairlie WD. Crystal structure of a BCL-W domain-swapped dimer: implications for the function of BCL-2 family proteins. Structure. 2011;19:1467–1476. doi: 10.1016/j.str.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. 2016a;540:139–143. doi: 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, Youle RJ, Cho H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem. 2007;282:22977–22983. doi: 10.1074/jbc.M700679200. [DOI] [PubMed] [Google Scholar]
- Lee WH, Higuchi H, Ikeda S, Macke EL, Takimoto T, Pattnaik BR, Liu C, Chu LF, Siepka SM, Krentz KJ, Rubinstein CD, Kalejta RF, Thomson JA, Mullins RF, Takahashi JS, Pinto LH, Ikeda A. Mouse Tmem135 mutation reveals a mechanism involving mitochondrial dynamics that leads to age-dependent retinal pathologies. Elife. 2016b;5 doi: 10.7554/eLife.19264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon FE, Cianci GC, Kanteti R, Riehm JJ, Arif Q, Poroyko VA, Lupovitch E, Vigneswaran W, Husain A, Chen P, Liao JK, Sattler M, Kindler HL, Salgia R. Unique fractal evaluation and therapeutic implications of mitochondrial morphology in malignant mesothelioma. Sci Rep. 2016;6:24578. doi: 10.1038/srep24578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AP, Cameron RB, Speiser JL, Wolf BJ, Peterson YK, Schnellmann RG, Beeson CC, Rohrer B. Quantitative analysis of mitochondrial morphology and membrane potential in living cells using high-content imaging, machine learning, and morphological binning. Biochim Biophys Acta. 2015;1853:348–360. doi: 10.1016/j.bbamcr.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE, Hardwick JM. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature. 1993;361:739–742. doi: 10.1038/361739a0. [DOI] [PubMed] [Google Scholar]
- Lewis J, Oyler GA, Ueno K, Fannjiang YR, Chau BN, Vornov J, Korsmeyer SJ, Zou S, Hardwick JM. Inhibition of virus-induced neuronal apoptosis by Bax. Nat Med. 1999;5:832–835. doi: 10.1038/10556. [DOI] [PubMed] [Google Scholar]
- Li H, Alavian KN, Lazrove E, Mehta N, Jones A, Zhang P, Licznerski P, Graham M, Uo T, Guo J, Rahner C, Duman RS, Morrison RS, Jonas EA. A Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat Cell Biol. 2013;15:773–785. doi: 10.1038/ncb2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MV, Pypaert M, Hickman JA, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Xu S, Roelofs BA, Boyman L, Lederer WJ, Sesaki H, Karbowski M. Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J Cell Biol. 2015;208:109–123. doi: 10.1083/jcb.201404050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Liao C, Zhang Z, Kale J, Andrews DW, Lin J, Li J. Conformational Heterogeneity of Bax Helix 9 Dimer for Apoptotic Pore Formation. Sci Rep. 2016;6:29502. doi: 10.1038/srep29502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon LA, Steward O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol. 2000;427:351–361. doi: 10.1002/1096-9861(20001120)427:3<351::aid-cne3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Lihavainen E, Makela J, Spelbrink JN, Ribeiro AS. Mytoe: automatic analysis of mitochondrial dynamics. Bioinformatics. 2012;28:1050–1051. doi: 10.1093/bioinformatics/bts073. [DOI] [PubMed] [Google Scholar]
- Lin MY, Sheng ZH. Regulation of mitochondrial transport in neurons. Exp Cell Res. 2015;334:35–44. doi: 10.1016/j.yexcr.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Golden JA, Zong WX, Minarcik J, Harris MH, Thompson CB. The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. J Neurosci. 2003;23:11112–11119. doi: 10.1523/JNEUROSCI.23-35-11112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hajnoczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011;18:1561–1572. doi: 10.1038/cdd.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weaver D, Shirihai O, Hajnoczky G. Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009;28:3074–3089. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mejia IC, Fajas L. Cell cycle regulation of mitochondrial function. Curr Opin Cell Biol. 2015;33:19–25. doi: 10.1016/j.ceb.2014.10.006. [DOI] [PubMed] [Google Scholar]
- Loson OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, Chan DC. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure. 2014;22:367–377. doi: 10.1016/j.str.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low HH, Sachse C, Amos LA, Lowe J. Structure of a bacterial dynamin-like protein lipid tube provides a mechanism for assembly and membrane curving. Cell. 2009;139:1342–1352. doi: 10.1016/j.cell.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Zheng X, Li F, Yu Y, Chen Z, Liu Z, Wang Z, Xu H, Yang W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget. 2017 doi: 10.18632/oncotarget.14695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Rolland SG, Conradt B. A molecular switch that governs mitochondrial fusion and fission mediated by the BCL2-like protein CED-9 of Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2011;108:E813–822. doi: 10.1073/pnas.1103218108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112. doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci. 2010;123:917–926. doi: 10.1242/jcs.059246. [DOI] [PubMed] [Google Scholar]
- Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta. 2006;1763:542–548. doi: 10.1016/j.bbamcr.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, Spudich J, Lippincott-Schwartz J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife. 2015;4 doi: 10.7554/eLife.08828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M. Neuronal Mitophagy in Neurodegenerative Diseases. Front Mol Neurosci. 2017;10:64. doi: 10.3389/fnmol.2017.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- McClatchey PM, Keller AC, Bouchard R, Knaub LA, Reusch JE. Fully automated software for quantitative measurements of mitochondrial morphology. Mitochondrion. 2016;26:58–71. doi: 10.1016/j.mito.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott CJ, Grierson AJ, Wood JD, Bingley M, Wharton SB, Bushby KM, Shaw PJ. Hereditary spastic paraparesis: disrupted intracellular transport associated with spastin mutation. Ann Neurol. 2003;54:748–759. doi: 10.1002/ana.10757. [DOI] [PubMed] [Google Scholar]
- McLelland GL, Lee SA, McBride HM, Fon EA. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016;214:275–291. doi: 10.1083/jcb.201603105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally MA, Soane L, Roelofs BA, Hartman AL, Hardwick JM. The N-terminal helix of Bcl-xL targets mitochondria. Mitochondrion. 2013;13:119–124. doi: 10.1016/j.mito.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S, McCaffery JM, Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–1752. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- Melentijevic I, Toth ML, Arnold ML, Guasp RJ, Harinath G, Nguyen KC, Taub D, Parker JA, Neri C, Gabel CV, Hall DH, Driscoll M. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. 2017;542:367–371. doi: 10.1038/nature21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Liu XA, Puthanveettil SV. Automated measurement of fast mitochondrial transport in neurons. Front Cell Neurosci. 2015;9:435. doi: 10.3389/fncel.2015.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19:630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Wong YC, Simpson CL, Holzbaur EL. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat Commun. 2016;7:12886. doi: 10.1038/ncomms12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran M, Rivera H, Sanchez-Arago M, Blazquez A, Merinero B, Ugalde C, Arenas J, Cuezva JM, Martin MA. Mitochondrial bioenergetics and dynamics interplay in complex I-deficient fibroblasts. Biochim Biophys Acta. 2010;1802:443–453. doi: 10.1016/j.bbadis.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104(Pt 3):917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Muller M, Mironov SL, Ivannikov MV, Schmidt J, Richter DW. Mitochondrial organization and motility probed by two-photon microscopy in cultured mouse brainstem neurons. Exp Cell Res. 2005;303:114–127. doi: 10.1016/j.yexcr.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H, Taneike M, Misaka T, Omiya S, Shah AM, Yamamoto A, Nishida K, Ohsumi Y, Okamoto K, Sakata Y, Otsu K. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. doi: 10.1038/ncomms8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med. 2001;7:934–940. doi: 10.1038/90976. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Swahari V, Plestant C, Smith I, McCoy E, Smith S, Moy SS, Anton ES, Deshmukh M. Bcl-xL Is Essential for the Survival and Function of Differentiated Neurons in the Cortex That Control Complex Behaviors. J Neurosci. 2016;36:5448–5461. doi: 10.1523/JNEUROSCI.4247-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanareddy BR, Vartiainen S, Hariri N, O’Dowd DK, Gross SP. A biophysical analysis of mitochondrial movement: differences between transport in neuronal cell bodies versus processes. Traffic. 2014;15:762–771. doi: 10.1111/tra.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah CM, Horvath TL. Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol. 2014;10:650–658. doi: 10.1038/nrendo.2014.160. [DOI] [PubMed] [Google Scholar]
- Nikolaisen J, Nilsson LI, Pettersen IK, Willems PH, Lorens JB, Koopman WJ, Tronstad KJ. Automated quantification and integrative analysis of 2D and 3D mitochondrial shape and network properties. PLoS One. 2014;9:e101365. doi: 10.1371/journal.pone.0101365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon-Abell J, Obara CJ, Weigel AV, Li D, Legant WR, Xu CS, Pasolli HA, Harvey K, Hess HF, Betzig E, Blackstone C, Lippincott-Schwartz J. Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science. 2016;354 doi: 10.1126/science.aaf3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi T, Koizumi M, Hayashi S. Sustained elongation of sperm tail promoted by local remodeling of giant mitochondria in Drosophila. Curr Biol. 2011;21:805–814. doi: 10.1016/j.cub.2011.04.016. [DOI] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki T, Kagaya Y, Ishide N, Kitada S, Miura M, Nawata J, Ohno I, Watanabe J, Shirato K. Interaction between sarcomere and mitochondrial length in normoxic and hypoxic rat ventricular papillary muscles. Cardiovasc Pathol. 2001;10:125–132. doi: 10.1016/s1054-8807(01)00071-0. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill JW, Manion MK, Maguire B, Hockenbery DM. BCL-XL dimerization by three-dimensional domain swapping. J Mol Biol. 2006;356:367–381. doi: 10.1016/j.jmb.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Oettinghaus B, D’Alonzo D, Barbieri E, Restelli LM, Savoia C, Licci M, Tolnay M, Frank S, Scorrano L. DRP1-dependent apoptotic mitochondrial fission occurs independently of BAX, BAK and APAF1 to amplify cell death by BID and oxidative stress. Biochim Biophys Acta. 2016;1857:1267–1276. doi: 10.1016/j.bbabio.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Ofengeim D, Chen YB, Miyawaki T, Li H, Sacchetti S, Flannery RJ, Alavian KN, Pontarelli F, Roelofs BA, Hickman JA, Hardwick JM, Zukin RS, Jonas EA. N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat Neurosci. 2012;15:574–580. doi: 10.1038/nn.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Onfelt B, Nedvetzki S, Benninger RK, Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MA, French PM, Davis DM. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J Immunol. 2006;177:8476–8483. doi: 10.4049/jimmunol.177.12.8476. [DOI] [PubMed] [Google Scholar]
- Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet. 2001;28:272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Stojanovski D, Ryan MT. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal. 2011;23:1534–1545. doi: 10.1016/j.cellsig.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun. 2015;6:6768. doi: 10.1038/ncomms7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendin D, McNew JA, Daga A. Balancing ER dynamics: shaping, bending, severing, and mending membranes. Curr Opin Cell Biol. 2011;23:435–442. doi: 10.1016/j.ceb.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JY, Lin CC, Chen YJ, Kao LS, Liu YC, Chou CC, Huang YH, Chang FR, Wu YC, Tsai YS, Hsu CN. Automatic morphological subtyping reveals new roles of caspases in mitochondrial dynamics. PLoS Comput Biol. 2011;7:e1002212. doi: 10.1371/journal.pcbi.1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Picard M, McManus MJ, Csordas G, Varnai P, Dorn GW, 2nd, Williams D, Hajnoczky G, Wallace DC. Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat Commun. 2015;6:6259. doi: 10.1038/ncomms7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletjushkina OY, Lyamzaev KG, Popova EN, Nepryakhina OK, Ivanova OY, Domnina LV, Chernyak BV, Skulachev VP. Effect of oxidative stress on dynamics of mitochondrial reticulum. Biochim Biophys Acta. 2006;1757:518–524. doi: 10.1016/j.bbabio.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Popkov VA, Plotnikov EY, Lyamzaev KG, Silachev DN, Zorova LD, Pevzner IB, Jankauskas SS, Zorov SD, Babenko VA, Zorov DB. Mitodiversity. Biochemistry (Mosc) 2015;80:532–541. doi: 10.1134/S000629791505003X. [DOI] [PubMed] [Google Scholar]
- Popov V, Medvedev NI, Davies HA, Stewart MG. Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: a three-dimensional ultrastructural study. J Comp Neurol. 2005;492:50–65. doi: 10.1002/cne.20682. [DOI] [PubMed] [Google Scholar]
- Posakony JW, England JM, Attardi G. Mitochondrial growth and division during the cell cycle in HeLa cells. J Cell Biol. 1977;74:468–491. doi: 10.1083/jcb.74.2.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A. 2006;103:5379–5384. doi: 10.1073/pnas.0601026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafelski SM. Mitochondrial network morphology: building an integrative, geometrical view. BMC Biol. 2013;11:71. doi: 10.1186/1741-7007-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafelski SM, Marshall WF. Building the cell: design principles of cellular architecture. Nat Rev Mol Cell Biol. 2008;9:593–602. doi: 10.1038/nrm2460. [DOI] [PubMed] [Google Scholar]
- Rafelski SM, Viana MP, Zhang Y, Chan YH, Thorn KS, Yam P, Fung JC, Li H, Costa Lda F, Marshall WF. Mitochondrial network size scaling in budding yeast. Science. 2012;338:822–824. doi: 10.1126/science.1225720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006;13:1378–1386. doi: 10.1038/sj.cdd.4401975. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23:7881–7888. doi: 10.1523/JNEUROSCI.23-21-07881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo G, Chamorro M, Salas ML, Vinuela E, Cuezva JM, Salas J. Migration of mitochondria to viral assembly sites in African swine fever virus-infected cells. J Virol. 1998;72:7583–7588. doi: 10.1128/jvi.72.9.7583-7588.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth KA, Kuan C, Haydar TF, D’Sa-Eipper C, Shindler KS, Zheng TS, Kuida K, Flavell RA, Rakic P. Epistatic and independent functions of caspase-3 and Bcl-X(L) in developmental programmed cell death. Proc Natl Acad Sci U S A. 2000;97:466–471. doi: 10.1073/pnas.97.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Reddy PH, Iijima M, Sesaki H. Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol. 2015;33:111–118. doi: 10.1016/j.ceb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci. 2003;116:2763–2774. doi: 10.1242/jcs.00479. [DOI] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholkmann F. Long range physical cell-to-cell signalling via mitochondria inside membrane nanotubes: a hypothesis. Theor Biol Med Model. 2016;13:16. doi: 10.1186/s12976-016-0042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Kner P, Rego EH, Gustafsson MG. Super-resolution 3D microscopy of live whole cells using structured illumination. Nat Methods. 2011;8:1044–1046. doi: 10.1038/nmeth.1734. [DOI] [PubMed] [Google Scholar]
- Shim SH, Xia C, Zhong G, Babcock HP, Vaughan JC, Huang B, Wang X, Xu C, Bi GQ, Zhuang X. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc Natl Acad Sci U S A. 2012;109:13978–13983. doi: 10.1073/pnas.1201882109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindler KS, Yunker AM, Cahn R, Zha J, Korsmeyer SJ, Roth KA. Trophic support promotes survival of bcl-x-deficient telencephalic cells in vitro. Cell Death Differ. 1998;5:901–910. doi: 10.1038/sj.cdd.4400421. [DOI] [PubMed] [Google Scholar]
- Shutt T, Geoffrion M, Milne R, McBride HM. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012;13:909–915. doi: 10.1038/embor.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci. 2001;26:23–29. doi: 10.1016/s0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–358. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One. 2012;7:e52830. doi: 10.1371/journal.pone.0052830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavru F, Palmer AE, Wang C, Youle RJ, Cossart P. Atypical mitochondrial fission upon bacterial infection. Proc Natl Acad Sci U S A. 2013;110:16003–16008. doi: 10.1073/pnas.1315784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen TL, Higgs NF, Sheehan DF, Al Awabdh S, Lopez-Domenech G, Arancibia-Carcamo IL, Kittler JT. Miro1 Regulates Activity-Driven Positioning of Mitochondria within Astrocytic Processes Apposed to Synapses to Regulate Intracellular Calcium Signaling. J Neurosci. 2015;35:15996–16011. doi: 10.1523/JNEUROSCI.2068-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepkowski TM, Meczynska-Wielgosz S, Kruszewski M. mitoLUHMES: An Engineered Neuronal Cell Line for the Analysis of the Motility of Mitochondria. Cell Mol Neurobiol. 2016 doi: 10.1007/s10571-016-0438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, Vizcay-Barrena G, Lin WL, Xu YF, Lewis J, Dickson DW, Petrucelli L, Mitchell JC, Shaw CE, Miller CC. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun. 2014;5:3996. doi: 10.1038/ncomms4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Stuart K. Mitochondrial DNA of an African trypanosome. J Cell Biochem. 1983;23:13–26. doi: 10.1002/jcb.240230103. [DOI] [PubMed] [Google Scholar]
- Sugiura A, Mattie S, Prudent J, McBride HM. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature. 2017;542:251–254. doi: 10.1038/nature21375. [DOI] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142–2156. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung TC, Li CY, Lai YC, Hung CL, Shih O, Yeh YQ, Jeng US, Chiang YW. Solution Structure of Apoptotic BAX Oligomer: Oligomerization Likely Precedes Membrane Insertion. Structure. 2015;23:1878–1888. doi: 10.1016/j.str.2015.07.013. [DOI] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Tang HL, Tang HM, Fung MC, Hardwick JM. In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci Rep. 2015;5:9015. doi: 10.1038/srep09015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu Q, Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. 2000;151:353–366. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, 2nd, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- Valero JG, Sancey L, Kucharczak J, Guillemin Y, Gimenez D, Prudent J, Gillet G, Salgado J, Coll JL, Aouacheria A. Bax-derived membrane-active peptides act as potent and direct inducers of apoptosis in cancer cells. J Cell Sci. 2011;124:556–564. doi: 10.1242/jcs.076745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Blerkom J, Davis P. High-polarized (Delta Psi m(HIGH)) mitochondria are spatially polarized in human oocytes and early embryos in stable subplasmalemmal domains: developmental significance and the concept of vanguard mitochondria. Reprod Biomed Online. 2006;13:246–254. doi: 10.1016/s1472-6483(10)60622-0. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM. Fussy mitochondria fuse in response to stress. EMBO J. 2009;28:1533–1534. doi: 10.1038/emboj.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet. 2011;20:927–940. doi: 10.1093/hmg/ddq531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar VS, Roy N, Liu A, Rajprohat S, Arnold B, Dukes AA, Holbein CD, Berman SB. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol Dis. 2015;74:180–193. doi: 10.1016/j.nbd.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanstone JR, Smith AM, McBride S, Naas T, Holcik M, Antoun G, Harper ME, Michaud J, Sell E, Chakraborty P, Tetreault M, Care4Rare C, Majewski J, Baird S, Boycott KM, Dyment DA, MacKenzie A, Lines MA. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur J Hum Genet. 2016;24:1084–1088. doi: 10.1038/ejhg.2015.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinegoni C, Leon Swisher C, Fumene Feruglio P, Giedt RJ, Rousso DL, Stapleton S, Weissleder R. Real-time high dynamic range laser scanning microscopy. Nat Commun. 2016;7:11077. doi: 10.1038/ncomms11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelz K, Johnston SA, Smith LM, Hall RA, Idnurm A, May RC. ‘Division of labour’ in response to host oxidative burst drives a fatal Cryptococcus gattii outbreak. Nat Commun. 2014;5:5194. doi: 10.1038/ncomms6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vowinckel J, Hartl J, Butler R, Ralser M. MitoLoc: A method for the simultaneous quantification of mitochondrial network morphology and membrane potential in single cells. Mitochondrion. 2015;24:77–86. doi: 10.1016/j.mito.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Wang C, Du W, Su QP, Zhu M, Feng P, Li Y, Zhou Y, Mi N, Zhu Y, Jiang D, Zhang S, Zhang Z, Sun Y, Yu L. Dynamic tubulation of mitochondria drives mitochondrial network formation. Cell Res. 2015;25:1108–1120. doi: 10.1038/cr.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Naruse K, Stamenovic D, Fredberg JJ, Mijailovich SM, Tolic-Norrelykke IM, Polte T, Mannix R, Ingber DE. Mechanical behavior in living cells consistent with the tensegrity model. Proc Natl Acad Sci U S A. 2001;98:7765–7770. doi: 10.1073/pnas.141199598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe W, Shimada T, Matsunaga S, Kurihara D, Fukui K, Shin-Ichi Arimura S, Tsutsumi N, Isobe K, Itoh K. Single-organelle tracking by two-photon conversion. Opt Express. 2007;15:2490–2498. doi: 10.1364/oe.15.002490. [DOI] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- Weaver D, Eisner V, Liu X, Varnai P, Hunyady L, Gross A, Hajnoczky G. Distribution and apoptotic function of outer membrane proteins depend on mitochondrial fusion. Mol Cell. 2014;54:870–878. doi: 10.1016/j.molcel.2014.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R, Jr, Mishra P, Levine B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell. 2017;168:224–238. e210. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta. 2012;1817:1833–1838. doi: 10.1016/j.bbabio.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Westphal D, Kluck RM, Dewson G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014;21:196–205. doi: 10.1038/cdd.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrate LM, Drocco JA, Martin KR, Hlavacek WS, MacKeigan JP. Mitochondrial morphological features are associated with fission and fusion events. PLoS One. 2014;9:e95265. doi: 10.1371/journal.pone.0095265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemerslage L, Lee D. Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters. J Neurosci Methods. 2016;262:56–65. doi: 10.1016/j.jneumeth.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TJ, Slupe AM, Strack S. Cell signaling and mitochondrial dynamics: Implications for neuronal function and neurodegenerative disease. Neurobiol Dis. 2013;51:13–26. doi: 10.1016/j.nbd.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Zhao W, Zhou L, Chang DC. Alpha 5/6 helix domains together with N-terminus determine the apoptotic potency of the Bcl-2 family proteins. Apoptosis. 2016;21:1214–1226. doi: 10.1007/s10495-016-1283-9. [DOI] [PubMed] [Google Scholar]
- Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, Ferguson AN, Kashatus DF, Bao S, Rich JN. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501–510. doi: 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, Saigusa T, Yoshimori T, Sakai Y, Mihara K, Kanki T. Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol. 2016;215:649–665. doi: 10.1083/jcb.201605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Ishii T, Suda H, Akatsuka A, Hartman PS, Goto S, Miyazawa M, Ishii N. Age-related changes of mitochondrial structure and function in Caenorhabditis elegans. Mech Ageing Dev. 2006;127:763–770. doi: 10.1016/j.mad.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, Van der Heiden MG, Yang R, Li F, Locasale JW, Sharfi H, Zhai B, Rodriguez-Mias R, Luithardt H, Cantley LC, Daley GQ, Asara JM, Gygi SP, Wagner G, Liu CF, Yuan J. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146:607–620. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Weaver D, Hajnoczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol. 2004;167:661–672. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zaidi AU, D’Sa-Eipper C, Brenner J, Kuida K, Zheng TS, Flavell RA, Rakic P, Roth KA. Bcl-X(L)-caspase-9 interactions in the developing nervous system: evidence for multiple death pathways. J Neurosci. 2001;21:169–175. doi: 10.1523/JNEUROSCI.21-01-00169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen YB, Hardwick JM, Miller MI, Plachez C, Richards LJ, Yarowsky P, van Zijl P, Mori S. Magnetic resonance diffusion tensor microimaging reveals a role for Bcl-x in brain development and homeostasis. J Neurosci. 2005;25:1881–1888. doi: 10.1523/JNEUROSCI.4129-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Iqbal S, O’Leary MF, Menzies KJ, Saleem A, Ding S, Hood DA. Altered mitochondrial morphology and defective protein import reveal novel roles for Bax and/or Bak in skeletal muscle. Am J Physiol Cell Physiol. 2013;305:C502–511. doi: 10.1152/ajpcell.00058.2013. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SG, Lapolla SM, Hays FA, Ding J, He F, Zhang XC, Li J, Senes A, Andrews DW, Lin J. BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 2016;35:208–236. doi: 10.15252/embj.201591552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Anderson RE, Agbaga MP, Rucker EB, 3rd, Le YZ. Loss of BCL-XL in rod photoreceptors: Increased susceptibility to bright light stress. Invest Ophthalmol Vis Sci. 2006;47:5583–5589. doi: 10.1167/iovs.06-0163. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–1113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsmaier KE, Babic M, Russo GJ. Mitochondrial transport dynamics in axons and dendrites. Results Probl Cell Differ. 2009;48:107–139. doi: 10.1007/400_2009_20. [DOI] [PubMed] [Google Scholar]