

Abstract
Purpose
A major mechanism of castration-resistant prostate cancer (CRPC) involves intratumoral biosynthesis of dihydrotestosterone (DHT) from adrenal precursors. We have previously shown that adrenal-derived androstenedione (AD) is the preferred substrate over testosterone (T) for 5α-reductase expressed in metastatic CRPC, bypassing T as an obligate precursor to DHT. However, the metabolic pathway of adrenal-derived DHT biosynthesis has not been rigorously investigated in the setting of primary disease in the prostate.
Experimental Design
Seventeen patients with clinically localized prostate cancer were consented for fresh tissues after radical prostatectomy. Prostate tissues were cultured ex vivo in media spiked with an equimolar mixture of AD and T, and stable isotopic tracing was employed to simultaneously follow the enzymatic conversion of both precursor steroids into nascent metabolites, detected by liquid chromatography-tandem mass spectrometry. CRPC cell line models and xenograft tissues were similarly assayed for comparative analysis. A tritium-labeled steroid radiotracing approach was used to validate our findings.
Results
Prostatectomy tissues readily 5α-reduced both T and AD. Furthermore, 5α-reduction of AD was the major directionality of metabolic flux to DHT. However, AD and T were comparably metabolized by 5α-reductase in primary prostate tissues, contrasting the preference exhibited by CRPC in which AD was favored over T. 5α-reductase inhibitors effectively blocked the conversion of AD to DHT.
Conclusions
Both AD and T are substrates of 5α-reductase in prostatectomy tissues, resulting in two distinctly non-redundant metabolic pathways to DHT. Furthermore, the transition to CRPC may coincide with a metabolic switch towards AD as the favored substrate.
Keywords: androgen metabolism, localized prostate cancer, 5α-reductase, adrenal steroids, prostatectomy
Introduction
Androgen deprivation therapy by medical or surgical castration is a mainstay of treatment for advanced prostate cancer. The primary action of androgen deprivation is attributable to the disruption of androgen signaling by depleting gonadal testosterone (T), a modest androgen receptor (AR) agonist and canonical precursor to the more potent agonist dihydrotestosterone (DHT) (1). Although initially effective, androgen deprivation therapy is invariably followed by the onset of castration-resistant prostate cancer (CRPC). CRPC is largely typified by an inappropriate restoration of the AR signaling axis (2–4), and in this setting, intratumoral biosynthesis of DHT from adrenal precursor steroids plays a major role to sustain AR activation (4–6). Thus, a detailed interrogation of androgen metabolism and enzymology is imperative towards understanding the pathophysiology of this disease.
In the absence of gonadal T, one route of DHT biosynthesis involves enzymatic modification of dehydroepiandrosterone/dehydroepiandrosterone sulfate (DHEA/DHEA-S), a primarily adrenal-derived androgen and the most abundant 19-carbon steroid in serum (5,7). Three enzymatic steps are required to convert DHEA to DHT, consisting of 1) 3β-hydroxyl oxidation and Δ5→Δ4 isomerization by 3β-hydroxysteroid dehydrogenase/isomerase (3β-HSD), 2) 17-keto reduction by 17β-hydroxysteroid dehydrogenase (17β-HSD), and 3) 5α-reduction by 5α-reductase (7,8). In normal male physiology, this sequence chiefly occurs within the testes and first requires the conversion of DHEA to its Δ4-3-keto derivative androstenedione (AD), which is subsequently 17-keto reduced to T (8). T is then secreted into circulation, where it may undergo irreversible 5α-reduction to DHT in the prostate (Figure 1a, left). This pathway (DHEA→AD→T→DHT) was also surmised to be the metabolic route of DHT biosynthesis from circulating DHEA by CRPC, in which T serves as an obligate precursor. Notably though, AD and T share the same Δ4-3-keto structure that is amenable to 5α-reduction (9,10). We have previously shown that AD is the preferred substrate relative to T for 5α-reductase expressed in CRPC (11). This results in generation of a 5α-reduced metabolite, 5α-androstanedione (5α-dione), which can be 17-keto reduced to DHT to bypass T via a different metabolic route (Figure 1a, right). Surprisingly, this “5α-dione pathway” (DHEA→AD→5α-dione→DHT) appears to be the predominant directionality of DHEA metabolism in the majority of metastatic CRPC cell lines, as well as in sampled metastatic CRPC patient tissues (11–14). We therefore sought to determine whether the same metabolic phenotype is operational in the eugonadal setting of clinically localized prostate cancer, where both gonadal T and adrenal steroids are potential precursors of DHT.
Figure 1.
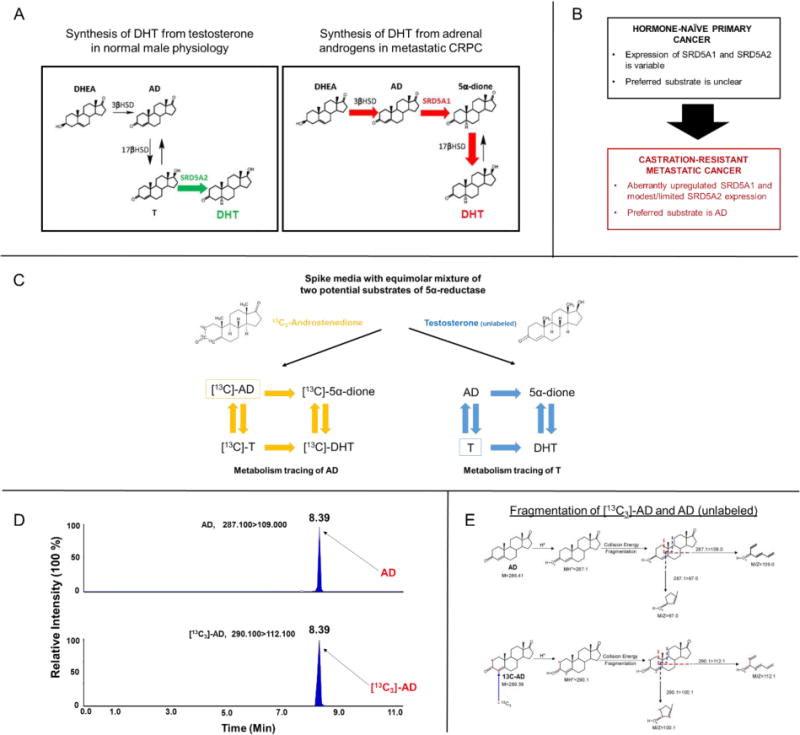
A mixed substrate approach to delineate multiple potential pathways of steroid metabolism in primary prostate tissues. A) The 5α-dione pathway bypasses T as an obligate precursor in metastatic CRPC. In normal male physiology, DHT biosynthesis first involves gonadal conversion of DHEA to T, which is subsequently 5α-reduced by the prostate (“conventional” pathway; left), primarily via 5α-reductase isoenzyme-2 (SRD5A2). In contrast, DHT biosynthesis from adrenal DHEA by CRPC likewise requires conversion to AD, but AD is 5α-reduced to 5α-dione via 5α-reductase isoenzyme-1 (SRD5A1), which is then 17-keto reduced to DHT (5α-dione pathway; right). Enzymatic steps occurring in target tissues are denoted by colored arrows. B) Expression of the two 5α-reductase isoenzymes differs between primary disease and CRPC. Although AD is the preferred substrate of 5α-reductase in CRPC, the preferred substrate and directionality of adrenal steroid metabolism in primary prostate tissues remains unclear. C-E) 13C3 labeling enables precise determination and comparison of metabolic flux from two potential substrates of 5α-reductase. Metabolites from the 13C3 isotopologue of AD can be differentiated from that of unlabeled T based on enrichment of an M+3 (13C3) fragment detected by mass spectrometry.
A number of studies have previously attempted to characterize androgen synthesis in clinically localized disease. These efforts have relied mostly on steroidogenic enzyme expression to infer patterns of metabolism or sampling of androgen content in frozen tissues, which provides a snapshot at one point in time (15–21). These methods yield informative observations but cannot definitively ascertain the directionality of metabolic flux from precursors into their respective metabolites. Thus, the true metabolic pathway of adrenal steroid metabolism in primary prostatic tissue remains unclear. Stable isotopes are valuable in the study of metabolism because isotopic tagging of a substrate permits direct identification of isotope-labeled enzymatic products over time, thus offering unambiguous and temporally-encoded information on pathway accessibility (22). Here, we report on a novel approach to assay androgen metabolism ex vivo in human prostatectomy tissues harboring localized cancer. Using liquid chromatography-mass spectrometry (LC-MS/MS) and capitalizing on stable isotopic tracing to follow the concurrent metabolism of two potential precursor steroids of DHT under mixed substrate conditions, our method demonstrates that primary prostate tissues harboring cancer metabolically utilize both AD and T as substrates of 5α-reductase. Consistent with observations in CRPC, the 5α-dione pathway appears to be the major directionality of flux from adrenal steroids to DHT in this context. However, AD and T appear to be comparably metabolized by 5α-reductase in primary prostate tissues, contrasting the preference exhibited by CRPC in which AD is clearly favored over T. This finding provides direct evidence that the transition of eugonadal primary disease to eventual CRPC may coincide with a metabolic switch towards AD as the preferred substrate of 5α-reductase to thereby augment adrenal steroid utilization in the biosynthesis of DHT and enable therapeutic resistance.
Materials and Methods
Cells and culture conditions
Human prostate cancer cell lines LNCaP and 22Rv1 were purchased from American Type Culture Collection (ATCC) and maintained in RPMI 1640 with 10% fetal bovine serum (FBS). VCaP and DU145 cells were purchased from ATCC and maintained in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS. MDA-PCa-2b cells were purchased from ATCC and cultured in BRFF-HPC1 (Athena ES) containing 20% FBS. RWPE-1 cells were obtained from ATCC and maintained in Keratinocyte Growth Medium with 0.05 mg/ml bovine pituitary extract and 5 ng/ml human recombinant epidermal growth factor according to ATCC recommendations. LAPC4 cells were generously provided by Charles Sawyers (Memorial Sloan Kettering Cancer Center, New York, NY) and were cultured in Iscove’s Modified Dulbecco’s Medium (IMEM) with 10% FBS. C4-2 cells were generously provided by J. T. Hsieh (University of Texas Southwestern, Dallas, TX) and maintained in 10% FBS. All cell lines were incubated in a 5% CO2 humidified incubator and 1% penicillin/streptomycin (P/S) was added to all culture media. FBS and charcoal:dextran stripped FBS were purchased from Gemini Bio Products. All cell lines were authenticated prior to use and routinely tested for mycoplasma contamination.
Human primary prostate tissue procurement
Institutional review board approval and patient written informed consent was obtained at the Cleveland Clinic prior to procurement of de-identified human tissue samples from surgical specimens of study participants undergoing radical prostatectomy for clinically localized prostate cancer. Following surgical removal of the prostate, the fresh surgical specimen was transported to pathology for processing. One 5 mm diameter core (approximately 0.03–0.10 g) was taken by punch biopsy from the peripheral and transition zone of the whole gland specimen, placed in separate sterile containers on ice, and brought to the laboratory by tissue procurement personnel for immediate ex vivo experimentation. The remaining prostate gland was then fixed in formalin and paraffin-embedded, slide-mounted, and assessed by the collaborating expert prostate cancer pathologist (CMG). The region surrounding the site of the core biopsy was reviewed by the pathologist for any identifiable prostate cancer. Clincopathologic data of study patients were retrospectively obtained by review of the electronic medical record.
Generation of mouse xenografts
Mouse studies were performed under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the Cleveland Clinic Lerner Research Institute (protocol number 2015–1549). NSG male mice (6–8 weeks old) were purchased from the Jackson Laboratory. A total of 107 LNCaP cells (re-suspended in 50 μl of matrix gel) were subcutaneously injected into mice (n=3). As previously described, mice were then surgically orchiectomized and implanted with DHEA pellets to mimic human adrenal DHEA production in patients with CRPC (11,13,23). When tumors reached 300–500 mm3, mice were sacrificed for harvesting of tumor tissue.
Steroid metabolism assays
Cell line metabolism
Cells were seeded into 12-well plates at 400,000 cells/well in triplicate and incubated overnight in respective phenol red-free media containing 10% charcoal:dextran stripped FBS and 1% P/S prior to the experiment. LAPC-4 cells and MDA-PCa-2b cells were seeded specifically on plates coated with poly-DL-ornithine (Sigma-Aldrich). On the day of the experiment, phenol red-free, serum-free media was spiked with 1 μM of 13C3-[2,3,4]-androstenedione (Cerilliant, Round Rock, Texas) and 1 μM of unlabeled testosterone (Steraloids, Newport, RI) dissolved in 100% ethanol. Cells were incubated in 1 ml of the steroid-spiked media over 48 hours, with aliquots of media obtained at indicated times. Samples were subsequently treated with 1,000 units of β-glucuronidase (Helix Pomatia, Sigma-Aldrich) at 37° C in a 5% CO2 incubator for 2 hours to de-conjugate all steroids.
Primary prostate tissue metabolism
Following procurement, peripheral and transition zone tissue samples were grossly minced and manually homogenized using razor blades in a 10 cm petri dish under sterile conditions and divided into four replicates per core of approximately equal weight. Given limited tissue availability, more replicates could not be performed. Each replicate was transferred to a 12-well plate, and two replicates were subsequently incubated in 1 ml of phenol red-free, serum-free DMEM with 1% P/S spiked with 1 μM of 13C3-[2,3,4]-androstenedione and 1 μM of testosterone dissolved in 100% ethanol, while the other two replicates were incubated in 1 ml of serum-free DMEM spiked with 1 μM of 13C3-[2,3,4]-androstenedione alone. Samples were incubated at 37° C in a 5% CO2 humidified incubator for 48 hours. The feasibility of culturing primary prostate tissues ex vivo and longevity of measurable enzyme activity during a time course experiment was previously established (24). Aliquots of media were obtained at indicated times and frozen at −20° C until the day of sample analysis.
Xenograft tumor metabolism
After removal of necrotic regions, xenograft tumor tissues (n=3, 0.4–0.5 g each) were pooled, grossly minced, and manually homogenized using scissors in a 10 cm petri dish under sterile conditions and divided into triplicates. Each replicate was transferred to a 12-well plate and incubated in serum-free media spiked with 1 μM of 13C3-[2,3,4]-androstenedione and 1 μM of testosterone or 13C3-[2,3,4]-androstenedione alone as previously described, with aliquots of media obtained at indicated times. Samples were subsequently treated with β-glucuronidase and analyzed as previously described.
LC-MS/MS analysis of steroid content in media
Extraction protocol
Aliquot samples were thawed from storage at −20° C and subjected to centrifugation at 5000 RPM for 5 minutes at 4° C to spin down insoluble debris, after which an equal volume (200 μl) of media supernatant from each aliquot was transferred to a new 1.5 ml Eppendorf tube. Samples were spiked with 20 μl of internal standard (50 ng/ml D4-[9,11,12,12]-cortisol; [D4]-cortisol; CDN Isotopes, Point-Claire, Quebec, Canada), briefly vortexed, and transferred to glass test tubes for liquid-liquid extraction. After addition of 2 ml of methyl-tert-butyl ether (MTBE, Across), glass tubes were vortexed for 5 minutes using a multi-tube vortexer (Fisher) and centrifuged for 5 minutes at 2500 RPM at 4°C. Samples were placed on dry ice for 20 minutes, and the top organic solvent with extracted steroid was transferred to a new glass tube. This fraction was evaporated under nitrogen gas at 40° C and then reconstituted in 110 μl of 50% methanol/water (v/v) and briefly vortexed again. The reconstituted samples were transferred into 1.5ml microcentrifuge tubes, centrifuged at 13,000 RPM for 10 mins at 4° C, and the supernatants were collected for mass spectrometry injections in high-performance liquid chromatography (HPLC) vials. To analyze glucuronidated steroids separately, the aqueous phase of the liquid-liquid extraction was separately collected, washed twice with MTBE extraction again to remove residual free steroids, and subjected to an overnight dry period to evaporate remaining MTBE. Samples were treated with β-glucuronidase as previously described. The samples were analyzed by LC-MS/MS to calculate the conjugated fraction of steroid metabolites.
Instrumentation and data analysis
Extracted steroids from samples were quantified using LC/MS/MS. Briefly, the LC-MS/MS system consists of an ultra-pressure liquid chromatography system (UPLC; Shimadzu Corporation, Japan), comprising of two LC-30AD pumps, a DGU-20A5R vacuum degasser, a CTO-30A column oven, SIL-30AC autosampler, and a system controller CBM-20A and coupled with a Qtrap 5500 mass spectrometer (AB Sciex, Redwood City, CA). Extracted steroids were injected onto the Shimadzu UPLC system, and the steroids were separated through a C18 column (Zorbax Eclipse Plus C18 column 150 mm × 2.1 mm, 3.5 μm, Agilent, Santa Clara, CA) using a gradient starting from 20% solvent B (acetonitrile/methanol [90/10, v/v] containing 0.2% formic acid) over 4 min and then to 65% solvent B over 15 min, followed by 90% solvent B for 3 min. Steroids were quantified on a Qtrap 5500 mass spectrometer using ESI in positive ion mode and multiple reaction monitoring (MRM) using characteristic parent → daughter ion transitions for specific molecular species monitored. D4-[9,11,12,12]-cortisol was used as an internal standard for calibration of steroids in each sample. Data acquisition and processing were performed using Analyst® software (AB Sciex, version 1.6.2). The peak area ratio of the analyte over the internal standard was used for quantification purposes. Each sample run included calibration curves with standards for data quantification using the analyte/internal standard peak area ratio. The specific transitions for molecular species were as follows: 13C3-[2,3,4]-androstenedione [290.1>112.1], androstenedione [287.1>109.0], 13C3-[2,3,4]-testosterone [292.1>112.1], testosterone [289.1>109.0], 13C3-[2,3,4]-5α-dione [292.2>274.2], 5α-dione [289.2>271.2], 13C3-[2,3,4]-DHT [294.1>162.1], DHT [291.1>255.2].
To calculate the rate of metabolic flux, a baseline precursor steroid concentration was first obtained from a sample draw of media spiked with 1 μM of 13C3-[2,3,4]-androstenedione and 1 μM of testosterone prior to tissue incubation. Steroid metabolite concentrations in media samples drawn at different time points of tissue incubation were normalized against this initial precursor concentration to derive a percent conversion value. Duplicate values for each target metabolite were averaged, and the SD was determined for each individual patient sample. For pooled analyses across the entire patient cohort, target metabolite concentrations for individual patient samples were averaged, and SEM was reported.
Separate validation of steroid content in media by radiotracing analysis
Primary prostate tissue metabolism by radiotracing
Following procurement, tissues were minced, homogenized, and transferred into a 12-well plate in triplicate, as previously described. Samples were incubated in serum-free DMEM spiked with [3H]-androstenedione dissolved in 100% ethanol (300,000–600,000 cpm, PerkinElmer) and 100 nM of unlabeled androstenedione (Steraloids) at 37° C in a 5% CO2 incubator over 48 hours. Aliquots of media were obtained, stored, and treated with β-glucuronidase, as previously described.
Steroid extraction, separation, and quantitation by HPLC
Steroids were extracted with 1:1 ethyl acetate:isooctane, and concentrated by evaporation of the solvent under nitrogen gas in glass tubes. Dried samples were reconstituted in 50% methanol and injected on a Breeze 1525 system equipped with a model 717 plus autoinjector (Waters Corp.) and a Kinetix 100 × 2.1-mm, 2.6 μM C18 reverse-phase column (Phenomenex) and methanol/water gradients at 30° C. The column effluent was analyzed using a β-RAM model 3 in-line radioactivity detector (IN/US Systems, Inc.) using Liquiscint scintillation mixture (National Diagnostics). Steroid metabolites were identified using tritiated standards of AD, T, DHEA, and DHT (PerkinElmer). The expected retention time for 5α-dione was previously validated by synthesizing tritium-labeled 5α-dione (11).
Results
Patient characteristics and case selection
Seventeen eugonadal patients presenting with clinically localized prostate cancer were consented for fresh tissue collection and subsequently underwent open or robotic-assisted radical prostatectomy. The first 12 samples were analyzed by LC-MS/MS, while the last 5 samples were used for independent validation through a radiotracing method. Clinicopathologic features of patients are reported (Table 1). The majority of patients had organ-confined disease, although final pathology revealed 3/17 patients with nodal metastases discovered upon pelvic lymph node dissection. Medication lists were also obtained from electronic medical records to determine whether any patients were on treatment with 5α-reductase inhibitors (5-ARI). Two patients were documented to be on finasteride and one patient on dutasteride at the time of surgery. Although these patients were included in the final patient series, data points from these cases are separately reported and were excluded from all pooled analyses to avoid any potential confounding from perturbed metabolism. Pathology slides were also assessed to determine whether sample cores obtained were involved with cancer. If the tissue surrounding the original core biopsy was found to harbor prostate cancer on review of histology, the core was designated as “possible cancer”. In total, 10/17 peripheral zone cores and 4/17 transition zone cores were deemed to potentially involve cancerous tissue, while the rest contained non-neoplastic tissue (Table 1).
Table 1.
Patient characteristics
| Patient # | Age at diagnosis | Pathologic Staging | Gleason Score | 5-ARI Status | PZ Location | PZ Pathology | TZ Location | TZ Pathology |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| 29 | 63 | pT3bN1Mx | 4+4 | N/A | L Basal | Negative | R Basal | Negative |
| 30 | 56 | pT3aN1Mx | 3+4 | N/A | L Apical | Possible | R Apical | Possible |
| 31 | 60 | pT2cN0Mx | 3+3 | N/A | R Basal | Possible | L Basal | Negative |
| 32* | 67 | pT3bN1Mx | 4+5 | Dutasteride | R Basal | Possible | L Apical | Negative |
| 33* | 68 | pT2cN0Mx | 4+3 | Finasteride | L Apical | Negative | R Basal | Negative |
| 34 | 65 | pT3bN0Mx | 3+4 | N/A | R Basal | Unavailable | L Basal | Unavailable |
| 35 | 60 | pT2cNxMx | 3+4 | N/A | R Apical | Negative | L Apical | Negative |
| 36 | 66 | pT2cN0Mx | 4+3 | N/A | R Basal | Possible | R Basal | Negative |
| 37* | 72 | pT2cN0Mx | 4+3 | Finasteride | L Apical | Negative | R Apical | Negative |
| 38 | 58 | pT2cN0Mx | 3+4 | N/A | L Apical | Possible | R Basal | Negative |
| 39 | 65 | pT2cN0Mx | 3+4 | N/A | L Apical | Possible | R Apical | Negative |
| 40 | 52 | pT3aN0Mx | 3+4 | N/A | R Apical | Unavailable | L Basal | Possible |
| 114 | 56 | pT2cN0Mx | 3+4 | N/A | L Apical | Possible | L Basal | Negative |
| 115 | 54 | pT3aNxMx | 3+4 | N/A | L Basal | Negative | L Basal | Possible |
| 116 | 67 | pT3bN0Mx | 4+5 | N/A | L Basal | Possible | R Basal | Negative |
| 117 | 61 | pT3aN0Mx | 4+5 | N/A | L Basal | Possible | L Basal | Negative |
| 118 | 43 | pT3aNxMx | 4+4 | N/A | R Basal | Possible | L Basal | Possible |
PZ = peripheral zone, TZ = transition zone
Patients who were determined to be on a 5-ARI at time of surgery (n=3) were excluded from all pooled analyses (cases are denoted by “*”).
Both androstenedione and testosterone are substrates of 5α-reductase for the biosynthesis of DHT by human primary prostate tissue samples
In order to simultaneously monitor the metabolism of both AD and T, which may compete for 5α-reductase and share common downstream metabolites, we employed a stable isotopic labeling method to definitively distinguish the enzymatic conversion of each Δ4-3-keto precursor steroid by prostatic tissue into their respective steroid products. To do this, prostate tissue samples were incubated in media spiked with an equimolar mixture of an 13C isotopologue of androstenedione (13C3-[2,3,4]-androstenedione; [13C3]-AD) and unlabeled T (Tunl). The generation of nascent metabolites in media was followed over time using LC-MS/MS. The origin of these metabolites was determined based on enrichment or absence of an incorporated M+3 13C3 fragment on multiple reaction monitoring (MRM) transitions (Figure 1c–e). To confirm the validity of this approach, we verified the concordance of our measurements with a previously validated radiotracing method (11) in three different prostate cancer cell lines (LAPC4, C4-2, DU145) (Figure S1).
Metabolic tracing revealed that the majority of [13C3]-AD and Tunl was enzymatically converted by primary prostate tissues to downstream metabolites within 48 hours. The principal metabolite that was detected arising from [13C3]-AD was [13C3]-5α-dione (Figure 2a), whereas Tunl was converted to DHTunl as well as ADunl to a lesser degree (Figure 2b). Furthermore, [13C3]-DHT originating from [13C3]-AD was detected in media as early as 7 hours of incubation in all incubated samples and was more abundant than [13C3]-T throughout the time course experiment (Figure 2c). We additionally confirmed these findings via radiotracing experiments (Figure 2d–e). Following treatment of prostate tissue samples with [3H]-AD, both [3H]-5α-dione and [3H]-DHT were successfully separated by HPLC and identified via radio flow detector. Notably, no [3H]-T signal was detectable (Figure 2f). Taken together, these findings suggest that the 5α-dione pathway is likely the preferred metabolic route to DHT from the adrenal source. Interestingly, we did not observe a difference in 5α-dione pathway utilization between tissue samples possibly harboring cancer and non-neoplastic samples (Figure 3). Thus, the 5α-dione pathway of DHT biosynthesis from adrenal steroids is probably the metabolic phenotype of prostatic tissue under normal physiologic conditions.
Figure 2.
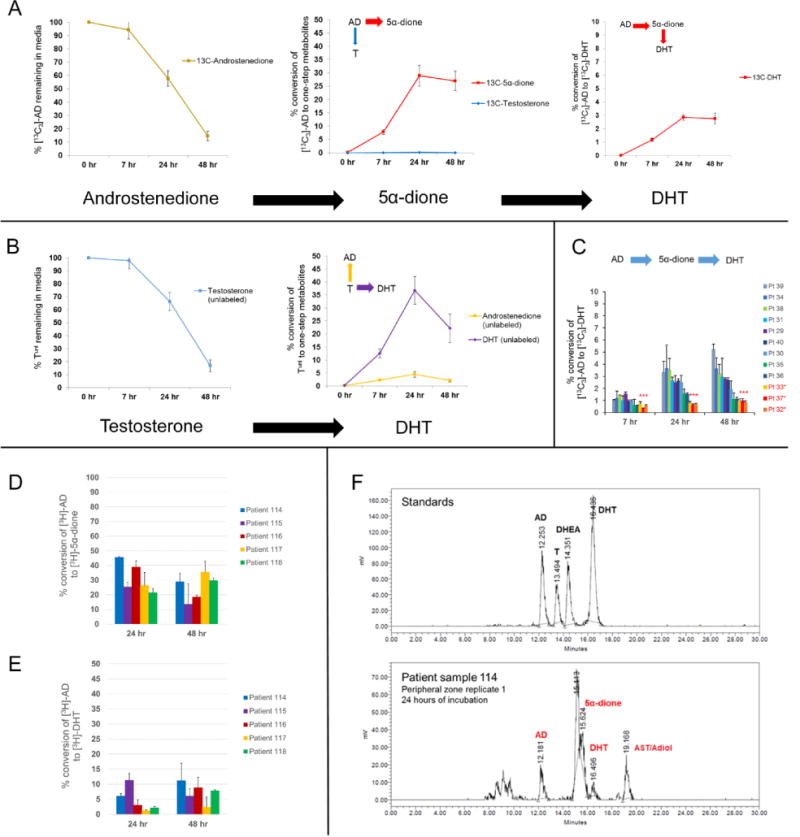
The 5α-dione pathway is the major route of DHT biosynthesis from the adrenal source in primary prostatic tissue. A) Metabolic tracing experiments indicate that [13C3]-AD is predominantly converted to [13C3]-5α-dione by 5α-reductase rather than to [13C3]-T by 17β-HSD. B) Tunl primarily undergoes 5α-reduction to DHTunl, as well as 17β-oxidation to ADunl to a lesser degree. Peripheral zone samples (n=9 patients) were incubated in an equimolar (1 μM) mixture of [13C3]-AD and Tunl in duplicate per patient sample. The rate of metabolic flux is reported as % conversion of original precursor, which was determined by averaging free steroid metabolite concentrations at each time point and normalizing to the concentration of precursor steroid initially spiked in media. Error bars represent the SEM across patients. C) [13C3]-DHT is observed in all patient samples after treatment with [13C3]-AD, although minimal conversion was observed in patients on a 5-ARI (denoted by red asterisks). Error bars denote the SD of patient sample duplicates. D-E) A previously validated radiotracing method confirms that [3H]-AD is converted to [3H]-5α-dione and subsequently to [3H]-DHT in five independent patient samples. Error bars indicate the SD across three replicates for each patient sample. F) A representative example is shown of [3H] peak signals obtained by HPLC indicating robust 5α-dione formation and minimal detection of T after 24 hours of incubation.
Figure 3.
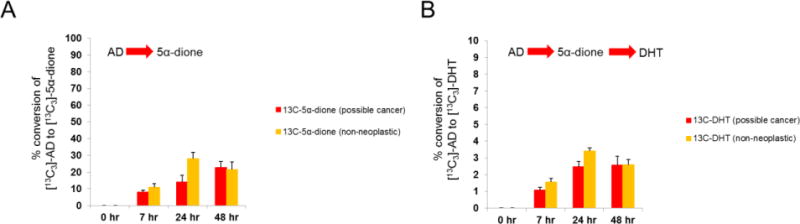
A comparison of samples possibly involved with cancer (n=5 peripheral zone samples, n=2 transition zone samples; total n=7) and confirmed non-neoplastic samples (n=4 peripheral zone samples, n=6 transitional zone samples; total n=10) reveals minimal consistent differences in 5α-dione pathway accessibility between the two groups. A and B represent the amount of [13C3]-5α-dione and [13C3]-DHT detected over 48 hours, respectively. Error bars represent the SEM across patients.
5α-reductase inhibitors restrict DHT biosynthesis from both androstenedione and testosterone in primary prostate tissue samples cultured ex vivo
To investigate whether the pharmacologic effect of orally administered 5-ARIs in patients is appreciable in primary prostate tissues cultured ex vivo several hours after radical prostatectomy, we similarly incubated tissue samples from three patients confirmed to be on a 5-ARI at time of surgery. Following treatment of these tissues with [13C3]-AD and Tunl, blockade of both [13C3]-AD→[13C3]-5α-dione and Tunl→DHTunl was observed, consistent with the expected inhibitory effect of a 5-ARI (Figure 4a–b). Furthermore, inhibition of 5-reductase enhanced the conversion of Tunl→ADunl, which subsequently accumulated in media. However, in contrast to total blockade of Tunl→DHTunl, [13C3]-AD→[13C3]-5α-dione conversion was reduced but not completely restricted, leading to residual [13C3]-DHT formation (Figure 4c–d). Furthermore, despite accumulation of [13C3]-AD from partial blockade of 5-reductase, only a marginal increase in alternative flux from [13C3]-AD to [13C3]-T was observed (Figure 4e), suggesting that 17-keto reduction of AD→T is overall limited in prostatic tissue. This observation, as well as the fact that AD can be converted to DHT despite total pharmacologic blockade of T→DHT, provides further evidence that the primary metabolic pathway from adrenal steroids to DHT may not require conversion of AD→T→DHT but rather involves the 5α-dione pathway. These findings also indicate that the dominantly accumulating Δ4-3-keto precursor steroid from blockade of 5α-reductase is AD rather than T, given the propensity of T to be oxidized back to AD in this context.
Figure 4.
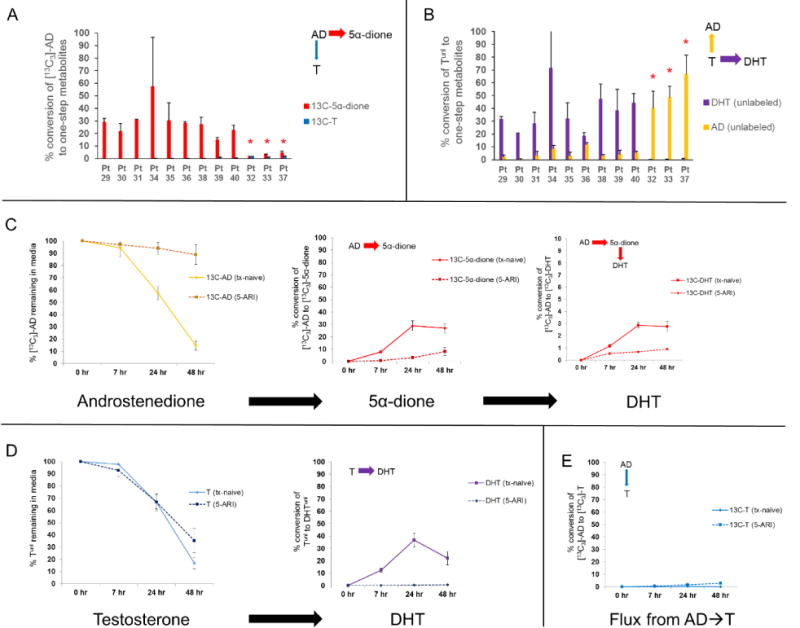
The pattern of observed metabolic flux differs between samples from patients on a 5-ARI at time of surgery (n=3) versus patients who were treatment-naïve (n=9). A) [13C3]-5α-dione is the predominant metabolite arising from [13C3]-AD in all patients at 24 hours of incubation. However, formation of [13C3]-5α-dione is notably reduced in patients confirmed to be on a 5-ARI at the time of surgery (denoted by red asterisks). B) DHTunl is the predominant metabolite arising from Tunl at 24 hours except in patients on a 5-ARI, in which case Tunl was rapidly converted to ADunl instead (red asterisks). C-D) Although no Tunl→DHTunl conversion was observed in any samples from patients on a 5-ARI, residual [13C3]-AD→[13C3]-5α-dione→[13C3]-DHT conversion remained. E) Only a modest increase in alternative metabolic flux of [13C3]-AD to [13C3]-T is observed despite pharmacologic inhibition of 5α-reduction and accumulation of [13C3]-AD, indicating that 17-keto reduction of AD is overall limited in prostatic tissue. Error bars represent the SEM.
The preferred substrate for 5α-reductase differs between primary prostatic tissue and metastatic CRPC
Since AD was observed to undergo relatively robust 5α-reduction, we also sought to determine whether AD is favored over T by 5α-reductase in primary prostate tissues. The conversion efficiency of [13C3]-AD→[13C3]-5α-dione was thus compared against Tunl→DHTunl under equimolar mixed substrate conditions, permitting a quantitative assessment of precursor utilization by 5α-reductase within the same biological system. Although AD was capably 5α-reduced, T appeared to be a comparable substrate in primary prostate tissue samples (Figure 5a). Since the 5α-reduced products of AD and T can be further metabolized by 17β-HSD via a second-step reaction to potentially confound conclusions drawn from tracking only the immediate metabolite, a secondary analysis was also performed by totaling the signal intensity from both the immediate 5α-reduced product and subsequent 5α-reduced, additionally C17-modified metabolite (Figure 5b). This comparison further suggested that T is a comparable, if not slightly preferred, substrate to AD in primary prostate tissue samples. We also considered the possibility that conjugation of DHT by uridine 5′-diphospho-glucuronosyltransferase (UGT) enzymes at its 17-position might potentially bias the ratio of free [13C3]-5α-dione/DHTunl detected by our method. To exclude this, we separately analyzed the conjugated steroid content in media but found no evidence of UGT activity in incubated primary prostate tissues (Figure S2). This observation of 5α-reductase slightly favoring T was generally consistent across individual malignant and non-neoplastic samples, as reflected by a [13C3]-5α-dione/DHTunl generation ratio of <1 (Figure 5c–d).
Figure 5.
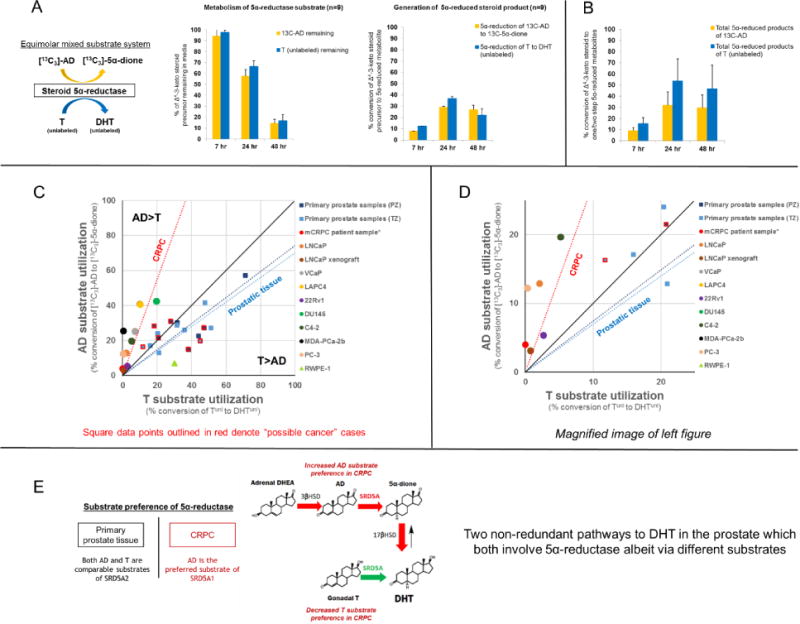
The preferred substrate of 5α-reductase differs between primary prostatic tissue and metastatic CRPC. A) A comparison of 5α-reduction efficiency of [13C3]-AD→[13C3]-5α-dione versus Tunl→DHTunl in a mixed substrate system enables direct characterization of substrate preference by 5α-reductase. [13C3]-AD and Tunl are 5α-reduced at comparable rates (n=9 patients). B) A secondary summative comparison of one- and two-step 5α-reduced metabolites of [13C3]-AD ([13C3]-5α-dione + [13C3]-DHT) versus Tunl (DHTunl + 5α-dioneunl) suggests that T is a comparable, if not slightly preferred, substrate to AD in primary tissues. Error bars represent the SEM. C) T versus AD utilization is depicted for each individual patient sample, where the x-axis and y-axis reflect rates of Tunl→DHTunl and [13C3]-AD→[13C3]-5α-dione conversion over 24 hours, respectively, and the black line indicates hypothetical equal proclivity of 5α-reductase for both substrates (slope = 1). 5α-reductase in primary tissues may exhibit a slight preference for T over AD, indicated by a linear fit across samples with a slope <1 (dark blue = peripheral zone [PZ]; light blue = transition zone [TZ]). Square data points outlined in red denote “possible cancer” cases. In contrast, AD is clearly preferred in 8 tested CRPC cell lines (average of biological triplicates), LNCaP xenograft tissue (average of biological triplicates), and tissue from a CRPC patient (data point obtained previously; Chang et al. PNAS 2011), as indicated by a linear fit with slope >1 (red). The triangular data point indicates RWPE-1, a benign immortalized prostate epithelial cell line. D) A magnification of the left plot is provided. E) In the eugonadal state, conversion of either gonadal T or adrenal DHEA into DHT, although non-redundant and sequentially distinct, must both engage 5α-reductase, albeit via different substrates. A shift towards AD as the preferred substrate likely reflects an increase in efficiency of adrenal steroid utilization for DHT biosynthesis.
Remarkably, this phenotype in primary prostate tissues contrasts with that of the majority of human CRPC cell lines. Our prior radiotracing experiments have shown in CRPC that AD is 5α-reduced more expeditiously than T (11). To directly compare AD and T utilization by 5α-reductase between primary prostate tissues and CRPC, we re-applied our mixed substrate tracing method using 8 available CRPC cell lines, LNCaP xenograft tumor tissue, as well as an immortalized benign prostate epithelial cell line (RWPE-1). In all CRPC cell lines and xenograft tissue, [13C3]-AD→[13C3]-5α-dione was more efficient than Tunl→DHTunl ([13C3]-5α-dione/DHTunl generation ratio of >1), confirming the clear preference for AD over T by 5α-reductase expressed in CRPC (Figure 5c–d). This metabolic pattern is also consistent with observations in tissues previously obtained from a metastatic CRPC patient, suggesting that this difference is not simply experimental artifact (11). Interestingly, the substrate preference of RWPE-1 aligned more closely with primary prostate tissue samples, supporting the notion that T may be the preferred substrate in non-neoplastic prostatic tissue. Overall, these findings indicate that the preference of Δ4-3-keto steroid utilization for DHT biosynthesis in metastatic CRPC differs from that of primary prostatic tissue.
Androstenedione and testosterone do not exhibit substrate competition for 5α-reductase in prostate tissue samples cultured ex vivo
Since AD and T are potentially competing substrates of 5α-reductase, we also considered the possibility that the enzyme kinetics observed under mixed substrate conditions might differ from that of mono-substrate conditions. To investigate potential effects of mixing alternative substrates, two conditions were established in which prostate tissues were incubated with [13C3]-AD, with or without an equimolar concentration of Tunl. However, no appreciable difference was observed in the rate of [13C3]-AD metabolism when Tunl was added to the substrate mix (Figure S3). The directionality of metabolic flux from AD is thus unlikely to be affected by competition for 5α-reductase under multiple substrate conditions in prostate tissues.
Discussion
Despite castrate levels of serum T after androgen deprivation therapy, intraprostatic androgen levels generally remain incompletely depleted, even prior to the onset of CRPC (25–28). Furthermore, recent studies strongly suggest that targeting the adrenal contribution in castration-naïve prostate cancer is a viable and effective therapeutic strategy (29,30). Although the modification of circulating adrenal steroids has long been recognized as a putative source of intraprostatic DHT requiring only a limited complement of steroidogenic enzymes expressed in the prostate (8,26), this metabolic pathway has not been rigorously defined. In this study, we show that the 5α-dione pathway, which was previously determined to be the major metabolic pathway to DHT from circulating adrenal steroids in metastatic CRPC, is also the predominant sequence occurring in primary prostate tissues from eugonadal patients with clinically localized disease. Notably, this pathway circumvents T as a precursor, resulting in two different and distinctly non-redundant metabolic routes to derive DHT via circulating steroids present in a eugonadal state (gonadal T→DHT, versus adrenal DHEA→AD→5α-dione→DHT). This observation deviates from the general view that 5α-reduction of T is the principal enzymatic step to generate intraprostatic DHT, regardless of the source (8). The absence of an appreciable difference in metabolism between non-neoplastic and cancerous samples in our study further suggests that this pattern is probably the metabolic phenotype assumed by physiologic prostatic tissue in the eugonadal setting.
While the 5α-dione pathway appears to be the consistent directionality of adrenal steroid metabolism in both primary tissue and CRPC, we also observe that AD and T are comparable substrates of 5α-reductase in prostatic tissue, juxtaposing findings in CRPC where AD is the clearly preferred substrate. This is noteworthy since the conversion of either gonadal T or adrenal DHEA into DHT, although sequentially distinct, must both engage 5α-reductase, albeit via different substrates (Figure 5e). Thus, this shift towards AD as the more favorable substrate may represent a critical metabolic switch in CRPC that heralds a compensatory increase in adrenal steroid utilization for DHT biosynthesis. This is supported by other independent observations illustrating the impact of enhanced adrenal steroid metabolism in CRPC—for instance the clinical consequence of a gain-of-function missense variant in 3β-HSD isoenzyme-1, which dramatically increases the rate of DHEA→AD conversion (13,31,32). Remarkably, this metabolic switch towards AD also appears to coincide with a conspicuous loss in the relative ability of 5α-reductase to harness T. In fact, the majority of prostate cancer cell lines and sampled patient metastases demonstrate very low capacity to convert T→DHT and thus exhibit an idiosyncratic dependence on adrenal precursor steroids (11).
The 5α-dione pathway is probably the major route of adrenal steroid metabolism in the prostate due to the strong catalytic activity of 5α-reductase, which dictates the overall directionality of metabolic flux from AD. The notion that AD can be 17-keto reduced to T originally derives from observations in gonadal tissue, where the expression of 17β-HSD isoenzyme-3 is essential towards driving this reductive process. This is evidenced by genetic deficiencies in 17β-HSD isoenzyme-3, which manifest in the characteristic metabolic restriction of AD→T (33). In contrast to the testes, the prostate is devoid of this particular enzyme but instead expresses 17β-HSD isoenzyme-5 (AKR1C3). AKR1C3 also demonstrates the capacity to reduce 17-keto steroids, including both AD and 5α-dione (34–37), but in the prostate, 17-keto reduction and 5α-reduction are competing reactions due to the abundant expression of 5α-reductase. We observe that the relatively high rate of 5α-reduction in prostatic tissue appears to considerably exceed that of 17-keto reduction, so that the majority of AD is rapidly converted by 5α-reductase to 5α-dione prior to 17-keto reduction.
Like 17β-HSD, more than one isoenzyme of 5α-reductase has been confirmed in humans. 5α-reductase isoenzyme-2 (SRD5A2) is dominantly expressed in the prostate and is primarily responsible for the robust 5α-reduction of T to DHT (19,38–40). However, prior cell-free enzyme kinetic studies suggest that this isoenzyme actually exhibits a similar kinetic profile for AD, explaining why the 5α-dione pathway is also observed in non-neoplastic tissue prostate samples in our study (10). In contrast, 5α-reductase isoenzyme-1 (SRD5A1) is expressed to a lesser degree in non-neoplastic prostatic tissue but appears to be the major isoenzyme found in CPRC. Unlike SRD5A2, the kinetic profile of SRD5A1 strongly favors AD (10) and appears to be main enzyme responsible for adrenal steroid metabolism in CRPC cell lines (11). Aberrantly high expression of SRD5A1 with relatively low expression of SRD5A2 is a fairly consistent pattern seen across human CRPC cell lines and sampled tissue from CRPC patients (11,15,17,21,41). It is probably this combination together that determines the overall reliance on adrenal steroids rather than T to generate DHT.
Our effort to determine the actual metabolic phenotype of prostate tissue samples obtained from patients adds direct evidence to pre-existing data from gene expression and cell-free enzyme kinetics studies. Importantly, this approach goes a step beyond inference, which should be acknowledged since enzyme transcript and protein expression levels do not always correlate well with extent of metabolic activity (13), and because kinetic information may not capture the overall metabolic picture within a complex, multiple substrate/multiple enzyme biological system. Furthermore, previous studies in this area have unfortunately been limited by the lack of available in vitro models which properly recapitulate the biology of primary disease. Likewise, primary disease of the prostate generally persists in a eugonadal state, where the interaction of several circulating steroids as potentially competing substrates is challenging to recapitulate. Our mixed substrate approach with stable isotopic labeling and mass spectrometry enables a direct comparison of substrate preference for 5α-reductase when both steroids are introduced into the same ex vivo biological system.
It is important to note that our study does not aim to ascertain the absolute contribution of adrenal steroids to DHT, which has been previously explored from in vivo studies on intraprostatic DHT concentrations in castrated men (42). Our study rather attempts to define the directionality of adrenal versus gonadal DHT biosynthesis and the comparative utilization of one metabolic pathway versus the other. However, while our approach serves as a proof-of-concept for the determination of androgen metabolism, we cannot conclude with certainty whether clinically localized cancer within our primary samples metabolically resembles the non-neoplastic phenotype. It is possible that steroid metabolites detected in media are not necessarily representative of localization of steroids across different tissue compartments. Our study also does not tease apart more nuanced differences in metabolism between epithelium and stroma within the tissue parenchyma or any possible paracrine effects which may contribute to regional differences in metabolism. Despite technical limitations, we nevertheless attempted to classify samples into tumor or non-neoplastic categories based on the pathology of the surrounding tissue, which did not reveal any appreciable differences in metabolism.
In summary, our findings posit that adrenal steroid metabolism to DHT in prostatic tissue harboring localized prostate cancer is dictated in large part by the catalytic activity of 5α-reductase to drive conversion via the 5α-dione pathway. This results in two distinctly non-redundant metabolic pathways through which gonadal T and adrenal DHEA are converted to DHT, both requiring 5α-reductase, albeit via different substrates. Under physiologic conditions of the eugonadal state where both T and DHEA are present, adrenal steroids are probably a comparatively inefficient source for DHT requiring multiple enzymatic modifications; however, pathway accessibility may increase in CRPC, in part due to a metabolic switch towards AD as the preferred substrate of 5α-reductase. This change is likely driven by differential expression of two 5α-reductase isoenzymes found in humans, which in the process creates an idiosyncratic dependence on adrenal steroids, while eschewing T as the major source of DHT. Ultimately, these findings underscore the fundamental need to better define steroid metabolic pathways of prostate cancer in its various stages, in the effort to develop more effective AR-directed therapies.
Supplementary Material
Translational relevance.
We show that DHT biosynthesis from adrenal steroids in the prostate of men with clinically localized prostate cancer primarily occurs through 5α-reductase utilizing AD. Notably, this results in two distinctly non-redundant metabolic pathways to DHT from circulating adrenal steroids versus gonadal T. This observation deviates from the general view that 5α-reduction of T is the principal enzymatic step to generate intraprostatic DHT, regardless of the source. Additionally, using a novel mixed substrate approach to assay relative substrate utilization, we provide direct evidence that the transition of eugonadal primary disease to eventual CRPC may coincide with a metabolic switch towards AD as the preferred substrate of 5α-reductase, thereby augmenting adrenal steroid utilization towards DHT biosynthesis and enabling therapeutic resistance. Ultimately, these findings underscore the need to better define steroidogenic pathways of prostate cancer in its various stages, in the effort to develop more effective androgen-directed therapies for CRPC.
Acknowledgments
None
Financial support
This work was supported by funding from the Howard Hughes Medical Institute Medical Fellows Program (to CD), a Howard Hughes Medical Institute Physician-Scientist Early Career Award (to NS), a Prostate Cancer Foundation Challenge Award (to NS) and National Cancer Institute grants (R01CA168899, R01CA172382, and R01CA190289) to NS.
Footnotes
Author conflicts of interest
The authors declare no potential conflicts of interest.
References
- 1.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 3.Mohler JL. Castration-recurrent prostate cancer is not androgen-independent. Adv Exp Med Biol. 2008;617:223–34. doi: 10.1007/978-0-387-69080-3_21. [DOI] [PubMed] [Google Scholar]
- 4.Ferraldeschi R, Welti J, Luo J, Attard G, de Bono JS. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene. 2015;34:1745–57. doi: 10.1038/onc.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mostaghel EA. Steroid hormone synthetic pathways in prostate cancer. Transl Androl Urol. 2013;2:212–27. doi: 10.3978/j.issn.2223-4683.2013.09.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharifi N. Mechanisms of androgen receptor activation in castration-resistant prostate cancer. Endocrinology. 2013;154:4010–7. doi: 10.1210/en.2013-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharifi N, Auchus RJ. Steroid biosynthesis and prostate cancer. Steroids. 2012;77:719–26. doi: 10.1016/j.steroids.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Luu-The V, Bélanger A, Labrie F. Androgen biosynthetic pathways in the human prostate. Best Pract Res Clin Endocrinol Metab. 2008;22:207–21. doi: 10.1016/j.beem.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 9.Luu-The V. Assessment of steroidogenic pathways that do not require testosterone as intermediate. Horm Mol Biol Clin Investig. 2011;5:161–5. doi: 10.1515/HMBCI.2011.007. [DOI] [PubMed] [Google Scholar]
- 10.Thigpen AE, Cala KM, Russell DW. Characterization of Chinese hamster ovary cell lines expressing human steroid 5 alpha-reductase isozymes. J Biol Chem. 1993;268:17404–12. [PubMed] [Google Scholar]
- 11.Chang K-H, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728–33. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharifi N. The 5α-Androstanedione Pathway to Dihydrotestosterone in Castration-Resistant Prostate Cancer. J Investig Med. 2012;60:504–7. doi: 10.231/JIM.0b013e31823874a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang K-H, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–84. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powell K, Semaan L, Conley-LaComb MK, Asangani I, Wu Y-M, Ginsburg KB, et al. ERG/AKR1C3/AR Constitutes a Feed-Forward Loop for AR Signaling in Prostate Cancer Cells. Clin Cancer Res. 2015;21:2569–79. doi: 10.1158/1078-0432.CCR-14-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Titus MA, Gregory CW, Ford OH, Schell MJ, Maygarden SJ, Mohler JL. Steroid 5alpha-reductase isozymes I and II in recurrent prostate cancer. Clin Cancer Res. 2005;11:4365–71. doi: 10.1158/1078-0432.CCR-04-0738. [DOI] [PubMed] [Google Scholar]
- 16.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res American Association for Cancer Research. 2005;11:4653–7. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 17.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res American Association for Cancer Research. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geller J, Albert J, Loza D, Geller S, Stoeltzing W, de la Vega D. DHT concentrations in human prostate cancer tissue. J Clin Endocrinol Metab The Endocrine Society. 1978;46:440–4. doi: 10.1210/jcem-46-3-440. [DOI] [PubMed] [Google Scholar]
- 19.Thomas LN, Douglas RC, Lazier CB, Too CKL, Rittmaster RS, Tindall DJ. Type 1 and type 2 5alpha-reductase expression in the development and progression of prostate cancer. Eur Urol. 2008;53:244–52. doi: 10.1016/j.eururo.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 20.Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–27. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 22.Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, Clish CB, et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Alyamani M, Zhang A, Chang K-H, Berk M, Li Z, et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. Elife. 2017 doi: 10.7554/eLife.20183. (Accepted for publication) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ercole C, Chang KH. Dehydroepiandrosterone metabolism in fresh human prostate: A feasibility study. J Clin Oncol. 2014;32 abstra 225. [Google Scholar]
- 25.Forti G, Salerno R, Moneti G, Zoppi S, Fiorelli G, Marinoni T, et al. Three-month treatment with a long-acting gonadotropin-releasing hormone agonist of patients with benign prostatic hyperplasia: effects on tissue androgen concentration, 5 alpha-reductase activity and androgen receptor content. J Clin Endocrinol Metab. 1989;68:461–8. doi: 10.1210/jcem-68-2-461. [DOI] [PubMed] [Google Scholar]
- 26.Labrie F, Dupont A, Simard J, Luu-The V, Bélanger A. Intracrinology: the basis for the rational design of endocrine therapy at all stages of prostate cancer. Eur Urol. 1993;24(Suppl 2):94–105. doi: 10.1159/000474399. [DOI] [PubMed] [Google Scholar]
- 27.Page ST, Lin DW, Mostaghel EA, Hess DL, True LD, Amory JK, et al. Persistent intraprostatic androgen concentrations after medical castration in healthy men. J Clin Endocrinol Metab. 2006;91:3850–6. doi: 10.1210/jc.2006-0968. [DOI] [PubMed] [Google Scholar]
- 28.Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin Cancer Res. 2004;10:7121–6. doi: 10.1158/1078-0432.CCR-04-0913. [DOI] [PubMed] [Google Scholar]
- 29.James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N Engl J Med. 2017 doi: 10.1056/NEJMoa1702900. NEJMoa1702900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N Engl J Med. 2017 doi: 10.1056/NEJMoa1704174. NEJMoa1704174. [DOI] [PubMed] [Google Scholar]
- 31.Hearn JWD, AbuAli G, Reichard CA, Reddy CA, Magi-Galluzzi C, Chang K-H, et al. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multicohort study. Lancet Oncol. 2016;17:1435–44. doi: 10.1016/S1470-2045(16)30227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal N, Hahn AW, Gill DM, Farnham JM, Poole AI, Cannon-Albright L. Independent Validation of Effect of HSD3B1 Genotype on Response to Androgen-Deprivation Therapy in Prostate Cancer. JAMA Oncol. 2017 doi: 10.1001/jamaoncol.2017.0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonca BB, et al. Male pseudohermaphroditism caused by mutations of testicular 17 beta-hydroxysteroid dehydrogenase 3. Nat Genet. 1994;7:34–9. doi: 10.1038/ng0594-34. [DOI] [PubMed] [Google Scholar]
- 34.Dufort I, Rheault P, Huang X-F, Soucy P, Luu-The V. Characteristics of a Highly Labile Human Type 5 17β-Hydroxysteroid Dehydrogenase 1. Endocrinology. 1999;140:568–74. doi: 10.1210/endo.140.2.6531. [DOI] [PubMed] [Google Scholar]
- 35.Azzarello J, Fung K-M, Lin H-K. Tissue distribution of human AKR1C3 and rat homolog in the adult genitourinary system. J Histochem Cytochem Histochemical Society. 2008;56:853–61. doi: 10.1369/jhc.2008.951384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin H-K, Steckelbroeck S, Fung K-M, Jones AN, Penning TM. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3α-hydroxysteroid dehydrogenase/type 5 17β-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids. 2004;69:795–801. doi: 10.1016/j.steroids.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 37.Penning TM, Steckelbroeck S, Bauman DR, Miller MW, Jin Y, Peehl DM, et al. Aldo-keto reductase (AKR) 1C3: role in prostate disease and the development of specific inhibitors. Mol Cell Endocrinol. 2006;248:182–91. doi: 10.1016/j.mce.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 38.Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 39.Bartsch G, Rittmaster RS, Klocker H. Dihydrotestosterone and the concept of 5alpha-reductase inhibition in human benign prostatic hyperplasia. World J Urol. 2002;19:413–25. doi: 10.1007/s00345-002-0248-5. [DOI] [PubMed] [Google Scholar]
- 40.Andersson S, Berman DM, Jenkins EP, Russell DW. Deletion of steroid 5 alpha-reductase 2 gene in male pseudohermaphroditism. Nature. 1991;354:159–61. doi: 10.1038/354159a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Audet-Walsh É, Yee T, Tam IS, Giguère V. Inverse regulation of DHT synthesis enzymes 5α Reductase Types 1 and 2 by the androgen receptor in prostate cancer. Endocrinology. 2017;158:1015–21. doi: 10.1210/en.2016-1926. [DOI] [PubMed] [Google Scholar]
- 42.Taplin M-E, Montgomery B, Logothetis CJ, Bubley GJ, Richie JP, Dalkin BL, et al. Intense Androgen-Deprivation Therapy With Abiraterone Acetate Plus Leuprolide Acetate in Patients With Localized High-Risk Prostate Cancer: Results of a Randomized Phase II Neoadjuvant Study. J Clin Oncol. 2014;32:3705–15. doi: 10.1200/JCO.2013.53.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.