

Abstract
Purpose
Adenoid cystic carcinoma (ACC) is a rare cancer arising from the major or minor salivary gland tissues of the head and neck. There are currently no approved systemic agents or known radiosensitizers for ACC. Unlike the more common head and neck squamous cell carcinomas that frequently harbor TP53 mutations, ACC contain TP53 mutations at a rate of <5%, rendering them an attractive target for MDM2 inhibition.
Experimental Design
We report the successful establishment and detailed characterization of a TP53-WT ACC patient derived xenograft (PDX) which retained the histologic features of the original patient tumor. We evaluated this model for response to the MDM2 inhibitor AMG 232 as monotherapy and in combination with radiation (RT).
Results
AMG 232 monotherapy induced modest tumor growth inhibition and RT monotherapy induced a transient tumor growth delay in a dose dependent fashion. Strikingly, combination treatment of AMG 232 with RT (including low dose RT of 2 Gy/fraction) induced dramatic tumor response and high local tumor control rates three months following treatment. Post treatment analysis revealed that while both AMG 232 and RT alone induced TP53 tumor suppressive activities, combination therapy amplified this response with potent induction of apoptosis after combination treatment.
Conclusions
These data identify that MDM2 inhibition can provide potent radiosensitization in TP53-WT ACC. In light of the absence of effective systemic agents for ACC, the powerful response profile observed here suggests that clinical trial evaluation of this drug/RT combination may be warranted to improve local control in this challenging malignancy.
Keywords: radiation, radiosensitization, MDM2, adenoid cystic carcinoma
Introduction
Malignancies of the salivary glands are a relatively rare and diverse group of tumors, accounting for roughly 3% of all head and neck cancers (1). Adenoid cystic carcinoma (ACC) comprises approximately 10% of salivary gland tumors and is characterized by a generally slow but unpredictable growth rate and a high rate of eventual local and distant metastasis (1). These tumors are categorized histologically as solid, tubular, or cribriform patterns, with the solid form typically demonstrating more aggressive disease than the other types (2–6). Despite modern surgical treatments for patients with newly diagnosed ACC, both local and distant recurrences remain common, highlighting the need for improved therapy options (7). Despite dozens of clinical trials examining drug therapies, there are no FDA approved systemic agents for ACC with conventional and molecular therapeutics eliciting crude response rates typically less than 10–20% (8–15). The most widely utilized regimen of cisplatin plus vinorelbine has significant toxicities and a modestly higher response rate (16). The current standard of care remains surgical resection and postoperative radiotherapy (1,8); however, with a high propensity for local and distant failure. The addition of cisplatin to adjuvant radiotherapy is currently being investigated in a randomized RTOG study enrolling multiple salivary gland histologies including ACC, but these results are not anticipated for several years.
The hallmark tumor suppressor protein p53 is a transcription factor that regulates cell cycle progression, senescence, DNA repair, and apoptosis to control tumor cell growth (17). TP53 is mutated in roughly 50% of all tumors (18,19) but the majority of ACCs express wild-type p53. This is in stark contrast to head and neck squamous cell carcinoma in which TP53 mutations are seen in over 50% of cases. In ACC, TP53 mutations have been identified in approximately 5% of cases (20,21). p53 protein levels are regulated by MDM2, an E3 ubiquitin ligase, which targets p53 for proteasomal degradation (22). MDM2 inhibitors are known to exhibit anti-tumor effects and are currently being studied in clinical trials (23–25). In ACC, the promising efficacy of an MDM2 inhibitor alone or in combination with cisplatin, has recently been demonstrated in the treatment of ACC patient derived xenografts (26,27). In this study, we investigated the radiosensitizing effect of AMG 232, a picomolar affinity piperidinone inhibitor of MDM2 that is currently in clinical trials for several tumor types. AMG 232 appears to synergize with p53 activating therapies such as DNA damaging agents, likely potentiating p53 signaling, leading to increased apoptosis and decreased cell proliferation (28–30).
Among many anti-tumor effects, ionizing RT activates p53 to induce its array of tumor suppressive functions. AMG 232 and other MDM2 inhibitors have been shown to radiosensitize non-small cell lung cancer, breast cancer, colon cancer, melanoma, and prostate cancer cell lines that are p53 wild type, with more modest efficacy in models that harbor p53 mutations (31,32). Given the low rate of TP53 mutation in ACC and promising preclinical data demonstrating a role for MDM2 inhibition in ACC, we hypothesized that the combination of AMG 232 and RT would result in improved tumor control than either treatment alone. Herein, we describe the characterization of an ACC PDX model, the potent radiosensitizing effect of AMG 232 on this tumor, and analysis investigating molecular mechanisms underlying this radiosensitization.
Materials and Methods
Mice
Six to eight-week-old female NOD-SCID gamma (NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice (Jackson Laboratories) were used for PDX establishment and tissue amplification. Six to eight-week-old female Hsd: athymic Nude-Foxn1nu (Harlan Laboratories) were used for therapy studies. Mice were kept in the Association for Assessment and Accreditation of Laboratory Animal Care-approved Wisconsin Institute for Medical Research (WIMR) Animal Care Facility, housed in specific pathogen free rooms, and had their clinical health evaluated at least twice weekly. All studies involving mice were carried out in accordance with a University of Wisconsin Institutional Animal Care and Use Committee approved protocol.
PDX propagation and tumor harvesting
The UW-ACC-60 PDX was established and propagated as described previously for head and neck squamous cell carcinomas (33,34). TP53 sequencing was performed on total genomic DNA using the Illumina TruSeq Cancer Amplicon panel and has been deposited with the sequence read archive under BioProject ID: PRJNA381909.
Genetic testing of ACC PDX and patient donor tumor
The identity of the first and fifth passages of the ACC PDX was confirmed to match its original patient donor tumor via short tandem repeat (STR) testing. Total genomic DNA was isolated from flash frozen tissue for all instances of the tumor using the Qiagen DNeasy kit. An 18 loci STR assay (Promega PowerPlex 16 HS System #DC2100) was performed by the UW Translational Initiatives in Pathology CORE facility. The same DNA samples were used to test for presence of the MYB-NFIB gene fusion frequently detected in ACC via a PCR based approach (35) using the following primer sets: MYB-1910F 5′-AGCTCCGTTTTAATGGC-3′/NFIB-1096R 5′-GGGTATAAATGCCTGCCGTT-3′; MYB-1910F 5′-AGCTCCGTTTTAATGGC-3′/ NFIB-1096R 5′-GGGTATAAATGCCTGCCGTT-3′, MYB- 1693F 5′-GCAGGATGTGATCAAACAGG-3′/ NFIB-1197R 5′-CCGGTAAGATGGGTGTCCTA-3′. A ~750 bp DNA fragment containing all sequences involved in or adjacent to known fusion regions in MYB and NFIB was synthesized and served as a positive control. Primer specific for ß-actin were used as a PCR reaction control.
Treatment and measurement
When established tumors had reached sufficient size, tumor tissue was harvested from NSG mice and passaged into nude mice via subcutaneous implantation in bilateral rear flanks. Tumor volume was assessed weekly with Vernier Calipers and calculated according to the equation V = (π/6) × (large diameter) × (small diameter)2. These slow growing tumors required approximately 3 months until average volume reached 250 mm3 at which time mice (n=6/group) were randomized into treatment groups: control (vehicle), AMG 232, RT, or combination of RT + AMG 232. Randomization resulted in a starting tumor size that was not statistically different across all groups at the start of treatment. RT at the specified dose (2Gy, 5Gy or 8 Gy per fraction) was delivered twice per week for four weeks for a total of eight fractions. AMG 232 was delivered every day concurrent with RT for four weeks by oral gavage at 50mg/kg, a level that fell within the range of doses used in prior studies in the same mouse strain (31). Additional earlier unpublished work had highlighted the clinical relevance of this dose showing that 50mg/kg produced drug exposure level in mice that is similar to the exposure level observed in human trials at well tolerated doses. RT was performed with a Precision Xray XRAD 320 with 1 Gy/min delivered at 320KV/12.5mA at 50cm SSD with a beam hardening filter with HVL≈4mm Cu. The delivered dose rate was confirmed monthly by ionization chamber. Mice were shielded with custom built lead jigs to isolate exposure to the rear quarter of the body. Two hours following the initial treatment, additional tumor-bearing mice were sacrificed and tumors were harvested for biomarker analysis. Each tumor was divided in half for formalin-fixed paraffin-embedded (FFPE) preservation and flash freezing in liquid nitrogen. At the end of the study, two mice from either the vehicle or AMG 232 alone groups were retreated with vehicle or drug and 0, 2 or 8 Gy and tumors were harvested 48 hours post treatment to investigate additional post-treatment effects (36).
Statistical analysis of PDX treatment study
A one-way analysis of variance (ANOVA) model was utilized, with the null hypothesis that the group means of control, AMG, AMG+2Gy, AMG+5Gy and AMG+8Gy are equal was tested globally. We investigated how well the model fit the data by reporting goodness-of-fit statistics, including R-squared measures, which describe the percentage of variation in the response explained by the model. Finally, we performed post-hoc analysis to assess the effects of pairwise comparisons between the groups (i.e., AMG vs. Control, AMG vs AMG+2Gy, AMG vs AMG+5Gy, AMG vs AMG+8Gy), using Tukey’s honestly significant difference (HSD) post-hoc test. All statistical analyses were performed using the 2-sided alpha significance level of 0.05, with STATA v14 software.
The dose of RT required to result in 50% tumor control (TCD50) was calculated at day 125. The proportion of injection sites with no palpable tumor (tumor volume = 0) was calculated. The confidence intervals of the proportion were used to set the range, the RT dose was converted to Log10 format, and the resulting data points fit to a Log(treatment) vs. response curve using a least squares fit. The Log(TCD50) was compared by the extra sum-of-squares F test using GraphPad Prism v7.0a.
To assess antagonistic, additive, or synergistic effects, we used the fractional product method as previously described(37) one week after the end of treatment. Briefly, the observed fractional tumor volume (FTV) is equal to the mean tumor volume of each treated group (AMG 232, radiation, or AMG 232 + radiation) divided by the mean tumor volume of the control group. The expected FTV from the combined treatment (FTVAMG 232 + radiation) is calculated by multiplying the observed FTVAMG 232 by the observed FTVradiation. Dividing the expected FTVAMG 232 + radiation by the observed FTVAMG 232 + radiation yields a synergy assessment ratio in which a value >1 suggests that the combined treatments are effectively synergistic, <1 antagonistic, and =1 additive.”
Histology and Immunohistochemistry
Hematoxylin and eosin (H&E) staining was used for formal pathologic analysis and to assess tumor quality. Additional slides were processed by the UW Translational Research in Pathology lab to assess for expression of p63, CD117, and CK5/6 by immunohistochemistry (IHC). Staining for expression of p53, p-p53, and Ki-67 was performed by standard IHC techniques as described previously(12,38). See Table S1 for reagent details. A no primary antibody control slide was stained with each condition to ensure specificity of the reaction. Briefly, slides were baked in an 80°C oven for 20 minutes to melt paraffin and deparaffinized by soaking in xylene three times for 5 minutes each. Slides were then rehydrated in ethanol through soaking in decreasing concentrations of ethanol (3× 100%, 2×95%, 1×80%, 1×50%). Heat-induced epitope retrieval followed rehydration. Slides were blocked for endogenous peroxidase activity and nonspecific antibody interactions using 10% goat serum in phosphate-buffered saline (PBS). Primary antibody prepared in 1% goat serum was incubated overnight at 4°C (primary antibody dilutions per Supplemental Table S1). The next day anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies were applied to the slides for 30 minutes followed by a 2-minute development with 3,3′-diaminobenzidine (DAB) (#SK-4100; Vector Laboratories, Burlingame, CA). Finally, slides were counterstained with hematoxylin, dipped in fresh xylene and coverslipped with Cytoseal XYL (Thermo Fisher Scientific, Waltham, MA) and a number 1.5 coverslip. Where conducted, positive cell counts were performed using ImageJ with statistical analysis using GraphPad Prism. Semi-quantitative analysis of IHC staining intensity was performed using ImageJ with the Colour Deconvolution plugin.
Immunoblotting
Lysates were prepared from flash frozen tumor by homogenization with Dounce homogenizer in NP40 buffer supplemented with protease/phosphatase inhibitor cocktail (Cell Signaling Technologies #5872) followed by sonication. Equal amounts of protein were analyzed by SDS-PAGE (50μg/well) and transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon-FL). Protein targets of interest were analyzed by specific primary antibodies, detected via incubation with anti-mouse or anti-rabbit (as appropriate) near infrared-conjugated secondary antibodies (LiCOR), and imaged on a LiCOR Odyssey FC. Antibodies and sources are listed in Table S1.
In situ hybridization by RNAScope
RNA levels were examined using the RNAscope 2.5 HD Detection Reagent – Brown (Advanced Cell Diagnostics) according to manufacturer’s protocol. Slides were baked at 60°C for an hour and then deparaffinized by incubating in Xylene twice for 5 minutes. Slides were then treated in 100% ethanol twice, for 1 minute each, at room temperature and then air dried. Heated antigen retrieval was carried out using RNAscope 1× Target Retrieval Reagents. RNAscope hydrogen peroxide was added to the tissue sections and the slides were incubated for 10 minutes and dried. Slides were then baked in the HybEZ Oven at 40°C for 30 minutes. After baking, the Hs-CDKN1A hybridization probe (ACD 311401) was applied to the slides and incubated in the HybEZ oven for 2 hours at 40°C. The probe was followed by the application of Hybridization Amps 1 – 6. Slides were washed between the application of the probe and amps using 1× RNAscope wash buffer. Signal detection was carried out using DAB, slides were counterstained using 50% Hematoxylin, and washed with 0.02% ammonia water. Slides were then dehydrated using 70% ethanol, 95% ethanol (2×), incubated in xylene for 5 minutes and then cover-slipped using Cytoseal XYL (Thermo Fisher Scientific, Waltham, MA) and a number 1.5 coverslip.
Apoptotic body count
The xenograft specimens were fixed in 10% buffered formalin and processed for routine paraffin sections. The 5-micron sections were stained by hematoxylin and eosin. The number of apoptotic bodies per field was counted with a 60× objective and 10× eyepiece under light microscopy on hematoxylin-eosin stained slides by a blinded pathologist (RH). Areas adjacent to necrosis were avoided. Apoptotic bodies were identified as shrunken cells with compact, segregated and sharply delineated chromatin with a deeply eosinophilic cytoplasm. Five to ten random fields were examined in each specimen depending on the size of tissue and average was taken to represent the count for each condition.
Results
Establishment and characterization of adenoid cystic carcinoma PDX
The PDX was established in NSG mice from a portion of a surgical specimen from a patient with a regional recurrence of ACC arising over 10 years after initial diagnosis. The PDX appeared approximately 3 months after implantation and was able to be successfully passaged for additional analyses. Comparison of H&E and IHC staining between the patient tumor and subsequent passages was performed by a board-certified pathologist specializing in head and neck cancer (RH). Evaluation of the patient tumor (Figure 1A) demonstrated an infiltrative tumor with collagenous and fibrotic stroma that was infiltrated by lymphocytes and plasma cells. Blood vessels were present in the stroma and invaginated into the tumor lobules. The tumor had a predominantly cribriform growth pattern with smaller areas displaying a solid growth pattern. Intracystic spaces filled with basophilic material were also identified. Comedonecrosis was present in less than 5% of the tumor mass. Evaluation of the 1st and 5th passages of the PDX tumor (Figure 1A) demonstrated a similar growth pattern and morphology to the original tumor with a predominantly cribriform pattern. The PDX tumors were well circumscribed with scant collagenous stroma infiltrated by a small number of macrophages and mast cells. Blood vessels were observed only in the stroma. The NSG mice do not generate B and T cells accounting for the lack of lymphocytes. The PDX tissue also demonstrated greater comedonecrosis (25–30%) relative to the patient tissue. A biphasic appearance typical of ACC was observed in both the patient primary and PDX tumors. The tumor was comprised of a luminal epithelium forming scattered ductules and abluminal (myoepithelial) cells, with abluminal cells forming the predominant cell type. This biphasic feature is highlighted by immunohistochemical studies of CD117, CK5/6, and p63: CD117 stains the luminal lining cells but not abluminal cells; p63 stains the abluminal cells only; CK5/6 stains abluminal cells weakly and stains luminal epithelium strongly. This luminal/abluminal pattern remained consistent across multiple passages of the PDX. STR profiling of the patient tumor and the same two passages of PDX confirmed the genetic identity of the patient and xenograft tumors (Table S2). A PCR based approach was used to test for the MYB-NFIB fusion commonly observed in ACC: both the original patient tumor and both PDX passages were found to be negative for this genetic lesion (Figure S1).
Figure 1. Characterization of Adenoid Cystic Carcinoma PDX.
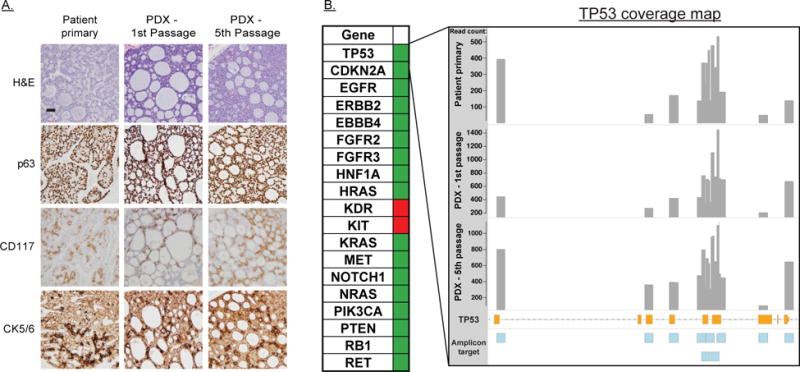
A: H&E and immunohistochemical (IHC) staining of the primary patient tumor, the first passage PDX and the most recent (fifth passage) PDX. IHC diagnostic markers for ACC included CD117, stains the luminal lining cells but not abluminal cells; p63 stains the abluminal cells only; CK5/6 stains abluminal cells weakly and stains luminal epithelium strongly. B: (left) Summary of non-synonymous mutations identified by hotspot sequencing. Green-no oncogenic mutations identified. Red-potentially oncogenic mutation identified. (right) Coverage map of TP53 sequencing. Grey bars are histogram of counts for each target amplicon. Orange bars are TP53 coding DNA sequence with intervening non-coding regions indicated by dashed line. Blue boxes are target amplicons included in the sequencing panel.
As part of the standard characterization of our PDXs, tumor tissue was isolated, gDNA was prepared, and Illumina HotSpot sequencing was performed as previously described (38) to determine the TP53 status of this tumor. With over 50× coverage over the most common sites of TP53 mutation, no oncogenic TP53 SNPs or INDELs were observed in either the patient tumor or any subsequent passage (Figure 1B). A cancer risk associated P72R alteration was identified in greater than 95% of reads for the patient and both PDX passages, consistent with a germline alteration (39). Given the fidelity of this PDX model to a typical TP53 WT ACC tumor we hypothesized that it would be a good candidate for MDM2 inhibition and RT combination therapy and proceeded to test this treatment regimen.
Combination of AMG 232 with escalating RT dose
We investigated the ability of AMG 232 to radiosensitize ACC in a pilot study using a single RT dose (5 Gy) delivered twice weekly over 4 weeks with concurrent daily AMG 232 (Figure S2A). Tracking of mouse weights indicated that this treatment combination was well tolerated by the animals (Figure S2B). The promising results of this study led us to conduct a dose escalation experiment combining a single dose level of AMG 232 (50 mg/kg) with an escalating dose of RT of 2, 5, or 8 Gy/fraction for a total of 8 fractions (Figure 2A). AMG 232 alone had minimal sustained effect on tumor growth, while the 3 RT alone groups slowed tumor growth in a dose dependent manner, but did not cause prolonged tumor regression. Strikingly, with as little as 2Gy/fraction (16 Gy total), the combination of AMG 232 and RT resulted in rapid and sustained tumor regression. To assess the extent of tumor control, mice were followed over 100 days after the end of treatment and still no tumor regrowth was observed. Animals treated with either 5 or 8 Gy/fraction demonstrated more rapid tumor shrinkage and also had no apparent tumor regrowth at the end of the study.
Figure 2. RT and AMG 232 combination therapy of ACC PDX.
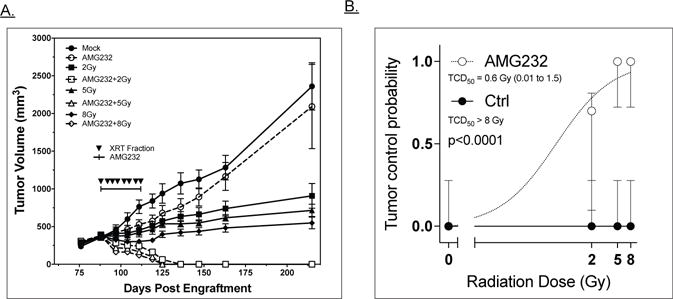
A: Growth curves representing tumor volume of the 8 treatment groups as a function of days post engraftment. Filled symbols represent mock/RT only groups; open symbols are AMG 232/RT combination groups. RT fractions were delivered on days indicated by inverted triangles. AMG 232 or vehicle was delivered by oral gavage 1×/day over time period shown. B: TCD50 analysis of ACC PDX +/− AMG 232; data were fit to a log(treatment) vs. response curve using a least squares fit. TCD50 with RT alone was not reached and can only be stated as > 8Gy. In combination with AMG 232, the TDC50 was calculated at 0.6Gy with a 95% CI of 0.01 to 1.5 Gy. Error bars are 95% confidence intervals.
Statistical analysis was performed to determine whether increased RT dose produced an anti-tumor benefit. One-way analysis of variance (ANOVA) model was conducted to determine whether the mean volumes were different between five treatment groups: Control, AMG 232, AMG 232+2Gy, AMG 232+5Gy, and AMG 232+8Gy. There was a statistically significant difference between groups as determined by ANOVA (F(4,45) = 14.47, p<0.0001), with approximately 56% of the variance being explained by the model. In addition, a Tukey’s post-hoc test shows that mean scores were significantly lower in AMG+2Gy group compared to AMG alone group (−0.657.54 ± 160.118 (SD), p=0.002). Similarly, a statistically significant difference revealed a lower mean score for all the combination therapy groups compared to either the control or AMG 232 only groups. However, there was no statistically significant difference between AMG+2Gy vs AMG+5Gy (p=0.998) or AMG+5Gy vs AMG+8Gy (p=0.999). These data suggest that increasing doses from 2Gy to 8Gy within the combination arms were not statistically different from each other. To further assess the ability of AMG 232 to improve tumor control, a TCD50 calculation was performed (Figure 2B). RT alone failed to control any tumors (TCD50 > 8Gy/fraction) while combination with AMG 232 significantly decreased the TCD50 (<0.6 Gy/fraction, p<0.0001) consistent with radiosensitization. We used the fractional product methods to calculate a synergy assessment ratio for AMG 232 and each radiation dose. The ratio’s calculated (2.1, 3.2, and 3.3 for 2 Gy, 5 Gy, and 8Gy, respectively) are consistent with a finding of synergy between radiation and AMG 232.
RT and AMG 232 activate p53 and downstream signaling
We next investigated the molecular response to this treatment regimen. The effect on p53 and p-p53 levels was examined by western blotting and immunohistochemistry on xenograft tissues harvested 2 hours post treatment. Total p53 levels were fairly stable with some increase at higher RT doses, while both AMG 232 or RT alone did activate p53 as shown by higher p-p53 levels. The combination of RT and AMG 232 showed increased p53 and p-p53 activation relative to RT or AMG 232 alone (Fig. 3A and 3B). Semi-quantitative analysis of the IHC images confirmed increased p53 and p-p53 activation in treated tumors (Fig S3). DNA damage was detected by immunoblot for ϒH2AX and KU80 two hours post RT; expression of ϒH2AX was RT dose dependent, but the addition of AMG 232 had no effect. Little effect on KU80 was observed. Tumor samples harvested 48 hours post treatment revealed that AMG 232 treatment promoted elevated levels of MDM2 consistent with feedback that is typical after prolonged exposure to an inhibitor, with elevated levels of the P53 effector protein PUMA observed in all treated tumors (Figure 3A).
Figure 3. TP53 pathway activation in response to RT and MDM2 inhibition.
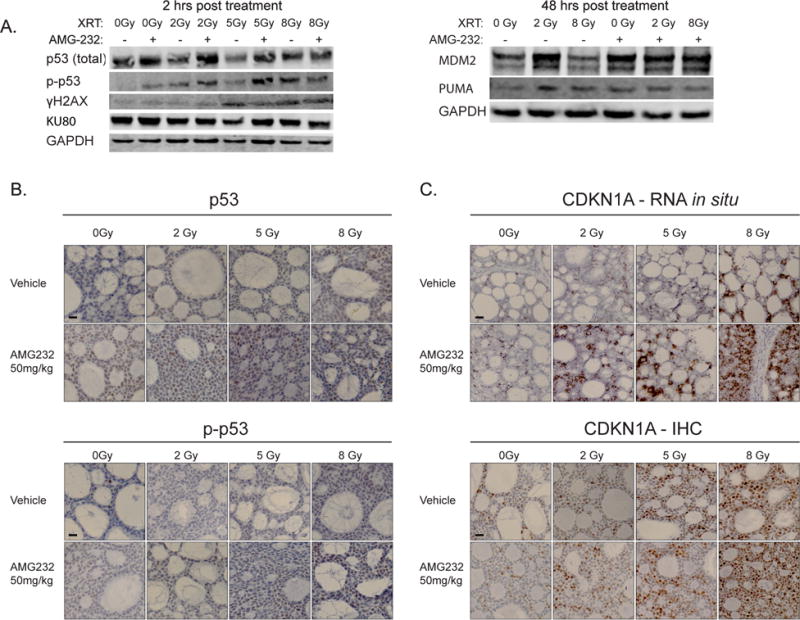
A: Left panel -immunoblot for p53, p-p53, ϒ-H2AX, and Ku80 of ACC PDX tissue two hours following AMG 232, RT, or combination treatment. Right panel -immunoblot of MDM2 and PUMA 48 hours following AMG 232, RT, or combination treatment. B: Representative fields of immunohistochemistry staining for p53 and p-p53 of ACC tumor samples two hours post RT and AMG 232 treatment. Scale bars are 20 μm. C: Representative fields of in situ RNA staining for CDKN1A (top) and CDKN1A protein (a.k.a. p21) IHC (bottom) of ACC tumor samples two hours post RT and AMG 232 treatment. Scale bars are 20 μm.
Downstream effects of p53 activation were evaluated by CDKN1A response (Figure 3C). At 2 hours post treatment, elevated CDKN1A RNA was identified by in situ hybridization (RNAScope) in both the AMG 232 and RT single modality treatments with the combination presenting even greater expression. This effect was confirmed at the protein level where the combination produced elevated CDKN1A as seen by IHC. Semi-quantitative analysis of the IHC images showed a dose dependent elevation of CDKN1A with increasing radiation (Fig S3). Together the results confirmed that the treatment regimen was inducing expected DNA damage and TP53 mediated tumor suppressive pathways.
RT and AMG 232 cooperatively induce anti-proliferative and pro-apoptotic responses
We next evaluated the growth and cell death impact of these treatments on the ACC PDX. Using tumor tissue harvested 48 hours post a single dose of drug and or RT, we immunostained for the proliferation marker Ki-67 and counted positive nuclei from multiple fields for each condition (Figure 4A). Relative to mock treated animals, all treatments reduced cell proliferation, with a greater reduction and higher level of significance for the high dose RT+AMG 232 combination. We next immunostained for cleaved caspase 3, a marker of early stages of apoptosis, in tumors 48 hours post retreatment with a single dose of AMG 232 or RT (Figure 4B). Based on counting of cleaved caspase 3 positive nuclei, neither AMG 232 nor 2Gy RT alone increased apoptosis in these tissues. While the 8Gy fraction did have higher counts, it did not reach statistical significance. Consistent with the rapid shrinkage of the tumors in the AMG 232+RT arms, both of these treatments presented significantly elevated caspase staining in a dose dependent manner. We analyzed later stages of apoptosis by counting apoptotic bodies in multiple high-powered fields for the same samples. In this analysis, only the AMG 232 + 8Gy treatment produced a statically significant increase in apoptosis, with the AMG 232+2Gy counts slightly elevated after this single fraction (Figure 4C). Overall these results are consistent with an anti-proliferative, growth delay impact of the AMG 232 and RT single modality treatments, but a cell death inducing, tumor shrinking effect of the combination.
Figure 4. Proliferation and apoptosis markers following RT and AMG 232 treatment.
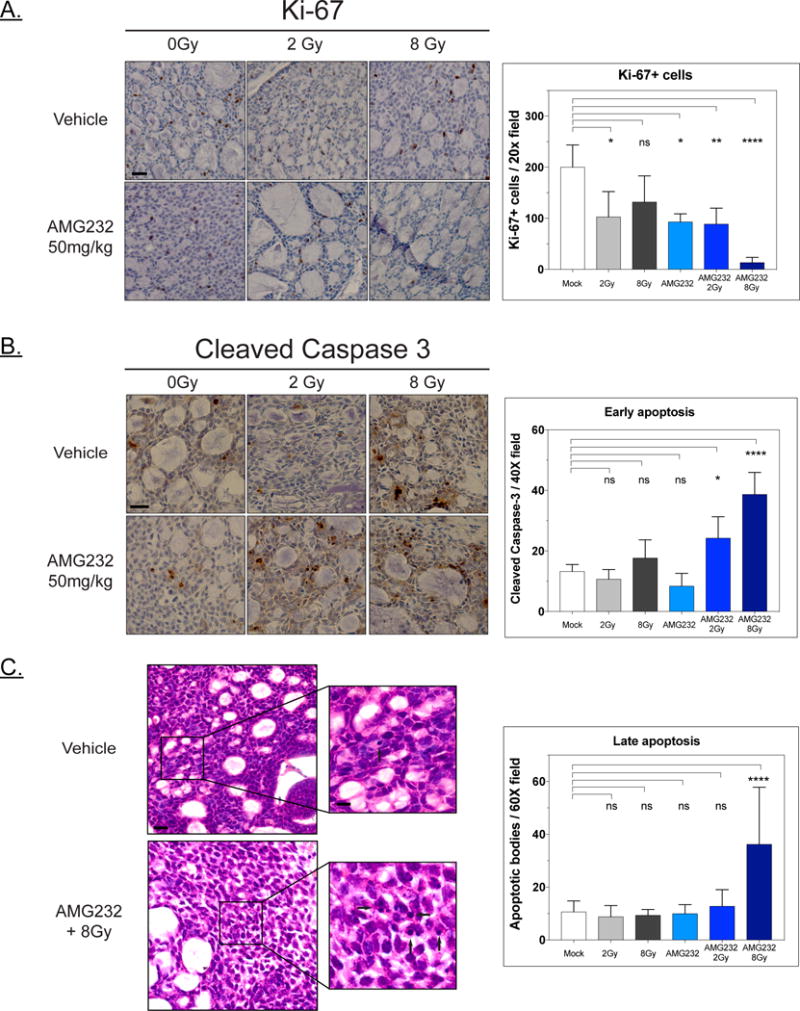
A. Ki-67 immunostaining of PDX samples harvested 48 hrs post treatment. Images are representative 20× fields, scale bars are 10 μm. Treatment was scored by counting Ki-67 positive nuclei from 3 20× fields per condition. Bar plots present mean counts with SD; statistical analysis by one-way ANOVA with Tukey’s multiple comparisons (ns – not significant, *<0.05, **<0.01, ****<0.0001). B: Cleaved caspase 3 staining and quantification of 48-hour post treatment tumor samples. Images are representative 40× fields, scale bars are 20 μm. Cl-caspase3 positive nuclei within the tumor were counted for 3 40× fields for duplicate tumor samples, stromal regions were ignored. Cells presenting cytoplasmic or background staining were not included in counts. Bars are mean of all fields scored for a given condition with SD; statistical analysis by one-way ANOVA with Tukey’s multiple comparisons (ns – not significant, *<0.05, ****<0.0001). C: Quantification of apoptotic bodies in 48-hour post treatment tumor samples in H&E stained slides. Five to ten 60× fields were counted for duplicate tumor samples. Representative images are shown for mock and AMG 232+8Gy treatments (scale bars 20 μm) with inset images with arrows highlighting apoptotic bodies (scale bars 5 μm). Bars are mean of all fields scored for a given condition with SD; statistical analysis by one-way ANOVA with Tukey’s multiple comparisons (ns – not significant, ****<0.0001).
Discussion
ACC patients have limited treatment options beyond surgery and RT and long-term prognosis remains poor. There is a need for novel therapeutic options to improve care, particularly for patients with metastatic disease. The rarity of the disease, the lack of robust preclinical models, and the absence of effective systemic therapies to date remains a major challenge for ACC patients. We developed and characterized a PDX model (UW-ACC-60) representing one of only a handful available for sustained preclinical investigation of ACC (27,40). This model represents the most prevalent type of ACC seen in patients, the cribriform sub-type, and maintains its histologic features over multiple passages.
Targeted sequencing demonstrated that this model, like the majority of ACCs, does not contain a TP53 function altering mutation, making it amenable for treatment approaches that rely on functional p53. The low rate of TP53 mutations seen in ACC has led other groups to investigate the efficacy of MDM2 inhibitors as potential systemic therapies given alone, or in combination with cisplatin. Nor and colleagues used PDXs established at their institution to demonstrate that a MDM2 inhibitor, MI-773, slowed growth of ACC both alone and when combined with cisplatin (26,27). They showed that MDM2 inhibition led to p53 activation, induction of apoptosis, tumor growth delay, and could prevent the recurrence of surgically resected xenografted tumors.
Based on the critical need for innovative approaches to improve local control for patients both newly diagnosed with ACC and dealing with local-regional recurrences, we began this project to investigate potential radiosensitizers of ACC. Building on work the work by Nor and colleagues (26,27), and our success using AMG 232 to radiosensitize lung squamous cell carcinoma in addition to cell lines derived from colon, breast, sarcoma, and melanoma (31), we combined AMG 232 with RT in ACC. We hypothesized that radiation induced p53 activity would be enhanced by inhibiting MDM2, a negative regulator of p53. This combination would thus take advantage of the low rate of TP53 mutation seen in ACC. The combination of RT and AMG 232 resulted not only in growth delay typically seen in xenograft studies, but also complete regression of the tumors that persisted for months after the conclusion of treatment. While both MDM2 inhibition or RT alone were sufficient to activate p53 signaling, as single modalities they had modest effects on tumor growth. Combination treatment decreased Ki67 staining and induced apoptosis consistent with not only growth delay but also the observed tumor regression. AMG 232 + RT resulted in greater p53 pathway induction than either treatment alone as demonstrated by upregulation of p53 target genes and increased protein expression. Given the key role of p53 as a tumor suppressor these findings are not unexpected and in fact complement what both we, and Nor and colleagues, have previously demonstrated regarding the mechanism of MDM2 inhibition as an anti-cancer therapy.
Radiosensitization and xenograft cures were observed with total RT doses as low as 16 Gy delivered over 4 weeks. While additional studies confirming the efficacy of AMG 232 in combination with low dose RT would be needed, these results are encouraging. Many ACC patients live years after completion of adjuvant RT and often are faced with the difficult decision to undergo repeated operations and/or reirradiation due to local and/or regional tumor progression. This work raises the possibility that a radiosensitizer could be combined with focal radiation to improve local tumor control. Using modern techniques, highly conformal radiation can be easily delivered to sites of measureable disease while limiting the volume of normal tissues receiving appreciable RT dose. While this combination would need to be carefully studied to ensure reasonable normal tissue toxicity; we did not identify any significant toxicity in the mice treated with combination therapy.
We acknowledge several limitations of this study. As with most current PDX studies, we completed these experiments in immunocompromised animals thus limiting our ability to identify immunologic mechanisms and focused the study on intrinsic radiosensitivity. It is possible that the addition of immune mediated cell death driven by RT and AMG 232 would require even lower doses to achieve similar results. While there are several other described ACC PDX models (27,28), the studies in this manuscript clearly utilize only a single in vivo model developed from a single patient. The fact that the histology of this model (cribiform subtype) is the most common type of ACC and that the mutational profile is consistent with recently published sequencing of ACC patients suggests that these results may have more broad implications, but additional work is needed to confirm these findings. Finally, because no ACC immortalized cell lines are publicly available (41), and we have thus far been unable to establish a reliable in vitro model, our experiments were limited to in vivo studies. We have an active tissue donation protocol on-going and are seeking to generate additional PDXs and derive cell lines or 3D culture models of ACC to further these and future investigations.
In conclusion, we have demonstrated robust radiosensitization of an ACC PDX by combining the MDM2 inhibitor AMG 232 with relatively low doses of RT. Physicians who care for ACC patients know all too well the desperate need for both new systemic agents as well as improved local control for these patients. Given the low frequency of TP53 mutation in ACC, tumor characterization or patient selection criteria to identify patients with wild-type TP53 may be unnecessary for a clinical trial. This work adds to the growing evidence that targeting MDM2, alone or in combination with another anti-cancer therapy, in patients with ACC may be a successful treatment strategy. Through continued work we may yet be able to improve quality of life, and potentially improve survival, in patients living with ACC.
Supplementary Material
Statement of Translational Relevance.
Adenoid cystic carcinoma is a rare form of salivary gland cancer in which localized tumor progression can significantly harm quality of life. Adjuvant radiation is commonly used for newly diagnosed disease, and many patients eventually undergo reirradiation for palliation. Here, we demonstrate robust radiosensitization of ACC using a MDM2 inhibitor in a patient-derived xenograft model. The combination of MDM2 inhibition and low-dose radiation causes tumor regression by reactivating p53 leading to growth arrest and apoptosis. These data add to the growing literature suggesting that MDM2 inhibition is a promising approach in ACC and suggest that patients with ACC could benefit from combination therapy of AMG 232 and radiation.
Acknowledgments
Financial support and disclosures: Supported in part by a Department of Human Oncology Seed Grant and the Karl Harter Scholarship Fund (RJK), CA160639 (RJK), a PhRMA Foundation Postdoctoral Fellowship in Translational Medicine and Therapeutics (ADS), University of Wisconsin Carbone Cancer Center Support Grant (P30 CA014520) and Wisconsin Head and Neck SPORE Grant (NIH P50 DE026787).
We would like to thank Ella Ward and University of Wisconsin Research Pathology facility for tissue processing and histology services. We also would like to acknowledge the University of Wisconsin Translational Research Initiatives in Pathology laboratory, in part supported by the UW Department of Pathology and Laboratory Medicine and UWCCC grant P30 CA014520, for use of its facilities and services including STR analysis. The author(s) thank the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing the hotspot sequencing facilities and services.
References
- 1.Bell RB, Dierks EJ, Homer L, Potter BE. Management and outcome of patients with malignant salivary gland tumors. J Oral Maxillofac Surg. 2005;63(7):917–28. doi: 10.1016/j.joms.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Spiro RH, Huvos AG, Strong EW. Adenoid cystic carcinoma of salivary origin. A clinicopathologic study of 242 cases. Am J Surg. 1974;128(4):512–20. doi: 10.1016/0002-9610(74)90265-7. [DOI] [PubMed] [Google Scholar]
- 3.Matsuba HM, Simpson JR, Mauney M, Thawley SE. Adenoid cystic salivary gland carcinoma: a clinicopathologic correlation. Head Neck Surg. 1986;8(3):200–4. doi: 10.1002/hed.2890080312. [DOI] [PubMed] [Google Scholar]
- 4.Spiro RH. Salivary neoplasms: overview of a 35-year experience with 2,807 patients. Head Neck Surg. 1986;8(3):177–84. doi: 10.1002/hed.2890080309. [DOI] [PubMed] [Google Scholar]
- 5.Adelstein DJ, Koyfman SA, El-Naggar AK, Hanna EY. Biology and management of salivary gland cancers. Semin Radiat Oncol. 2012;22(3):245–53. doi: 10.1016/j.semradonc.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Fordice J, Kershaw C, El-Naggar A, Goepfert H. Adenoid cystic carcinoma of the head and neck: predictors of morbidity and mortality. Arch Otolaryngol Head Neck Surg. 1999;125(2):149–52. doi: 10.1001/archotol.125.2.149. [DOI] [PubMed] [Google Scholar]
- 7.van der Wal JE, Becking AG, Snow GB, van der Waal I. Distant metastases of adenoid cystic carcinoma of the salivary glands and the value of diagnostic examinations during follow-up. Head Neck. 2002;24(8):779–83. doi: 10.1002/hed.10126. [DOI] [PubMed] [Google Scholar]
- 8.Dodd RL, Slevin NJ. Salivary gland adenoid cystic carcinoma: a review of chemotherapy and molecular therapies. Oral Oncol. 2006;42(8):759–69. doi: 10.1016/j.oraloncology.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Milano A, Longo F, Basile M, Iaffaioli RV, Caponigro F. Recent advances in the treatment of salivary gland cancers: emphasis on molecular targeted therapy. Oral Oncol. 2007;43(8):729–34. doi: 10.1016/j.oraloncology.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Laurie SA, Ho AL, Fury MG, Sherman E, Pfister DG. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: a systematic review. Lancet Oncol. 2011;12(8):815–24. doi: 10.1016/S1470-2045(10)70245-X. [DOI] [PubMed] [Google Scholar]
- 11.Agulnik M, Siu LL. An update on the systemic therapy of malignant salivary gland cancers: role of chemotherapy and molecular targeted agents. Curr Med Chem Anticancer Agents. 2004;4(6):543–51. doi: 10.2174/1568011043352641. [DOI] [PubMed] [Google Scholar]
- 12.Locati LD, Perrone F, Cortelazzi B, Bergamini C, Bossi P, Civelli E, et al. A phase II study of sorafenib in recurrent and/or metastatic salivary gland carcinomas: Translational analyses and clinical impact. Eur J Cancer. 2016;69:158–65. doi: 10.1016/j.ejca.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 13.Ho AL, Dunn L, Sherman EJ, Fury MG, Baxi SS, Chandramohan R, et al. A phase II study of axitinib (AG-013736) in patients with incurable adenoid cystic carcinoma. Ann Oncol. 2016;27(10):1902–8. doi: 10.1093/annonc/mdw287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong SJ, Karrison T, Hayes DN, Kies MS, Cullen KJ, Tanvetyanon T, et al. Phase II trial of dasatinib for recurrent or metastatic c-KIT expressing adenoid cystic carcinoma and for nonadenoid cystic malignant salivary tumors. Ann Oncol. 2016;27(2):318–23. doi: 10.1093/annonc/mdv537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keam B, Kim SB, Shin SH, Cho BC, Lee KW, Kim MK, et al. Phase 2 study of dovitinib in patients with metastatic or unresectable adenoid cystic carcinoma. Cancer. 2015;121(15):2612–7. doi: 10.1002/cncr.29401. [DOI] [PubMed] [Google Scholar]
- 16.Airoldi M, Pedani F, Succo G, Gabriele AM, Ragona R, Marchionatti S, et al. Phase II randomized trial comparing vinorelbine versus vinorelbine plus cisplatin in patients with recurrent salivary gland malignancies. Cancer. 2001;91(3):541–7. doi: 10.1002/1097-0142(20010201)91:3<541::aid-cncr1032>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 17.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 18.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soussi T, Ishioka C, Claustres M, Béroud C. Locus-specific mutation databases: pitfalls and good practice based on the p53 experience. Nat Rev Cancer. 2006;6(1):83–90. doi: 10.1038/nrc1783. [DOI] [PubMed] [Google Scholar]
- 20.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137(3):413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015;58(3):1038–52. doi: 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khoo KH, Hoe KK, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13(3):217–36. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- 25.Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016;6:7. doi: 10.3389/fonc.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nor F, Warner K, Zhang Z, Acasigua G, Pearson AT, Kerk S, et al. Therapeutic inhibition of the MDM2-53 interaction prevents recurrence of adenoid cystic carcinomas. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warner KA, Nör F, Acasigua GA, Martins MD, Zhang Z, McLean SA, et al. Targeting MDM2 for Treatment of Adenoid Cystic Carcinoma. Clin Cancer Res. 2016;22(14):3550–9. doi: 10.1158/1078-0432.CCR-15-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rew Y, Sun D. Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J Med Chem. 2014;57(15):6332–41. doi: 10.1021/jm500627s. [DOI] [PubMed] [Google Scholar]
- 29.Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014;57(4):1454–72. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- 30.Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol Cancer Ther. 2015;14(3):649–58. doi: 10.1158/1535-7163.MCT-14-0710. [DOI] [PubMed] [Google Scholar]
- 31.Werner LR, Huang S, Francis DM, Armstrong EA, Ma F, Li C, et al. Small Molecule Inhibition of MDM2-p53 Interaction Augments Radiation Response in Human Tumors. Mol Cancer Ther. 2015;14(9):1994–2003. doi: 10.1158/1535-7163.MCT-14-1056-T. [DOI] [PubMed] [Google Scholar]
- 32.Feng FY, Zhang Y, Kothari V, Evans JR, Jackson WC, Chen W, et al. MDM2 Inhibition Sensitizes Prostate Cancer Cells to Androgen Ablation and Radiotherapy in a p53-Dependent Manner. Neoplasia. 2016;18(4):213–22. doi: 10.1016/j.neo.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sjöblom B, Polentarutti M, Djinovic-Carugo K. Structural study of X-ray induced activation of carbonic anhydrase. Proc Natl Acad Sci U S A. 2009;106(26):10609–13. doi: 10.1073/pnas.0904184106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindskog S. Structure and mechanism of carbonic anhydrase. Pharmacol Ther. 1997;74(1):1–20. doi: 10.1016/s0163-7258(96)00198-2. [DOI] [PubMed] [Google Scholar]
- 35.Brill LB, Kanner WA, Fehr A, Andrén Y, Moskaluk CA, Löning T, et al. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod Pathol. 2011;24(9):1169–76. doi: 10.1038/modpathol.2011.86. [DOI] [PubMed] [Google Scholar]
- 36.Li C, Huang S, Armstrong EA, Francis DM, Werner LR, Sliwkowski MX, et al. Antitumor Effects of MEHD7945A, a Dual-Specific Antibody against EGFR and HER3, in Combination with Radiation in Lung and Head and Neck Cancers. Mol Cancer Ther. 2015;14(9):2049–59. doi: 10.1158/1535-7163.MCT-15-0155. [DOI] [PubMed] [Google Scholar]
- 37.Kimple RJ, Vaseva AV, Cox AD, Baerman KM, Calvo BF, Tepper JE, et al. Radiosensitization of epidermal growth factor receptor/HER2-positive pancreatic cancer is mediated by inhibition of Akt independent of ras mutational status. Clin Cancer Res. 2010;16(3):912–23. doi: 10.1158/1078-0432.CCR-09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swick AD, Stein AP, McCulloch TM, Hartig GK, Ong IM, Sampene E, et al. Defining the boundaries and expanding the utility of neck cancer patient derived xenografts. Oral Oncology. 2017;64:65–72. doi: 10.1016/j.oraloncology.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moskaluk CA, Baras AS, Mancuso SA, Fan H, Davidson RJ, Dirks DC, et al. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Lab Invest. 2011;91(10):1480–90. doi: 10.1038/labinvest.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLoS One. 2009;4(6):e6040. doi: 10.1371/journal.pone.0006040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.