

Abstract
Purpose
NRAS mutations in malignant melanoma are associated with aggressive disease requiring rapid antitumor intervention, but there is no approved targeted therapy for this subset of patients. In clinical trials, the MEK inhibitor (MEKi) binimetinib displayed modest antitumor activity, making combinations a requisite. In a previous study, the BRAF inhibitor (BRAFi) vemurafenib was shown to induce endoplasmic reticulum (ER) stress that together with inhibition of the RAF-MEK-ERK (MAPK) pathway amplified its pro-apoptotic activity in BRAF-mutant melanoma. The present study investigated whether this effect might extent to NRAS-mutant melanoma, in which MAPK activation would be expected.
Experimental design and results
BRAFi increased pERK, but also significantly increased growth inhibition and apoptosis induced by the MEKi in monolayer, spheroids, organotypic and patient-derived tissue slice cultures of NRAS-mutant melanoma. BRAFi such as encorafenib induced an ER stress response via the PERK pathway, as detected by phosphorylation of eIF2α and upregulation of the ER stress-related factors ATF4, CHOP and NUPR1 and the pro-apoptotic protein PUMA. MEKi such as binimetinib induced the expression of the pro-apoptotic protein BIM and activation of the mitochondrial pathway of apoptosis, the latter of which was enhanced by combination with encorafenib. The increased apoptotic rates caused by the combination treatment were significantly reduced through siRNA knockdown of ATF4 and BIM, confirming its critical roles in this process.
Conclusion
The data presented herein encourage further advanced in vivo and clinical studies to evaluate MEKi in combination with ER stress inducing BRAFi as a strategy to treat rapidly progressing NRAS-mutant melanoma.
Keywords: Melanoma, NRAS-mutant, encorafenib, binimetinib, endoplasmic reticulum stress, apoptosis
Introduction
15–25% of all melanoma harbor activating NRAS mutations. Although not all studies support this notion (1–4), NRAS mutations generally appear to be an independent predictor of poor survival and are associated with rapid tumor progression and an increased incidence of brain metastases. Treatment for patients with NRAS mutations is currently limited to immune checkpoint inhibitors or chemotherapy. Unfortunately, chemotherapy produces only low response rates and no durable responses, while immunotherapy achieves durable responses, but tend to be slow acting. Thus, there is a high unmet medical need for fast-acting, first-line therapy options, and also effective second-line therapies after failure of immune checkpoint inhibitors in these patients.
In vitro, some NRAS-mutant melanoma cell lines are sensitive to MEK inhibition (5). In a phase II trial, the MEK inhibitor binimetinib showed activity in patients with NRAS-mutant melanoma with an overall response rate of 21% and a median progression-free survival of 3.65 months (6). In a recent phase III study, patients with NRAS-mutant metastatic melanoma without previous therapy or after prior immunotherapy were treated with the MEKi binimetinib or chemotherapy (DTIC) (7). Overall response rates were 15% for binimetinib vs. 7% for DTIC. Even though an overall survival benefit was not observed for binimetinib vs. DTIC, binimetinib displayed a clear benefit over DTIC in patients with prior immunotherapy (median progression-free survival 5.5 vs. 1.6 months). Moreover, in a phase Ib/II study of the MEKi binimetinib in combination with the CDK4/6 inhibitor LEE011 partial responses were achieved in 33% of patients with NRAS-mutant metastatic melanoma. However, this combination was associated with frequent adverse events (8). Altogether, these clinical data suggest that the MEKi binimetinib provides a backbone for combination treatments with other targeted therapies or immunotherapies.
Several groups observed that the BRAFi vemurafenib induces both, inhibition of the MAPK signaling pathway and induction of ER stress in the setting of activating BRAF mutations (9,10). ER stress is caused by multiple factors such as Ca2+ imbalance, hypoxia, nutrient deprivation or perturbation of protein glycosylation, leading to the accumulation of unfolded proteins in the ER that can then trigger the unfolded protein response (UPR) (11–13). The UPR pathway is initiated by three sensors, the PKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) that are normally kept in check by binding to the chaperone protein glucose-regulated protein 78 (GRP78), but are released upon ER stress. The UPR reduces the accumulation of unfolded proteins by inducing UPR gene transcription, reducing global protein synthesis, and stimulating ER-associated protein degradation in order to restore normal ER function. If normal ER function cannot be restored, the UPR switches modes from pro-survival to pro-apoptosis (13–15).
In our previous study, we demonstrated that the BRAFi vemurafenib raised cytosolic Ca2+ levels, suppressed the ER chaperone protein GRP78 and induced phosphorylation of the α-subunit of the eukaryotic initiation factor 2 (eIF2α) downstream of PERK. These effects led to an increased expression of ER stress-related genes, including nuclear protein 1 (NUPR1), activating transcription factor 4 (ATF4), and growth arrest and DNA-damage-inducible transcript 3 (DDIT3/CHOP) (9). In cooperation with increased expression of pro-apoptotic BIM, these activities induced intrinsic apoptosis.
Next to inhibition of the MAPK pathway, ER stress induction thus appeared to be a secondary event of vemurafenib that remarkably enhances its pro-apoptotic activity in BRAFV600-mutant melanoma.
In this study we explored whether BRAFi-induced ER stress could be utilized to amplify the pro-apoptotic activity of MEKi in NRAS-mutant melanoma, despite the paradoxical activation of the MAPK pathway in this setting.
Materials and Methods
Isolation and culture of human cells
The use of human skin tissues was approved by the Medical Ethics committee of the University of Tübingen. Experiments were performed in accordance with the Declaration of Helsinki.
Melanoma cell lines were kindly provided by M. Herlyn (451Lu, Mel1617, WM1346, WM1366), A. Bosserhoff (MelJuso) or purchased from ATCC (SKMel19, SKMel147). Melanoma cell lines, cells isolated from excised melanoma metastases, melanocytes, keratinocytes and fibroblasts were isolated and cultured as described previously (16–18). All melanoma cell lines were used within 3 months of thawing the frozen stock. Mycoplasma infection in the cells was regularly checked using a Venor GeM Classic Mycoplasma Detection Kit (Minerva Biolabs).
Melanoma spheroids were generated and prepared via the “hanging drop” method as described previously (19).
Establishment of resistant cell lines
The resistant cell lines were generated by continuous treatment with increasing concentrations of encorafenib or/and binimetinib for 5 month, starting at 0.1 μM and increasing the concentration with every passage up to 10 μM or 5 μM, respectively.
Signaling pathway inhibitors and treatments
Thapsigargin and QVD-OPH were purchased from Sigma. Dabrafenib, encorafenib, binimetinib, trametinib and Raf265 were purchased from Selleck Chemicals, while vemurafenib was from Roche. The combination indices (CI’s) showing the synergism between various BRAF and MEK inhibitor combinations were calculated using COMPUSYN based on the methodology and stratification of T.C. Chou (20) and displayed in Table S1. All chemicals were dissolved in dimethylsulfoxide (DMSO) and added directly to the cell culture medium.
Anchorage-independent (long-term) growth assay
Anchorage-independent growth was assessed as described earlier (21).
Growth inhibition assay
Melanoma cell lines and cells from melanoma biopsies were tested for growth inhibition using the 4-methylumbelliferyl heptanoate (MUH) assay as described previously (21). Briefly, cells were treated with inhibitors or with DMSO (at the highest combined DMSO concentration of the inhibitors) for 72 h and were then incubated with 100 μg/ml MUH in PBS for 1 h at 37°C. The absolute fluorescence intensity at λex of 355 nm and λem of 460 nm was measured using a Fluoroskan II (Labsystems).
Cell cycle analysis to measure apoptotic cells
Cell cycle analyses were described previously (21).
Western Blotting
Total protein was extracted from cell pellets, separated by SDS-PAGE (15–60 μg/lane), blotted onto PVDF membranes (Roche) and probed with antibodies listed in Table S2. Proteins were visualized with secondary peroxidase-conjugated antibodies (Cell Signaling) and the ECL detection system (Thermo). Band intensities for peIF2a in comparison to eIF2 were quantified using the Image J software.
Cell Death ELISA
Cell lysates were prepared from monolayer and spheroid cultures. Nucleosomal enrichment in the cytosolic fraction during apoptotic cell death was determined with the Cell Death Detection ELISA kit (Roche) according to manufacturer’s instructions and was calculated as: absorbance of sample cells/absorbance of control cells. The enrichment factor of 2 corresponds to 10% apoptotic cells as determined by FACS analysis.
Gene expression
Quantitative real-time PCR (RT-PCR) was performed with the LightCycler® 480 (Roche) using the Kapa SYBR Fast System (VWR). RNA extraction (MACHERY-NAGEL) and reverse transcription (Invitrogen) were performed according to manufacturer’s instructions. 25 ng cDNA was used for PCR analyses. Primer sequences are listed in Table S3. PCR product specificity was verified by melting curve analysis.
Organotypic skin culture
Melanoma cells were seeded onto human dermal reconstructs as described previously (18). Reconstructs were cultured for 4 days, treated with the inhibitors for another 10 days before fixation with 4% formalin and embedment in paraffin. Sections were stained with either hematoxylin and eosin (H&E) or a α-Ki67 antibody (DAKO #M7240) and the UltraView Universal Alkaline Phosphatase Red Detection kit from Ventana (Tucson).
Tumor slice cultures
Patient tissue samples were expanded in a patient-derived xenograft (PDX) mouse model by implanting the digested tumor tissue subcutaneously, as described previously (22). After expansion, tumors were excised and cut with the Leica Microtome VT1200S into 400 μm slices. The slices were treated for 4 days with BRAF and MEK inhibitors in quadruplicates and then analyzed by confocal microscopy. H&E stainings of the two NRAS-mutant human and corresponding mouse tumors were performed (Fig. S6A), revealing a similar appearance and structure of the matching tumor tissues.
Immunofluorescence staining and confocal microscopy
Immunofluorescence staining was performed as described previously (23) using a confocal laser scanning microscope (Leica) and the antibodies listed in Table S2. Cell nuclei were stained with YO-PRO-1 (Thermo).
Transmission electron microscopy
Electron microscopy was performed as described previously (9).
Transfection experiments
Genes were knocked down with pre-validated, small interfering RNAs (siRNAs) against ATF4 (Qiagen, SI03019345) and BIM (Cell Signaling, 6518). As control siRNA a non-silencing, luciferase-specific siRNA (Biomers) was used. Cells were transfected with 25 nM siRNA using RiboxxFectTM (Riboxx) according to manufacturer’s instructions and were cultured for 6 h prior to drug treatment.
Statistical analysis
GraphPad Prism version 7.0 (GraphPad Software) was used for statistical analysis. P-value calculation and significance determination were performed using one-way ANOVA followed by Tukey’s multiple comparisons test. P-values <0.05 were considered statistically significant, with * for p<0.05, ** for p<0.01, *** for p<0.001 and **** for p<0.0001.
Results
MEKi combined with BRAFi inhibit ERK phosphorylation as well as cell growth and induce apoptosis in NRAS-mutant melanoma cells
To investigate whether BRAFi increased the antitumor activity of MEKi in NRAS-mutant melanoma, we tested a range of melanoma cell lines for ERK phosphorylation, growth inhibition and apoptosis induction following treatment with MEKi or/and BRAFi.
As described previously (24), BRAFi (encorafenib, dabrafenib, vemurafenib) and the panRAFi (Raf265) increased ERK phosphorylation in NRAS-mutant melanoma cells (Fig S1A and S2A). However, the MEKi binimetinib was able to counteract this paradoxical activation of the MAPK pathway, resulting in complete inhibition of ERK phosphorylation (Fig. 1A and S2A).
Figure 1. The MEKi binimetinib combined with the BRAFi encorafenib leads to strong growth inhibition and apoptosis induction in NRAS-mutant melanoma cells.

(A) Whole cell lysates from NRAS- or BRAF-mutant melanoma cells treated with encorafenib or/and binimetinib or DMSO as a control for 24 h were subjected to Western blot analysis to detect pERK, ERK and β-Actin. Experiment shown is a representative of three independent experiments. (B) Melanoma cells were treated with encorafenib or/and binimetinib for 72 h. The percentage of growth inhibition was calculated, normalized to the DMSO-treated control. One representative experiment of two is shown (mean ± SD of quadruplicates). (C) Melanoma cells were treated with the inhibitors or DMSO as a control for 72 h. Cell cycle analysis was performed and displayed in a bar graph with apoptotic cells (sub-G1 fraction) in black (mean ± SD of duplicates from three independent experiments). (D; E) Melanoma cells grown in monolayer (D) or spheroids (E) were treated with the inhibitors or DMSO for 48 h. Nucleosomal enrichment in cytosolic fractions (apoptotic cell death) was calculated and displayed in a bar graph (mean ± SD of triplicates from three independent experiments).
In short-term proliferation assays, encorafenib treatment had a minimal effect on the growth of NRAS-mutant melanoma cell lines (SKMel147, WM1366, MelJuso, WM1346) or patient-derived NRAS-mutant melanoma cells, while binimetinib caused moderate growth inhibition (Fig. 1B and S1B). The combination of encorafenib plus binimetinib achieved synergistic growth inhibition (Table S1) with growth-inhibitory rates of >80% (Fig. 1B and S1B). In BRAF-mutant melanoma cells (SKMel19, Mel1617, 451Lu), encorafenib or binimetinib alone led to a marked growth inhibition of 75% that was slightly enhanced by combination of both inhibitors (Fig. 1B and S1C). Of note, consistent with the paradoxical hyperactivation of the MAPK kinase pathway, BRAFi alone caused acceleration of growth in NRAS-mutant melanoma cell lines in a long-term growth assay, which could be inhibited by addition of a MEK inhibitor (Fig S1D).
Cell cycle analysis of NRAS-mutant melanoma cells revealed that binimetinib but not encorafenib increased the sub-G1 fraction (apoptotic fraction) (Fig. 1C and S1E). The combination of binimetinib plus encorafenib further enhanced the number of apoptotic cells up to 40%. A nucleosomal enrichment assay of cytosolic fractions during apoptosis showed similar results (Fig. 1D): moderate apoptosis after binimetinib treatment alone (4-fold enrichment) that was further enhanced (nearly 10-fold enrichment) with the combination of binimetinib plus encorafenib. Both apoptosis assays revealed high apoptosis rates in BRAF-mutant melanoma cells with encorafenib or binimetinib alone that were further increased with combination treatment (Fig. 1C–D and S1F).
Of note, combination of binimetinib with Raf265, also led to increased growth inhibition and apoptosis induction in NRAS- and BRAF-mutant melanoma cells compared to the MEKi and panRAFi single treatments (Fig S2B–C).
Of relevance for clinical translation of these findings, other BRAF and MEK inhibitors such as dabrafenib and trametinib caused similar growth inhibition and apoptosis in NRAS-mutant and BRAF-mutant melanoma cells, respectively (Fig. S3A–C and Table S1).
Together, these data illustrate that BRAFi augment the growth inhibiting and apoptosis-inducing effects of MEKi in NRAS-mutant melanoma cells.
Binimetinib plus encorafenib does not induce apoptosis in cells from normal human skin
In fibroblasts, melanocytes and keratinocytes, encorafenib treatment increased ERK phosphorylation, which was partially, and in keratinocytes completely, inhibited with the combination treatment (Fig. S4A). In contrast to melanoma cells (Fig. 1B and S1B), growth inhibition rates of normal skin cells after binimetinib or/and encorafenib treatment remained below 40% (Fig. S4B). Furthermore, cell cycle analyses indicated that apoptosis was not induced in these cells with neither treatment (Fig. S4C). Our data together with the corresponding clinical data (25) shows that the effects of BRAFi and MEKi treatment on normal skin cells are tolerable.
MEKi combined with BRAFi display antitumor activities in spheroids, organotypic and tissue slice cultures
To test whether binimetinib plus encorafenib also affects survival of NRAS-mutant melanoma cells in a more physiological context, we first assessed apoptotic cell death of melanoma cells in a 3D melanoma spheroid model. As observed in the NRAS-mutant monolayer culture (Fig. 1D), in the spheroid culture, binimetinib but not encorafenib induced apoptosis that was further enhanced by the combination treatment (Fig. 1E). In BRAF-mutant cells, binimetinib and encorafenib alone induced apoptosis that was further increased by the combination treatment.
Melanoma cells were seeded onto human dermal reconstructs and treated with encorafenib or/and binimetinib. Histological sections of these reconstructs were stained with H&E to identify melanoma cells (cells with dense, blue nucleus and small cytoplasm) invading the dermis. In addition, sections were stained for Ki67 to visualize proliferating melanoma cells. In NRAS-mutant melanoma cells, encorafenib did not inhibit proliferation or invasive tumor growth. Binimetinib inhibited proliferation but not invasion of the tumor cells into the skin reconstructs, while binimetinib plus encorafenib completely abolished proliferation and invasive tumor growth (Fig. 2A). In BRAF-mutant melanoma cells, encorafenib and binimetinib were effective as mono- or combination treatment.
Figure 2. MEKi combined with BRAFi inhibit invasive tumor growth in organotypic skin cultures and lead to apoptosis induction and reduction in proliferation in patient-derived tumor slice cultures.

(A) Melanoma cells were seeded onto organotypic skin reconstructs, incubated for 4 days and then treated for 10 days with DMSO (control) or 0.1 (BRAF-mutant cells) and 1 μM (NRAS-mutant cells) encorafenib or/and binimetinib. Fixed tissue sections were stained with H&E or Ki67. An orange line marks the tumor front. T = tumor cells, D = dermis, Scale bar = 10 μm. (B) Tumor tissue from patients with NRAS-mutant melanoma was expanded in a PDX model, sliced and then treated with the indicated inhibitor combinations. After 4 days the slices were stained with YO-PRO-1 (nuclei marker), Ki67 (proliferation marker) or cleaved PARP (apoptosis marker) and analyzed by confocal microscopy. Images of one representative per treatment group are shown. The percentage of apoptotic and proliferating cells was quantified and displayed in a bar graph (error bars represent SD of 4 random sites in each tumor per treatment group). (C) NRAS-mutant patient-derived melanoma cells were treated with the indicated inhibitors for 72 h. The percentage of growth inhibition was calculated, normalized to the DMSO-treated control (mean ± SD of quadruplicates).
To demonstrate the efficacy of this combination treatment on NRAS-mutant melanoma metastases in tissue context (Fig. S6A), tumor tissue excised from two patients with NRAS-mutant melanoma was expanded in a mouse PDX model and then cut into slices. After treatment of the slices with MEKi and BRAFi, the tumor cells in these tissue slice cultures displayed proliferation inhibition and apoptosis induction compared to the untreated controls (Fig. 2B). Furthermore, in monolayer cell cultures derived from the same tumor tissue, MEKi combined with BRAFi significantly enhanced growth inhibition (>80%) compared to MEKi alone (Fig. 2C). These results underscore the therapeutic potential of this combination treatment for patients with NRAS-mutant melanoma.
BRAFi induces ER swelling and upregulation of ER stress-related transcription factors in NRAS-mutant melanoma cells
We next determined whether BRAFi caused ER stress in NRAS-mutant melanoma, as we previously described for BRAF-mutant melanoma (9).
Using electron microscopy, we found that dabrafenib, vemurafenib and in particular encorafenib induced morphological features of ER stress with significant dilation of the ER in both NRAS-mutant and BRAF-mutant cells (Fig. 3A). Similar effects on the ER were observed with the combination of encorafenib plus binimetinib, while binimetinib alone did not appear to affect the ER.
Figure 3. BRAF inhibitors alone and in combination with MEK inhibitors induce ER swelling, eIF2α phosphorylation and expression of NUPR1, ATF4 and CHOP.

(A) EIectron microscopy of melanoma cells treated with dabrafenib (1 μM), vemurafenib (6 μM), encorafenib (1 μM) or/and binimetinib (1 μM) or DMSO for 6 h. The cell nuclei are shown in blue. The endoplasmic reticulae are shown in green and additionally marked by red arrows. Scale bar = 0.5 μm, n = 2. (B) Whole cell lysates from melanoma cells treated with the inhibitors or DMSO as a control for the indicated times were subjected to Western blot analysis to detect peIF2α, eIF2α and β-Actin. Experiment shown is a representative of two independent experiments. Quantification of the peIF2α band intensities in comparison to eIF2α is displayed in the bottom row. (C) Melanoma cells were treated with the inhibitors for 18 h. Real-time PCR results show levels of NUPR1, ATF4 and CHOP mRNA expression in comparison with DMSO treated cells. All samples were normalized to TBP mRNA (mean ± SD of triplicates from three independent experiments). (D) Melanoma cells were treated with the inhibitors or DMSO for 24 h. Whole cell lysates were subjected to Western blot analysis of NUPR1, ATF4 and CHOP in comparison to β-Actin as loading control. Experiment shown is a representative of three independent experiments.
Upon ER stress, the sensor PERK is activated, leading to phosphorylation of eIF2α. Encorafenib alone or in combination with binimetinib caused a 1.5 to 2-fold increase in peIF2α in both NRAS-mutant melanoma cell lines, while peIF2α was increased 7 to 12-fold in the BRAF-mutant melanoma cell line (Fig. 3B).
Encorafenib and more pronounced encorafenib plus binimetinib also induced expression of the ER stress-related transcription factor genes ATF4, CHOP and NUPR1 downstream of eIF2α phosphorylation in the NRAS-mutant melanoma cells (Fig. 3C). In BRAF-mutant cells, treatment with encorafenib alone or in combination increased expression of these genes. Similar results were obtained with dabrafenib and trametinib (Fig. S3D). In addition, combination of Raf265 and binimetinib also led to increased expression of the ER stress-related transcription factors (Fig. S2D). Expression of NUPR1, ATF4 and CHOP proteins was elevated with encorafenib and encorafenib plus binimetinib in NRAS- and BRAF-mutant melanoma cells, while binimetinib alone only minimally affected levels of these ER stress factors (Fig. 3D).
In addition to activation of the PERK pathway, ER stress also induces activation of the ATF6 and IRE1 pathway, which can be monitored by cleavage of ATF6 and by phosphorylation of IRE1α. Interestingly, in contrast to the classical ER stress inducer thapsigargin, encorafenib and/or binimetinib treatment did not cause phosphorylation of IRE1α or cleavage of ATF6 (Fig. S5).
Altogether, these data indicate that BRAFi trigger an ER stress response via the PERK pathway in melanoma cells.
Apoptosis induction by binimetinib combined with encorafenib is mediated by caspase 9 and caspase 3 activation in NRAS-mutant melanoma cells
So far, we have determined that BRAFi cause ER stress and boost apoptosis induced by MEKi in NRAS-mutant melanoma cells. We thus wanted to further investigate the underlying mechanisms involved in apoptosis induction.
Inhibition of caspase activity with the pan-caspase inhibitor QVD almost completely abrogated apoptosis induction caused by treatment with binimetinib alone or in combination with encorafenib in the NRAS-mutant cells, and also inhibited apoptosis induced by any treatment in the BRAF-mutant cells (Fig. 4A).
Figure 4. Binimetinib and encorafenib induced apoptosis is mediated by the pro-apoptotic BH3-only protein BIM and ATF4.

(A) Melanoma cells were pretreated for 1 h without or with 1 μM pan-caspase inhibitor QVD-OPH and then treated with encorafenib or/and binimetinib or DMSO as control for 72 h. Cell cycle analysis was performed and displayed in a bar graph with apoptotic cells (sub-G1 fraction) in black (mean ± SD of duplicates from three independent experiments). (B) Melanoma cells were treated with the inhibitors or DMSO for the indicated time points. Western Blot analysis of whole cell lysates was performed to detect cleavage of caspase 3 and 9 as well as PARP compared to β-Actin as a loading control. Experiment shown is a representative of two independent experiments. (C) Melanoma cells were treated with the inhibitors or DMSO for 24 h. Western Blot analysis of whole cell lysates was performed to detect BIM isoforms (EL, L, S) and PUMA compared to β-Actin as a loading control. Experiment shown is a representative of two independent experiments. (D) Melanoma cells were transfected with siCtrl or siRNA directed against BIM prior to treatment with encorafenib plus binimetinib or DMSO. Western blot analysis of whole cell lysates was performed after 24 h to detect BIM isoforms compared to β-Actin as a loading control. Experiment shown is a representative of three independent experiments. (E; F) Melanoma cells were transfected with siCtrl or siRNA directed against ATF4 prior to treatment with or without (DMSO) encorafenib plus binimetinib. (E) Western blot analysis of whole cell lysates was performed after 24 h to detect ATF4 compared to β-Actin as a loading control. Experiment shown is a representative of three independent experiments. (F) After 18 h, real-time PCR was performed to measure ATF4, NUPR1, CHOP and PUMA mRNA expression in comparison with DMSO treated cells. All samples were normalized to TBP mRNA (one representative experiment of three is shown, mean ± SD of triplicates). (G) Melanoma cells were transfected with control, siRNA directed against BIM or siRNA directed against ATF4 prior to treatment with encorafenib plus binimetinib or DMSO. Cell cycle analysis was performed and the sub-G1 fraction was displayed in a bar graph (mean ± SD of duplicates from three independent experiments).
Performing Western blot analysis we next characterized the activation of members of the intrinsic caspase cascade. In NRAS-mutant melanoma cells, processing of caspase 9, caspase 3 and the caspase product PARP was observed in response to binimetinib treatment alone, but was much more distinct when combined with encorafenib (Fig. 4B). In contrast, encorafenib treatment alone did not show processing of these proteins. In BRAF-mutant melanoma cells, all 3 treatment strategies caused cleavage of caspase 9, caspase 3 and PARP. These results indicate that the combination of binimetinib plus encorafenib induces apoptosis via the intrinsic mitochondrial pathway in NRAS-mutant melanoma cells.
Binimetinib combined with encorafenib upregulate the pro-apoptotic proteins BIM and PUMA in NRAS-mutant melanoma cells
Pro-apoptotic Bcl-2 proteins like BIM, PUMA and NOXA mediate the activation of the intrinsic mitochondrial apoptosis pathway. Induction of BIM by MEKi has been reported (26), prompting us to investigate whether BIM is induced in NRAS-mutant melanoma cells following MEKi or/and BRAFi. In NRAS-mutant melanoma cells, binimetinib alone and in combination with encorafenib increased the expression of the three BIM isoforms, while encorafenib alone showed no effect on any BIM isoform (Fig. 4C). In BRAF-mutant melanoma cells, binimetinib or/and encorafenib resulted in an increase of the three BIM isoforms. Analogous with BIM, binimetinib alone and in combination with encorafenib caused down-regulation of the anti-apoptotic proteins MCL-1 and BCL-XL and the pro-apoptotic protein NOXA in NRAS-mutant cells, while encorafenib had a minimal effect on these Bcl-2 proteins (Fig. S7).
PUMA has been reported to mediate ER stress-induced apoptosis (27,28). In both NRAS- and BRAF-mutant melanoma cells, elevated levels of PUMA were detected following treatment with encorafenib or encorafenib plus binimetinib but not with binimetinib alone, suggesting that PUMA is upregulated by encorafenib through ER stress induction (Fig. 4C). In accordance with this, PUMA expression was also increased in NRAS-mutant tissue slice cultures after treatment with BRAFi and MEKi compared to the untreated controls (Fig. S6B).
Together these data indicate that in NRAS-mutant melanoma cells the MEKi binimetinib induces the pro-apoptotic protein BIM and the BRAFi encorafenib induces the pro-apoptotic protein PUMA, possibly causing the marked apoptosis seen in NRAS-mutant cells after combination treatment.
ATF4 and BIM play important roles in apoptosis induction by binimetinib and encorafenib
We previously showed that both, the pro-apoptotic protein BIM and the ER stress-related factor ATF4 play relevant roles in apoptosis induction by the vemurafenib in BRAF-mutant melanoma cells (9).
In NRAS-mutant melanoma cells treated with binimetinib plus encorafenib, knockdown of BIM by siRNA inhibited up-regulation of all three BIM isoforms (Fig. 4D) and significantly suppressed apoptosis (Fig. 4G).
In NRAS-mutant melanoma cells treated with binimetinib plus encorafenib, siRNA-mediated knockdown of ATF4 reduced not only ATF4 itself (Fig. 4E), but also other ER-stress related factors such as CHOP and NUPR1 as well as the pro-apoptotic protein PUMA (Fig. 4F). Furthermore, ATF4 knockdown significantly inhibited apoptosis in these cells (Fig. 4G).
These findings indicate that the pro-apoptotic protein BIM and the ER stress-related factor ATF4 are important factors in NRAS-mutant melanoma cells during induction of apoptosis by binimetinib plus encorafenib.
NRAS-mutant cells resistant to encorafenib plus binimetinib show hyperactivation of the MAP kinase pathway and adaptation to ER stress
We established NRAS-mutant WM1366 melanoma cell lines resistant to encorafenib, binimetinib or the combination of both drugs.
Double-drug resistant cells showing neither growth inhibition nor apoptosis following treatment with encorafenib or/and binimetinib were obtained after 5–6 months of treatment (Fig. 5A and B). Interestingly, single-drug resistant cells were partially sensitive to the combination treatment in terms of growth inhibition and apoptosis induction (Fig. S8A and B), proposing the use of upfront combination therapy in NRAS-mutant melanoma.
Figure 5. NRAS-mutant cells resistant to the combination of encorafenib and binimetinib show hyperactivation of the MAP kinase pathway and adaptation to ER stress.

Melanoma cells resistant or sensitive to encorafenib plus binimetinib were treated with or without (DMSO) this combination. (A) After 72 h, the percentage of growth inhibition was calculated, normalized to the DMSO-treated control. One representative experiment of two is shown (mean ± SD of quadruplicates). (B) After 72 h, cell cycle analysis was performed and displayed in a bar graph with apoptotic cells (sub-G1 fraction) in black (mean ± SD of duplicates from three independent experiments). (C) After 24 h, Western blot analysis of whole cell lysates was performed to detect pERK, ERK, ATF4, BIM EL, L, S, PUMA, cleaved PARP and PARP compared to β-Actin as a loading control. Experiment shown is a representative of two independent experiments. (D) After 18 h, real-time PCR was performed to measure ATF4, NUPR1, CHOP mRNA expression in comparison with DMSO treated cells. All samples were normalized to TBP mRNA (one representative experiment of two is shown, mean ± SD of triplicates).
Double-drug resistant cells displayed a strong increase in phosphorylated ERK, demonstrating hyperactivation of the MAPK pathway that could not be counteracted by treatment with binimetinb plus encorafenib (Fig. 5C). In double-drug resistant cells treated with binimetinib plus encorafenib, expression of ATF4, CHOP and NUPR was only slightly increased and BIM and PUMA were only marginally altered (Fig. 5C, 5D and S8C). Furthermore, combination treatment hardly led to PARP cleavage in these cells (Fig. 5C), consistent with the observed lack of apoptotic cell death.
These data illustrate the relevance and interplay of significant ER stress induction by BRAFi and robust MAP kinase pathway inhibition by MEKi to induce efficient apoptosis in NRAS-mutant melanoma cells.
Discussion
Finding effective therapies for patients with aggressive NRAS-mutant melanoma is one of the current major aims in the field of melanoma research. In a recent phase III study (7), the MEKi binimetinib was superior to chemotherapy with dacarbazine but failed to establish an overall survival benefit. However, the demonstrated antitumor activity of binimetinib provides a solid basis for combination treatment strategies in patients with NRAS-mutant metastatic melanoma.
The present study shows for the first time that BRAF inhibitors can augment the antitumor effects of MEK inhibitors in NRAS-mutant melanoma. While BRAF inhibitors like encorafenib produce MAPK activation, co-administration of a MEK inhibitor can not only overcome this effect, but also lead to greater cell killing than observed with MEK inhibition alone. The potent growth-inhibiting and pro-apoptotic effects of BRAFi combined with MEKi on tissue slice cultures of metastases derived from NRAS-mutant melanoma patients further supports the potential clinical relevance of the combination strategy in this patient group given the inclusion of additional components of the tumor microenvironment.
This study also provides the first insight into the mechanisms of BRAF and MEK inhibitor activity in NRAS-mutant melanoma cells. ER stress induction by BRAFi together with MAPK pathway inhibition by MEKi culminate in the activation of the mitochondrial apoptosis inducing pathway (Fig. 6). The decisive role of the ER stress and MAPK pathways during BRAFi and MEKi treatment is further strengthened by the observed hyperactivation of the MAPK pathway, abrogation of ER stress induction and lack of apoptosis in treated double-drug resistant NRAS-mutant melanoma cells.
Figure 6. Proposed mechanism of MEK and BRAF inhibitor dependent induction of apoptosis in NRAS-mutant melanoma cells.
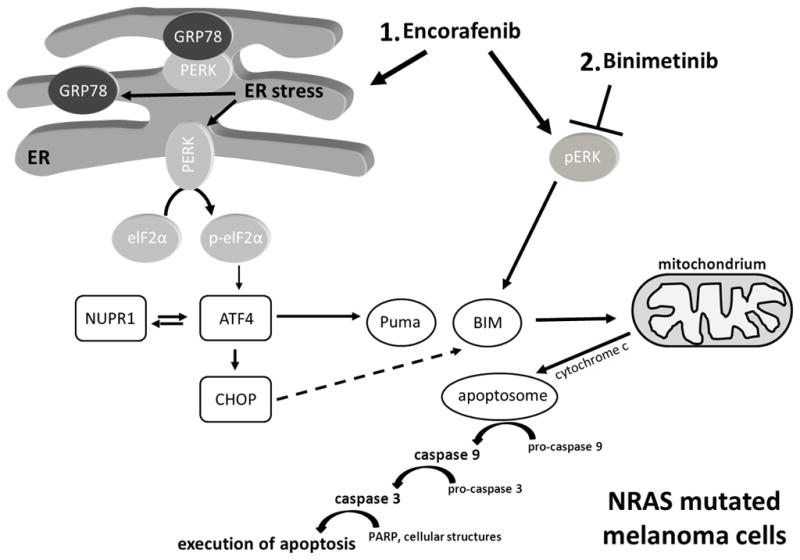
BRAF inhibitors such as encorafenib activate the MAPK pathway (increase of pERK) in NRAS-mutant melanoma cells. Additionally, they induce ER stress, thereby activating the downstream ER stress pathway, including phosphorylation of eIF2α and expression of ER stress-related genes and proteins such as ATF4, NUPR and CHOP as well as the pro-apoptotic Bcl-2 family member PUMA. Inhibitors of the MAPK pathway such as binimetinib are able to counteract the increased ERK phosphorylation by BRAFi and also cause upregulation of the pro-apoptotic Bcl-2 family member BIM. Both Bcl-2 proteins affect the mitochondrial pathway of apoptosis, causing an activation of caspases to induce PARP cleavage and ultimately apoptosis in NRAS-mutant melanoma cells.
First data on the mechanisms that underlie ER stress induction by BRAFi were provided by Ma and colleagues. They reported that BRAFi enhance binding of the mutant BRAFV600E protein to the chaperone protein GRP78, thereby reducing GRP78 binding to PERK and promoting activation of PERK (10). The data from our study demonstrate that BRAFi also cause activation of the PERK sub-pathway in NRAS-mutant melanoma, indicating that ER stress may be initiated in a similar manner in a BRAFwt setting. The fact that BRAFi such as vemurafenib can also bind to BRAFwt (29) would support this hypothesis.
Cerezo et al. (30) reported on the development of the small molecule HA15 that displayed antitumor activity in both BRAF-mutant and NRAS-mutant melanoma cell lines and in melanoma xenograft mouse models. HA15 appears to interact directly with the chaperone protein GRP78, leading to melanoma cell death by significantly increasing ER stress and activating the UPR. Analogous to our findings with BRAFi in BRAF-mutant (9) and NRAS-mutant melanoma (this study), HA15 caused rapid phosphorylation of the downstream target of PERK, eIF2α, and expression of the downstream transcription factors ATF4 and CHOP. In addition to activating the PERK pathway, HA15 also caused activation of the ATF6 and IRE1 branches of the ER stress pathway, while the action of the BRAFi encorafenib was confined to the PERK branch. Activation of individual arms of the UPR has also been described for other cellular stresses, indicating that different factors can trigger unique ER stress pathway profiles (31).
Altogether, these findings highlight the key role of the ER stress axis/UPR pathway in melanoma and strengthen the idea that ER stress inducers could be useful in melanoma treatment.
Interestingly, HA15 induced both apoptosis and autophagy, culminating in cell death that could be blocked by QVD and siRNA against the autophagy protein LC3 (30). In our system, BRAFi alone was not capable of inducing apoptosis in NRAS-mutant melanoma. However, combinations of BRAFi and MEKi displayed apoptosis rates higher than those achieved by the MEKi alone, suggesting that ER stress induced by BRAFi leads to apoptosis when combined with additional cell death triggers. Long-term ER stress has been shown to cause apoptotic cell death through ATF4-CHOP-mediated induction of pro-apoptotic or suppression of anti-apoptotic members of the Bcl-2 family (28,32), the guardians of the mitochrondrial pathway of apoptosis. The pro-apoptotic Bcl-2 proteins BIM, PUMA and NOXA are known to play a role in ER stress-mediated apoptosis (27). In our study, encorafenib treatment increased the expression of ATF4, CHOP and NUPR1 and induced the expression of PUMA. Furthermore, silencing ATF4 using siRNA reduced transcription of CHOP, NUPR1 and PUMA, resulting in diminished apoptosis following treatment with encorafenib plus binimetinib. This is consistent with two reports showing that siRNA against ATF4 reduces NUPR1 expression and that ATF4-CHOP-mediated ER-stress causes neuronal apoptosis via PUMA induction (28,33).
Our data demonstrate that binimetinib induces cell death through the mitochondrial pathway of apoptosis in NRAS-mutant melanoma. Binimetinib caused activation of the caspase 9 and caspase 3 and cleavage of PARP. The apoptotic activity of binimetinib was enhanced by combination treatment with encorafenib, possibly via increased expression of PUMA, as discussed above. Binimetinib did not induce PUMA, but increased expression of BIM (all three isoforms). Importantly, knockdown of BIM using siRNA diminished apoptosis induction by the combination treatment.
In agreement with our data, inhibition of the MAPK pathway was found to increase the expression of BIM and to induce apoptosis in melanoma cells (26). In accordance, Jiang and co-workers showed that increased expression of the short BIM isoform, BIM S, is a key mechanism underlying apoptosis induction by the BRAFi PLX4720 (34).
Altogether, we have shown that MEKi induces pro-apoptotic BIM and that BRAFi induces pro-apoptotic PUMA, which together promote apoptosis in NRAS-mutant melanoma.
In summary, we demonstrate that BRAFi cause ER stress in NRAS-mutant melanoma cells, thereby sensitizing these cells to MEKi-induced apoptosis. This study thus provides a rationale for investigating MEKi and ER stress inducing BRAFi in further advanced in vivo studies followed by clinical studies on patients with NRAS-mutant melanoma. ER stress inducers such as thapsigargin have been shown to induce severe skin irritation, salivary hypersecretion, gastroenteritis, vomiting, and in severe cases lead to death (35). Instead, we observed no detrimental effects on the viability of normal human fibroblasts or melanocytes with BRAFi and MEKi combinations. Additionally, hyperactivation of the MAPK pathway in keratinocytes caused by encorafenib was abolished in combination with the binimetinib. These findings are consistent with data from a phase III trial showing that in BRAF-mutant melanoma patients the frequency of hyperproliferative skin lesions including keratoacanthomas or squamous cell carcinomas is lower under combined encorafenib and binimetinib therapy compared with encorafenib monotherapy (36). Additionally, grade 3/4 events and the discontinuation rate due to adverse events are also lower in the combination compared to the monotherapy. Numerous clinical trials and real-world experience confirm that combinations of BRAFi (dabrafenib, vemurafenib, encorafenib) with MEKi (trametinib, cobimetinib, binimetinib) have a favourable safety profile and are generally well tolerated in the long run (37–39).
Supplementary Material
Translational relevance.
Patients with NRAS-mutant metastatic melanoma often have aggressive disease requiring a fast acting, effective therapy. The MEK inhibitor binimetinib shows an overall response rate of 15% in patients with NRAS-mutant melanoma, providing a backbone for combination strategies. Our studies demonstrate that in NRAS-mutant melanoma the antitumor activity of the MEK inhibitor binimetinib is significantly potentiated by the BRAF inhibitor encorafenib through induction of ER stress, leading to melanoma cell death by apoptotic mechanisms. Encorafenib combined with binimetinib was well tolerated in a phase III trial showing potent antitumor activity in BRAF-mutant melanoma, making rapid evaluation in NRAS-mutant melanoma imminently feasible. These data provide a mechanistic rationale for evaluation of binimetinib combined with encorafenib in clinical studies on patients with NRAS-mutant metastatic melanoma.
Acknowledgments
We would like to thank Theresia Schneider, Renate Nordin, Astrid Odon and Birgit Fehrenbacher for outstanding assistance in electron microscopy.
Financial support: The authors of this publication were supported by the University of Tübingen (fortüne 2224-0-0, Heike Niessner), the Federal Ministry of Education and Research (FKZ 031A423B; Christian Praetorius, Dagmar Kulms, Friedegund Meier) and the National Center for Cancer/NCT Program and Infrastructure Grant (Friedegund Meier). Keiran Smalley is supported by NIH grants R01 CA161107-01 and P50 CA168536.
Footnotes
Authors’ Contributions
Conception and design: Heike Niessner, Ines Wanke, Daniela Beck, Tobias Sinnberg, Keith Flaherty, Keiran Smalley, Friedegund Meier.
Development of methodology: Heike Niessner, Ines Wanke, Daniela Beck, Tobias Sinnberg, Dagmar Kulms, Friedegund Meier.
Acquisition of data: Heike Niessner, Ines Wanke, Tobias Sinnberg, Corinna Kosnopfel, Christian Praetorius, Dana Westphal, Marion Mai, Dagmar Kulms.
Analysis and interpretation of data: Heike Niessner, Ines Wanke, Tobias Sinnberg, Christian Praetorius, Martin Schaller, Dagmar Kulms, Dana Westphal, Friedegund Meier.
Writing and review or/and revision of the manuscript: Heike Niessner, Ines Wanke, Tobias Sinnberg, Christian Praetorius, Stefan Beissert, Dagmar Kulms, Martin Schaller, Keith Flaherty, Keiran Smalley, Claus Garbe, Dana Westphal, Friedegund Meier.
Administrative, technical and material support (e.g. providing tumor material, funding support): Heike Niessner, Tobias Sinnberg, Martin Schaller, Claus Garbe, Stefan Beissert, Dagmar Kulms, Dana Westphal, Friedegund Meier.
Uncategorized References
- 1.Jakob JA, Bassett RL, Jr, Ng CS, Curry JL, Joseph RW, Alvarado GC, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118(16):4014–23. doi: 10.1002/cncr.26724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thumar J, Shahbazian D, Aziz SA, Jilaveanu LB, Kluger HM. MEK targeting in N-RAS mutated metastatic melanoma. Mol Cancer. 2014;13:45. doi: 10.1186/1476-4598-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Devitt B, Liu W, Salemi R, Wolfe R, Kelly J, Tzen CY, et al. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011;24(4):666–72. doi: 10.1111/j.1755-148X.2011.00873.x. [DOI] [PubMed] [Google Scholar]
- 4.Carlino MS, Haydu LE, Kakavand H, Menzies AM, Hamilton AL, Yu B, et al. Correlation of BRAF and NRAS mutation status with outcome, site of distant metastasis and response to chemotherapy in metastatic melanoma. Br J Cancer. 2014;111(2):292–9. doi: 10.1038/bjc.2014.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–56. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 7.Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(4):435–45. doi: 10.1016/S1470-2045(17)30180-8. [DOI] [PubMed] [Google Scholar]
- 8.Sosman JA, Kittaneh M, Lolkema MP, Postow M, Schwartz G, Franklin C, et al. A Phase 1b/2 Study of LEE011 in Combination With Binimetinib (MEK162) in Patients With Advanced NRAS-Mutant Melanoma: Early Encouraging Clinical Activity. J Clin Oncol. 2014:32. [Google Scholar]
- 9.Beck D, Niessner H, Smalley KS, Flaherty K, Paraiso KH, Busch C, et al. Vemurafenib potently induces endoplasmic reticulum stress-mediated apoptosis in BRAFV600E melanoma cells. Sci Signal. 2013;6(260):ra7. doi: 10.1126/scisignal.2003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124(3):1406–17. doi: 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. The Journal of biological chemistry. 2004;279(25):25935–8. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- 12.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annual review of biochemistry. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 13.Schoenthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Frontiers in bioscience (Scholar edition) 2012;4:412–31. doi: 10.2741/s276. [DOI] [PubMed] [Google Scholar]
- 14.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. The Journal of clinical investigation. 2005;115(10):2656–64. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell death and differentiation. 2006;13(3):363–73. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 16.Lasithiotakis KG, Sinnberg TW, Schittek B, Flaherty KT, Kulms D, Maczey E, et al. Combined inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. The Journal of investigative dermatology. 2008;128(8):2013–23. doi: 10.1038/jid.2008.44. [DOI] [PubMed] [Google Scholar]
- 17.Mancianti ML, Herlyn M, Weil D, Jambrosic J, Rodeck U, Becker D, et al. Growth and phenotypic characteristics of human nevus cells in culture. The Journal of investigative dermatology. 1988;90(2):134–41. doi: 10.1111/1523-1747.ep12462099. [DOI] [PubMed] [Google Scholar]
- 18.Meier F, Nesbit M, Hsu MY, Martin B, Van Belle P, Elder DE, et al. Human melanoma progression in skin reconstructs : biological significance of bFGF. Am J Pathol. 2000;156(1):193–200. doi: 10.1016/S0002-9440(10)64719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vorsmann H, Groeber F, Walles H, Busch S, Beissert S, Walczak H, et al. Development of a human three-dimensional organotypic skin-melanoma spheroid model for in vitro drug testing. Cell Death Dis. 2013;4:e719. doi: 10.1038/cddis.2013.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 21.Sinnberg T, Menzel M, Ewerth D, Sauer B, Schwarz M, Schaller M, et al. beta-Catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS One. 2011;6(8):e23429. doi: 10.1371/journal.pone.0023429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krepler C, Xiao M, Sproesser K, Brafford PA, Shannan B, Beqiri M, et al. Personalized Preclinical Trials in BRAF Inhibitor-Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin Cancer Res. 2016;22(7):1592–602. doi: 10.1158/1078-0432.CCR-15-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niessner H, Schmitz J, Tabatabai G, Schmid AM, Calaminus C, Sinnberg T, et al. PI3K Pathway Inhibition Achieves Potent Antitumor Activity in Melanoma Brain Metastases In Vitro and In Vivo. Clin Cancer Res. 2016;22(23):5818–28. doi: 10.1158/1078-0432.CCR-16-0064. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez-Laorden B, Viros A, Girotti MR, Pedersen M, Saturno G, Zambon A, et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal. 2014;7(318):ra30. doi: 10.1126/scisignal.2004815. [DOI] [PubMed] [Google Scholar]
- 25.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386(9992):444–51. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 26.Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res. 2007;13(16):4934–42. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- 27.Hersey P, Zhang XD. Adaptation to ER stress as a driver of malignancy and resistance to therapy in human melanoma. Pigment cell & melanoma research. 2008;21(3):358–67. doi: 10.1111/j.1755-148X.2008.00467.x. [DOI] [PubMed] [Google Scholar]
- 28.Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30(50):16938–48. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerezo M, Lehraiki A, Millet A, Rouaud F, Plaisant M, Jaune E, et al. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer Cell. 2016;30(1):183. doi: 10.1016/j.ccell.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Sanderson TH, Gallaway M, Kumar R. Unfolding the unfolded protein response: unique insights into brain ischemia. Int J Mol Sci. 2015;16(4):7133–42. doi: 10.3390/ijms16047133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–70. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin HO, Seo SK, Woo SH, Choe TB, Hong SI, Kim JI, et al. Nuclear protein 1 induced by ATF4 in response to various stressors acts as a positive regulator on the transcriptional activation of ATF4. IUBMB Life. 2009;61(12):1153–8. doi: 10.1002/iub.271. [DOI] [PubMed] [Google Scholar]
- 34.Jiang CC, Lai F, Tay KH, Croft A, Rizos H, Becker TM, et al. Apoptosis of human melanoma cells induced by inhibition of B-RAFV600E involves preferential splicing of bimS. Cell Death Dis. 2010;1:e69. doi: 10.1038/cddis.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foufelle F, Fromenty B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol Res Perspect. 2016;4(1):e00211. doi: 10.1002/prp2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dummer R, Ascierto PA, Gogas HJ, Arance AM, Mandala M, Liszkay G, et al. Results of COLUMBUS Part 1: A Phase 3 Trial of Encorafenib (ENCO) Plus Binimeitnib (BINI) Versus Vemurafenib (VEM) or ENCO in BRAF-Mutant Melanoma. PCMR. 2016 [Google Scholar]
- 37.Rauschenberg R, Garzarolli M, Dietrich U, Beissert S, Meier F. Systemic therapy of metastatic melanoma. J Dtsch Dermatol Ges. 2015;13(12):1223–35. doi: 10.1111/ddg.12891. quiz 36–7. [DOI] [PubMed] [Google Scholar]
- 38.Eroglu Z, Ribas A. Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther Adv Med Oncol. 2016;8(1):48–56. doi: 10.1177/1758834015616934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Margolin K. The Promise of Molecularly Targeted and Immunotherapy for Advanced Melanoma. Curr Treat Options Oncol. 2016;17(9):48. doi: 10.1007/s11864-016-0421-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.