

Abstract
Under basal conditions, postnatal skeletal muscle displays little cell turnover. With injury, muscle initiates a rapid repair response to reseal damaged membrane, reactivating many developmental pathways to facilitate muscle regeneration and prevent tissue loss. Muscle precursor cells become activated accompanied by differentiation and fusion during both muscle growth and regeneration; inter-cellular communication is required for successful completion of these processes. Cellular communication is mediated by lipids, fusogenic membrane proteins, and exosomes. Muscle-derived exosomes carry proteins and micro RNAs as cargo. Secreted factors such as IGF-1, TGFβ, and myostatin are also released by muscle cells providing local signaling cues to modulate muscle fusion and regeneration. Proteins that regulate myoblast fusion also participate in membrane repair and regeneration. Here we will review methods of muscle cell communication focusing on proteins that mediate membrane fusion, exosomes, and autocrine factors.
Keywords: fusion, repair, membrane, myoblast, muscle, development, cell communication, exosome
Introduction
Skeletal muscle is the largest tissue in the human body comprising approximately 30-40% of total body weight [1]. Due to the elongated nature of individual myofibers and their role in muscle contraction, skeletal muscle is prone to injury. Muscle continuously adapts to environmental and physical challenges through regeneration and membrane repair to ensure tissue survival [2,3]. Muscle growth and regeneration are multi-step processes requiring cellular activation and cell-cell fusion. Myoblast and myofiber differentiation occur both before and after cellular fusion (Figure 1), including the expression of cell surface markers and secreted factors that coordinate these activities. Membrane lipids, exosome-borne proteins/microRNAs (miRNAs), and autocrine cytokines help orchestrate differentiation and fusion. This review will evaluate the current knowledge of muscle cell communication during growth, repair and regeneration.
Figure 1. Muscle fusion.
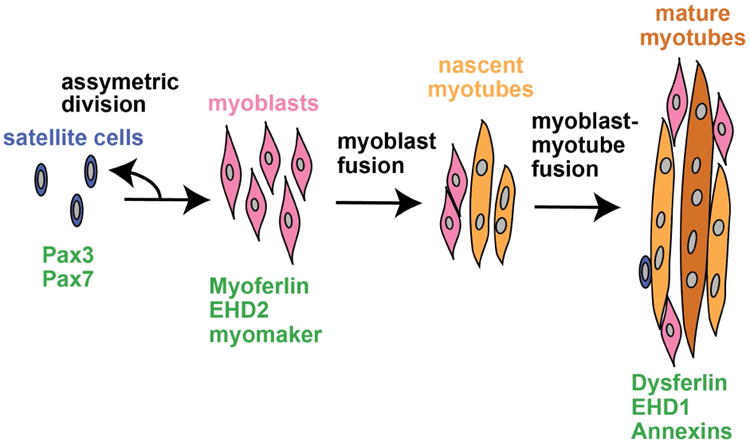
During muscle growth and regeneration satellite cells (blue) asymmetrically divide giving rise to mono-nucleated muscle precursor cells, myoblasts (pink). Myoblasts are activated and fuse with one-another to generate nascent myotubes (orange). Myoblasts fuse with existing myotubes to promote muscle growth and regeneration (dark orange). Pax3 and Pax7 are highly expressed in satellite cells. Fusogenic proteins myoferlin, EHD2, and myomaker are highly expressed in myoblasts with expression decreasing as differentiation proceeds. Dysferlin, EHD1, and annexins have low-level expression in myoblasts with increased expression in mature muscle.
Membrane Phospholipids
The muscle plasma membrane, known as the sarcolemma, is a lipid bilayer containing phospholipids such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and phosphatidic acid (PA). The composition of lipids varies along the sarcolemma, clustering in lipid rafts and forming sites with a specific enrichment of PS and a reduction in PC at the specialized membrane of transverse tubules (T-tubules) [4,5]. As in other cells, phospholipid clustering is temporally dynamic and depends on the cell state. PS is normally found within the inner lipid bilayer and flips to the outer bilayer with plasma membrane disruption during injury, and this configuration serves as a signal to invading macrophages to remove the damaged myofiber [6]. However, external PS need not always signaling injury, as PS also localizes to the site of cell-cell contact during myoblast fusion and at the site of membrane repair [7-11]. Blocking PS exposure with an anti-PS antibody inhibits muscle cell fusion [9,12] indicating the necessity of external PS in cell-cell communication during membrane fusion.
Phosphoinositides (PI), specifically phosphatidylinositol (4,5)-bisphosphate (PIP2) and phosphatidylinositol (3,4,5)-trisphosphate (PIP3) comprise approximately 1% of the lipid content within the membrane, yet are critical components that direct membrane and cytoskeleton reorganization [13]. PIP2 is enriched at the site of muscle cell contact providing a local cue for fusogenic proteins and actin regulators [14]. Disruption of PIP2 expression in C2C12 myoblasts, a myogenic cell line, with calcimycin, LiCl, neomycin, or genetic reduction of PIP5KIγ, reduced myoblast fusion suggesting PIP2 contributes to the fusogenic signaling pathway [14]. Additionally, manipulation of PIP3 levels improved the severe muscular dystrophy phenotype in myotubularin (Mtm1) deficient mice and zebrafish [15]. Therefore, expression and localization of phosphoinositides serve as a lipid code to direct cellular activity, and a unique balance of lipids is required by muscle for proper function.
Regulators of membrane fusion in development and repair
Ferlins
The Ferlin family of proteins is a family of six related proteins that regulate membrane fusion, vesicle trafficking and membrane repair [16-21]. Dysferlin (also known as Fer1L1) was the first mammalian ferlin family member identified and is broadly expressed, including in mature skeletal muscle where it localizes to the plasma membrane and T-tubules [22-24]. Loss-of-function mutations in the dysferlin gene result in muscle disease, specifically muscular dystrophy [25,26]. Dysferlin contains seven C2 domains and a carboxyl-terminal transmembrane domain [27]. These C2 domains are highly related to the C2 domains found in synaptotagmin, a Ca2+-sensing regulator of membrane fusion. C2A, the most amino-terminal C2 domain, binds phospholipids in the presence of Ca2+ likely contributing to the localization and fusogenic potential of dysferlin [16,28,29].
After membrane damage, it is thought that membrane patches, derived from intracellular vesicles, form to reseal the disrupted membrane [30]. This model does not preclude that resealing also uses additional sources of membrane-bilayer. For example, labeling studies have suggested that plasma membrane components adjacent to the disruption contribute to resealing in what has been referred as “lateral contribution” of membranes to repair [10,23]. Dysferlin's role in muscle membrane repair was first observed utilizing laser-ablation to induce membrane injury [16]. Dysferlin-null myofibers reseal membrane disruptions much more slowly than normal myofibers, illustrated by an increase in fluorescent FM 1-43 dye uptake after sarcolemmal disruption [16,31]. Immunofluorescence microscopy was used to demonstrate dysferlin's recruitment to the site of membrane insult [16]. With high-resolution live-cell imaging of multiple components mediating the repair process, dysferlin was observed to localize in the membrane immediately adjacent to sites of disruption [10]. This position may indicate that dysferlin regulates membrane incorporation at the site of damage through its interaction with phospholipids and other repair proteins [11].
In addition to a role in mediating plasma membrane resealing, dysferlin regulates vesicle trafficking. Dysferlin strongly localizes to discrete cytoplasmic puncta ranging from 200nm to >1μm in size. Dysferlin specifically colocalizes with the early endosomal marker Rab5 and the late endosomal marker Rab7. Furthermore, dysferlin colocalizes with endocytosed transferrin [32,33]. Pulse chase studies of Alexa-488 labeled transferrin in dysferlin-null myoblasts demonstrate a delay in recycling [17], consistent with a broader membrane and vesicle trafficking role within cells.
Myoferlin is highly homologous to dysferlin, but myoferlin is most highly expressed during early muscle development and expression decreases as muscle matures [19,34]. Myoferlin is upregulated after muscle injury, and myoferlin is enriched at the site of cell-cell contact during myoblast fusion [19]. Similar to dysferlin, myoferlin regulates endocytic recycling of multiple receptors including transferrin and the insulin like growth factor receptor (IGF1R) [18]. Indicative of decreased IGF-1 signaling and ineffective myoblast fusion, myoferlin-null mice exhibit reduced myofiber size and delayed muscle regeneration [18,19]. Myoferlin-null myofibers reseal more slowly than normal fibers after laser-induced membrane injury [10], consistent with a role in mature myofibers. Myofibers lacking both myoferlin and dysferlin have enhanced repair defects compared to either single mutation. Curiously, transgenic overexpression of myoferlin in dysferlin-null mice was sufficient to improve resealing, however, dystrophic pathology remained [35].
Myomaker
Myomaker (also known as TMEM8C) is enriched in developing muscle; the myomaker protein contains seven transmembrane domains and is localized to the plasma membrane [36]. Consistent with the reactivation of developmental gene programs in response to muscle injury, myomaker is re-expressed after muscle insult while it is minimally expressed in mature uninjured muscle. Similar to myoferlin, myomaker localizes to the site of cell-cell contact during myoblast fusion. Through co-culture experiments, it was found that myomaker-null myoblasts fuse more efficiently with wildtype myoblasts than myomaker-null myoblasts, consistent with a model where at least one cellular partner must expose myomaker for membrane coalescence [36,37]. Correspondingly, myomaker-null mice die during gestation with a severe defect in skeletal muscle formation [37]. Overexpression of myomaker in the C2C12 myoblast cell line or even in fibroblasts results in enhanced fusion potential. Deletion of myomaker's carboxy-terminal 8 amino acids (aa214-221) interferes with its fusogenic potential. Expression of the related proteins TMEM8A and TMEM8B had no effect on fusion, suggesting distinct cellular roles compared to TMEM8C [36].
Exosomal proteins and miRNAs in muscle communication
Exosomes are small membranous vesicles, 50–150-nm in diameter, derived from the late endosomal system, specifically the multivesicular bodies (MVBs). Exosomes are released from cells through exocytic fusion with the plasma membrane. Multiple cell types, including skeletal muscle, shed exosomes into the extracellular space or circulation under both normal and pathological conditions [38,39]. Exosomes, as high as ∼ 1010/ml, can be found in serum of healthy subjects suggesting a biological function beyond disease [40]. Released exosomes contain cargo proteins, miRNAs and lipids that are thought to be involved in trans-cellular and probably even trans-tissue communication (Figure 2). Efforts have been directed at characterizing the cargo contained within exosomes to better understand their biological function.
Figure 2. Exosome biogenesis and release.
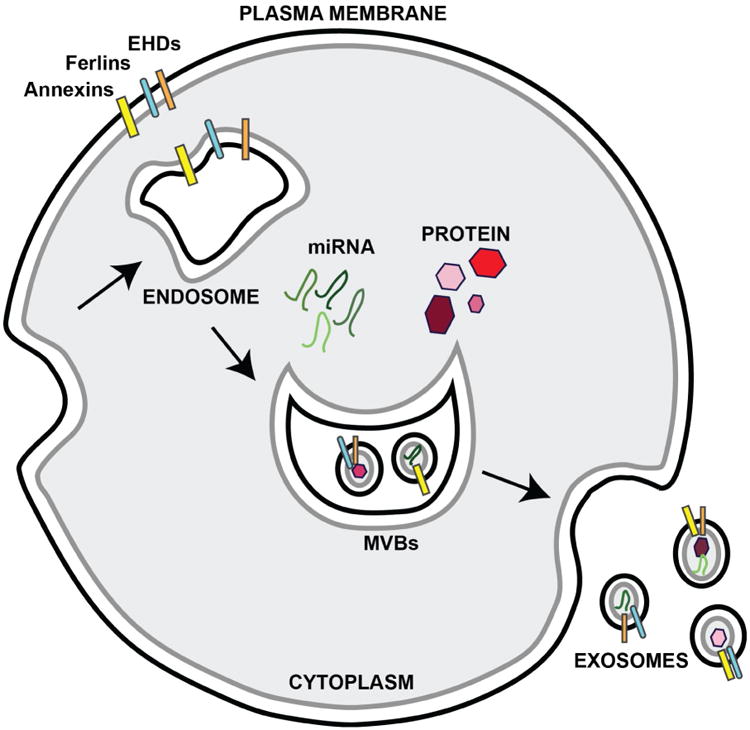
Exosomes are formed from multi-vescular bodies (MVBs), which are formed from endosomes. Exosomes are released as MVBs fuse with the plasma membrane. Exosome membranes are enriched with fusogenic proteins annexins (yellow), ferlins (teal), and EHDs (orange). Exosomes contain both miRNA (green) and cytosolic proteins (red) thought to participate in cell-cell communication.
Proteomic analysis of exosome-like vesicles released from C2C12 myoblasts and myotubes revealed distinct specific subsets of proteins released throughout muscle differentiation [41]. Vesicles from both cultures showed enrichment in annexins, EHDs, LAMPs, Rabs, VAMPs, and VPSs (ESCRT complex members) consistent with the proposed multi-vesicular body / endosomal origin. Exosome-like vesicles derived from myotube cultures were also enriched in proteins involved in muscle contraction including DAG1, FLNC, TLN1, TTN, VCL, and VIM. Incubation of myoblasts with exosomes from myotubes inhibited myoblast proliferation and promoted differentiation. To determine if this increase in fusion was due to uptake of exosomal contents by myoblasts, GFP-positive myotube exosomes were incubated with non-fluorescent myoblasts [41]. After 24hrs, GFP fluorescence was detected within the myoblasts confirming myotube-to-myoblast transfer of exosomal cargo. These data support a role for exosomes actively participating in muscle cell communication through dispersion of released contents. In addition, differentiation in the presence of exosome-depleted serum, reduces the efficiency of myoblast differentiation implicating exosomes in cross-talk between mature muscle and myoblasts [42].
The mechanism by which exosomes interact with and are taken up by target cells is not well understood. This is further complicated by the idea that tissue type specificity may exist between the target cell and released exosome. Multiple mechanisms between exosomes and recipient cells have been documented including interaction with cell surface receptors, plasma membrane fusion, and phagocytosis [43-45]. Both myoferlin and dysferlin interact with Eps15 homology domain containing proteins, EHD1 and EHD2, in muscle and loss of EHD1 results in deficient myoblast fusion [20,46,47]. Proteins identified in exosomes derived from myoblasts and myotubes include myoferlin, annexins and the EHD proteins [41,48]. EHDs bind phospholipids and have been implicated in endocytic recycling in multiple cell types, acting as membrane “pinchases” [49,50]. Annexins have also been implicated in muscle cell fusion, membrane repair, and ferlin binding [10,51-53]. The annexin family includes twelve different members, and the annexins are implicated in Ca2+-dependent phospholipid binding with the actin cytoskeleton [54]. Annexin expression increases during muscle differentiation and inhibition of annexin A1 or A5 decreases myoblast fusion [12,55]. Additionally, annexins participate in membrane repair and can be observed to aggregate at the membrane lesion [10,53]. Enrichment of EHD proteins and annexins within exosomes may reflect a role for these proteins in exosome release or docking on target membranes. Alternatively, these proteins may simply passively enrich in the exosomal cargo due to their natural abundance on the sarcolemma. Koumangoye et. al. found that shRNA-mediated reduction of annexin A2 and A6 in a breast cancer cell line was sufficient to reduce the internalization of exosomes, suggesting a functional role for the annexin proteins in exosomal uptake [56].
microRNAs (miRNAs) are small non-coding RNAs approximately 19-24 nucleotides in length that function as negative regulators of gene expression through binding mRNA [57-59]. miRNAs are evolutionarily conserved and global loss of miRNA production is inconsistent with life [60]. miRNAs are temporally regulated with unique tissue and/or disease signatures facilitating their role as biomarkers. Dysregulation of miRNA expression has been noted in many disease states including muscular dystrophy [61]. Forterre et al. tested whether miRNA incorporation into exosomes could act as a viable means of cellular communication regulating gene transcription, as miRNAs modulate muscle development [62,63]. Using C2C12 myoblasts and myotubes, miRNAs contained within exosomes were analyzed [63]. Over 170 miRNAs were found within muscle exosomes including miR-1, miR-133a, miR-133b miR-206, which regulate muscle differentiation [64-66]. To demonstrate physical transfer of miRNAs as means of muscle cell communication, the C.elegans miRNA, cel-miRNA-238, which is not normally expressed in C2C12 myoblasts, was expressed in myotubes. After incubation with exosomes from transfected myotubes, cel-miRNA-238 miRNA was detected within myoblasts, illustrating miRNA transfer by muscle cells [63]. Additionally, Fry et. al. demonstrated that co-culture of exosomes isolated from muscle precursor cells (MPCs) with fibrogenic muscle cells was sufficient to downregulate Col1a2, Col3a1, Col6a2 and Fibronectin [67]. MPC depletion of the myo-miR, miR-206, resulted in an increase in Rrbp1, a master regulator of collagen synthesis, and increased fibrogenic collagen expression. In vivo, miR-206 is highly expressed in activated satellite cells. Using the Pax7/DTA mouse model, satellite cells were depleted and upregulation of miR-206 after mechanical overload was lost. This correlated with an increase in Rrbp1 and upregulation of Col1a2, Col3a1, and Col12a1. These findings are consistent with the idea that exosomes release miRNAs which regulate gene expression in target cells. Engineered exosomal miRNAs are now being considered as a circulating tool to promote myogenesis and regeneration.
Autocrine signaling during skeletal muscle development and regeneration
Skeletal muscle secretes cytokines to regulate its own growth and regeneration. These factors are referred to as myokines and include IGF-1 and transforming growth factor beta (TGFβ) superfamily members TGFβ and myostatin (also known as GDF8) (Figure 3). IGF-1 modulates muscle growth promoting muscle cell activation, differentiation and hypertrophy [68-70]. IGF-1 binds the IGF1 receptor (IGF1-R) initiating AKT activation promoting protein synthesis and inhibiting protein degradation [71]. In mature muscle, TGFβ negatively regulates muscle differentiation inhibiting fusion through binding its receptor, TGFβR, and activating SMAD signaling, which inhibits AKT-induced muscle growth. TGFβ inhibition through neutralizing antibodies or nonspecific drugs like losartan improve muscle performance and disease pathology in mouse models of muscular dystrophy [72,73]. Myostatin is a negative regulator of muscle growth suppressing muscle maturation. Overexpression of myostatin, in mice, results in decreased myofiber area and muscle mass [74,75]. Inhibition of myostatin or genetic loss of myostatin results in muscle hyperplasia and hypertrophy, with a profound increase in muscle mass [76,77]. Myostatin binds the activin receptor IIβ (ActRIIB) inhibiting myogenesis through SMAD activation and inhibition of AKT induced hypertrophy [78]. miRNAs have been identified that modulate these signaling pathways or are modulated by these autocrine factors providing an additional layer of regulation [79-81]. Manipulating the IGF-1, TGFβ, and myostatin signaling pathways to promote muscle growth and improve regenerative capacity are currently being investigated for treating muscle disease.
Figure 3. Autocrine factors regulating muscle growth and regeneration.
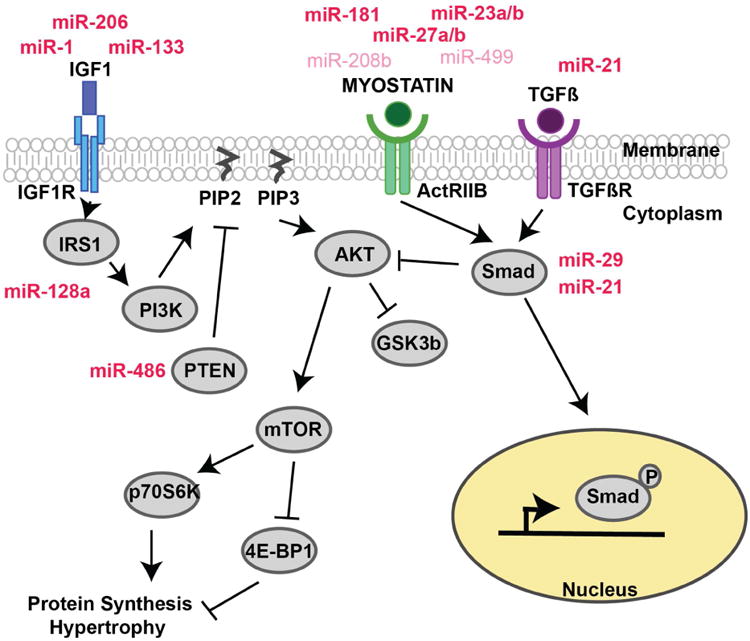
IGF1 is a potent muscle growth factor that binds IGF1-R activating PI3K/AKT signaling to promote protein synthesis and muscle hypertrophy. Myostatin and TGFβ are negative regulators of muscle growth inhibiting AKT induced hypertrophy through SMAD activation. miRNAs (mi-R) both regulate and are regulated by these signaling pathways adding additional layers of control to the system. mi-Rs (dark red) have been isolated from skeletal muscle derived exosomes, suggesting a mechanism by which cells can communicate and control muscle growth in a synchronous fashion. mi-Rs (pink) have been implicated in autocrine regulation but have yet to be isolated from skeletal muscle derived exosomes.
Conclusion
Modulation of membrane lipids, localization and expression of fusogenic proteins, and secretion of exosomal proteins / miRNAs are key modes of communication utilized by muscle cells to regulate skeletal muscle growth, regeneration, and repair. Understanding how muscle cells communicate will allow the development of better therapeutics to promote muscle growth and regeneration.
Highlights.
Cell-cell communication facilitates fusion and regulates muscle growth, repair, and regeneration
Exosomes contain protein and micro RNAs that facilitate communication among muscle cells
Autocrine factors including IGF-1, TGFβ, and myostatin modulate muscle growth and regeneration
Phospholipid composition within the membrane influences cell fusion
Myoferlin and myomaker localize to the site of muscle cell contact promoting fusion
Acknowledgments
This work was supported by National Institutes of Health grants NS047726, NS072027, and HL061322.
Abbreviations
- EHD
Eps15 Homology Domain
- PIP2
Phosphatidylinositol 4,5-bisphosphate
- PS
phosphatidylserine
- T-tubule
transverse tubule
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PA
phosphatidic acid
- IGF1
insulin like growth factor 1
Footnotes
Conflict of Interest: There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Janssen I, Heymsfield SB, Wang ZM, Ross R. Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J Appl Physiol (1985) 2000;89:81–88. doi: 10.1152/jappl.2000.89.1.81. [DOI] [PubMed] [Google Scholar]
- 2.Cooper ST, McNeil PL. Membrane Repair: Mechanisms and Pathophysiology. Physiol Rev. 2015;95:1205–1240. doi: 10.1152/physrev.00037.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grounds MD. The need to more precisely define aspects of skeletal muscle regeneration. Int J Biochem Cell Biol. 2014;56:56–65. doi: 10.1016/j.biocel.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Pediconi MF, Donoso P, Hidalgo C, Barrantes FJ. Lipid composition of purified transverse tubule membranes isolated from amphibian skeletal muscle. Biochim Biophys Acta. 1987;921:398–404. doi: 10.1016/0005-2760(87)90042-7. [DOI] [PubMed] [Google Scholar]
- 5.Lau YH, Caswell AH, Brunschwig JP, Baerwald R, Garcia M. Lipid analysis and freeze-fracture studies on isolated transverse tubules and sarcoplasmic reticulum subfractions of skeletal muscle. J Biol Chem. 1979;254:540–546. [PubMed] [Google Scholar]
- 6.Fadeel B, Xue D. The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Crit Rev Biochem Mol Biol. 2009;44:264–277. doi: 10.1080/10409230903193307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taulet N, Comunale F, Favard C, Charrasse S, Bodin S, Gauthier-Rouviere C. N-cadherin/p120 catenin association at cell-cell contacts occurs in cholesterol-rich membrane domains and is required for RhoA activation and myogenesis. J Biol Chem. 2009;284:23137–23145. doi: 10.1074/jbc.M109.017665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sessions A, Horwitz AF. Differentiation-related differences in the plasma membrane phospholipid asymmetry of myogenic and fibrogenic cells. Biochim Biophys Acta. 1983;728:103–111. doi: 10.1016/0005-2736(83)90442-x. [DOI] [PubMed] [Google Scholar]
- 9.van den Eijnde SM, van den Hoff MJ, Reutelingsperger CP, van Heerde WL, Henfling ME, Vermeij-Keers C, Schutte B, Borgers M, Ramaekers FC. Transient expression of phosphatidylserine at cell-cell contact areas is required for myotube formation. J Cell Sci. 2001;114:3631–3642. doi: 10.1242/jcs.114.20.3631. [DOI] [PubMed] [Google Scholar]
- 10**.Demonbreun AR, Quattrocelli M, Barefield DY, Allen MV, Swanson KE, McNally EM. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J Cell Biol. 2016;213:705–718. doi: 10.1083/jcb.201512022. Live cell imaging and high resolution micrscopy were used to visualize the repair complex in muscle identifying annexin rich caps supported by dysferlin and EHD containing ring proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11**.Middel V, Zhou L, Takamiya M, Beil T, Shahid M, Roostalu U, Grabher C, Rastegar S, Reischl M, Nienhaus GU, et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat Commun. 2016;7:12875. doi: 10.1038/ncomms12875. Identifies the role of dysferlin in externalizing phosphatidylserine to repair patches in muscle cells as a signal for macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leikina E, Melikov K, Sanyal S, Verma SK, Eun B, Gebert C, Pfeifer K, Lizunov VA, Kozlov MM, Chernomordik LV. Extracellular annexins and dynamin are important for sequential steps in myoblast fusion. J Cell Biol. 2013;200:109–123. doi: 10.1083/jcb.201207012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–606. doi: 10.1016/s0092-8674(00)80696-0. [DOI] [PubMed] [Google Scholar]
- 14.Bach AS, Enjalbert S, Comunale F, Bodin S, Vitale N, Charrasse S, Gauthier-Rouviere C. ADP-ribosylation factor 6 regulates mammalian myoblast fusion through phospholipase D1 and phosphatidylinositol 4,5-bisphosphate signaling pathways. Mol Biol Cell. 2010;21:2412–2424. doi: 10.1091/mbc.E09-12-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15**.Sabha N, Volpatti JR, Gonorazky H, Reifler A, Davidson AE, Li X, Eltayeb NM, Dall'Armi C, Di Paolo G, Brooks SV, et al. PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J Clin Invest. 2016;126:3613–3625. doi: 10.1172/JCI86841. Documents the role of of PI3 kinases and suggests that PI3K inhibtion is a target for treating myotubular myopath. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 17.Demonbreun AR, Fahrenbach JP, Deveaux K, Earley JU, Pytel P, McNally EM. Impaired muscle growth and response to insulin-like growth factor 1 in dysferlin-mediated muscular dystrophy. Hum Mol Genet. 2011;20:779–789. doi: 10.1093/hmg/ddq522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demonbreun AR, Posey AD, Heretis K, Swaggart KA, Earley JU, Pytel P, McNally EM. Myoferlin is required for insulin-like growth factor response and muscle growth. Faseb J. 2010;24:1284–1295. doi: 10.1096/fj.09-136309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, Hadhazy M, McNally EM. Normal myoblast fusion requires myoferlin. Development. 2005;132:5565–5575. doi: 10.1242/dev.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doherty KR, Demonbreun AR, Wallace GQ, Cave A, Posey AD, Heretis K, Pytel P, McNally EM. The Endocytic Recycling Protein EHD2 Interacts with Myoferlin to Regulate Myoblast Fusion. J Biol Chem. 2008;283:20252–20260. doi: 10.1074/jbc.M802306200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Posey AD, Jr, Demonbreun A, McNally EM. Ferlin proteins in myoblast fusion and muscle growth. Curr Top Dev Biol. 2011;96:203–230. doi: 10.1016/B978-0-12-385940-2.00008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kerr JP, Ziman AP, Mueller AL, Muriel JM, Kleinhans-Welte E, Gumerson JD, Vogel SS, Ward CW, Roche JA, Bloch RJ. Dysferlin stabilizes stress-induced Ca2+ signaling in the transverse tubule membrane. Proc Natl Acad Sci U S A. 2013;110:20831–20836. doi: 10.1073/pnas.1307960110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23**.McDade JR, Archambeau A, Michele DE. Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repair. FASEB J. 2014;28:3660–3670. doi: 10.1096/fj.14-250191. Documents the role of actin in supporting effective resealing of myofibers and the contribution of sarcolemmal membrane adjacent to the wound site as important for resealing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piccolo F, Moore SA, Ford GC, Campbell KP. Intracellular accumulation and reduced sarcolemmal expression of dysferlin in limb--girdle muscular dystrophies. Ann Neurol. 2000;48:902–912. [PubMed] [Google Scholar]
- 25.Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 27.Lek A, Evesson FJ, Sutton RB, North KN, Cooper ST. Ferlins: regulators of vesicle fusion for auditory neurotransmission, receptor trafficking and membrane repair. Traffic. 2012;13:185–194. doi: 10.1111/j.1600-0854.2011.01267.x. [DOI] [PubMed] [Google Scholar]
- 28.Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–213. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Davis DB, Doherty KR, Delmonte AJ, McNally EM. Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J Biol Chem. 2002;277:22883–22888. doi: 10.1074/jbc.M201858200. [DOI] [PubMed] [Google Scholar]
- 30.McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nat Rev Mol Cell Biol. 2005;6:499–505. doi: 10.1038/nrm1665. [DOI] [PubMed] [Google Scholar]
- 31*.Demonbreun AR, Allen MV, Warner JL, Barefield DY, Krishnan S, Swanson KE, Earley JU, McNally EM. Enhanced Muscular Dystrophy from Loss of Dysferlin Is Accompanied by Impaired Annexin A6 Translocation after Sarcolemmal Disruption. Am J Pathol. 2016;186:1610–1622. doi: 10.1016/j.ajpath.2016.02.005. Mice lacking dysferlin have an intensified muscle phenotype in the C57Bl6/J background which carries a dominant negative annexin A6 genotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evesson FJ, Peat RA, Lek A, Brilot F, Lo HP, Dale RC, Parton RG, North KN, Cooper ST. Reduced plasma membrane expression of dysferlin mutants is attributed to accelerated endocytosis via a syntaxin-4-associated pathway. J Biol Chem. 2010;285:28529–28539. doi: 10.1074/jbc.M110.111120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Redpath GM, Sophocleous RA, Turnbull L, Whitchurch CB, Cooper ST. Ferlins Show Tissue-Specific Expression and Segregate as Plasma Membrane/Late Endosomal or Trans-Golgi/Recycling Ferlins. Traffic. 2016;17:245–266. doi: 10.1111/tra.12370. This study examined the intracellular trafficking pathways of ferlins identifying that dysferlin and myoferlin cycle to Rab7-positive late endosomes. [DOI] [PubMed] [Google Scholar]
- 34.Davis DB, Delmonte AJ, Ly CT, McNally EM. Myoferlin, a candidate gene and potential modifier of muscular dystrophy. Hum Mol Genet. 2000;9:217–226. doi: 10.1093/hmg/9.2.217. [DOI] [PubMed] [Google Scholar]
- 35.Lostal W, Bartoli M, Roudaut C, Bourg N, Krahn M, Pryadkina M, Borel P, Suel L, Roche JA, Stockholm D, et al. Lack of correlation between outcomes of membrane repair assay and correction of dystrophic changes in experimental therapeutic strategy in dysferlinopathy. PLoS One. 2012;7:e38036. doi: 10.1371/journal.pone.0038036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36**.Millay DP, Gamage DG, Quinn ME, Min YL, Mitani Y, Bassel-Duby R, Olson EN. Structure-function analysis of myomaker domains required for myoblast fusion. Proc Natl Acad Sci U S A. 2016;113:2116–2121. doi: 10.1073/pnas.1600101113. This paper demonstrates the importance of the intracellular C-terminus of myomaker for its ability to induce and regulate cell fusion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Millay DP, O'Rourke JR, Sutherland LB, Bezprozvannaya S, Shelton JM, Bassel-Duby R, Olson EN. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature. 2013;499:301–305. doi: 10.1038/nature12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38*.He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci U S A. 2014;111:4525–4529. doi: 10.1073/pnas.1402714111. This study characterizes the miRNA cargo in muscle derived microvesicles including miR-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henningsen J, Rigbolt KT, Blagoev B, Pedersen BK, Kratchmarova I. Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol Cell Proteomics. 2010;9:2482–2496. doi: 10.1074/mcp.M110.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJ, Hole P, Carr B, Redman CW, Harris AL, Dobson PJ, et al. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomedicine. 2011;7:780–788. doi: 10.1016/j.nano.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forterre A, Jalabert A, Berger E, Baudet M, Chikh K, Errazuriz E, De Larichaudy J, Chanon S, Weiss-Gayet M, Hesse AM, et al. Proteomic analysis of C2C12 myoblast and myotube exosome-like vesicles: a new paradigm for myoblast-myotube cross talk? PLoS One. 2014;9:e84153. doi: 10.1371/journal.pone.0084153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aswad H, Jalabert A, Rome S. Depleting extracellular vesicles from fetal bovine serum alters proliferation and differentiation of skeletal muscle cells in vitro. BMC Biotechnol. 2016;16:32. doi: 10.1186/s12896-016-0262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng D, Zhao WL, Ye YY, Bai XC, Liu RQ, Chang LF, Zhou Q, Sui SF. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11:675–687. doi: 10.1111/j.1600-0854.2010.01041.x. [DOI] [PubMed] [Google Scholar]
- 44.Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, Baty CJ, Gibson GA, Erdos G, Wang Z, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119:756–766. doi: 10.1182/blood-2011-02-338004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Segura E, Guerin C, Hogg N, Amigorena S, Thery C. CD8+ dendritic cells use LFA-1 to capture MHC-peptide complexes from exosomes in vivo. J Immunol. 2007;179:1489–1496. doi: 10.4049/jimmunol.179.3.1489. [DOI] [PubMed] [Google Scholar]
- 46.Posey AD, Jr, Pytel P, Gardikiotes K, Demonbreun AR, Rainey M, George M, Band H, McNally EM. Endocytic recycling proteins EHD1 and EHD2 interact with fer-1-like-5 (Fer1L5) and mediate myoblast fusion. J Biol Chem. 2011;286:7379–7388. doi: 10.1074/jbc.M110.157222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Posey AD, Jr, Swanson KE, Alvarez MG, Krishnan S, Earley JU, Band H, Pytel P, McNally EM, Demonbreun AR. EHD1 mediates vesicle trafficking required for normal muscle growth and transverse tubule development. Dev Biol. 2014;387:179–190. doi: 10.1016/j.ydbio.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guescini M, Guidolin D, Vallorani L, Casadei L, Gioacchini AM, Tibollo P, Battistelli M, Falcieri E, Battistin L, Agnati LF, et al. C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp Cell Res. 2010;316:1977–1984. doi: 10.1016/j.yexcr.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 49.Daumke O, Lundmark R, Vallis Y, Martens S, Butler PJ, McMahon HT. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007;449:923–927. doi: 10.1038/nature06173. [DOI] [PubMed] [Google Scholar]
- 50.Naslavsky N, Caplan S. EHD proteins: key conductors of endocytic transport. Trends Cell Biol. 2011;21:122–131. doi: 10.1016/j.tcb.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH., Jr Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003 doi: 10.1074/jbc.M307247200. [DOI] [PubMed] [Google Scholar]
- 52.McNeil AK, Rescher U, Gerke V, McNeil PL. Requirement for annexin A1 in plasma membrane repair. J Biol Chem. 2006;281:35202–35207. doi: 10.1074/jbc.M606406200. [DOI] [PubMed] [Google Scholar]
- 53.Roostalu U, Strahle U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev Cell. 2012;22:515–529. doi: 10.1016/j.devcel.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 55.Bizzarro V, Fontanella B, Franceschelli S, Pirozzi M, Christian H, Parente L, Petrella A. Role of Annexin A1 in mouse myoblast cell differentiation. J Cell Physiol. 2010;224:757–765. doi: 10.1002/jcp.22178. [DOI] [PubMed] [Google Scholar]
- 56.Koumangoye RB, Sakwe AM, Goodwin JS, Patel T, Ochieng J. Detachment of breast tumor cells induces rapid secretion of exosomes which subsequently mediate cellular adhesion and spreading. PLoS One. 2011;6:e24234. doi: 10.1371/journal.pone.0024234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T. New microRNAs from mouse and human. RNA. 2003;9:175–179. doi: 10.1261/rna.2146903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 59.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 60.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 61.Alexander MS, Kunkel LM. Skeletal Muscle MicroRNAs: Their Diagnostic and Therapeutic Potential in Human Muscle Diseases. J Neuromuscul Dis. 2015;2:1–11. doi: 10.3233/JND-140058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ge Y, Chen J. MicroRNAs in skeletal myogenesis. Cell Cycle. 2011;10:441–448. doi: 10.4161/cc.10.3.14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63*.Forterre A, Jalabert A, Chikh K, Pesenti S, Euthine V, Granjon A, Errazuriz E, Lefai E, Vidal H, Rome S. Myotube-derived exosomal miRNAs downregulate Sirtuin1 in myoblasts during muscle cell differentiation. Cell Cycle. 2014;13:78–89. doi: 10.4161/cc.26808. This paper describes the role of miRNAs derived from exosomes in muscle differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Townley-Tilson WH, Callis TE, Wang D. MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac muscle development, function, and disease. Int J Biochem Cell Biol. 2010;42:1252–1255. doi: 10.1016/j.biocel.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCarthy JJ. MicroRNA-206: the skeletal muscle-specific myomiR. Biochim Biophys Acta. 2008;1779:682–691. doi: 10.1016/j.bbagrm.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67**.Fry CS, Kirby TJ, Kosmac K, McCarthy JJ, Peterson CA. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell. 2017;20:56–69. doi: 10.1016/j.stem.2016.09.010. This paper shows intercellular communication by myogenic precursor cells on fibroblasts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Semsarian C, Sutrave P, Richmond DR, Graham RM. Insulin-like growth factor (IGF-I) induces myotube hypertrophy associated with an increase in anaerobic glycolysis in a clonal skeletal-muscle cell model. Biochem J. 1999;339(Pt 2):443–451. [PMC free article] [PubMed] [Google Scholar]
- 69.Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A. 1998;95:15603–15607. doi: 10.1073/pnas.95.26.15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995;270:12109–12116. doi: 10.1074/jbc.270.20.12109. [DOI] [PubMed] [Google Scholar]
- 71.Schiaffino S, Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle. 2011;1:4. doi: 10.1186/2044-5040-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elbaz M, Yanay N, Aga-Mizrachi S, Brunschwig Z, Kassis I, Ettinger K, Barak V, Nevo Y. Losartan, a therapeutic candidate in congenital muscular dystrophy: studies in the dy(2J)/dy(2J) mouse. Ann Neurol. 2012;71:699–708. doi: 10.1002/ana.22694. [DOI] [PubMed] [Google Scholar]
- 73.Nelson CA, Hunter RB, Quigley LA, Girgenrath S, Weber WD, McCullough JA, Dinardo CJ, Keefe KA, Ceci L, Clayton NP, et al. Inhibiting TGF-beta activity improves respiratory function in mdx mice. Am J Pathol. 2011;178:2611–2621. doi: 10.1016/j.ajpath.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Durieux AC, Amirouche A, Banzet S, Koulmann N, Bonnefoy R, Pasdeloup M, Mouret C, Bigard X, Peinnequin A, Freyssenet D. Ectopic expression of myostatin induces atrophy of adult skeletal muscle by decreasing muscle gene expression. Endocrinology. 2007;148:3140–3147. doi: 10.1210/en.2006-1500. [DOI] [PubMed] [Google Scholar]
- 75.Reisz-Porszasz S, Bhasin S, Artaza JN, Shen R, Sinha-Hikim I, Hogue A, Fielder TJ, Gonzalez-Cadavid NF. Lower skeletal muscle mass in male transgenic mice with muscle-specific overexpression of myostatin. Am J Physiol Endocrinol Metab. 2003;285:E876–888. doi: 10.1152/ajpendo.00107.2003. [DOI] [PubMed] [Google Scholar]
- 76.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- 79.Butz H, Racz K, Hunyady L, Patocs A. Crosstalk between TGF-beta signaling and the microRNA machinery. Trends Pharmacol Sci. 2012;33:382–393. doi: 10.1016/j.tips.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 80.Jung HJ, Suh Y. Regulation of IGF -1 signaling by microRNAs. Front Genet. 2014;5:472. doi: 10.3389/fgene.2014.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hitachi K, Tsuchida K. Role of microRNAs in skeletal muscle hypertrophy. Front Physiol. 2013;4:408. doi: 10.3389/fphys.2013.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]