

Abstract
The interaction of β-lactamase inhibitory protein II (BLIP-II) with β-lactamases serves as a model system to investigate the principles underlying protein-protein interactions. Previous studies have focused on identifying the determinants of binding affinity and specificity between BLIP-II and class A β-lactamases. However, interactions between BLIP-II and other bacterial proteins have yet to be explored. Here, we provide evidence that BLIP-II binds penicillin binding protein 2a (PBP2a) from methicillin resistant Staphylococcus aureus (MRSA) with a KD in the low micromolar range. In comparison to the binding constants for the potent interaction between BLIP-II and TEM-1 β-lactamase (KD= 0.5 pM), the on-rate for BLIP-II binding PBP2a is 44,000 times slower and the off-rate is 170 times faster. Therefore, a slow association rate is a limiting factor for the potency of the interaction between BLIP-II and PBP2a. Results from alanine scanning mutagenesis of the predicted interface residues of BLIP-II indicate that charged residues on the periphery of the BLIP-II interface play a critical role for binding PBP2a in contrast to previous findings that aromatic residues at the center of the BLIP-II interface are critical for the interaction with β-lactamases. Interestingly, many of the alanine mutants at the BLIP-II interface increase kon for binding PBP2a, consistent with the association rate being a limiting factor for affinity. In summary, the results of the study reveal that BLIP-II binds PBP2a, although weakly compared to binding of β-lactamases, and provides insights into the different binding strategies used for these targets.
Keywords: protein-protein interactions, β-lactamase, penicillin binding protein, binding specificity, hotspots, enzyme inhibition
TOC Graphic
Numerous diseases caused by single amino acid substitutions have demonstrated that the ability of proteins to discriminate between binding partners is essential for proper cell functioning(1–3). Despite the crucial role that proteins play in the cell, our understanding of the principles that govern the fine-tuned process of binding specificity is limited. Numerous studies have shown that although the binding interface between protein binding partners ranges from hundreds to several thousand square Angstroms, only a subset of residues contribute the majority of the binding energy(4–9).
This subset of residues, termed hotspots, is identified by alanine scanning mutagenesis of the predicted interface whereupon residues that, when substituted with alanine, exhibit a greater than 10-fold loss in binding affinity(4, 5, 10). The binding specificity of a protein, defined as variation in affinity between binding partners, can be achieved by employing a different set of hotspot residues for different binding partners(11). The development of protein interaction prediction programs that accurately predict binding affinity and specificity between protein binding partners would obviate the time-consuming task of identifying hotspots and could, thereby, accelerate the drug development process and unveil new biological signaling networks. As the pool of sequencing data continues to expand so, too, does the demand for understanding how mutations in proteins affect binding and cell biology.
Model systems of protein-protein interactions are used to provide insight into the determinants of binding affinity(12). One such system is the BLIP-β-lactamase interaction. β-lactamase inhibitory proteins (BLIPs), such as BLIP and BLIP-II, are potent inhibitors of class A β-lactamases(4–6, 11–16). BLIP-II is a 28 kDa protein secreted by Streptomyces exfolatius and has been used as a model to study principles of binding affinity and specificity because of its ability to bind various class A β-lactamases with femtomolar to picomolar affinity(11, 13, 17, 18). BLIP-II has a seven-bladed β-propeller structure and employs its numerous β-loops to bind β-lactamases and sterically block the active site to inhibit activity(19). Structurally, the binding interface of BLIP-II can be separated into an outer and inner ring of residues, resembling a bullseye target. Previous studies indicate that the majority of the binding energy in BLIP-II-β-lactamase complexes stems from interactions with the inner ring of residues.
β-lactamases are bacterial enzymes that provide resistance to the β-lactam antibiotics(20). There are four classes of β-lactamases (A-D) with class A β-lactamases being the most prevalent among clinical strains of bacteria(20). Class A β-lactamases exhibit 30–50% amino acid sequence identity and share the same overall three-dimensional structure. As a group, the class A enzymes display a wide range of substrate profiles for β-lactam antibiotics(20, 21). Despite different substrate profiles and differences in sequence identity, BLIP-II potently binds and inhibits class A β-lactamases using a similar set of hotspot residues and exhibits a less than 10-fold difference in binding affinity across the class of enzymes(13). This result suggests that BLIP-II preferentially binds to the β-lactamase structure and is able to accommodate sequence variation at the interface.
Two common mechanisms of bacterial resistance to β-lactam antibiotics are the production of β-lactamases (Gram-negative) and acquisition of penicillin binding proteins (PBPs) that exhibit a low affinity for β-lactams (Gram-positive). Penicillin binding proteins (PBPs) are transpeptidase enzymes that catalyze cross-linking of penta-peptides in the peptidoglycan layer of the cell wall. β-lactam antibiotics are covalent inhibitors of PBPs that block the cross-linking reaction mediated by the transpeptidase domain and lead to bacterial cell death.
Staphylococcus aureus is a Gram-positive bacterium that is a common cause of skin and respiratory infections and is frequently treated with β-lactam antibiotics. The first methicillin-resistant Staphylococcus aureus (MRSA) strain was identified in 1961 in the United Kingdom. Two years later, MRSA could be found globally and today, it remains a significant public health threat(22, 23). Resistance to β-lactam antibiotics in MRSA strains is due to the acquisition of the mecA gene from a non-S. aureus origin(22). The mecA gene encodes PBP2a, providing these strains with an additional PBP. PBP2a is a unique protein, in that its transpeptidase activity is poorly inhibited by β-lactam antibiotics(22). Consequently, PBP2a is able to resume crosslinking the cell wall despite the presence of β-lactam antibiotics and thereby provides resistance. Thus, PBP2a and class A β-lactamases are two distinct and widespread modes of resistance to β-lactam antibiotics.
BLIP-II binds and inhibits the class A β-lactamase TEM-1 with a Ki of 0.48 pM(13). The transpeptidase domain of PBP2a is structurally similar to TEM-1 with an RMSD of 1.18 Å (Figure 1A). Based on this structural homology, we hypothesized that BLIP-II would also bind and inhibit PBP2a. A previous study showed that the majority of the binding energy in the BLIP-II-β-lactamase interaction is located in the loop-helix region on class A β-lactamases and the inner ring of residues on BLIP-II (Figure 1A)(13). This loop helix region on class A β-lactamases shows large structural differences from the analogous region in PBP2a, and the electrostatics in this region are basic in PBP2a and acidic in TEM-1 β-lactamase (Figure 1B). For these reasons, BLIP-II may employ a different set of hotspot residues for binding PBP2a versus β-lactamases.
Figure 1.
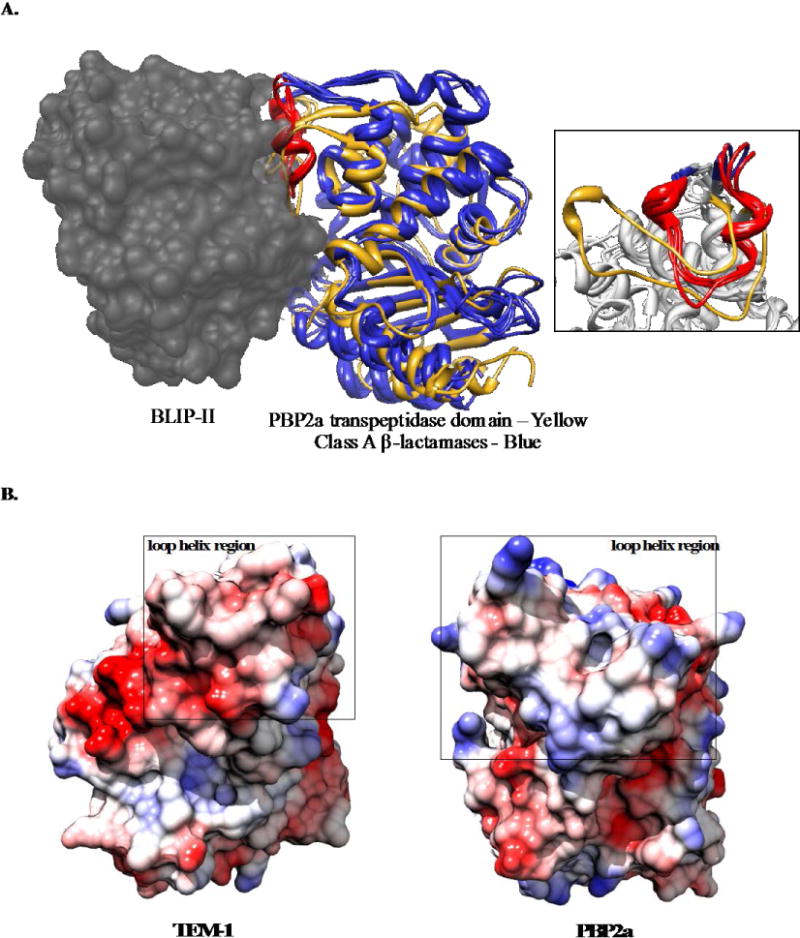
Comparison of the structure and electrostatic surface of PBP2a and class A β-lactamases. A: Structurally aligned class A β-lactamases [TEM-1 (PDB ID: 1JTD) Bla1 (PDB ID: 3QHY) and KPC-2 (PDB ID: 2OV5] are shown as blue ribbons with the loop-helix region that is responsible for majority of binding energy to BLIP-II highlighted in red. BLIP-II bound to the class A β-lactamases is shown in gray spacefill (PDB ID: 1JTD). The transpeptidase domain of PBP2a is shown as a yellow ribbon and aligned with the class A β-lactamase structures. A zoomed-in view of the loop-helix region is shown in the inset on the right with the β-lactamase loop-helix region shown as a red ribbon and PBP2a shown as a yellow ribbon. B: A map of the electrostatic potential of TEM-1 β-lactamase (at left) and the transpeptidase domain of PBP2a (at right) is shown with blue and red representing basic and acidic residues, respectively. The loop-helix region (depicted in A) is surrounded by a black box for comparison. Coulombic surface coloring completed in Chimera with default settings and a scale of −10 (red) to 10 (blue) kcal/(mol*e).
In this study, we show that BLIP-II binds and inhibits PBP2a with low micromolar affinity. Alanine-scanning mutagenesis of BLIP-II was used to identify the energetic contribution of each BLIP-II residue at the predicted interface with PBP2a. Surface plasmon resonance was used to determine the association and dissociation rate constants for each BLIP-II alanine mutant, providing a detailed view of the BLIP-II-PBP2a interaction. Comparison of the results of this study to previous work detailing the interaction of BLIP-II with class A β-lactamases reveals that BLIP-II is able to bind a wide range of target proteins by utilizing different combinations of hotspot residues on the same binding interface.
Materials and Methods
Site-directed mutagenesis
The BLIP-II alanine mutants were designed and constructed as previously reported (13). In short, residues at the interface were identified based on structural analyses of BLIP-II complexes with the TEM-1 and Bla1 β-lactamases and mutated to alanine using QuikChange site-directed mutagenesis (Stratagene). The BLIP-II gene in the pET-BLIP-II plasmid was sequenced for each mutant to confirm the presence of the desired alanine mutation and that no extraneous mutations were present on the gene.
Protein purification
The C-terminal His-tagged wild-type and alanine mutant BLIP-II proteins were purified using a Talon metal affinity resin (Clontech) as previously described(13, 18). PBP2a was expressed from E. coli strain BL21(DE3) containing a PBP2a-pET plasmid encoding PBP2a lacking the N-terminal 40 amino acid membrane-spanning domain. The strain was grown in 1.5 L of LB at 37°C to an OD600 ~ 0.6 and PBP2a expression was induced with a final concentration of 0.2 mM IPTG and incubated with shaking at 18°C for at least 24 hours. Cells were then lysed using a French press at 1250 psi. The cell lysate was centrifuged at 7,000 rpm for 45 minutes, filtered, and dialyzed overnight in 0.1 M NaAc, 0.4 M NaCl pH 8.0 buffer. The cell lysate was then loaded onto a Zn-chelating column and eluted on a gradient with 0.1 M NaAc, 0.4 M NaCl pH 4.0 buffer. Fractions with greater than 85% purity as determined by SDS-PAGE gel were combined and purified further on a size exclusion chromatography Superdex S75 column using 10 mM HEPES pH 7.4, 150 mM NaCl. Fractions with greater than 90% homogeneity after sizing were combined and used for binding analyses. The concentration of the BLIP-II and PBP2a protein preparations were determined using extinction coefficients calculated by the ExPASY protparam tool.
Determination of binding by isothermal titration calorimetry
Isothermal Titration Calorimetry (ITC) was performed on a VP-ITC instrument at 25°C. 140 μM BLIP-II was injected into 10 μM PBP2a. Values were calculated using Origin 7.0 software with an ITC customized module. Proteins were dialyzed into 25 mM HEPES, 1.0 M NaCl for 24 hours before ITC was performed.
PBP2a enzymatic activity inhibition assay
The catalytic activity of PBP2a can be monitored at 482 nm using nitrocefin, a colorimetric substrate(24). 10 μM BLIP-II was incubated with 2 μM PBP2a in 25 mM HEPES, 1.0 M NaCl for 45 minutes at room temperature before nitrocefin was added. The hydrolysis of nitrocefin was monitored in 20 second time intervals at 37°C in a 96-well plate format using a Tecan Infinite Pro 200 plate reader.
Determination of binding constants by surface plasmon resonance
Kinetic parameters for binding of PBP2a to wild-type BLIP-II and the alanine mutants were obtained using a Biacore3000 SPR instrument. Binding data for wild-type BLIP-II and the N50A, N51A, W152A, D167A, D170A, Y208A, Y248A and R286A variants was obtained from two different protein preparations by two different lab members. The remaining alanine mutants were assayed with a single protein preparation. PBP2a was immobilized by amine coupling to a CM5 sensor chip at pH 4.0 to total resonance units (RU) of ~2000. The values of binding constants determined in these experiments were not dependent on the amount of PBP2a coupled to the chip or on the flow rate suggesting the impact of mass action on the values was low(25). All measurements were baseline-corrected by subtracting the signal obtained from BLIP-II mutants injected over a channel with no protein attached. Experiments were performed at room temperature with the flow rate set at 10 μl/min using at least 3 BLIP-II concentrations. HBS-EP (0.01 M HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% v/v Surfactant P20) buffer prepared by GE was used for these experiments. Response curves were fit using the BiaEval program using a 1:1 Langmuir binding model with separate on- and off-rate determinations. These on- and off-rates were then used to calculate the standard error of the mean (SEM) using measurements from at least three concentrations in duplicate. The SEM reported for the KD is the summation of the SEM of the on- and off-rates.
ΔΔG calculations
ΔΔG values for the BLIP-II alanine mutants were calculated using the following equation:
Using this equation, a decrease in Ki upon mutation results in a negative ΔΔG value while an increase in KD yields a positive ΔΔG change. The error associated with the ΔΔG values was calculated using the following equation:
Where ‘SEM’ represents standard error of the mean, ‘WT’ represents wild-type BLIP and ‘MUT’ represents the mutant protein.
Results
Binding of BLIP-II to PBP2a
Based on the structural similarity between class A β-lactamases and the transpeptidase domain of PBP2a, we hypothesized that BLIP-II would also bind and inhibit PBP2a (Figure 1). To test this hypothesis, an enzymatic inhibition assay was performed in conditions where PBP2a turns over the substrate nitrocefin, albeit slowly. In this assay, PBP2a activity was monitored with the chromogenic substrate nitrocefin in the presence and absence of BLIP-II. When 2 μM PBP2a was incubated with 5-fold stoichiometric excess (10 μM) of BLIP-II for 45 minutes at 25°C, subsequent nitrocefin hydrolysis by PBP2a was noticeably decreased, suggesting inhibition by BLIP-II (Figure 2A).
Figure 2.
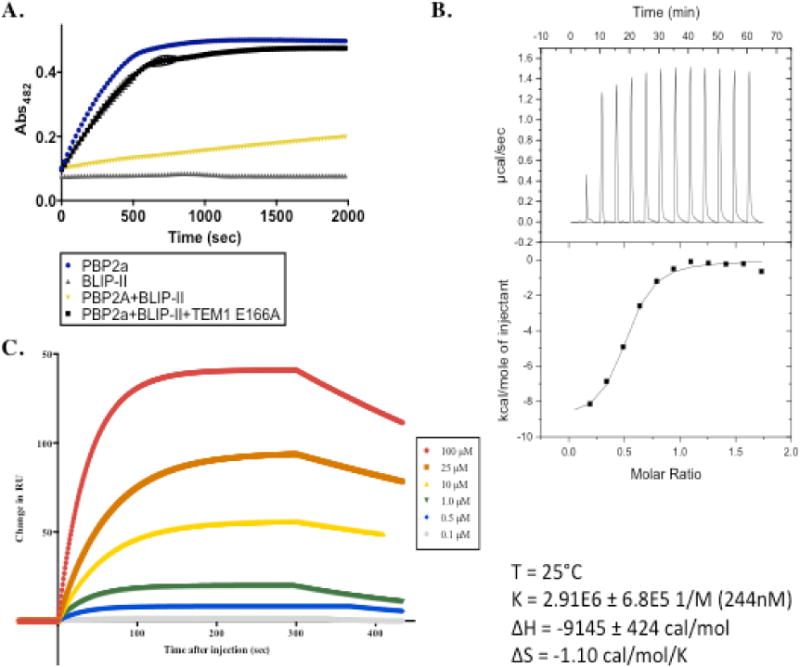
Binding of BLIP-II to PBP2a. A. Enzymatic inhibition assay is shown where inhibition was evaluated by monitoring hydrolysis of the colorimetric substrate, nitrocefin, by PBP2a at Abs482 over time. Nitrocefin hydrolysis by PBP2a alone is plotted as blue circles. Hydrolysis of nitrocefin in the presence of BLIP-II alone (negative control) is plotted as gray triangles. Nitrocefin hydrolysis by PBP2a in the presence of BLIP-II is shown as yellow triangles. Nitrocefin hydrolysis in a reaction containing PBP2a with BLIP-II and an excess of enzymatically inactive TEM-1 E166A is shown as black boxes. B. Isothermal titration calorimetry experiment with BLIP-II (140 μM) injected into PBP2a (10 μM) at 25°C over the course of an hour. The fit of the binding curve and the energy parameters are shown. C. Surface plasmon resonance binding curves with PBP2a fixed to a CM5 chip and BLIP-II injected at flow rate of 10 μl/min. Binding curves generated in the presence of various concentrations of BLIP-II are shown with the curve fitting plotted.
To test whether BLIP-II uses the same interface to bind PBP2a as for class A β-lactamases, BLIP-II was mixed with PBP2a and an excess of an enzymatically inactive TEM-1 E166A enzyme and the reaction was monitored for 45 minutes. BLIP-II has been shown to bind TEM-1 β-lactamase (and the inactive E166A variant) with picomolar affinity(13). Therefore, if BLIP-II uses the same interface to bind TEM-1 β-lactamase and PBP2a, the excess TEM-1 E166A will bind tightly to BLIP-II and binding of BLIP-II to PBP2a will no longer be detected. As seen in Fig. 2A, in the presence of BLIP-II and excess TEM-1 E166A β-lactamase PBP2a exhibited similar nitrocefin hydrolysis activity as the reaction with PBP2a and no BLIP-II (Figure 2A). This result suggests that BLIP-II uses a similar interface to bind β-lactamases and PBP2a.
Isothermal titration calorimetry (ITC) was used to confirm that BLIP-II binds PBP2a and to estimate the binding affinity of the interaction. In this experiment, 140 μM BLIP-II was injected into 10 μM PBP2a and analysis of the titration indicated a KD for the interaction of 244 nM (Figure 2B). This result is in agreement with the inhibition assay results indicating BLIP-II binds PBP2a.
Surface plasmon resonance (SPR) was used to further confirm binding and to determine on- and off-rates for this interaction. In this experiment, PBP2a was fixed to a chip and wild-type BLIP-II was injected at a flow rate of 10 μl/min in at least 3 different concentrations with each concentration performed in at least duplicate (Figure 2C). The KD of the BLIP-II-PBP2a interaction was determined to be 1.5 μM with an on-rate of 174 M−1s−1 and an off-rate of 2.62 × 10−4 s−1 (Table 1). The binding constant differs by 6-fold from that determined by ITC. This may be due to differences in the buffer (ITC was performed in high salt buffer) and temperature used for the experiments. Nevertheless, the SPR results provide further confirmation of binding between BLIP-II and PBP2a. Taken together, these experiments show that BLIP-II binds and inhibits PBP2a. Note, however, that the binding affinity of BLIP-II for PBP2a (~1 μM KD) is approximately one-million-fold weaker than the extremely potent binding to TEM-1 β-lactamase (0.48 pM KD). The weaker binding of BLIP-II to PBP2a versus TEM-1 is due to both a 44,000-fold slower on-rate (174 M−1s−1 vs. 5.4 × 106 M−1s−1) and a 170-fold faster off-rate (2.62 × 10−4 s−1 vs. 4.3 × 10−6 s−1)(17). Thus, a major factor in the weaker affinity of BLIP-II for PBP2a versus TEM-1 is the very slow association rate for binding PBP2a. The slow on-rate suggests that a large energy barrier may need to be overcome for complex association(26, 27).
Table 1.
Binding constants generated by SPR and ΔΔG calculations for BLIP-II alanine mutants for PBP2a
| BLIP-II Variants |
SPR binding constants | ΔΔG (kcal/mol) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| KD (μM) | ka (1/Ms) | kd (10−4/s) | KD | ka | kd | |
| WT | 1.5±0.3 | 174±13 | 2.62±0.3 | |||
| N50A | 0.45±0.09 | 1545±180 | 7.19±0.7 | −0.7±0.1 | −1.3±0.2 | 0.61±0.07 |
| N51A | 0.7±0.2 | 1274±215 | 8.6±0.9 | −0.5±0.1 | −1.2±0.2 | 0.72±0.07 |
| D52A | 1.8±0.5 | 354±73 | 6.1±0.6 | 0.12±0.03 | −0.43±0.09 | 0.51±0.06 |
| W53A | 0.93±0.04 | 1422±20 | 13.2±0.4 | −0.28±0.03 | −1.26±0.09 | 0.97±0.07 |
| T57A | 0.6±0.1 | 1753±296 | 9.5±0.9 | −0.5±0.1 | −1.4±0.3 | 0.78±0.09 |
| Y73A | 1.2±0.2 | 961±99 | 11.9±0.9 | −0.10±0.02 | −1.0±0.1 | 0.91±0.09 |
| F74A | 2.2±0.7 | 410±85 | 8.6±0.9 | 0.24±0.06 | −0.5±0.1 | 0.72±0.09 |
| L91A | 4±1 | 168±26 | 6.6±0.6 | 0.7±0.1 | 0.021±0.003 | 0.55±0.06 |
| N112A | 1.2±0.2 | 1188±139 | 14±1 | −0.13±0.02 | −1.2±0.2 | 1.0±0.1 |
| Y113A | 4±1 | 217±43 | 8±1 | −0.6±0.1 | −0.13±0.03 | 0.67±0.09 |
| D131A | 3.2±0.7 | 255±30 | 8.9±0.9 | 0.5±0.1 | −0.23±0.03 | 0.74±0.09 |
| W152A | 7±1 | 185±2 | 12±2 | 0.9±0.2 | −0.036±0.002 | 0.9±0.1 |
| D167A | >150 | ND | ND | ND | ND | ND |
| S169A | 1.4±0.4 | 504±100 | 7.4±0.9 | −0.029±0.006 | −0.6±0.1 | 0.63±0.09 |
| D170A | >150 | ND | ND | ND | ND | ND |
| Y191A | 4±1 | 466±100 | 16±1 | 0.05±0.01 | −0.6±0.1 | 1.1±0.1 |
| D206A | 1.56±0.4 | 239±33 | 3.7±0.5 | 0.036±0.008 | −0.19±0.3 | 0.21±0.03 |
| Y208A | 10±3 | 39±6 | 3.9±0.7 | 1.2±0.3 | 0.9±0.1 | 0.24±0.05 |
| F209A | 6±1 | 137±11 | 7.8±0.7 | 0.8±0.2 | 0.14±0.01 | 0.66±0.08 |
| I229A | 3.4±0.2 | 339±25 | 11.5±0.7 | 0.50±0.05 | −0.40±0.04 | 0.89±0.08 |
| F230A | 1.5±0.5 | 1435±244 | 21±3 | 0.012±0.003 | −1.3±0.2 | 1.3±0.2 |
| Y248A | 8±2 | 114±14 | 8.6±0.9 | 1.0±0.2 | 0.25±0.04 | 0.72±0.09 |
| E268A | 4.7±0.9 | 424±36 | 20±2 | 0.7±0.1 | −0.54±0.06 | 1.2±0.1 |
| W269A | 3.1±0.3 | 375±22 | 11.6±0.6 | 0.45±0.06 | −0.46±0.04 | 0.90±0.08 |
| R286A | >150 | ND | ND | ND | ND | ND |
| N304A | 0.75±0.08 | 2080±143 | 15.6±0.7 | −0.40±0.06 | −1.5±0.2 | 1.07±0.09 |
ND - binding was too weak to be determined
Impact of BLIP-II alanine mutations on association rate constants
It was of interest to determine the contribution of residues in the BLIP-II binding surface to the interaction with PBP2a. For this purpose, a set of 26 previously constructed alanine mutants of BLIP-II residues that were determined to be in contact with TEM-1 β-lactamase based on the TEM-1-BLIP-II crystal structure (1JTD) were purified and used to examine binding to PBP2a by SPR(13). These mutants are relevant for examining binding to PBP2a because the competition experiment described above indicated that the same surface of BLIP-II is used for binding both TEM-1 and PBP2a. The 26 BLIP-II interface residues examined form a variety of interactions including hydrogen bonds, electrostatic interactions, van der Waals contacts and/or noncanonical interactions (such as cation-π) based on the BLIP-II-β-lactamase crystal structure in complex with TEM-1(13). The results of the SPR experiments for binding PBP2a revealed a wide range of on-rates (39 to 2,080 M−1s−1) (Table 1, Figure 3). For ease of comparison, binding data was converted to changes in free energy of association (activation energy) using the following equation: . This results in a negative ΔΔG value for a mutation that increases the association rate and a positive ΔΔG for a mutation that decreases the association rate. A 10-fold change in association upon mutation to alanine would be equivalent to a ΔΔG value of 1.4 kcal mol−1 and is designated as a hotspot for association. The ΔΔG values are shown in Table 1 and plotted in Figure 3.
Figure 3.
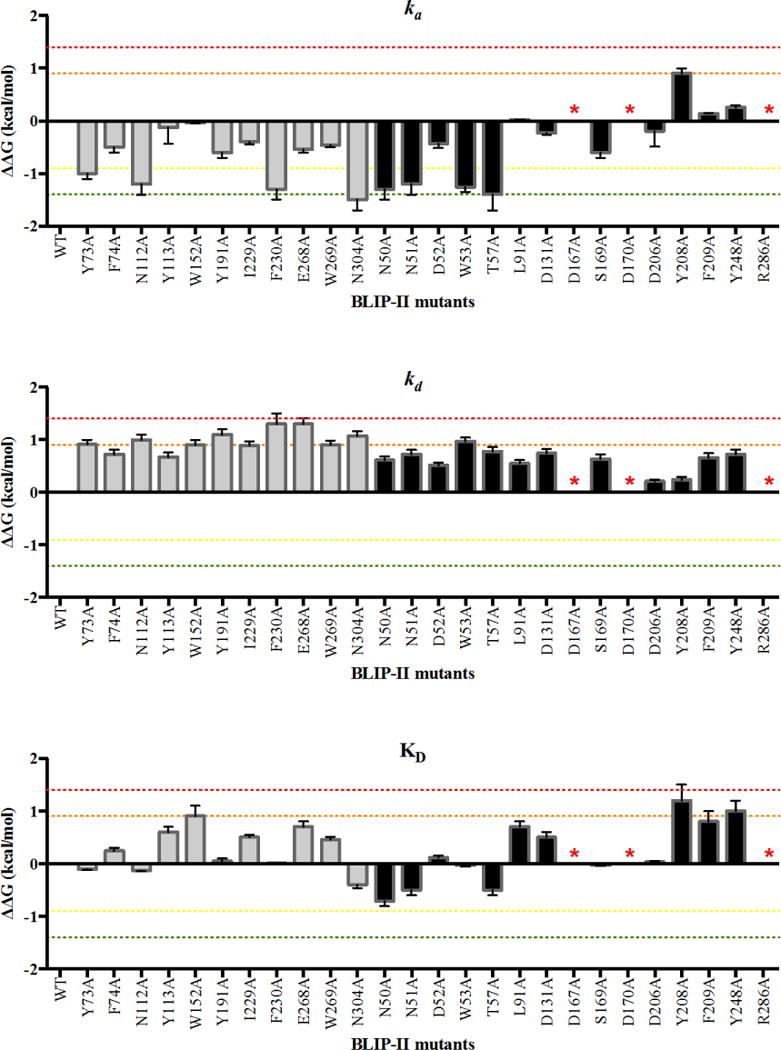
ka, kd, and KD ΔΔG values for BLIP-II alanine mutants. The ΔΔG value is depicted as a bar for each BLIP-II alanine mutant. The residues on the periphery of BLIP-II are shown as black bars while the inner ring of BLIP-II residues are shown as gray bars. Residues that reduced binding affinity to unmeasurable values when mutated to alanine are labeled with a red asterisk. The red dotted line marks a ΔΔG value of 1.4 kcal mol−1. The orange dotted line marks a ΔΔG of 0.9 kcal mol−1. The green dotted line marks a ΔΔG of −1.4 kcal mol−1. The yellow dotted line marks a ΔΔG of −0.9 kcal mol−1.
The majority of alanine mutants (18/26) resulted in an increased rate of association between BLIP-II and PBP2a (Table 1, Fig. 3). Alanine substitutions at T57 and N304 resulted in at least 10-fold faster association (<−1.4 kcal mol−1) while alanine at positions N50, N51, W53, Y73, N112 and F230 resulted in ΔΔG values < −1.0 kcal mol−1 (Table 1, Fig. 3). In addition, alanine substitutions at residues D52, F74, Y112, D131, S169, Y191, D206, I229, E268 and W269 resulted in ΔΔG values between −1.0 and −0.1 kcal/mol−1. In contrast, BLIP-II Y208A showed a five-fold decrease in kon and all other substitutions showed no change (Table 1, Fig. 3). Note that for D167, D170, and R286, the alanine substitutions abolish binding and therefore an accurate association rate could not be determined (Table 1).
A surprising aspect of the association rate determinations was the finding that the majority of the interface residues examined increased kon when mutated to alanine indicating that the wild-type amino acid at these positions impairs association of the complex. Therefore, the BLIP-II interface residues are not optimized for association with PBP2a. This is consistent with the finding that wild-type BLIP-II associates very slowly with PBP2a compared to TEM-1. A number of the residues that result in faster association when mutated to alanine are found clustered together on the BLIP-II binding interface suggesting this region contributes strongly to the slow association rate (Figure 4A).
Figure 4.
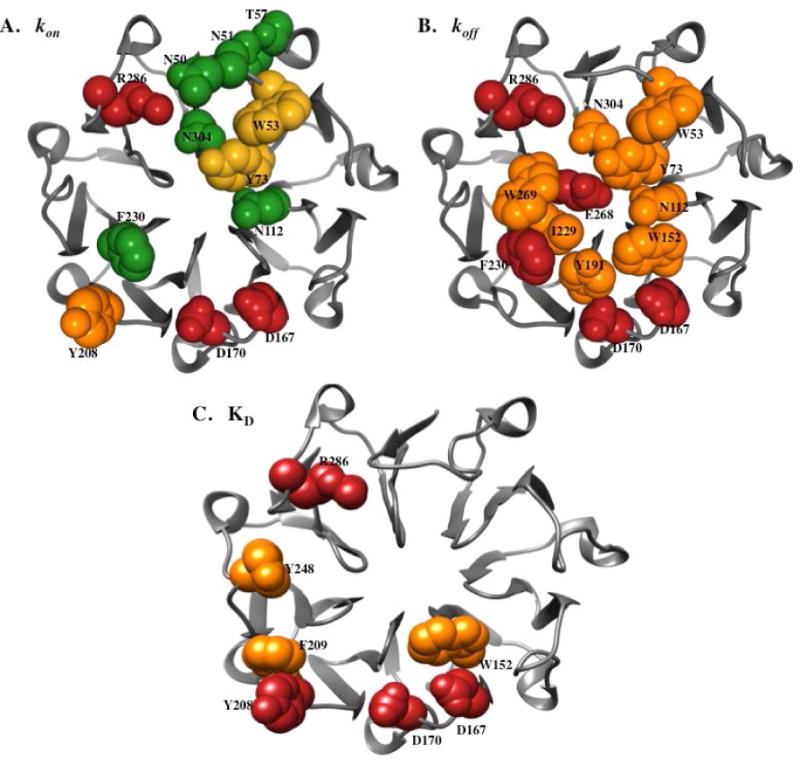
Energetic contributions of BLIP-II interface residues for binding PBP2a. The energetic contributions (ΔΔG) of the BLIP-II residues are shown on the BLIP-II structure for kon (A), koff (B), and KD (C). Residues that resulted in a ΔΔG value greater than 1.4 kcal mol−1 (within error) when mutated to alanine are shown as red spheres, those with a ΔΔG value greater than 0.9 kcal mol−1 (within error) are shown as orange spheres, those with a ΔΔG less than −1.4 kcal mol−1 (within error) are shown green spheres, and those with a ΔΔG less than −0.9 kcal mol−1 (within error) are shown as yellow spheres.
Impact of BLIP-II alanine substitutions on dissociation rate constants
In contrast to the effect of the alanine substitutions on association where the majority of mutants exhibited increased on-rates that favor binding, all of the BLIP-II alanine mutants for which koff could be determined exhibited increased dissociation rates (Table 1). The off-rates varied from 2.62 × 10−4 s−1 for the wild-type complex to 21 × 10−4 s−1 for the F230A mutant (Table 1). BLIP-II mutants F230A and E268A exhibited the largest increase in dissociation rate with an 8-fold increase compared to wild-type. Note, however, that the affinity of the D167A, D170A, and R286A mutants was too low to measure binding and so a dissociation constant could not be determined. It is likely, however, that these mutants have greatly increased dissociation rates. There were eight residues that increased koff by at least 5-fold when mutated to alanine including W53, Y73, N112, W152, Y191, I229, W269 and N304 while the alanine substitutions at the remaining positions had more modest effects (Table 1). The binding results for dissociation rates were converted to changes in free energy where a negative ΔΔG indicates a mutation decreases the dissociation rate and a positive ΔΔG increases the rate of dissociation. There were no mutations that exhibited a negative change in free energy upon mutation (Table 1, Figure 3). The residues that when mutated exhibited a ΔΔG of greater than 0.9 kcal mol−1 grouped together on the interface of BLIP-II. A comparison of the location of residues that, when mutated, increased the association rate with those that resulted in faster dissociation reveals about half of the residues, such as W53, Y73, N112, F230 and N304, overlap indicating many of the residues controlling dissociation also influence association (Figure 4B). Because mutations that led to a large increase in the on-rate were usually accompanied by an increase in the off-rate of the complex, the overall change in binding affinity was greatly reduced. Therefore, optimization of the interface for association is not necessarily associated with an increase in the overall binding affinity of the complex.
BLIP-II hotspots for binding PBP2a
Binding hotspots are residues that when mutated to alanine exhibit a greater than 10-fold decrease in binding affinity as defined by KD. In the BLIP-II-PBP2a interaction, there are only four hotspot residues – D167, D170, Y208 and R286. These are largely charged residues that likely participate in electrostatic interactions with PBP2a. The relative role of these charged residues in the association and dissociation reactions is unknown as we were not able to measure binding when any one of these residues was absent from the interface. All four residues, including the uncharged Y208, are located on the periphery of the BLIP-II interface (Figure 4). Three other residues, W152, F209 and Y248, resulted in an at least 5-fold decrease in binding affinity when mutated to alanine. These residues are all aromatic residues and are also located on the periphery of the BLIP-II interface. This study is in agreement with previous studies that have shown that aromatic and charged residues commonly make large contributions the binding energy of protein-protein complexes; however, the BLIP-II-PBP2a results are unique in that the hotspot residues are dispersed around the periphery of the interface as opposed to clustered in the center of the interface(5, 13, 28).
Overall, most BLIP-II residues were found to contribute modestly to the energy of the BLIP-II-PBP2a interaction (Table 1). Electrostatic interactions appear to play an important role in the binding of BLIP-II and PBP2a as three residues on the outer ring of BLIP-II that were found to be hotspots for binding were charged residues. The slow on-rate (103 M−1s−1) of the interaction contributes to the weak affinity and, based on the results, the wild-type residues are detrimental and, thus, there are numerous ways to increase the association rate for complex formation. In contrast, the alanine substitutions at BLIP-II residues in the interface increase the dissociation rate suggesting the wild-type residues contribute to the stability of the complex. In conclusion, BLIP-II primarily uses charged residues on its outer ring to bind PBP2a a million-fold weaker than class A β-lactamases and employs a separate group of hotspot residues, likely because of the differences in both structure and sequence at the binding interface between the PBP2a and class A β-lactamases.
Discussion
This study determined that BLIP-II binds the Staphylococcus aureus PBP2a enzyme with low micromolar affinity and defined the residues on the BLIP-II interface that contribute to this interaction. The BLIP-II-PBP2a interaction is characterized by a slow on-rate (103 M−1sec−1) compared with BLIP-II binding to class A β-lactamases such as TEM-1 (106 M−1sec−1). The finding that the majority of the alanine substitution mutants exhibited increased association rates suggests the wild-type residues in the BLIP-II interface are detrimental compared to other residue types and multiple mutational pathways could optimize binding through improved association rates.
The dynamic nature and allosteric regulation of the transpeptidase domain of PBP2a has been established in crystal structures(29, 30). This structural plasticity may have implications in the slow rate of association and overall weak binding of BLIP-II with PBP2a. Although the illustration in figure 1 is not a docked pose, the alignment of PBP2a with class A β-lactamases and the resulting positioning of PBP2a relative to BLIP-II suggests there would be multiple clashes at the interface that would need to be overcome for binding. Before proteins can bind, they must first form an encounter complex that is composed of specific, long-range electrostatic interactions and nonspecific short-range interactions (hydrophobic and van der Waals) with large parts of the interface remaining solvated(31, 32). Multiple conformations are sampled in this process to facilitate optimal contacts, resulting in the most favorable conformation in the final complex(32). Kinetic data generated on colicin-immunity protein complexes also suggests that the proteins in the encounter complexes undergo rigid body rotation events to form the final bound states(33). The BLIP-II-PBP2a interaction could potentially employ both modes of binding by requiring a specific structural conformation of the transpeptidase domain of PBP2a and rotation of the rigid BLIP-II structure, significantly slowing the rate of association. Furthermore, previous studies indicate that long-range electrostatic interactions play an important role in steering of the protein binding partners for association(26, 27). It is possible that the charged hotspot residues on BLIP-II identified in this study (D167, D170 and R286) are essential for steering of these proteins into the correct orientation for successful association.
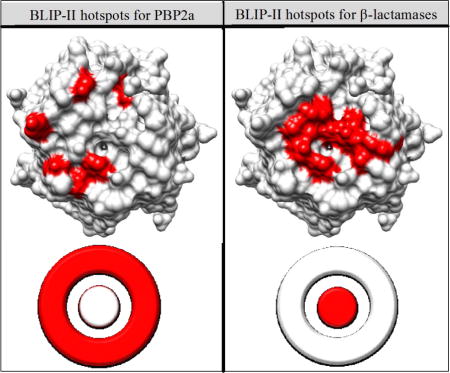
Previously, alanine scanning mutagenesis of the BLIP-II interface residues was performed to identify hotspots for binding various class A β-lactamases(13). The study revealed that the hotspots for BLIP-II binding to class A β-lactamases consists of aromatic residues clustered at the center of the interface(13). This is in agreement with the O-ring hypothesis, which states that hotspot residues are usually clustered at the center of the interface and surrounded a ring of largely hydrophilic residues(34). The interaction between BLIP-II and PBP2a, however, is facilitated in a different manner with hotspot residues on the periphery of the BLIP-II binding surface and an overall million-fold weaker affinity than the interaction between BLIP-II and class A β-lactamases(13). The structure of BLIP-II can be visualized as consisting of an inner ring and outer ring, much like a bullseye target. Brown et al. showed that BLIP-II uses its inner ring of aromatic residues for the majority of the energy to bind class A β-lactamases(13). Here, we show that BLIP-II primarily uses two clusters of charged and aromatic residues on the periphery or outer ring of the interface to bind PBP2a. The use of dispersed charged residues on the periphery of the binding interface is a less common mode of binding and could be why this interaction is in the micromolar range and shows numerous avenues for optimization(7, 34). Together, these results suggest that BLIP-II uses its inner ring of aromatic residues to bind class A β-lactamases with picomolar affinity and its outer ring of charged and aromatic residues to mediate its micromolar interaction with PBP2a (Figure 5).
Figure 5.
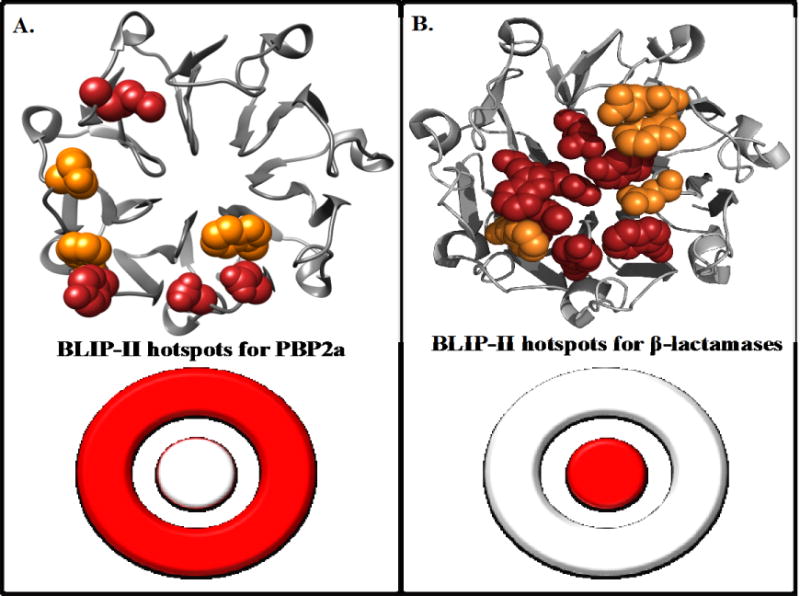
BLIP-II hotspot residues for binding PBP2a (A) and class A β-lactamases (B). BLIP-II is shown in grey ribbon with hotspot residues shown as spheres. Red residues indicate a greater than 10-fold decrease in binding upon mutation to alanine and orange indicates at least a 5-fold decrease in binding upon mutation. The hotspot residues for the class A β-lactamases (B) are hotspots for all class A β-lactamases tested in Brown et. al.(13). A bullseye representation of BLIP-II binding hotspots is shown on the bottom half of the figure.
Several other proteins have been shown to achieve binding specificity by using different combinations of hotspot residues for different binding partners(11, 35, 36). For example, alanine scanning of the common binding surface on TEM-1 β-lactamase that binds both BLIP and BLIP-II revealed that different combinations of TEM-1 hotspot residues are used to bind BLIP versus BLIP-II(11). In addition, computational design of 20 multi-specific proteins (engineered specificity) suggested that although the same binding interface is used to bind two different proteins, each partner prefers its own distinct set of hotspots residues(37). This was also shown to be the case in a recent study examining 16 different co-crystal structures of calmodulin and its target proteins(38). This study extends the observation that one binding interface (BLIP-II) can bind a range of interaction partners with varying affinities (class A β-lactamases and PBP2a) by employing distinct subsets of hotspot residues for each interaction. This binding strategy highlights the multifaceted nature of protein interfaces.
The biological role of BLIP-II remains unknown. BLIP-II was first identified as a secreted protein in the culture supernatant of Streptomyces exfoliates SMF19(39, 40). An S. exfoliates SMF19 BLIP-II null mutant strain exhibited defects in sporulation and septum formation and immune fluorescence studies indicate BLIP-II is associated with the cell envelope(39). Therefore, it is possible that the natural target of BLIP-II could be PBPs involved in peptidoglycan cross-linking during septation(19). However, our study with PBP2a is the first evidence that BLIP-II binds to a PBP and there is currently no information on BLIP-II binding to Streptomyces PBPs.
PBP2a and class A β-lactamases are widespread sources of resistance to the commonly prescribed β-lactam antibiotics and are therefore a serious public health concern. PBP2a, the protein responsible for MRSA, poses a serious challenge for current treatment regimens including all classes of antibiotics(23, 41). Therefore, the interaction between BLIP-II and PBP2a presents a potential route for development of inhibitors or diagnostics for clinical use. It is noteworthy that since PBP2a is anchored on the outside of the cytoplasmic membrane and Gram-positive bacteria such as S. aureus do not have an outer membrane suggesting exogenously added BLIP-II could potentially bind PBP2a. In addition, although the size of BLIP-II presents challenges for delivery, the β-loop structures on BLIP-II provide a promising scaffold for the development of cyclized peptide inhibitors. Future studies will aim to optimize the BLIP-II-PBP2a interaction and explore the binding of BLIP-II to other clinically relevant PBPs.
Acknowledgments
Funding sources
This work was supported by NIH grant AI32956 to T.P. and a training fellowship from the Houston Area Molecular Biophysics Training Program (NIGMS grant no. T32 GM008280) of the Gulf Coast Consortia to C.J.A.
References
- 1.White MB, Amos J, Hsu JM, Gerrard B, Finn P, Dean M. A frame-shift mutation in the cystic fibrosis gene. Nature. 1990;344:665–667. doi: 10.1038/344665a0. [DOI] [PubMed] [Google Scholar]
- 2.Morganella S, Alexandrov LB, Glodzik D, Zou X, Davies H, Staaf J, Sieuwerts AM, Brinkman AB, Martin S, Ramakrishna M, Butler A, Kim HYY, Borg Å, Sotiriou C, Futreal PA, Campbell PJ, Span PN, Van Laere S, Lakhani SR, Eyfjord JE, Thompson AM, Stunnenberg HG, van de Vijver MJ, Martens JW, Børresen-Dale ALL, Richardson AL, Kong G, Thomas G, Sale J, Rada C, Stratton MR, Birney E, Nik-Zainal S. The topography of mutational processes in breast cancer genomes. Nat Comm. 2016;7:11383. doi: 10.1038/ncomms11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326–328. doi: 10.1038/180326a0. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z, Palzkill T. Determinants of binding affinity and specificity for the interaction of TEM-1 and SME-1 beta-lactamase with beta-lactamase inhibitory protein. J Biol Chem. 2003;278:45706–45712. doi: 10.1074/jbc.M308572200. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Palzkill T. Dissecting the Protein-Protein Interface between β-Lactamase Inhibitory Protein and Class A β-Lactamases. J Biol Chem. 2004;279:42860–42866. doi: 10.1074/jbc.M406157200. [DOI] [PubMed] [Google Scholar]
- 6.Reichmann D, Cohen M, Abramovich R, Dym O, Lim D, Strynadka NC, Schreiber G. Binding hot spots in the TEM1-BLIP interface in light of its modular architecture. J Mol Biol. 2007;365:663–679. doi: 10.1016/j.jmb.2006.09.076. [DOI] [PubMed] [Google Scholar]
- 7.Keskin O, Ma B, Nussinov R. Hot Regions in Protein–Protein Interactions: The Organization and Contribution of Structurally Conserved Hot Spot Residues. J Mol Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 8.Kozakov D, Hall DR, Chuang GY, Cencic R, Brenke R, Grove LE, Beglov D, Pelletier J, Whitty A, Vajda S. Structural conservation of druggable hot spots in protein–protein interfaces. Proc Natl Acad Sci. 2011;108:13528–13533. doi: 10.1073/pnas.1101835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baskaran K, Duarte JM, Biyani N, Bliven S, Capitani G. A PDB-wide, evolution-based assessment of protein–protein interfaces. BMC Struct Biol. 2014;14:22. doi: 10.1186/s12900-014-0022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLaughlin RN, Jr, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fryszczyn BG, Adamski CJ, Brown NG, Rice K, Huang W, Palzkill T. Role of β-lactamase residues in a common interface for binding the structurally unrelated inhibitory proteins BLIP and BLIP-II. Prot Sci. 2014;23:1235–1246. doi: 10.1002/pro.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gretes M, Lim DC, de Castro L, Jensen SE, Kang SG, Lee KJ, Strynadka NC. Insights into positive and negative requirements for protein-protein interactions by crystallographic analysis of the beta-lactamase inhibitory proteins BLIP, BLIP-I, and BLP. J Mol Biol. 2009;389:289–305. doi: 10.1016/j.jmb.2009.03.058. [DOI] [PubMed] [Google Scholar]
- 13.Brown NG, Chow DC, Ruprecht KE, Palzkill T. Identification of the beta-lactamase inhibitor protein-II (BLIP-II) interface residues essential for binding affinity and specificity for class A beta-lactamases. J Biol Chem. 2013;288:17156–17166. doi: 10.1074/jbc.M113.463521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reynolds KA, Thomson JM, Corbett KD, Bethel CR, Berger JM, Kirsch JF, Bonomo RA, Handel TM. Structural and computational characterization of the SHV-1 beta-lactamase-beta-lactamase inhibitor protein interface. J Biol Chem. 2006;281:26745–26753. doi: 10.1074/jbc.M603878200. [DOI] [PubMed] [Google Scholar]
- 15.Reichmann D, Rahat O, Albeck S, Meged R, Dym O, Schreiber G. The modular architecture of protein-protein binding interfaces. Proc Natl Acad Sci. 2005;102:57–62. doi: 10.1073/pnas.0407280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potapov V, Reichmann D, Abramovich R, Filchtinski D, Zohar N, Halevy BD, Edelman M, Sobolev V, Schreiber G. Computational Redesign of a Protein–Protein Interface for High Affinity and Binding Specificity Using Modular Architecture and Naturally Occurring Template Fragments. J Mol Biol. 2008;384:109–119. doi: 10.1016/j.jmb.2008.08.078. [DOI] [PubMed] [Google Scholar]
- 17.Brown NG, Chow DC, Sankaran B, Zwart P, Prasad BV, Palzkill T. Analysis of the binding forces driving the tight interactions between beta-lactamase inhibitory protein-II (BLIP-II) and class A beta-lactamases. J Biol Chem. 2011;286:32723–32735. doi: 10.1074/jbc.M111.265058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown NG, Palzkill T. Identification and characterization of beta-lactamase inhibitor protein-II (BLIP-II) interactions with beta-lactamases using phage display. PEDS. 2010;23:469–478. doi: 10.1093/protein/gzq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim D, Park HU, De Castro L, Kang SG, Lee HS, Jensen S, Lee KJ, Strynadka NC. Crystal structure and kinetic analysis of beta-lactamase inhibitor protein-II in complex with TEM-1 beta-lactamase. Nat Struct Biol. 2001;8:848–852. doi: 10.1038/nsb1001-848. [DOI] [PubMed] [Google Scholar]
- 20.Fisher JF, Meroueh SO, Mobashery S. Bacterial Resistance to β-Lactam Antibiotics: Compelling Opportunism, Compelling Opportunity. Chem Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 21.Bush K, Fisher JF. Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from gram-negative bacteria. Ann Rev Microbiol. 2011;65:455–478. doi: 10.1146/annurev-micro-090110-102911. [DOI] [PubMed] [Google Scholar]
- 22.Fishovitz J, Hermoso JA, Chang M, Mobashery S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life. 2014;66:572–577. doi: 10.1002/iub.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stryjewski ME, Corey RG. Methicillin-Resistant Staphylococcus aureus: An evolving pathogen. Clin Infect Dis. 2014;58:S10. doi: 10.1093/cid/cit613. [DOI] [PubMed] [Google Scholar]
- 24.Roychoudhury S, Kaiser RE, Brems DN, Yeh WK. Specific interaction between beta-lactams and soluble penicillin-binding protein 2a from methicillin-resistant Staphylococcus aureus: development of a chromogenic assay. Antimicrob Agents Chemother. 1996;40:2075–2079. doi: 10.1128/aac.40.9.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jason-Moller L, Murphy M. Overview of Biacore systems and their applications. Curr Protocals Prot Sci. 2006 doi: 10.1002/0471140864.ps1913s45. Chap 19: Unit 19. [DOI] [PubMed] [Google Scholar]
- 26.Selzer T, Albeck S, Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nat Struct Biol. 2000;7:537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- 27.Selzer T, Schreiber G. Predicting the rate enhancement of protein complex formation from the electrostatic energy of interaction. J Mol Biol. 1999;287:409–419. doi: 10.1006/jmbi.1999.2615. [DOI] [PubMed] [Google Scholar]
- 28.Kang SG, Park HU, Lee HS, Kim HT, Lee KJ. New beta -lactamase inhibitory protein (BLIP-I) from Streptomyces exfoliatus SMF19 and its roles on the morphological differentiation. J Biol Chem. 2000;275:16851–16856. doi: 10.1074/jbc.M000227200. [DOI] [PubMed] [Google Scholar]
- 29.Fishovitz J, Rojas-Altuve A, Otero LH, Dawley M, Carrasco-López C, Chang M, Hermoso JA, Mobashery S. Disruption of allosteric response as an unprecedented mechanism of resistance to antibiotics. J Amer Chem Soc. 2014;136:9814–9817. doi: 10.1021/ja5030657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahasenan KV, Molina R, Bouley R, Batuecas MT, Fisher JF, Hermoso JA, Chang M, Mobashery S. Conformational dynamics in penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus, allosteric communication network and enablement of catalysis. J Amer Chem Soc. 2017 doi: 10.1021/jacs.6b12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schreiber G. Kinetic studies of protein-protein interactions. Curr Opin Struct Biol. 2002;12:41–47. doi: 10.1016/s0959-440x(02)00287-7. [DOI] [PubMed] [Google Scholar]
- 32.Taylor MG, Kirsch JF, Rajpal A. Kinetic epitope mapping of the chicken lysozyme. HyHEL-10 fab complex: Delineation of docking trajectories. Prot Sci. 1998;7:1857–1867. doi: 10.1002/pro.5560070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keeble AH, Kleanthous C. The kinetic basis for dual recognition in colicin endonuclease–immunity protein complexes. J Mol Biol. 2005;352:656–671. doi: 10.1016/j.jmb.2005.07.035. [DOI] [PubMed] [Google Scholar]
- 34.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 35.Ngan CH, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S. The structural basis of pregnane X receptor binding promiscuity. Biochemistry. 2009;48:11572–11581. doi: 10.1021/bi901578n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atkins WM. Biological messiness vs. HyHEL-10 fab complex: Delineation of docking trajectories. J Steroid Biochem Mol Biol. 2015;151:3–11. doi: 10.1016/j.jsbmb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphris EL, Kortemme T. Design of multi-specificity in protein interfaces. PLoS Comput Biol. 2007;3:e164. doi: 10.1371/journal.pcbi.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fromer M, Shifman JM. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Comput Biol. 2009;5:e1000627. doi: 10.1371/journal.pcbi.1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim ES, Song JY, Kim DW, Ko EJ, Jensen SE, Lee KJ. Involvement of beta-lactamase inhibitory protein, BLIP-II, in morphological differentiation of Streptomyces exfoliatus SMF19. J Microbiol and Biotech. 2008;18:1884–1889. [PubMed] [Google Scholar]
- 40.Park HU, Lee KJ. Cloning and heterologous expression of the gene for BLIP-II, a beta-lactamase-inhibitory protein from Streptomyces exfoliatus SMF19. Microbiology. 1998;144(Pt 8):2161–2167. doi: 10.1099/00221287-144-8-2161. [DOI] [PubMed] [Google Scholar]
- 41.Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother. 1997;40:135–136. doi: 10.1093/jac/40.1.135. [DOI] [PubMed] [Google Scholar]