

Abstract
Objective
Intestinal metabolism might play a greater role in regulating whole body metabolism than previously believed. We aimed to enhance enterocyte metabolism in mice and investigate if it plays a role in diet-induced obesity (DIO) and its comorbidities.
Methods
Using the cre-loxP system, we overexpressed the mitochondrial NAD+ dependent protein deacetylase SIRT3 in enterocytes of mice (iSIRT3 mice). We chronically fed iSIRT3 mice and floxed-SIRT3 control (S3fl) mice a low-fat, control diet (CD) or a high-fat diet (HFD) and then phenotyped the mice.
Results
There were no genotype differences in any of the parameters tested when the mice were fed CD. Also, iSIRT3 mice were equally susceptible to the development of DIO as S3fl mice when fed HFD. They were, however, better able than S3fl mice to regulate their blood glucose levels in response to exogenous insulin and glucose, indicating that they were protected from developing insulin resistance. This improved glucose homeostasis was accompanied by an increase in enterocyte metabolic activity and an upregulation of ketogenic gene expression in the small intestine.
Conclusion
Enhancing enterocyte oxidative metabolism can improve whole body glucose homeostasis.
Keywords: Insulin resistance, Oxidative metabolism, Ketone bodies, Enterocytes, Gut, Sirtuins
Highlights
-
•
Enterocyte SIRT3 overexpression (OE) increased enterocyte metabolic activity.
-
•
Enterocyte SIRT3 OE did not affect energy intake or body weight gain.
-
•
Enterocyte SIRT3 OE stimulated gut ketogenesis in high fat-fed mice.
-
•
Enterocyte SIRT3 OE protected high fat-fed mice from becoming insulin resistant.
1. Introduction
Type 2 diabetes (T2D), a leading global health concern, is a major cause of premature deaths worldwide [1]. Overweight and obesity massively increase the risk of developing T2D, which involves a progressive rise in insulin resistance (IR), due in part to ectopic fat accumulation. The pancreas tries to compensate for the rising IR by overproducing insulin, which ultimately leads to pancreatic β-cell dysfunction [2]. Current pharmacological treatments for T2D mainly involve a combination of insulin and hypoglycemic or insulin sensitizing agents. The most efficient treatment for T2D and morbid obesity so far is surgical intervention such as Roux-en-Y gastric bypass (RYGB) [3]. In fact, gastric bypass surgery leads to an immediate reversal of the diabetic phenotype, even before any substantial weight loss [4], [5]. A large part of this success is supposedly due to morphological and functional changes in the small intestine [6]. Data from gastric bypass rat and mouse models support this theory, i.e., postsurgical morphological and/or metabolic changes in the small intestine might play a role in improving glycemic control in these animals [7], [8], [9].
Genetic [10] and pharmacological [11], [12] inhibition of intestinal triacylglycerol (TAG) re-esterification prevented diet-induced obesity (DIO) and/or improved glycemic control in rodents. Pharmacological studies have also implicated enterocyte fatty acid oxidation (FAO) in the control of food intake and energy expenditure [13], [14]. Intestines of obesity resistant mice show an increase in FAO genes in response to HFD feeding compared to obesity prone mice [15]. A recent study also revealed that within three days of HFD exposure, mouse enterocytes increase their capacity to oxidize fat and to produce ketone bodies [16]. Together, these studies suggest that enterocyte metabolism might play an important role in the development of obesity and the metabolic syndrome.
In this study, we aimed to examine whether genetically enhancing enterocyte mitochondrial metabolic functions, such as FAO, affects DIO and whole body glucose homeostasis. We therefore overexpressed the mitochondrial protein Sirtuin 3 (SIRT3) in the enterocytes of mice and tested the effects of this manipulation on the development of DIO and IR. All sirtuins require NAD+ to be catalytically active, which directly links their function to the metabolic status of a cell [17]. SIRT3, primarily expressed in mitochondria, deacylates and activates global mitochondrial proteins leading to increased mitochondrial function, including FAO, tricarboxylic acid (TCA) cycle flux, and ketogenesis [18], [19].
We found that SIRT3 overexpression in the enterocytes did not produce any effects when the mice were on control diet (CD). On HFD, however, SIRT3 overexpression protected the mice from developing glucose intolerance and IR. This was accompanied by an increased gene expression of the ketogenic marker 3-hydroxy-3-methylglutaryl-CoA synthase 2 (Hmgcs2) in the duodenal and jejunal enterocytes of these mice. In addition, in control mice, post prandial systemic ketone body levels were lower on the HFD than on the CD, whereas in mice with the enterocyte-specific SIRT3 overexpression, this difference was not significant. Intestinal SIRT3 overexpression in mice chronically fed HFD led to an increase in small intestinal enterocyte metabolic activity in response to an oral load of oleic acid. SIRT3 overexpression did not affect body weight, body composition, fat distribution, or small intestinal morphology. These findings suggest that an increase in the metabolic flux of enterocytes is sufficient to improve whole body glucose homeostasis in DIO, independent of body weight, body composition, or fat distribution.
2. Materials and methods
2.1. Animals
Mice with an enterocyte-specific SIRT3 overexpression (iSIRT3) were generated by crossing transgenic mice with a floxed stop cassette preceding the SIRT3 gene (S3fl) [20] with mice expressing Cre recombinase under the enterocyte-specific promoter Villin (Vil-Cre) [21] (stock number 004586 from the Jax cre repository), so that an additional copy of SIRT3 was expressed in the epithelial cells of the intestine. The S3fl mice served as controls. All mice had a C57Bl6/J background. All breedings were carried out in our in-house specified and opportunistic pathogen free (SOPF) animal facility. At 8–10 weeks of age, male mice of the appropriate genotype were moved into the experimental room with controlled temperature and humidity (22 ± 2 °C, 55 ± 5%) and a 12 h/12 h dark/light cycle. All animals were group-housed (2–4 animals per cage) and had ad libitum access to food and water unless otherwise specified. Their body weights were monitored regularly as indicated. All animal experiments were approved by the Cantonal Veterinary Office of Zurich.
2.2. Diets
In the SOPF breeding facility, mice were fed standard chow diet (Kliba Nafag, Switzerland). In the experimental room, they were switched to either refined low-fat control diet (CD, #S9213-E001, 10 kJ% fat) or high-fat diet (HFD, #E15742-34, 60 kJ% fat) from ssniff Spezialdiäten GmbH, Germany.
2.3. Body composition
Awake mice were scanned at the start and after 2, 4, 8, and 11 weeks of CD and HFD feeding using the EchoMRI 3-in-1 analyzer (EchoMRI™, Singapore and Houston, USA) to assess fat and lean mass. After 16 weeks on the diets, mice were scanned under isoflurane anesthesia using a high-resolution micro computed tomography scanner (CT; La Theta LCT-100; Hitachi-Aloka Medical Ltd, Japan) to determine fat distribution in addition to body composition.
2.4. Energy intake and indirect calorimetry
Measurements were carried out in PhenoMaster/LabMaster metabolic cages (TSE systems, Bad Homburg, Germany). Mice were fed CD and adapted over 1 week to single housing in cages similar to the PhenoMaster cages before measurements. Data displayed were collected after an additional 2 days of habituation in the system. After 2 days of data collection on CD, mice were switched to HFD, and data were recorded for 2 more days. Food intake was measured manually every 12 h, at the beginning of the light or dark phases. Energy intake was calculated by multiplying the absolute food intake values with the energy density of the diets (1 g of CD = 15.25 kJ; 1 g of HFD = 21.53 kJ). Mice were returned to group caging at the end of the PhenoMaster experiments, and their body compositions were measured using the Echo-MRI analyzer [22], [23].
2.5. Insulin sensitivity test (IST)
After 8 weeks on the diets, mice were fasted for 4–5 h in the middle of the dark phase with ad libitum access to water. Actrapid HM human insulin (Novo Nordisk, Denmark) was injected intraperitoneally (IP), and tail blood glucose levels were monitored at the time points indicated using the Accu-Chek Aviva blood glucose monitor (Roche, Switzerland). The Insulin dose was 0.4 and 0.8 mU/g body weight for CD and HFD fed mice, respectively.
2.6. Oral glucose tolerance test (OGTT)
After 10 weeks on the diets, mice were fasted for 6 h at the onset of dark phase with ad libitum access to water [24]. They received a 20% glucose solution in water by gavage (glucose dose: 2 g/kg body weight). Tail blood glucose levels were monitored at the time points indicated.
2.7. Animal sacrifice and tissue collection
After 17 or 20 weeks on the diets, mice were food deprived for 2 h in the dark phase (post prandial) or overnight, respectively, with ad libitum access to water until sacrifice. All animals were sacrificed in the dark phase by decapitation; trunk blood was collected and processed as described later in the plasma metabolite analysis section (method 2.11). The intestine and liver were dissected out. Enterocytes were isolated as described below. Livers were flash frozen in liquid nitrogen and stored at −80 °C until required.
2.7.1. Intestinal epithelial cell isolation
Intestinal epithelial cells were isolated using a modified version of a protocol described earlier [16], [25]. The small intestines were dissected out, divided into duodenum and jejunum, flushed with ice-cold PBS, and then inverted. Each jejunal section was divided into 2 pieces and together with the duodenum, each intestinal section was tied to the end of a 12.5 mL Gilson DistriTip Maxi syringe with the plunger pulled out partially and the barrel filled with air. To secure the intestine in place, we inserted a piece of plastic tubing onto the end of the syringe and below the knotted string. To prevent the intestine from floating in the solution when inflated, we tied a small metal washer with string at the lower end of the intestinal section. These tissues were submerged in ice-cold Cell Recovery Solution (Corning #354253), in 5 mL polystyrene tubes and placed on ice. The intestinal sections were then inflated and deflated by pushing down and pulling up the plunger in a series of 4 inflations and deflations every 5 min (min). After 20 min, the intestine still attached to the syringe was laid out on a petri dish containing ice-cold PBS, inflated and the epithelium was scraped off carefully with a pair of thin blunt forceps to avoid pricking the intestinal tube. The cells were then transferred into Falcon tubes and pelleted. The pellets were flash frozen in liquid nitrogen and stored at −80 °C until subsequent analyses. Further mentions of the duodenum or jejunum in this manuscript (with the exception of the histological analysis) refer to the enterocytes isolated from these regions.
2.8. Histology
Two cm pieces of jejunum were collected into tissue processing/embedding cassettes and fixed overnight in 4% paraformaldehyde at 4 °C. They were transferred to 65% ethanol in an automatic paraffin-embedding processor (STP 120 spin Tissue Processor, Thermo Fischer, Waltham, MA, USA). The tissues were embedded in paraffin blocks with an automatic embedding system (Automatic Embedding System Bio-optica, Milano, Italy). Paraffin sections were cut with a motorized rotary microtome (Hyrax M55, Zeiss, Oberkochen, Germany) and disposable microtome razor blades (S-22 blades, Feather, Osaka, Japan).
2.8.1. Hematoxylin and eosin (H&E) stain
Paraffin sections were stained automatically using an H&E-staining machine (Shandon Varistain, Thermo Fischer Scientific, Waltham, MA, USA). The stained sections were mounted in embedding medium (Entellan® New, DPX mountant, Sigma–Aldrich, St. Louis, MO, USA). The sections were scanned with the Axio Scope.A1 (Zeiss, Oberkochen, Germany). The desired pictures were taken with an integrated camera and the pictures were analyzed with the software AxioVision (Zeiss, Oberkochen, Germany). Additionally, the villi lengths of the jejunal sections were measured using the software AxioVision.
2.9. Mitochondrial isolation and western blotting
Mitochondria were isolated from frozen duodenum and jejunum using a method described previously [26]. Mitochondria were lysed in RIPA buffer containing protease inhibitors and protein concentrations were determined using the DC Protein assay kit (BioRad # 5000112). Equal amounts of proteins were denatured with 6X Laemmli containing DTT and run on SDS PAGE gels. Alternatively, the samples were processed and run in precast BOLT-Bis-Tris Plus gels (Thermo Fisher Scientific, Switzerland) according to manufacturer's instructions. Gels were transferred to PVDF membranes, which were blocked with milk, probed with the appropriate primary antibody dilutions overnight at 4 °C. They were washed, incubated in the appropriate HRP linked secondary antibody dilutions, washed, and then developed using a homemade chemiluminescent developing substrate. The blots were imaged using ImageQuant software. Antibodies used are listed in Sup Table 1.
2.10. Real time quantitative polymerase chain reaction (RT-qPCR) analysis
RNA was extracted from the duodenum and jejunum using Trizol reagent (Life Technologies #15596018) following the manufacturer's protocol and treated with DNAse (Qiagen #79254). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems #4368813) and used for RT-qPCR reactions using FAST SYBR green and the Viia7 Real Time PCR system (Applied Biosystems). Each sample was run in triplicate and analysed using the 2−ΔΔCT method [27] with Peptidylprolyl Isomerase B (Ppib) as the reference gene [28]. Primers used are listed in Sup Table 2.
2.11. Plasma metabolite and insulin analysis
Post prandial trunk blood samples were collected in micro tubes containing 0.5 M EDTA after a 2 h fast in the dark phase; fasting blood samples (approximately 50 μL) were collected from the lateral tail vein in EDTA coated micro tubes (Sarstedt, Switzerland), after 6 h of fasting beginning at the onset of dark phase. Blood samples were centrifuged at 8700g for 10 min at 4 °C, and plasma were collected and stored at −80 °C until required. Plasma TAG, free glycerol, non-esterified fatty acids (NEFA), and β-hydroxybutyrate (BHB) concentrations were measured using standard colorimetric and enzymatic methods adapted for the Cobas MIRA auto analyzer (Hoffman LaRoche, Basel, Switzerland) [29]. Final TAG values were obtained by subtracting the free glycerol values from the measured TAG values. Plasma insulin levels were measured using the Mouse/Rat Insulin Kit (Meso Scale Discovery, USA #K152BZC-2) according to manufacturer's instructions.
2.12. Metabolomics
2.12.1. Sample preparation
After 20 weeks on HFD, mice were fasted overnight and gavaged with a 500 μL bolus of 150 mM 13C2 Oleic acid (SIGMA #646466) in a 0.5% methylcellulose solution. The mice were sacrificed 2 h post gavage and duodenum, jejunum, and livers were collected and frozen in liquid nitrogen. Tissue samples were pulverized in liquid nitrogen and pre-weighed samples (approximately 100 mg) were lysed in 1 mL of 80% methanol and incubated on a thermoshaker at 4 °C for 1 h at 1000 rpm. The samples were centrifuged at 14,000g and 4 °C for 15 min, and 900 μL of the supernatants were transferred to fresh glass vials and sealed with Teflon/PTFE caps. These 80% methanol extracts were stored at −20 °C until further processing. Fifty μL of each extract were transferred to an Eppendorf vial, dried completely under N2, re-dissolved with 20 μL water, and diluted with 80 μL of 50 mM ammonium acetate in acetonitrile/methanol/water/saturated aqueous ammonium hydroxide (900: 88: 10: 2, v/v, pH 9). The final dilutions were centrifuged at 16,100g and 4 °C for 15 min. The supernatants were transferred into glass vials for liquid chromatography–mass spectrometry (LC-MS) analyses [30].
2.12.2. LC-MS analyses
Hydrophilic interaction chromatography (HILIC) was performed on a nano-UPLC system (Waters Inc. Milford, USA) coupled by a nano-ESI source to a Synapt G2 HDMS (Waters, Manchester, UK) as described in [31] with slight modification in the flow rate. The initial flow rate was 3.5 μl/min and went down to 2.5 μl/min during the gradient for 10 min. A pooled sample consisting of a mix of every sample collected was repeatedly analyzed after every fifth sample in order to monitor the LC-MS performance and to assess the technical variance. Along with the pooled samples, a standard reference compound mix was analyzed containing the targeted metabolites glutamate, citrate, isocitrate, NAD+, ATP, ADP, acetyl CoA, malonyl CoA, and palmitoyl CoA, each at 5 μM concentration. Relative quantifications of the targeted metabolites were done by using the module QuanLynx of the mass spectrometry software MassLynx (version 4.1, Waters, United Kingdom).
2.13. Statistical analysis
All data are presented as mean ± standard error of the mean (SEM) of absolute values, % changes or fold changes as indicated in the figures. Data normality was verified using the Shapiro–Wilk test (when n ≥ 7) and Kolmogorov–Smirnov test (when 5 ≤ n ≤ 6). Non-parametric tests were used when the data were not normally distributed. Outliers were identified using the Grubb's test and removed. Statistical tests used are mentioned in the figure legends. When P < 0.05, the differences were considered statistically significant.
3. Results
3.1. iSIRT3 mice overexpressed SIRT3 in the small intestine
A western blot analysis of mitochondria isolated from the enterocytes of the duodenum and jejunum of S3fl and iSIRT3 mice showed that SIRT3 protein levels were higher in the small intestine of iSIRT3 mice (Figure 1), thus validating the intended transgenic manipulation. The mitochondrial proteins cytochrome-c oxidase subunit-IV (COX IV) and voltage-dependent anion channel (VDAC) were used as loading controls.
Figure 1.

iSIRT3 mice overexpressed SIRT3 in the small intestine. Western blot analysis of mitochondrial fractions isolated from the duodenum (lanes 1 and 2) and jejunum (lanes 3 and 4) of S3fl and iSIRT3 mice. Antibodies used are for sirtuin 3 (SIRT3), cytochrome-c oxidase subunit-IV (COX IV), and voltage-dependent anion channel (VDAC). The dotted line separates discontinuous blots. [Lanes 1 and 2, n = 3 (pooled samples); lanes 3 and 4, n = 1].
3.2. iSIRT3 and S3fl control mice had similar body weight, body composition, energy intake, and whole body energy metabolism
All animals irrespective of genotype gained weight similarly on the HFD over time (diet, P < 0.0001; time, P < 0.0001; time × diet, P < 0.0001) (Figure 2A). Monitoring the body composition of awake animals every few weeks using the EchoMRI scanner revealed that all animals showed similar increases in fat and lean mass over time (Sup Figure 1A). All animals on HFD gained more fat mass over time compared to those on CD (fat mass: diet, P < 0.0001; fat and lean mass: time, P < 0.0001; time × diet, P < 0.0001). After 16 weeks on the diets, a CT scan of anesthetized mice also revealed a diet effect, such that there was a similar increase in lean mass (P < 0.05), overall fat mass (P < 0.0001), and visceral fat mass (P < 0.0001) in S3fl and iSIRT3 mice on HFD compared to those on CD (Figure 2B). Both S3fl and iSIRT3 mice also showed an increase in subcutaneous fat on HFD versus (vs) CD (P < 0.0001), with an interaction between the independent parameters, genotype and diet (P < 0.05); however, a post hoc analysis did not reveal any significant differences between the genotypes on either diet.
Figure 2.

iSIRT3 and S3fl control mice had similar body weight, body composition, energy intake, and whole body energy metabolism. (A) Body weights of iSIRT3 and S3fl mice on control diet (CD) or high-fat diet (HFD) monitored over time. [n = 8–10; 3-way RM ANOVA (time × diet × genotype), time × diet, P < 0.0001: followed by a restricted 2-way ANOVA (diet × genotype) at each time point; diet, #P < 0.0001. (B) CT scan analyses of S3fl and iSIRT3 mice fed CD or HFD. [n = 6–8; 2-way ANOVA (diet × genotype) with Tukey's multiple comparisons post hoc test. For lean mass, fat mass, visceral fat, and subcutaneous fat; diet effects, ∗P < 0.05, #P < 0.0001. Subcutaneous fat; diet × genotype, $P < 0.05]. (C–F) Indirect calorimetry data. The grey and white bars in the background represent the dark and light phases, respectively. (C) Energy intake measured every 12 h. (D) Respiratory exchange ratio (RER) (E) Locomotor Activity and (F) Energy expenditure (EE) values plotted as mean values of 1 h time bins. (C–F) [n = 8–9; mixed RM ANOVA (genotype × diet × days × phases) with measurements over time compacted into diet (CD and HFD), days (day 1 and 2) and phases (dark and light)]. Data are presented as mean values ± SEM.
Both S3fl and iSIRT3 mice had lower energy intake (P < 0.01) (Figure 2C), respiratory exchange ratio (RER) (P < 0.0001) (Figure 2D), and locomotor activity (P < 0.001) (Figure 2E) when switched from CD to HFD. No genotype differences were observed. Also, no diet or genotype effects in energy expenditure were observed in these mice (Figure 2F and Sup Figure 1B).
Histological analyses of the jejunum of the mice revealed no visible differences between the genotypes. All animals on HFD showed an increase in villi length vs those on CD (P < 0.05) (Sup Figure 1C and D).
3.3. iSIRT3 mice on HFD showed improved glucose homeostasis compared to S3fl mice
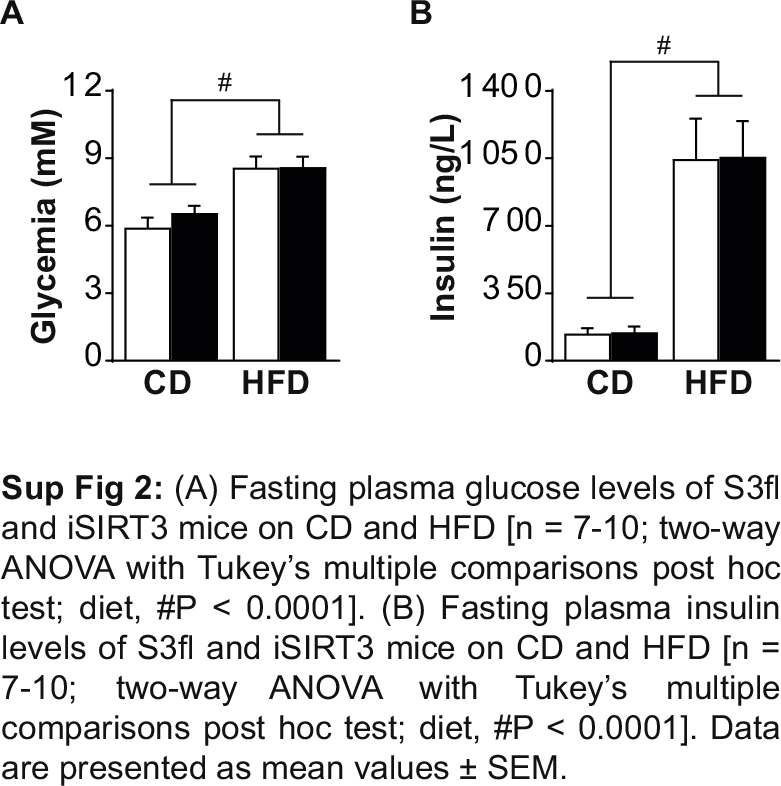
S3fl and iSIRT3 mice on CD were similarly sensitive to insulin given IP (Figure 3A) and similarly tolerant to an oral load of glucose (Figure 3E). iSIRT3 mice on HFD, however, showed an increased sensitivity to insulin (time × genotype, P < 0.01) (Figure 3B), with lower circulating glucose levels compared to controls 90 min post injection (P < 0.05). iSIRT3 mice on HFD also showed improved glucose clearance compared to S3fl mice, in response to an oral glucose load (time × genotype, P < 0.05) with lower levels of glucose 30 (P < 0.01) and 60 min (P < 0.05) post gavage (Figure 3F). The fasting levels of plasma glucose and insulin were increased in both S3fl and iSIRT3 mice on HFD (P < 0.0001), compared to those on CD, with no genotype differences on either diet (Sup Figure 2).
Figure 3.

iSIRT3 mice on HFD showed improved glucose homeostasis compared to S3fl mice. (A–B) Tail blood glucose values of insulin sensitivity test (IST) for S3fl and iSIRT3 mice on CD and HFD, respectively; (A) CD [n = 7–8; 2-way RM ANOVA (time × genotype)]. (B) HFD [n = 7–10; 2-way RM ANOVA (time × genotype) with Bonferroni's multiple comparisons post hoc test; time × genotype, P < 0.01; genotype effect at time point 90 min, ∗P < 0.05]. (C) Area above the curve (AAC) of IST curves in A [n = 7–8; Mann–Whitney test]. (D) AAC of IST curves in B [n = 7–10; Unpaired t test]. (E–F) Tail blood glucose values of oral glucose tolerance test (OGTT) for S3fl and iSIRT3 mice on CD and HFD respectively. (E) CD [n = 9–10; 2-way RM ANOVA (time × genotype)]. (F) HFD [n = 6–7; 2-way RM ANOVA (time × genotype) with Bonferroni's multiple comparisons post hoc test; time × genotype, P < 0.05; genotype effects at time point 30 min, ∗∗P < 0.01 and 60 min, ∗P < 0.05]. (G) Area under the curve (AUC) for OGTT curves in E [n = 9–10; Unpaired t test]. (H) AUC for OGTT curves in F [n = 6–7; Mann–Whitney test; genotype, ns]. Data are presented as mean values ± SEM.
3.4. iSIRT3 mice on HFD showed an induction of the ketogenic gene Hmgcs2 in the small intestine, but not in the liver
There was an induction of Hmgcs2 (P < 0.0001), carnitine palmitoyltransferase 1a (Cpt1a; P < 0.0001) and long chain acyl CoA dehydrogenase (Lcad; P < 0.001) mRNA and a downregulation of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1a) mRNA (P < 0.001) in the duodenum of all animals on HFD compared to those on CD (Figure 4A). A similar increase in Hmgcs2, Cpt1a, and Lcad, as well as a downregulation of Pgc1a was seen in the jejunum of all the HFD fed mice compared to CD fed mice, irrespective of genotype (P < 0.0001) (Figure 4B). iSIRT3 mice on HFD showed an even greater upregulation of Hmgcs2 expression in the duodenum and jejunum compared to all the other groups (duodenum, P < 0.01 vs S3fl on HFD; jejunum, P < 0.05 vs S3fl on HFD) (Figure 4A,B). Livers of S3fl and iSIRT3 mice also showed an upregulation of Hmgcs2, Cpt1a, ATP synthase (ATPsyn), and Lcad on HFD vs CD, with no genotypes differences.
Figure 4.

iSIRT3 mice on HFD showed an induction of the ketogenic gene Hmgcs2 in the small intestine but not in the liver. (A–C) Relative mRNA expression of Hmgcs2, Cpt1a, ATPsyn, Lcad, and Pgc1a in the duodenum, jejunum, and liver of S3fl and iSIRT3 mice on CD or HFD. (A) Duodenum [n = 6–8; 2-way ANOVA (diet × genotype) with Tukey's multiple comparison post hoc test. Hmgcs2, diet, #P < 0.0001; genotype, P < 0.05; diet × genotype, $P < 0.05; S3fl HFD vs iSIRT3 HFD, ∗∗P < 0.01. Cpt1a, diet, #P < 0.0001. Lcad, diet, ∗∗P < 0.001. Pgc1a, diet, ∗∗∗P < 0.001]. (B) Jejunum [n = 5–8; 2-way ANOVA (diet × genotype) with Tukey's multiple comparison post hoc test. Hmgcs2, genotype, P < 0.05; diet, #P < 0.0001; diet × genotype, P = 0.06, ns; S3fl HFD vs iSIRT3 HFD, ∗P < 0.05. Cpt1a, genotype, P < 0.05; diet, #P < 0.0001, diet × genotype, ns. ATPsyn, genotype, P < 0.05; diet, ns; diet × genotype, ns. Lcad, genotype, ns; diet, #P < 0.0001, diet × genotype, ns. Pgc1a, genotype, ns; diet, #P < 0.0001, diet × genotype, ns]. (C) Liver [n = 6–9; 2-way ANOVA (diet × genotype) with Tukey's multiple comparison post hoc test. Hmgcs2, diet, #P < 0.0001. Cpt1a, diet, #P < 0.0001. ATPsyn, diet, ∗∗∗P < 0.001. Lcad, diet, #P < 0.0001. Pgc1a, diet, ∗P < 0.05]. Data are presented as mean values ± SEM.
3.5. iSIRT3 mice on HFD did not show the lower systemic plasma ketone body levels compared to CD seen in S3fl mice
The plasma NEFA, TAG, and glucose levels were similar in S3fl and iSIRT3 mice on CD or HFD with all animals on HFD showing lower NEFA (P < 0.0001) and TAG levels (P < 0.0001) (Figure 5A,B) and higher plasma glucose levels (P < 0.0001) (Figure 5C) compared to mice on CD. Interestingly, the plasma BHB levels were lower in HFD fed mice compared to CD fed mice (P < 0.0001), but the post hoc analysis showed that this difference was significant only in S3fl and not in iSIRT3 mice.
Figure 5.

iSIRT3 mice on HFD did not show the lower systemic plasma ketone body levels compared to CD seen in S3fl mice. (A–D) Post prandial trunk blood plasma metabolite levels of S3fl and iSIRT3 mice on CD and HFD [n = 13–18; 2-way ANOVA (diet × genotype) with Tukey's multiple comparison post hoc test]. (A) Non-esterified fatty acids (NEFA); diet, #P < 0.0001. (B) Triacylglycerol (TAG); diet, P < 0.01; S3fl CD vs S3fl HFD and iSIRT3 CD vs iSIRT3 HFD, ∗∗P < 0.01; S3fl CD vs iSIRT3 HFD, ∗∗∗P < 0.001. (C) Glucose; diet, #P < 0.0001. (D) β-hydroxybutyrate (BHB); diet, P < 0.0001; S3fl CD vs S3fl HFD, ∗∗∗P < 0.001; iSIRT3 CD vs S3fl HFD, ∗∗P < 0.01. Data are presented as mean values ± SEM.
3.6. iSIRT3 mice showed a differentially regulated metabolic activity of jejunal enterocytes in response to an oral gavage of long-chain fatty acid
We could not detect the 13C2 label from the oleic acid gavage in any of the metabolites tested in the duodenum, jejunum, or liver of S3fl and iSIRT3 mice on HFD. Interestingly, the jejunum of iSIRT3 mice showed lower levels of NAD+, as well as palmitoyl CoA and malonyl CoA levels, compared to S3fl mice (P < 0.05) (Figure 6A,D, Sup Figure 3B). There were no differences in the levels of these compounds in the duodenum or the liver, or in the NADH levels and ATP/ADP ratios of all three regions tested (Figure 6A–E). We also saw no differences in acetyl CoA, glutamate, citrate, and isocitrate levels in the intestinal samples (Sup Figure 3A–B). Of these, only acetyl CoA and glutamate were detectable in the liver, with no differences between the two genotypes (Sup Figure 3C).
Figure 6.
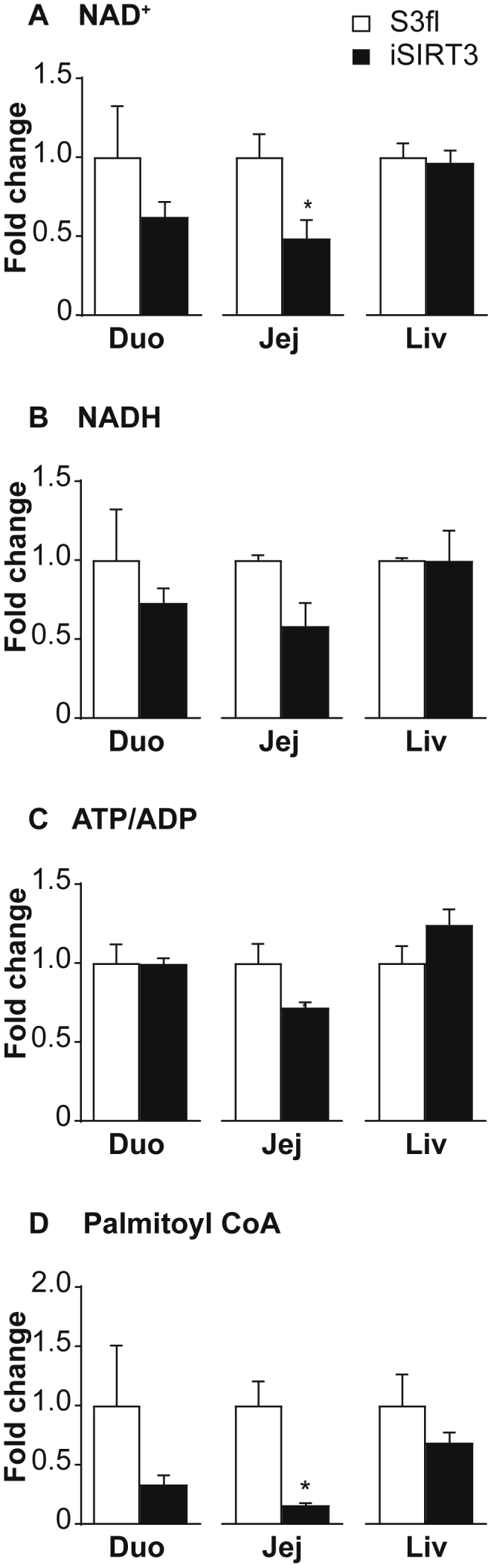
iSIRT3 mice showed a differentially regulated metabolic activity of jejunal enterocytes in response to an oral gavage of fatty acid. LC-MS analyses of metabolites from the duodenum, jejunum, and liver of S3fl and iSIRT3 mice after oleic acid gavage, represented as fold change compared to S3fl controls. (A) NAD+, (B) NADH, (C) ATP/ADP, (D) Palmitoyl CoA [n = 3–5; Mann–Whitney test, *p < 0.05]. Data are presented as mean values ± SEM.
4. Discussion
The major aim of this study was to examine if enhancing the metabolic flux of the enterocytes in mice could influence DIO and its comorbidities. Using the cre-loxP system, we genetically overexpressed the mitochondrial NAD+ dependent protein deacetylase SIRT3, in the epithelial cells of the intestine. We characterized both iSIRT3 and S3fl mice fed CD or HFD for several weeks and found that an overexpression of SIRT3 in the enterocytes did not lead to differences in any of the parameters tested when these mice were on CD. On HFD, both S3fl and iSIRT3 mice showed similar manifestations of DIO, but iSIRT3 mice were better able to regulate their blood glucose levels in response to exogenous insulin or glucose. This indicates that iSIRT3 mice on HFD were protected from developing IR, one of the major comorbidities of DIO. iSIRT3 mice also showed increased ketogenic gene expression in the small intestine as well increased enterocyte metabolic activity. Our findings suggest that enterocyte oxidative metabolism plays a role in the regulation of whole body glucose metabolism under conditions of DIO.
As post-translational regulator of several mitochondrial proteins, SIRT3 has been shown to upregulate multiple metabolic pathways including FAO, the TCA cycle, and ketogenesis [19]. Here we show that in chronic HFD fed iSIRT3 mice, after an oral bolus of oleic acid, SIRT3 activity was upregulated in the small intestine but not in the liver. The decrease in the NAD+ levels in the jejunum is indirect evidence for increased SIRT3 activity. NAD+ is the cosubstrate for several enzymes, one of which is SIRT3 [17]. SIRT3 breaks down a molecule of NAD+ during every deacetylation reaction [32], and an upregulation of SIRT3 activity would in turn reduce the NAD+ levels. The reverse is true when NAD+ consuming enzymes are inhibited or downregulated. For example, PARP1 inhibition leads to an increase in NAD+ levels as shown in [33]. iSIRT3 mice also showed a decrease in malonyl CoA and palmitoyl CoA levels in the jejunum. Malonyl CoA is an intermediate of fatty acid synthesis, while palmitoyl CoA is its end product. Malonyl CoA is also an inhibitor of the mitochondrial long-chain fatty acid transport protein CPT1a, and thereby an inhibitor of FAO. A decrease in the levels of these two metabolites implies a downregulation of de-novo fatty acid synthesis and an upregulation of FAO [34], [35]; further indicating that FAO is enhanced in the enterocytes of iSIRT3 mice on HFD. Together, these data demonstrate that an overexpression of SIRT3 in enterocytes of HFD fed mice enhanced the mitochondrial metabolic activity of small intestinal epithelial cells.
Data from whole body SIRT3 knock out mice showed that the absence of SIRT3 led to an accelerated development of DIO and metabolic syndrome on chronic HFD feeding [36]. This was attributed to increased mitochondrial dysfunction due to the hyperacetylation of mitochondrial proteins in the absence of SIRT3. Subsequent studies on SIRT3 overexpression mice showed that increased SIRT3 expression protected mice from developing noise induced hearing loss, as well as ageing associated tissue fibrosis [20], [37]; however, they did not study the effects of whole body or tissue specific SIRT3 overexpression on the development of DIO or metabolic syndrome in response to chronic HFD feeding. We here show that an increase in SIRT3 expression and function in the enterocytes had no impact on body weight, body composition, or fat distribution, and iSIRT3 mice on HFD developed DIO similar to S3fl mice. Both S3fl and iSIRT3 mice also showed increased jejunal villi length and had similarly high circulating levels of fasting glucose and insulin after chronic HFD feeding. These effects are consistent with previous observations in animals fed HFD that develop DIO [38], [39]. Indirect calorimetry data also showed no differences between S3fl and iSIRT3 mice on CD or when switched to HFD. The reductions in energy intake, RER, and activity, seen in both S3fl and iSIRT3 mice on exposure to HFD, were in line with previous studies about HFD feeding in mice [40], [41], [42].
Despite these similarities though, iSIRT3 mice showed distinct differences from S3fl controls in their ability to regulate systemic glucose. After IP insulin, iSIRT3 mice on HFD showed prolonged hypoglycemia and a slow rise in blood glucose levels. This indicates that iSIRT3 mice were less resistant to insulin than S3fl controls, even after several weeks on HFD. iSIRT3 mice were also able to rapidly uptake and clear the high circulating blood glucose levels in the OGTT, compared to S3fl mice. These results clearly indicate that iSIRT3 mice on HFD developed less IR than S3fl mice, despite similar DIO. Conversely, the whole body SIRT3 knock out mice on HFD developed IR and DIO faster than control mice; this was attributed mainly to hepatic dyslipidemia caused by increased lipogenesis [36]. Mice deficient in LCAD as well as malonyl CoA carboxylase showed reduced fatty acid oxidation in the liver and skeletal muscle, respectively, and developed IR [43], [44]. These data suggest that decreased fatty acid oxidation in the liver and muscle caused peripheral IR. Here we show that increased SIRT3 (and therefore increased FAO in the enterocytes), contributes to protection from developing IR.
At the molecular level, both S3fl and iSIRT3 mice showed an upregulation of genes involved in FAO (Cpt1a and Lcad) and ketogenesis (Hmgcs2) in the duodenum, jejunum, and liver on HFD compared to CD. Of note was the nearly 10 to 20-fold increase in Hmgcs2 mRNA expression in the duodenum and jejunum of all HFD fed mice vs CD fed mice, compared to the smaller, 3-fold increase in the liver. This is consistent with previous data showing that exposure to HFD for two weeks increased the expression of fat oxidation and mainly ketogenic genes in the duodenum [16]. Our data indicate that after 17 weeks of HFD feeding, these increases in gene expression occur in the duodenum and jejunum as well as in the liver. HFD fed iSIRT3 mice showed an even greater increase in Hmgcs2 expression in the duodenum and jejunum than S3fl mice (15–30-fold increase vs CD fed mice), but not in the liver, indicating that intestinal SIRT3 overexpression further upregulates ketogenic genes in the small intestine of mice fed HFD. SIRT3 is a posttranslational regulator of proteins that deacylates several genes in the FAO pathway, thereby enhancing fat oxidation [45]. Hence, it is not surprising that we see an upregulation of FAO in the enterocytes of iSIRT3 mice, with no changes at the gene expression level, compared to S3fl mice on HFD. SIRT3 is also known to deacylate and upregulate HMGCS2 and thereby ketogenesis [46]. The increased gene expression of Hmgcs2 in the enterocytes of iSIRT3 mice suggests that this might be an indirect mechanism of upregulation rather than an activation by direct deacetylation, though we cannot rule out the latter possibility based on our results.
Despite the upregulation of Hmgcs2 mRNA expression in the duodenum, jejunum, and liver of all mice on HFD, systemic post prandial plasma BHB levels in control S3fl mice on HFD were lower than on CD. The overall decrease in BHB levels in the trunk blood plasma of HFD fed mice is consistent with previous findings showing that long-term obesity or HFD feeding leads to reduced circulating ketone body levels in humans as well as rodents [47], [48], [49], [50]. These studies suggest that while mild IR leads to an upregulation of hepatic ketogenesis, severe IR leads to blunted hepatic ketogenesis even under conditions of high fat oxidation in the liver. The plasma BHB levels of iSIRT3 mice fed HFD were not different from those of mice fed CD and showed an increasing trend compared to S3fl mice on HFD, which did not reach statistical significance. This might be because the blood samples from our mice were trunk blood samples collected after decapitation, which represent a diluted pool of blood coming from the intestine. Analysis of plasma BHB levels from the hepatic portal vein would have allowed for a better estimate of the amount of ketone bodies generated in the small intestine [11], [51], [52]. Our results suggest that SIRT3 overexpression in the enterocytes led to an increase in ketone body production in the intestine but not in the liver of iSIRT3 mice on HFD. This is consistent with pharmacological studies in rats, where peripheral administration of FAO stimulators showed an upregulation of FAO and ketogenic markers in the small intestine, but not the liver, as well as increased BHB levels in the hepatic portal vein blood plasma [11], [51], [52]. Both S3fl and iSIRT3 mice on HFD also showed reduced NEFA and TAG levels under fed conditions compared to CD fed mice, with no genotype effect. These HFD-induced reductions in plasma NEFA and TAG levels are consistent with previous findings, which show that there is a species-specific decrease in circulating TAG and free fatty acids in mice with DIO [53].
Traditionally, ketone bodies were mainly thought of as an alternative fuel source for the brain and other peripheral organs, in conditions of prolonged fasting. More recently, BHB has been proposed as a signaling metabolite even under conditions of nutrient abundance (as reviewed in [54] and [55]) and thereby might protect against the development of IR and T2D. These reviews mainly focus on the liver as the main organ contributing to circulating ketone bodies. In line with previous findings [15], [16], our data suggest that the gut also has the capacity to contribute to circulating ketone levels and, in fact, might be an interesting extra-hepatic target to manipulate whole body glucose homeostasis, especially in DIO.
Studies addressing the effects of peripheral administration of FAO stimulators suggest that enhanced FAO and ketogenesis in the small intestine might reduce energy intake and body weight [51], [52]. Our results seem to indicate otherwise because enhanced FAO and ketogenesis in enterocytes did not lead to changes in energy intake or body weight, on CDor on HFD. This difference might be due to the fact that the pharmacological studies looked at the acute effects of peripherally administered FAO stimulators. Our transgenic mice overexpressed SIRT3 under the constitutive expression of Cre recombinase, downstream of the Villin promoter. This might have caused compensatory changes during development and, hence, blunted any changes in food intake. The use of an inducible Cre mouse model that would allow for the initiation of SIRT3 overexpression in a specific time window might help to answer this question. In addition, our mouse model expressed only one extra copy of the Sirt3 gene. A homozygous knock-in mouse model could presumably cause a greater overexpression of SIRT3 with stronger or different phenotypic effects.
Further studies should try to identify the exact mechanisms of the observed effects in HFD fed iSIRT3 mice. In any case, targeting intestinal metabolism in obese patients through non-surgical interventions might be an interesting new approach to fight IR and T2D.
Acknowledgements
This work was supported by the Swiss National Science Foundation, Research Grant 310030_153149 to WL. We are grateful to Prof. WD Hardt and P Kaiser for the Vil-Cre mice; E Karimian, F Mouttet, E Weber, F Mueller, L Michel, and M Heyner for assistance in experimentation or genotyping, to JP Krieger for his support in editing the manuscript and the SLA animal facility for contribution to animal husbandry.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molmet.2017.07.009.
Conflicts of interest
None declared.
Appendix A. Supplementary data
The following are the supplementary data related to this article:


References
- 1.World Health Organization . Geneva; 2016. Global report on diabetes.http://www.who.int/diabetes/global-report/en/ [Google Scholar]
- 2.Kahn S.E., Hull R.L., Utzschneider K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 3.Mingrone G., Cummings D.E. Changes of insulin sensitivity and secretion after bariatric/metabolic surgery. Surgery for Obesity and Related Diseases. 2016;12:1199–1205. doi: 10.1016/j.soard.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Pories W.J., Swanson M.S., Macdonald K.G., Long S.B., Morris P.G., Brown B.M. Who would have thought it – an operation proves to be the most effective therapy for adult-onset diabetes-mellitus. Annals of Surgery. 1995;222:339–352. doi: 10.1097/00000658-199509000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pories W.J., Albrecht R.J. Etiology of type II diabetes mellitus: role of the foregut. World Journal of Surgery. 2001;25:527–531. doi: 10.1007/s002680020348. [DOI] [PubMed] [Google Scholar]
- 6.Hickey M.S., Pories W.J., MacDonald K.G., Cory K.A., Dohm G.L., Swanson M.S. A new paradigm for type 2 diabetes mellitus – could it be a disease of the foregut? Annals of Surgery. 1998;227:637–644. doi: 10.1097/00000658-199805000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saeidi N., Meoli L., Nestoridi E., Gupta N.K., Kvas S., Kucharczyk J. Reprogramming of intestinal glucose metabolism and glycemic control in rats after gastric bypass. Science. 2013;341:406–410. doi: 10.1126/science.1235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan Y., Zhou Z., Kong F., Feng S., Li X., Sha Y. Roux-en-Y gastric bypass surgery suppresses hepatic gluconeogenesis and increases intestinal gluconeogenesis in a T2DM rat model. Obesity Surgery. 2016;26:2683–2690. doi: 10.1007/s11695-016-2157-5. [DOI] [PubMed] [Google Scholar]
- 9.Troy S., Soty M., Ribeiro L., Laval L., Migrenne S., Fioramonti X. Intestinal gluconeogenesis is a key factor for early metabolic changes after gastric bypass but not after gastric lap-band in mice. Cell Metabolism. 2008;8:201–211. doi: 10.1016/j.cmet.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 10.Nelson D.W., Gao Y., Yen M.I., Yen C.L. Intestine-specific deletion of acyl-CoA:monoacylglycerol acyltransferase (MGAT) 2 protects mice from diet-induced obesity and glucose intolerance. Journal of Biological Chemistry. 2014;289:17338–17349. doi: 10.1074/jbc.M114.555961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schober G., Arnold M., Birtles S., Buckett L.K., Pacheco-Lopez G., Turnbull A.V. Diacylglycerol acyltransferase-1 inhibition enhances intestinal fatty acid oxidation and reduces energy intake in rats. Journal of Lipid Research. 2013;54:1369–1384. doi: 10.1194/jlr.M035154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuda N., Kumadaki S., Higashi C., Ozawa M., Shinozaki M., Kato Y. Intestine-targeted DGAT1 inhibition improves obesity and insulin resistance without skin aberrations in mice. PLoS One. 2014;9 [Google Scholar]
- 13.Langhans W. Fatty acid oxidation in the energostatic control of eating–a new idea. Appetite. 2008;51:446–451. doi: 10.1016/j.appet.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 14.Mansouri A., Langhans W. Enterocyte-afferent nerve interactions in dietary fat sensing. Diabetes, Obesity and Metabolism. 2014;16(Suppl. 1):61–67. doi: 10.1111/dom.12339. [DOI] [PubMed] [Google Scholar]
- 15.Kondo H., Minegishi Y., Komine Y., Mori T., Matsumoto I., Abe K. Differential regulation of intestinal lipid metabolism-related genes in obesity-resistant A/J vs. obesity-prone C57BL/6J mice. American Journal of Physiology. Endocrinology and Metabolism. 2006;291:E1092–E1099. doi: 10.1152/ajpendo.00583.2005. [DOI] [PubMed] [Google Scholar]
- 16.Clara R., Schumacher M., Ramachandran D., Fedele S., Krieger J.P., Langhans W. Metabolic adaptation of the small intestine to short- and medium-term high-fat diet exposure. Journal of Cellular Physiology. 2017;232:167–175. doi: 10.1002/jcp.25402. [DOI] [PubMed] [Google Scholar]
- 17.Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 18.Rardin M.J., Newman J.C., Held J.M., Cusack M.P., Sorensen D.J., Li B. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osborne B., Bentley N.L., Montgomery M.K., Turner N. The role of mitochondrial sirtuins in health and disease. Free Radical Biology & Medicine. 2016;100:164–174. doi: 10.1016/j.freeradbiomed.2016.04.197. [DOI] [PubMed] [Google Scholar]
- 20.Brown K.D., Maqsood S., Huang J.Y., Pan Y., Harkcom W., Li W. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metabolism. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madison B.B., Dunbar L., Qiao X.T., Braunstein K., Braunstein E., Gumucio D.L. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. Journal of Biological Chemistry. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 22.Tschop M.H., Speakman J.R., Arch J.R., Auwerx J., Bruning J.C., Chan L. A guide to analysis of mouse energy metabolism. Nature Methods. 2012;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer K., Ruiz H.H., Jhun K., Finan B., Oberlin D.J., van der Heide V. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nature Medicine. 2017;23:623–630. doi: 10.1038/nm.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGuinness O.P., Ayala J.E., Laughlin M.R., Wasserman D.H. NIH experiment in centralized mouse phenotyping: the vanderbilt experience and recommendations for evaluating glucose homeostasis in the mouse. American Journal of Physiology. Endocrinology and Metabolism. 2009;297:E849–E855. doi: 10.1152/ajpendo.90996.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nik A.M., Carlsson P. Separation of intact intestinal epithelium from mesenchyme. Biotechniques. 2013;55:42–44. doi: 10.2144/000114055. [DOI] [PubMed] [Google Scholar]
- 26.Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nature Protocols. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 27.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Sirakov M., Borra M., Cambuli F.M., Plateroti M. Defining Suitable reference genes for RT-qPCR analysis on intestinal epithelial cells. Molecular Biotechnology. 2013 doi: 10.1007/s12033-012-9643-3. [DOI] [PubMed] [Google Scholar]
- 29.Langhans W. Hepatic and intestinal handling of metabolites during feeding in rats. Physiology & Behavior. 1991;49:1203–1209. doi: 10.1016/0031-9384(91)90352-o. [DOI] [PubMed] [Google Scholar]
- 30.Riera-Borrull M., Rodriguez-Gallego E., Hernandez-Aguilera A., Luciano F., Ras R., Cuyas E. Exploring the process of energy generation in pathophysiology by targeted metabolomics: performance of a simple and quantitative method. Journal of the American Society for Mass Spectrometry. 2016;27:168–177. doi: 10.1007/s13361-015-1262-3. [DOI] [PubMed] [Google Scholar]
- 31.Abplanalp J., Laczko E., Philp N.J., Neidhardt J., Zuercher J., Braun P. The cataract and glucosuria associated monocarboxylate transporter MCT12 is a new creatine transporter. Human Molecular Genetics. 2013;22:3218–3226. doi: 10.1093/hmg/ddt175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirschey M.D. Old enzymes, new tricks: sirtuins are NAD(+)-dependent de-acylases. Cell Metabolism. 2011;14:718–719. doi: 10.1016/j.cmet.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai P., Canto C., Oudart H., Brunyanszki A., Cen Y.N., Thomas C. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metabolism. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foster D.W. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. Journal of Clinical Investigation. 2012;122:1958–1959. doi: 10.1172/JCI63967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abu-Elheiga L., Matzuk M.M., Abo-Hashema K.A., Wakil S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- 36.Hirschey M.D., Shimazu T., Jing E., Grueter C.A., Collins A.M., Aouizerat B. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Molecular Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sundaresan N.R., Bindu S., Pillai V.B., Samant S., Pan Y., Huang J.Y. SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3beta. Molecular and Cellular Biology. 2015;36:678–692. doi: 10.1128/MCB.00586-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldassano S., Amato A., Cappello F., Rappa F., Mule F. Glucagon-like peptide-2 and mouse intestinal adaptation to a high-fat diet. The Journal of Endocrinology. 2013;217:11–20. doi: 10.1530/JOE-12-0500. [DOI] [PubMed] [Google Scholar]
- 39.Collins S., Martin T.L., Surwit R.S., Robidoux J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Physiology & Behavior. 2004;81:243–248. doi: 10.1016/j.physbeh.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Ellacott K.L., Morton G.J., Woods S.C., Tso P., Schwartz M.W. Assessment of feeding behavior in laboratory mice. Cell Metabolism. 2010;12:10–17. doi: 10.1016/j.cmet.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longo K.A., Charoenthongtrakul S., Giuliana D.J., Govek E.K., McDonagh T., DiStefano P.S. The 24-hour respiratory quotient predicts energy intake and changes in body mass. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2010;298:R747–R754. doi: 10.1152/ajpregu.00476.2009. [DOI] [PubMed] [Google Scholar]
- 42.Bjursell M., Gerdin A.K., Lelliott C.J., Egecioglu E., Elmgren A., Tornell J. Acutely reduced locomotor activity is a major contributor to western diet-induced obesity in mice. American Journal of Physiology. Endocrinology and Metabolism. 2008;294:E251–E260. doi: 10.1152/ajpendo.00401.2007. [DOI] [PubMed] [Google Scholar]
- 43.Zhang D., Liu Z.X., Choi C.S., Tian L., Kibbey R., Dong J. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koves T.R., Ussher J.R., Noland R.C., Slentz D., Mosedale M., Ilkayeva O. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metabolism. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 45.Hirschey M.D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D.B. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464 doi: 10.1038/nature08778. 121–U37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimazu T., Hirschey M.D., Hua L., Dittenhafer-Reed K.E., Schwer B., Lombard D.B. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metabolism. 2010;12:654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vice E., Privette J.D., Hickner R.C., Barakat H.A. Ketone body metabolism in lean and obese women. Metabolism. 2005;54:1542–1545. doi: 10.1016/j.metabol.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 48.Soeters M.R., Sauerwein H.P., Faas L., Smeenge M., Duran M., Wanders R.J. Effects of insulin on ketogenesis following fasting in lean and obese men. Obesity (Silver Spring) 2009;17:1326–1331. doi: 10.1038/oby.2008.678. [DOI] [PubMed] [Google Scholar]
- 49.Satapati S., He T., Inagaki T., Potthoff M., Merritt M.E., Esser V. Partial resistance to peroxisome proliferator-activated receptor-alpha agonists in ZDF rats is associated with defective hepatic mitochondrial metabolism. Diabetes. 2008;57:2012–2021. doi: 10.2337/db08-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Satapati S., Sunny N.E., Kucejova B., Fu X., He T.T., Mendez-Lucas A. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. Journal of Lipid Research. 2012;53:1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azari E.K., Leitner C., Jaggi T., Langhans W., Mansouri A. Possible role of intestinal fatty acid oxidation in the eating-inhibitory effect of the PPAR-alpha agonist Wy-14643 in high-fat diet fed rats. PLoS One. 2013:8. doi: 10.1371/journal.pone.0074869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Azari E.K., Ramachandran D., Weibel S., Arnold M., Romano A., Gaetani S. Vagal afferents are not necessary for the satiety effect of the gut lipid messenger oleoylethanolamide. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2014;307:R167–R178. doi: 10.1152/ajpregu.00067.2014. [DOI] [PubMed] [Google Scholar]
- 53.Gao S., He L., Ding Y., Liu G. Mechanisms underlying different responses of plasma triglyceride to high-fat diets in hamsters and mice: roles of hepatic MTP and triglyceride secretion. Biochemical and Biophysical Research Communications. 2010;398:619–626. doi: 10.1016/j.bbrc.2010.05.114. [DOI] [PubMed] [Google Scholar]
- 54.Newman J.C., Verdin E. beta-hydroxybutyrate: much more than a metabolite. Diabetes Research and Clinical Practice. 2014;106:173–181. doi: 10.1016/j.diabres.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Puchalska P., Crawford P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metabolism. 2017;25:262–284. doi: 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.