

Abstract
Objective
Hypothalamic arcuate nucleus-specific pro-opiomelanocortin deficient (ArcPomc−/−) mice exhibit improved glucose tolerance despite massive obesity and insulin resistance. We demonstrated previously that their improved glucose tolerance is due to elevated glycosuria. However, the underlying mechanisms that link glucose reabsorption in the kidney with ArcPomc remain unclear. Given the function of the hypothalamic melanocortin system in controlling sympathetic outflow, we hypothesized that reduced renal sympathetic nerve activity (RSNA) in ArcPomc−/− mice could explain their elevated glycosuria and consequent enhanced glucose tolerance.
Methods
We measured RSNA by multifiber recording directly from the nerves innervating the kidneys in ArcPomc−/− mice. To further validate the function of RSNA in glucose reabsorption, we denervated the kidneys of WT and diabetic db/db mice before measuring their glucose tolerance and urine glucose levels. Moreover, we performed western blot and immunohistochemistry to determine kidney GLUT2 and SGLT2 levels in either ArcPomc−/− mice or the renal-denervated mice.
Results
Consistent with our hypothesis, we found that basal RSNA was decreased in ArcPomc−/− mice relative to their wild type (WT) littermates. Remarkably, both WT and db/db mice exhibited elevated glycosuria and improved glucose tolerance after renal denervation. The elevated glycosuria in obese ArcPomc−/−, WT and db/db mice was due to reduced renal GLUT2 levels in the proximal tubules. Overall, we show that renal-denervated WT and diabetic mice recapitulate the phenotype of improved glucose tolerance and elevated glycosuria associated with reduced renal GLUT2 levels observed in obese ArcPomc−/− mice.
Conclusion
Hence, we conclude that ArcPomc is essential in maintaining basal RSNA and that elevated glycosuria is a possible mechanism to explain improved glucose tolerance after renal denervation in drug resistant hypertensive patients.
Keywords: Hypothalamic POMC, Renal denervation, Glucose tolerance, Glycosuria, GLUT2, Sympathetic nervous system
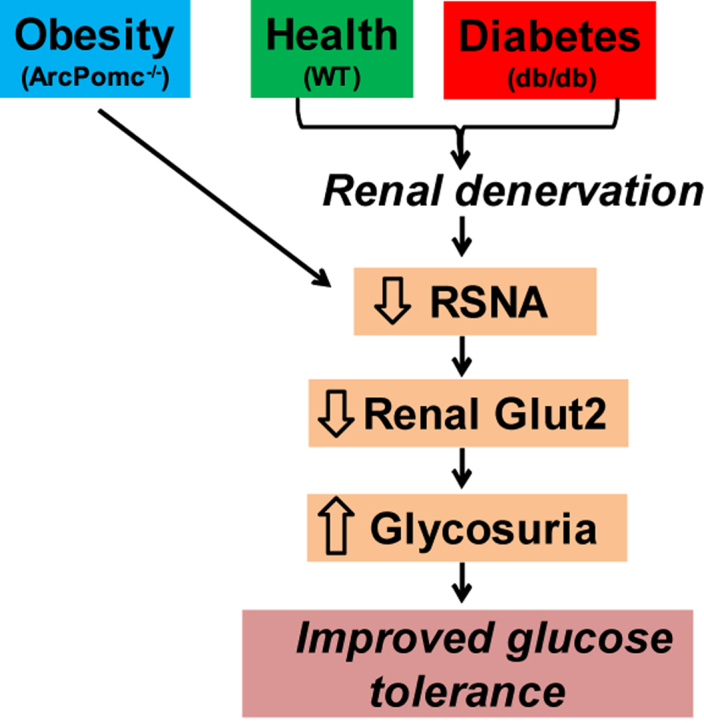
Graphical abstract
Highlights
-
•
Hypothalamic POMC is essential in maintaining basal renal sympathetic nerve activity.
-
•
Renal denervation improves glucose tolerance in wild-type and db/db mice by elevating their glycosuria.
-
•
Decreased renal GLUT2 is responsible for elevated glycosuria in mice with suppressed renal sympathetic nerve activity.
1. Introduction
The central melanocortin system is essential in maintaining energy balance [1]. Hypothalamic pro-opiomelanocortin (POMC) and agouti gene-related protein (AgRP) as well as their downstream receptor targets comprise the melanocortin system [2], [3], [4], [5], [6], [7], [8], [9], [10], [11]. POMC is a precursor polypeptide that is synthesized in the pituitary gland as well as the arcuate nucleus (Arc) of the hypothalamus. POMC is enzymatically cleaved to generate small bioactive peptides including α-melanocyte stimulating hormone (MSH), which acts through melanocortin receptors 3 and 4 (MC3/4R) to control energy homeostasis [12], [13]. Moreover, the melanocortin system is a downstream target for the hunger suppressing hormone leptin [14], [15], [16], [17], [18], [19]. Deficiency of either POMC, MC4R or leptin causes obesity in rodents and humans due to a combination of increased food intake and reduced energy expenditure [4], [10], [11], [20], [21], [22], [23], [24], [25], [26], [27]. MC3R deficiency increases fat mass, but not overall body weight, in mice [28] and is an important regulator of the fasting response [29]. Altogether, the components of the melanocortin system play a critical role in body weight regulation.
We recently identified the function of hypothalamic ArcPOMC in renal proximal tubular glucose reabsorption [30]. We demonstrated that obese and insulin resistant ArcPomc−/− mice exhibit improved glucose tolerance associated with a decreased blood glucose threshold for glycosuria and reduced renal GLUT2, but not SGLT2 glucose transporter levels. However, the molecular mechanisms that couple glucose reabsorption with hypothalamic POMC remain unclear. It is known that hypothalamic melanocortin signaling via MC4R is critical in maintaining sympathetic outflow in rodents [31], [32], [33], [34], [35], [36] and humans [37]. Consequently, MC4R-deficient mice and humans do not display hypertension despite obesity and insulin resistance [31], [34]. Similarly, pharmacological activation and inhibition of MC4R increases and reduces blood pressure, respectively [38], [39], [40]. Moreover, MC4R mediates a leptin-induced increase in renal sympathetic nerve activity (RSNA) in mice [39]. MC4R is also involved in early life programming of hypertension resulting either from maternal obesity or exposure to hyperleptinemia [41]. In addition to MC4R, POMC neurons are implicated in mediating leptin-induced hypertension [42], and administration of POMC peptides such as α-MSH and ACTH increases blood pressure [43]. Overall, these reports indicate a direct role of ArcPOMC in regulating sympathetic outflow.
Reduced RSNA, accomplished by renal denervation, improves glucose tolerance in drug-resistant hypertensive patients [44], [45]. However, the mechanisms underlying this beneficial effect are unclear. Our previous report [30] indicates that ArcPomc−/− mice exhibit improved glucose tolerance due to elevated glycosuria. Given the function of the central melanocortin system in the regulation of sympathetic outflow [31], [32], [33], [34], [35], [36], [37], [43], we hypothesized that reduced RSNA leads to elevated glycosuria in ArcPomc−/− mice. To test this hypothesis, we measured RSNA in ArcPomc−/− mice and determined glucose regulation in renal-denervated wild type (WT) and diabetic db/db mice. We also delineated the impact of renal denervation on levels of the renal proximal tubular glucose transporters GLUT2 and SGLT2 using western blotting and immunohistochemistry.
2. Methods
2.1. Animal care and high-fat diet protocol
All procedures were approved by the Institutional Animal Care and Use Committees at the University of Michigan and University of Iowa and followed the Public Health Service guidelines for the humane care and use of experimental animals. Mice were housed in ventilated cages under a controlled temperature (∼23 °C) and photoperiod (12 h light/dark cycle, lights on from 6:00 a.m. to 6:00 p.m.) and fed tap water and laboratory chow (5L0D; LabDiet) containing 28.5 kcal% protein, 13.5 kcal% fat, and 58 kcal% carbohydrate. A separate cohort of mice was fed with either the regular chow or high-fat diet (HFD, OpenSource Diets®, D12451 – 20 kcal% protein, 45 kcal% fat, and 35 kcal% carbohydrate) immediately after weaning for 16 weeks. Moreover, a group of female ArcPomc−/− mice was weight-matched to that of WT mice by calorie restriction as described previously [30].
The generation and breeding of ArcPomc−/− mice have been reported previously [11], [30]. ArcPomc−/− mice were backcrossed for at least ten generations onto the C57BL/6J genetic background, and these congenic mice were used throughout the study. Male db/db mice and their db/m littermate controls were purchased from The Jackson Laboratory (Stock number: 000642). Mice were randomly assigned to different experimental groups throughout the study.
We utilized female ArcPomc−/− mice and their littermates for blood pressure and renal sympathetic nerve activity (RSNA) measurements, kidney histology and urine analysis, and HFD study. Male mice were included for studies involving renal denervation and corresponding urine analysis.
2.2. Measurement of blood pressure and RSNA
Mice were anesthetized with an intraperitoneal injection of a ketamine (91 mg/kg)/xylazine (9.1 mg/kg) cocktail. Each mouse was intubated using polyethylene tubing (PE-50) to allow spontaneous breathing of oxygen-enriched air throughout the experimental procedure. Next, a tapered micro-renathane tubing (MRE-040; Braintree Scientific, Inc. Braintree, MA) was inserted into the right jugular vein from which α-chloralose was infused at an initial dose of 12 mg/kg during the surgical preparation then at a sustaining dose of 6 mg/kg/h; until completion of the study. Another tapered MRE-040 tubing was inserted into the left carotid artery for continuous measurement of arterial pressure. The carotid cannula was connected to a low-volume pressure transducer (BP-100; iWorks System, Inc., Dover, NH) that led to an ETH-250 Bridge/Bio Amplifier (CB Sciences; Milford, MA). The filtered, amplified pulsatile pressure signal was directed to an analog–digital acquisition system (MacLab 8S; see below) for continuous display of not only phasic arterial pressure and heart rate but also sympathetic nerve activity on a computer monitor. Core body temperature of the mouse was measured using a rectal probe (YSI 4000A Precision Thermometer; Yellow Springs, OH) and maintained constant at 37.5 °C using a custom-made heated surgical platform.
RSNA was measured by multifiber recording directly from the nerves innervating the kidneys as described previously [46], [47]. Briefly, nerves innervating the left kidney were identified, dissected free, and placed on a bipolar 36-gauge platinum–iridium electrode (A-M Systems; Carlsborg, WA). The electrode was connected to a high impedance probe (HIP-511; Grass Instruments Co., Quincy, MA), and the nerve signal was amplified 105 times with a Grass P5 AC pre-amplifier and filtered at low and high frequency cutoffs of 100 Hz and 1000 Hz, respectively. This nerve signal was directed to a speaker system and to an oscilloscope (54501A, Hewlett–Packard Co., Palo Alto, CA) for auditory and visual monitoring of the nerve activity. The signal was then directed to a resetting voltage integrator (B600C, University of Iowa Bioengineering) that sums the total voltage output in units of 1 V * sec before resetting to zero and counting the number of spikes per second. The final neurograms were continuously routed to a MacLab analogue–digital converter (8S, AD Instruments Castle Hill, New South Wales, Australia) for permanent recording and data analysis on a Macintosh computer. RSNA was corrected for post-mortem background activity to eliminate background electrical noise in the assessment of sympathetic outflow in the integrated voltage.
2.3. Renal denervation
Mice were placed in the prone position under general anesthesia with isoflurane. To minimize the invasive approach to the kidneys, a single skin incision was made in the dorsal midline. After exposing the kidneys, renal blood vessels were identified and all visible nerves along the vessels were severed. Furthermore, the vessels were painted with a solution of 10% phenol in absolute ethanol using a fine brush. This treatment did not affect the vessels. After surgery, the paravertebral muscle layers and skin were closed with nylon sutures. Carprofen was administered prior to skin incision and post-operatively for analgesia. Mice were allowed to recover for a week before subjecting them to any experiments. To confirm reduced RSNA post renal denervation procedure, kidney norepinephrine levels were measured as described previously [30] after the completion of all the experiments.
2.4. Glucose and insulin measurements
Oral glucose tolerance and intraperitoneal insulin tolerance tests were performed as described previously [30]. A fixed dose of glucose (G5767; Sigma, 60 mg/mouse in 300 μl water) was administered rather than a dose adjusted by body weight to eliminate the confounding effects of obesity. For insulin tolerance tests, insulin (Humulin R; Eli Lilly, 0.5 units/kg lean mass) i.p. was administered. The total area under the curve (AUC) was calculated using the trapezoidal rule.
A glucose-stimulated insulin secretion assay was performed according to our published protocol [48].
2.5. Urine glucose and electrolytes
For 24 h urine collections, mice were housed individually in metabolic cages (Tecniplast) and were allowed to adapt to the cages for 1 week prior to sample collection. Urine was collected for 24 h, and urine glucose was measured by a colorimetric assay as per manufacturer's instructions (81692; Crystal Chem). Urine sodium, potassium, and chloride levels were measured using indirect ion-selective electrodes on ADVIA 1800.
2.6. Western blotting and kidney histology
Western blots were carried out as per our published protocol [30]. Rabbit polyclonal anti-GLUT2 (600-401-GN3; Rockland Immunochemicals) and goat polyclonal anti-SGLT2 (sc-47402; Santa Cruz Biotechnology) primary antibodies were used at 1:2,000 and 1:1,000 dilutions, respectively, in tris-buffered saline with Tween 20 (TBST) containing 5% powdered milk. Secondary antibodies anti-rabbit (NA934; GE Healthcare) and anti-goat IgGs (sc-2768; Santa Cruz Biotechnology) coupled to horseradish peroxidase were used to detect the corresponding primary antibodies bound to their respective target proteins. Vinculin (ab73412; Abcam) levels were quantified on the same membranes as target proteins to confirm equal loading of samples. Luminescence was generated with the Amersham ECL Advance Western Blotting Detection Kit (GE Healthcare) and recorded on a Fotodyne Imaging System.
For immunohistochemistry, kidneys were removed 45 min after oral glucose administration, cut longitudinally into two equal halves, and fixed overnight in 10% neutral buffered formalin. The fixed kidneys were embedded in paraffin and 5 μm sections collected with a microtome. The sections were mounted onto slides, deparaffinized and rehydrated before an antigen retrieval procedure of incubating the slides in sodium citrate buffer (pH 6.0) at 80 °C in a water bath for 1 h. Following this antigen retrieval step, the slides were washed 3 times (10 min/wash) in TBST. The sections were blocked in 10% normal goat serum for 2 h before incubating them with primary antibodies, rabbit anti-GLUT2 (600-401-GN3; Rockland Immunochemicals, 1:500 dilution) or rabbit anti-SGLT2 (ab85626; Abcam, 1:500 dilution), overnight at 4 °C. Following the incubation period, the sections were washed 3 times (10 min/wash) in TBST and then incubated with secondary goat-anti-rabbit serum conjugated to Alexa Fluor 488 (A-11034; Invitrogen, 1:500 dilution) for 1 h at room temperature. The sections were washed again 3 times (10 min/wash) in TBST and counterstained with DAPI (D9542; Sigma, 0.1 μg/ml). After a 5 min wash, the slides were air-dried and coverslipped using ProLong Antifade mounting medium (P36930; Molecular Probes). Images were captured using a Nikon 90i upright microscope (Nikon, Tokyo, Japan) equipped with an X-Cite 120Q fluorescent light source (Lumen Dynamics, Mississauga, Ontario, Canada), and a CoolSNAP HQ2 CD camera (Photometrics, Tucson, AZ).
Hematoxylin and Eosin (H&E) staining was performed on mouse kidney sections to determine changes in glomerular morphology. The sections were mounted onto slides, deparaffinized and rehydrated as described above. The sections were air-dried and incubated in hematoxylin solution (MHS16; Sigma) for 15 min followed by a 15 min wash with running tap water. Thereafter, the slides were counterstained with alcoholic eosin Y solution (HT110116; Sigma) for 1 min, rinsed with tap water, and coverslipped using ProLong Antifade mounting medium.
The quantitative analysis of histological images was performed using Nikon software NIS-Elements AR. Investigators were blinded to the identity of different samples during the analysis.
2.7. Statistics
All data are presented as mean ± SEM. The data were analyzed by Student's unpaired 2-tailed t-test, or by 2-way or repeated measures (RM) 2-way ANOVA followed by Tukey's multiple comparison test as appropriate with GraphPad Prism 7 software. P < 0.05 was considered significant.
3. Results
3.1. Obese ArcPomc−/− mice have normal blood pressure and reduced RSNA concomitantly with improved glucose tolerance and elevated glycosuria
First, we performed OGTT and measured 24 h urine glucose on two separate occasions in obese ArcPomc−/− mice (Bodyweight: ArcPomc−/− mice, 55.4 ± 1.7 vs. WT mice, 22.9 ± 0.7 g, P < 0.01). In agreement with our previous study [30], ArcPomc−/− mice exhibited improved glucose tolerance (Figure 1A) and elevated glycosuria (Figure 1B). Moreover, to determine a mechanism responsible for elevated glycosuria in ArcPomc−/− mice, we measured RSNA in ArcPomc−/− mice and littermate controls. Obese ArcPomc−/− mice had normal blood pressure (Figure 1C) and reduced RSNA (Figure 1D), suggesting an essential role of ArcPOMC in maintaining basal renal sympathetic tone. Together, these data indicate an association of RSNA with glucose reabsorption.
Figure 1.

Improved glucose tolerance and elevated glycosuria associated with reduced renal sympathetic nerve activity in obese 22–24-wk old female ArcPomc−/− mice. A) Improved glucose tolerance in ArcPomc−/− mice; B) Elevated glycosuria in ArcPomc−/− mice; C) Normal blood pressure in ArcPomc−/− mice; D) Reduced renal sympathetic nerve activity (RSNA) in ArcPomc−/− mice. *P < 0.05, ***P < 0.001, ****P < 0.0001, 2-tailed Student's t-test or 2-way RMANOVA followed by Tukey's multiple comparison test was used for comparisons as appropriate; n = 6. Error bars reflect mean ± SEM.
3.2. Obese ArcPomc−/− mice have decreased proximal tubular GLUT2 levels without any changes in subcellular distribution
Using a western blotting approach in our previous report [30], we attributed elevated glycosuria to reduced renal cortical GLUT2, but not SGLT2, levels in ArcPomc−/− mice. However, it was unclear if changes in proximal tubular subcellular GLUT2 distribution also accounted for decreased glucose reabsorption in ArcPomc−/− mice. Even though we did not see any changes in total SGLT2 levels, there was a possibility that translocation of SGLT2 away from the apical membrane brush border of the proximal tubular cells led to reduced glucose reabsorption. To address these possibilities, we performed immunohistochemistry on kidney sections from WT as well as ArcPomc−/− mice. Consistent with our previous report, ArcPomc−/− mice exhibited decreased GLUT2 (Figure 2A), but not SGLT2 (Figure 2B), immunofluorescence. However, there were no changes in the distribution pattern or location of either GLUT2 or SGLT2 transporters. These data indicate that elevated glycosuria is due to overall decreased GLUT2 levels in proximal tubular cells and not translocation of the glucose transporters away from the plasma membrane in ArcPomc−/− mice.
Figure 2.

Immunofluorescence detection of GLUT2 and SGLT2 levels in 24-wk ArcPomc−/− mice. A) Reduced GLUT2 levels in proximal tubular cells, but no change in its distribution or location, in ArcPomc−/− mice; B) No change in proximal tubular SGLT2 levels in ArcPomc−/− mice. *P < 0.05, 2-tailed Student's t-test was used for comparisons; n = 6. Error bars reflect mean ± SEM. Scale bar = 33 μm; images were taken under 40× objective lens field.
3.3. Obese ArcPomc−/− mice exhibit glomerular hypertrophy, natriuresis, kaliuresis, and chloriuresis
Obese ArcPomc−/− mice exhibited increased glomerular surface area (Figure 3A) compared to their WT littermates. However, weight-matched ArcPomc−/− mice had normal glomerular surface area (ArcPomc−/− mice, 3305 ± 321 vs. WT, 2852 ± 308 μm2, n = 4, no statistical difference). These data suggest that the glomerular hypertrophy was a secondary consequence of obesity and possibly increased blood volume, but not a direct effect of ArcPomc deficiency. This observation is in accordance with that from MC4R−/− mice [49], and data showing that obesity itself can lead to glomerular hypertrophy [50].
Figure 3.

Glomerular morphology and urine electrolytes in 24-wk old ArcPomc−/− mice. A) Glomerular hypertrophy in obese ArcPomc−/− mice (H&E staining); Elevated B) Natriuresis; C) Kaliuresis; D) Chloriuresis in obese and weight-matched female ArcPomc−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001, 2-tailed Student's t-test was used for comparisons; n = 6. Error bars reflect mean ± SEM. Scale bar = 33 μm; images were taken under 40× objective lens field. The red arrows point out the glomeruli.
Both obese and weight-matched ArcPomc−/− mice had elevated natriuresis (Figure 3B), kaliuresis (Figure 3C) and chloriuresis (Figure 3C). Consequently, obese ArcPomc−/− mice exhibited polydipsia (Water intake per day: ArcPomc−/−, 7.4 ± 0.3 ml vs. WT, 6.4 ± 0.2 ml, n = 7–9, P < 0.05) and polyuria (Urine volume per day: ArcPomc−/−, 6.9 ± 0.4 ml vs. WT, 2.7 ± 0.3 ml, n = 7–9, P < 0.001). However, these latter changes were absent in weight-matched ArcPomc−/− mice (data not shown) possibly due to their calorie restriction. The electrolyte excretion results clearly indicate a function of ArcPomc in the regulation of sodium, potassium and chloride reabsorption, possibly via RSNA.
3.4. Obese ArcPomc−/− mice preserve their β-cell function and the mice are protected from HFD-mediated impaired glucose tolerance
Generally, the β-cell compensatory response of enhanced insulin secretion, to overcome the insulin resistance associated with obesity, is decreased over time and leads to hyperglycemia. However, massively obese and insulin resistant aged ArcPomc−/− mice do not manifest hyperglycemia [30]. To elucidate this atypical phenotype, we performed a glucose-stimulated insulin secretion test on isolated islets from 26-wk old ArcPomc−/− mice. Remarkably, the magnitude of insulin secretion was higher in the islets from ArcPomc−/− mice in response to 25 mM glucose compared to that from WT mice (26 wk: ArcPomc−/− mice, 0.13 ± 0.01 vs. WT mice, 0.07 ± 0.01 insulin ng/mg protein, n = 4, P < 0.05). Basal insulin secretion from the islets in response to 3.3 mM glucose was not different between the two groups (data not shown). These results indicate that the function of β-cells of ArcPomc−/− mice is preserved despite chronic insulin resistance and obesity.
ArcPomc−/− mice were fed a HFD to determine their propensity to develop diet-induced impaired glucose tolerance. The mice consumed a significantly lower mass of HFD compared to that of regular chow per day (Figure 4A). However, there was no change in the total calorie intake in HFD-fed ArcPomc−/− mice relative to regular chow fed mice (Figure 4B). The body weight of HFD-fed ArcPomc−/− mice was significantly increased compared to that of the regular chow fed mice (Figure 4C). Interestingly, despite this further increase in body weight, ArcPomc−/− mice remained protected from HFD-induced fasting hyperglycemia or impaired glucose tolerance (Figure 4D, E) probably due to their elevated glycosuria (Figure 4F).
Figure 4.

Impact of high-fat diet (HFD) on glycemia in female ArcPomc−/− mice. A) Reduced mass of daily food intake in HFD fed WT and ArcPomc−/− mice relative to their regular chow (RC) fed controls, *P < 0.05 for RC vs. HFD; ##P < 0.01 for RC ArcPomc−/− vs. WT (n = 6), 2-way ANOVA followed by Tukey's multiple comparisons test (F1, 17 = 3.3, P < 0.05, interaction of genotype and diet); B) No change in daily calorie intake between HFD and RC fed WT or ArcPomc−/− mice. Increased calorie intake in ArcPomc−/− mice relative to WT mice, ∗∗P < 0.01 for ArcPomc−/− vs. WT (n = 6), 2-way ANOVA followed by Tukey's multiple comparisons test; C) HFD increases body weight in WT and ArcPomc−/− mice, *P < 0.05 for ArcPomc−/− RC vs. HFD, #P < 0.01 for WT RC vs HFD, 2-way RMANOVA followed by Tukey's multiple comparisons test (F24, 112 = 91.6, P < 0.05, interaction of time and genotype); D) No change in fasting blood glucose levels in HFD fed ArcPomc−/− mice relative to their RC controls, but an increase in fasting blood glucose levels in HFD fed WT mice compared to their RC controls, *P < 0.05 for WT RC vs. HFD, 2-way ANOVA followed by Tukey's multiple comparison test; E) No impairment in glucose tolerance in HFD fed ArcPomc−/− mice; bar-graph shows AUC for the glucose tolerance test, *P < 0.05 for WT RC vs. HFD, ##P < 0.05 for ArcPomc−/− RC or HFD vs WT, 2-way ANOVA followed by Tukey's multiple comparison test; F) Elevated glycosuria in HFD fed ArcPomc−/− mice. ∗∗P < 0.01 for ArcPomc−/− vs. WT, 2-way ANOVA followed by Tukey's multiple comparisons test. KO, ArcPomc−/− mice; RC, regular chow; HFD, high-fat diet; AUC, area under the curve. Error bars reflect mean ± SEM.
3.5. Renal-denervated WT and db/db mice recapitulate the phenotype of ArcPomc−/− mice
To further elucidate the function of RSNA in glucose reabsorption, we denervated the kidneys of WT and db/db mice. Kidney norepinephrine levels were about 80% lower in the denervated mice relative to their sham-operated controls (WT: 0.06 ± 0.02 vs. 0.28 ± 0.01; db/db: 0.08 ± 0.02 vs. 0.35 ± 0.03, ng/mg tissue, renal-denervated vs. sham), thereby validating the renal denervation procedure. We found that renal-denervated WT mice exhibited improved glucose tolerance (Figure 5A) and elevated glycosuria (Figure 5B) compared to their WT sham-operated littermates. Insulin sensitivity was not affected by the renal denervation procedure in WT mice (Figure 5C). These results suggest a critical role of RSNA in glucose reabsorption, independently of insulin action.
Figure 5.

Effects of renal denervation on glycemia in male WT and db/db mice. A) Renal denervation improves glucose tolerance in 8–10-wk old WT mice; B) Elevated glycosuria in renal-denervated WT mice; C) No change in insulin sensitivity after renal denervation in WT mice; D) Renal denervation improves glucose tolerance in db/db mice; E) Elevated glycosuria in renal-denervated 9-wk old db/db mice; F) Reduced glycosuria in renal-denervated 12-wk old db/db mice. *P < 0.05, **P < 0.01, ***P < 0.001, 2-tailed Student's t-test or 2-way RMANOVA followed by Tukey's multiple comparison test was used for comparisons as appropriate; n = 6. RD, renal denervation; AUC, area under the curve. Error bars reflect mean ± SEM.
The renal-denervation procedure in diabetic db/db mice prevented an age-associated increase in their fasting blood glucose levels (Baseline: designated sham group, 288 ± 16 vs. designated renal-denervated group, 334 ± 30, no statistical difference; 9-wk: 451 ± 6 vs 309 ± 10; 12-wk: 459 ± 14 vs 310 ± 8 mg/dl, sham vs. renal-denervated, Student's unpaired 2-tailed t-test, P < 0.05). Moreover, renal-denervated db/db mice had improved glucose tolerance at different ages compared to their sham-operated littermates (Figure 5D). This improvement in glucose tolerance was associated with elevated glycosuria in 9-wk old db/db mice (Figure 5E). However, with no further increase in fasting glycemia at 12-wk of age, renal-denervated db/db mice showed reduced glycosuria compared to sham-operated db/db mice (Figure 5F). It is important to acknowledge that renal-denervation in db/db mice did not completely restore normal glycemia or glucose tolerance; however, the procedure prevented further deterioration of glucose tolerance and hyperglycemia. Overall, our data indicate a direct role of RSNA in glucose reabsorption in normal as well as diabetic mice.
To understand the mechanism underlying elevated glycosuria, we determined the levels of the major renal glucose transporters, GLUT2 and SGLT2. Consistent with the results obtained from ArcPomc−/− mice, we observed reduced renal GLUT2 (Figure 6A), but not SGLT2 (Figure 6B), levels in renal-denervated WT and db/db mice. These data further reinforce that the sympathetic nervous system controls glucose reabsorption via renal GLUT2.
Figure 6.

Representative western blots showing renal cortical GLUT2 and SGLT2 levels in renal-denervated WT and db/db mice. A) Decreased renal cortical GLUT2 levels in renal-denervated WT and db/db mice; B) No change in renal cortical SGLT2 levels in renal-denervated WT and db/db mice. *P < 0.05, 2-tailed Student's t-test was used for comparisons; n = 8 (4 samples per group on each blot). Error bars reflect mean ± SEM.
Like ArcPomc−/− mice, renal-denervated WT and db/db mice had elevated natriuresis (Figure 7A), kaliuresis (Figure 7B) and chloriuresis (Figure 7C). Altogether, renal-denervated WT and db/db mice recapitulated the phenotype observed in ArcPomc−/− mice. Hence, our study strongly suggests a role of RSNA in glucose homeostasis via modulation of glucose reabsorption.
Figure 7.

Effects of renal denervation on urine electrolytes in male WT and db/db mice. Elevated A) Natriuresis; B) Kaliuresis; C) Chloriuresis in renal-denervated WT and db/db mice. *P < 0.05, **P < 0.01, 2-tailed Student's t-test was used for comparisons; n = 6. Error bars reflect mean ± SEM.
4. Discussion
In this study, we report the function of ArcPomc in maintaining basal RSNA. ArcPomc−/− mice are protected from hypertension, despite obesity and insulin resistance, because of reduced RSNA. Moreover, utilizing normal and diabetic db/db mice, we demonstrate that RSNA contributes to glucose reabsorption via GLUT2 in renal proximal tubular cells. Hence, renal denervation improves glucose tolerance in mice by elevating their glycosuria.
Generally, obesity and insulin resistance are major risk factors for cardiovascular disorders including hypertension [33]. Obesity stimulates sympathetic nervous system activity leading to hypertension [33]. Leptin is the primary mediator for obesity-induced increase in sympathetic nervous system activity and consequent hypertension [40], [51], [52], [53], [54]. Moreover, insulin resistance is associated with hypertension [55], [56], [57]. However, in this study, we demonstrate that despite obesity, insulin resistance and hyperleptinemia, ArcPomc−/− mice remain protected from hypertension due to suppressed RSNA. This phenotype is reminiscent of that of MC4R knockout mice. MC4R-deficient mice do not exhibit increases in RSNA or hypertension despite obesity, insulin resistance and hyperleptinemia [31]. Moreover, MC4R is critical in mediating the leptin-induced increase in RSNA [39]. Hence, our results from ArcPomc−/− mice are in line with that obtained from MC4R knockout mice. Altogether, these data indicate that POMC/MC4R signaling is essential in causing obesity-induced hypertension via regulating RSNA.
We recently reported that ArcPomc−/− mice do not exhibit hyperglycemia despite obesity and insulin resistance [30]. Similarly, it is known that obese and insulin resistant POMC-deficient humans do not develop diabetes [24], [58]. Moreover, neither MC4R knockout mice nor MC4R-deficient humans manifest hyperglycemia in spite of obesity and insulin resistance [20], [23], [59]. We attributed the improvement in glucose tolerance in ArcPomc−/− mice to elevated glycosuria mediated by reduced proximal tubular GLUT2 levels [30]. However, the mechanism that links glucose reabsorption with ArcPOMC is unclear. We have previously observed that epinephrine increases renal GLUT2 levels in mice [30]. Moreover, we showed that ArcPomc−/− mice have reduced epinephrine levels in their kidneys, indicating the role of ArcPOMC in regulating sympathetic nervous system activity. In this study, we directly showed by electrophysiological measurements that ArcPomc−/− mice do have reduced RSNA, consistent with our hypothesis that it is the mechanism for their elevated glycosuria.
To further validate the function of RSNA in glucose reabsorption using alternative mouse models, we denervated kidneys in WT and db/db mice. Renal denervation improves glucose metabolism in drug-resistant hypertensive patients [44], [45]. However, the mechanism remains unclear. We found that, like ArcPomc−/− mice, renal-denervated WT and db/db mice exhibited improved glucose tolerance in association with elevated glycosuria. As fasting hyperglycemia and glucose tolerance continued to improve in db/db mice after renal denervation, glycosuria was subsequently decreased in 12-wk old db/db mice. This observation corroborates a previous study that reported a decrease in glycosuria in db/db mice after chronic treatment with an SGLT2 inhibitor [60]. Collectively, our data from renal-denervated mice confirm a role of RSNA in glucose reabsorption.
In contrast, a recent study reported that renal denervation does not improve glucose tolerance in obese hypertensive mice [61]. It is very likely that the authors of that report did not see any improvement in glucose tolerance because the mice were administered glucose based on their body weight. Hence, the obese mice received a higher glucose dose compared to lean mice, which might have affected their response to glucose tolerance tests. Moreover, the mice in that study [61] were fasted for 14 h prior to glucose tolerance tests, which is different than our protocol of fasting the mice for only 6 h [30]. These variations, in addition to different routes of administration of glucose, could have led to inconsistent results and conclusions between our present study and that by Asirvatham-Jeyaraj et al. [61].
Interestingly, a link between RSNA and glycosuria was also recently suggested using Otsuka Long-Evans Tokushima fatty rats [62]. The authors removed the right kidney before denervating the left kidney at a later age to determine the role of renal sympathetic nerves in glucose reabsorption. The authors stated that right uninephrectomy was performed to prevent reno-renal reflexes. However, consistent with clinical protocols in humans [44], we suggest that a bilateral renal denervation procedure would have been a more powerful physiological approach to test the impact of renal sympathetic nerves on glucose reabsorption. Moreover, unilateral nephrectomy might have led to compensatory changes in the contralateral kidney in the rats [63]. Besides species differences, these different methodologies might explain why we, unlike Rafiq et al. [62], did not observe any changes in SGLT2 levels post renal denervation. Nevertheless, conclusions from our current study support the main findings from the report by Rafiq et al. [62] that reduced RSNA elevates glycosuria leading to improvement in glucose tolerance.
We previously demonstrated using a western blotting method that elevated glycosuria in ArcPomc−/− mice is due to reduced renal proximal tubular GLUT2, but not SGLT2, levels [30]. Both GLUT2 and SGLT2 are involved in glucose reabsorption. While SGLT2 is a major glucose transporter in the apical brush border membrane of proximal tubular cells, GLUT2 is located at the basolateral membrane of the cells. In the current study, we performed immunohistochemistry to verify the location and distribution of these transporters in ArcPomc−/− mice. GLUT2 in epithelial cells of the intestine is translocated from basolateral membrane to apical brush border membrane in response to meals or elevated glucose [64], [65]. We wanted to determine whether this translocation-phenomenon in proximal tubule cells of the kidney could explain the elevated glycosuria in ArcPomc−/− mice. Consistent with our previous report [30], we observed reduced GLUT2, but not SGLT2, immunofluorescence in renal proximal tubular cells. However, we did not see any change in the location or distribution pattern of either transporter in ArcPomc−/− mice. Nonetheless, differences in SGLT2 or GLUT2 activity between WT and ArcPomc−/− mice cannot be excluded and might further clarify elevated glycosuria in the latter.
Deficiency or inhibition of only SGLT2 [66], [67] or GLUT2 [68], [69] can lead to elevated glycosuria in rodents as well as humans. The elevated glycosuria protects them from hyperglycemia. Moreover, SGLT2 knockout mice remain protected from HFD mediated hyperglycemia and impaired glucose tolerance despite obesity and insulin resistance [70]. Similarly, we demonstrate that obese and insulin resistant ArcPomc−/− mice are protected from HFD mediated impaired glucose tolerance. We further show that islets isolated from aged ArcPomc−/− mice release higher insulin compared to that isolated from their WT littermates in a glucose-stimulated insulin secretion test. During obesity, hyperinsulinemia compensates for insulin resistance to maintain normal blood glucose levels. With time, this compensation fails to overcome insulin resistance leading to overt hyperglycemia due to β-cell failure. However, in ArcPomc−/− mice, glucose-stimulated insulin secretion is not impaired, probably due to elevated glycosuria. These data are in agreement with a report indicating the beneficial effects of elevated glycosuria on β-cell function in SGLT2 knockout mice [70].
In addition to elevated glycosuria, ArcPomc−/− mice manifested increased natriuresis, kaliuresis and chloriuresis. The function of RSNA in sodium reabsorption is well documented [71]. Reduced RSNA leads to elevated natriuresis. Moreover, epinephrine is also involved in potassium and chloride homeostasis. Epinephrine treatment increases potassium and chloride reabsorption [72]. Hence, suppressed RSNA may also explain the elevated kaliuresis and chloriuresis in ArcPomc−/− mice. Renal denervation in WT and diabetic db/db mice recapitulated the phenotype of increased natriuresis, kaliuresis, and chloriuresis observed in ArcPomc−/− mice (Figure 7).
In summary, we report that ArcPOMC is essential in maintaining basal RSNA in mice. We have also demonstrated the critical function of RSNA in glucose reabsorption. Reduced RSNA in ArcPomc−/− mice as well as renal denervation in WT and diabetic db/db mice improves their glucose tolerance by elevating glycosuria via reduced proximal tubular GLUT2 levels (see Graphical Abstract). Therefore, elevated glycosuria is likely a mechanism for improving glucose tolerance after renal denervation in drug resistant hypertensive patients.
Author contributions
K.H.C. conceived the study, designed and performed experiments, analyzed results and wrote the manuscript. M.J.L. conceived the study, designed experiments, analyzed results and edited the manuscript. D.A.M. and K.R. performed renal nerve activity experiments and blood pressure measurements, discussed results and edited the manuscript for intellectual content. B.P.T. performed western blot and analyzed results. J.M.A. conducted the metabolic study involving urine analysis in obese ArcPomc−/− mice. The authors have no conflict of interest relevant to this study. M.J.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This work was supported by NIH grants R01 DK066604 and DK068400 to M.J.L.; K01 DK113115 to K.H.C; and P01 HL084207 and the American Heart Association 14EIA18860041 to K.R. This work utilized core services provided by the University of Michigan Animal Phenotyping Core and Chemistry Core supported by the Michigan Diabetes Research Center and the Michigan Nutrition and Obesity Research Center (NIH grants P30 DK020572 and P30 DK089503).
Acknowledgements
We thank T. Sutton for mouse colony management and genotyping, G. Jones for his help designing the graphical abstract and members of the Low laboratory for their valuable suggestions on this project.
Conflict of interest
None declared.
References
- 1.Cone R.D. The central melanocortin system and its role in energy homeostasis. Annales D’endocrinologie (Paris) 1999;60(1):3–9. [PubMed] [Google Scholar]
- 2.Rosenfeld R.D., Zeni L., Welcher A.A., Narhi L.O., Hale C., Marasco J. Biochemical, biophysical, and pharmacological characterization of bacterially expressed human agouti-related protein. Biochemistry. 1998;37(46):16041–16052. doi: 10.1021/bi981027m. [DOI] [PubMed] [Google Scholar]
- 3.Hagan M.M., Rushing P.A., Schwartz M.W., Yagaloff K.A., Burn P., Woods S.C. Role of the CNS melanocortin system in the response to overfeeding. Journal of Neuroscience. 1999;19(6):2362–2367. doi: 10.1523/JNEUROSCI.19-06-02362.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yaswen L., Diehl N., Brennan M.B., Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nature Medicine. 1999;5(9):1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- 5.Quillan J.M., Sadee W., Wei E.T., Jimenez C., Ji L., Chang J.K. A synthetic human Agouti-related protein-(83-132)-NH2 fragment is a potent inhibitor of melanocortin receptor function. FEBS Letters. 1998;428(1–2):59–62. doi: 10.1016/s0014-5793(98)00487-6. [DOI] [PubMed] [Google Scholar]
- 6.Nijenhuis W.A., Oosterom J., Adan R.A. AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor. Molecular Endocrinology. 2001;15(1):164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 7.Korner J., Wissig S., Kim A., Conwell I.M., Wardlaw S.L. Effects of agouti-related protein on metabolism and hypothalamic neuropeptide gene expression. Journal of Neuroendocrinology. 2003;15(12):1116–1121. doi: 10.1111/j.1365-2826.2003.01113.x. [DOI] [PubMed] [Google Scholar]
- 8.Gropp E., Shanabrough M., Borok E., Xu A.W., Janoschek R., Buch T. Agouti-related peptide-expressing neurons are mandatory for feeding. Nature Neuroscience. 2005;8(10):1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 9.Tolle V., Low M.J. In vivo evidence for inverse agonism of agouti-related peptide in the central nervous system of proopiomelanocortin-deficient mice. Diabetes. 2008;57(1):86–94. doi: 10.2337/db07-0733. [DOI] [PubMed] [Google Scholar]
- 10.Smart J.L., Low M.J. Lack of proopiomelanocortin peptides results in obesity and defective adrenal function but normal melanocyte pigmentation in the murine C57BL/6 genetic background. Annals of the New York Academy of Sciences. 2003;994:202–210. doi: 10.1111/j.1749-6632.2003.tb03181.x. [DOI] [PubMed] [Google Scholar]
- 11.Bumaschny V.F., Yamashita M., Casas-Cordero R., Otero-Corchon V., de Souza F.S., Rubinstein M. Obesity-programmed mice are rescued by early genetic intervention. Journal of Clinical Investigation. 2012;122(11):4203–4212. doi: 10.1172/JCI62543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cone R.D. Anatomy and regulation of the central melanocortin system. Nature Neuroscience. 2005;8(5):571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 13.Wardlaw S.L. Hypothalamic proopiomelanocortin processing and the regulation of energy balance. European Journal of Pharmacology. 2011;660(1):213–219. doi: 10.1016/j.ejphar.2010.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seeley R.J., Yagaloff K.A., Fisher S.L., Burn P., Thiele T.E., van Dijk G. Melanocortin receptors in leptin effects. Nature. 1997;390(6658):349. doi: 10.1038/37016. [DOI] [PubMed] [Google Scholar]
- 15.Enriori P.J., Evans A.E., Sinnayah P., Jobst E.E., Tonelli-Lemos L., Billes S.K. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metabolism. 2007;5(3):181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Cowley M.A., Smart J.L., Rubinstein M., Cerdan M.G., Diano S., Horvath T.L. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 17.Marsh D.J., Hollopeter G., Huszar D., Laufer R., Yagaloff K.A., Fisher S.L. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nature Genetics. 1999;21(1):119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 18.Mizuno T.M., Kleopoulos S.P., Bergen H.T., Roberts J.L., Priest C.A., Mobbs C.V. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47(2):294–297. doi: 10.2337/diab.47.2.294. [DOI] [PubMed] [Google Scholar]
- 19.Chhabra K.H., Adams J.M., Jones G.L., Yamashita M., Schlapschy M., Skerra A. Reprogramming the body weight set point by a reciprocal interaction of hypothalamic leptin sensitivity and Pomc gene expression reverts extreme obesity. Molecular Metabolism. 2016;5(10):869–881. doi: 10.1016/j.molmet.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huszar D., Lynch C.A., Fairchild-Huntress V., Dunmore J.H., Fang Q., Berkemeier L.R. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 21.Montague C.T., Farooqi I.S., Whitehead J.P., Soos M.A., Rau H., Wareham N.J. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 22.Yeo G.S., Farooqi I.S., Aminian S., Halsall D.J., Stanhope R.G., O'Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nature Genetics. 1998;20(2):111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 23.Farooqi I.S., Yeo G.S., Keogh J.M., Aminian S., Jebb S.A., Butler G. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. Journal of Clinical Investigation. 2000;106(2):271–279. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krude H., Biebermann H., Luck W., Horn R., Brabant G., Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nature Genetics. 1998;19(2):155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 25.Ingalls A.M., Dickie M.M., Snell G.D. Obese, a new mutation in the house mouse. The Journal of Heredity. 1950;41(12):317–318. doi: 10.1093/oxfordjournals.jhered.a106073. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y., Proenca R., Maffei M., Barone M., Leopold L., Friedman J.M. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 27.Coleman D.L. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14(3):141–148. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- 28.Butler A.A., Kesterson R.A., Khong K., Cullen M.J., Pelleymounter M.A., Dekoning J. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141(9):3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 29.Renquist B.J., Murphy J.G., Larson E.A., Olsen D., Klein R.F., Ellacott K.L. Melanocortin-3 receptor regulates the normal fasting response. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(23):E1489–E1498. doi: 10.1073/pnas.1201994109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chhabra K.H., Adams J.M., Fagel B., Lam D.D., Qi N., Rubinstein M. Hypothalamic POMC deficiency improves glucose tolerance despite insulin resistance by increasing glycosuria. Diabetes. 2016;65(3):660–672. doi: 10.2337/db15-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tallam L.S., Stec D.E., Willis M.A., da Silva A.A., Hall J.E. Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension. 2005;46(2):326–332. doi: 10.1161/01.HYP.0000175474.99326.bf. [DOI] [PubMed] [Google Scholar]
- 32.Tallam L.S., da Silva A.A., Hall J.E. Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension. 2006;48(1):58–64. doi: 10.1161/01.HYP.0000227966.36744.d9. [DOI] [PubMed] [Google Scholar]
- 33.Hall J.E., da Silva A.A., do Carmo J.M., Dubinion J., Hamza S., Munusamy S. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. Journal of Biological Chemistry. 2010;285(23):17271–17276. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stepp D.W., Osakwe C.C., Belin de Chantemele E.J., Mintz J.D. Vascular effects of deletion of melanocortin-4 receptors in rats. Physiological Reports. 2013;1(6):e00146. doi: 10.1002/phy2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li P., Cui B.P., Zhang L.L., Sun H.J., Liu T.Y., Zhu G.Q. Melanocortin 3/4 receptors in paraventricular nucleus modulate sympathetic outflow and blood pressure. Experimental Physiology. 2013;98(2):435–443. doi: 10.1113/expphysiol.2012.067256. [DOI] [PubMed] [Google Scholar]
- 36.Morgan D.A., McDaniel L.N., Yin T., Khan M., Jiang J., Acevedo M.R. Regulation of glucose tolerance and sympathetic activity by MC4R signaling in the lateral hypothalamus. Diabetes. 2015;64(6):1976–1987. doi: 10.2337/db14-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenfield J.R., Miller J.W., Keogh J.M., Henning E., Satterwhite J.H., Cameron G.S. Modulation of blood pressure by central melanocortinergic pathways. The New England Journal of Medicine. 2009;360(1):44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 38.Kuo J.J., da Silva A.A., Tallam L.S., Hall J.E. Role of adrenergic activity in pressor responses to chronic melanocortin receptor activation. Hypertension. 2004;43(2):370–375. doi: 10.1161/01.HYP.0000111836.54204.93. [DOI] [PubMed] [Google Scholar]
- 39.Rahmouni K., Haynes W.G., Morgan D.A., Mark A.L. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. Journal of Neuroscience. 2003;23(14):5998–6004. doi: 10.1523/JNEUROSCI.23-14-05998.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haynes W.G., Morgan D.A., Djalali A., Sivitz W.I., Mark A.L. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33(1 Pt 2):542–547. doi: 10.1161/01.hyp.33.1.542. [DOI] [PubMed] [Google Scholar]
- 41.Samuelsson A.S., Mullier A., Maicas N., Oosterhuis N.R., Eun Bae S., Novoselova T.V. Central role for melanocortin-4 receptors in offspring hypertension arising from maternal obesity. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(43):12298–12303. doi: 10.1073/pnas.1607464113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.do Carmo J.M., da Silva A.A., Cai Z., Lin S., Dubinion J.H., Hall J.E. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension. 2011;57(5):918–926. doi: 10.1161/HYPERTENSIONAHA.110.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunbar J.C., Lu H. Proopiomelanocortin (POMC) products in the central regulation of sympathetic and cardiovascular dynamics: studies on melanocortin and opioid interactions. Peptides. 2000;21(2):211–217. doi: 10.1016/s0196-9781(99)00192-8. [DOI] [PubMed] [Google Scholar]
- 44.Mahfoud F., Schlaich M., Kindermann I., Ukena C., Cremers B., Brandt M.C. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation. 2011;123(18):1940–1946. doi: 10.1161/CIRCULATIONAHA.110.991869. [DOI] [PubMed] [Google Scholar]
- 45.Witkowski A., Prejbisz A., Florczak E., Kadziela J., Sliwinski P., Bielen P. Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension. 2011;58(4):559–565. doi: 10.1161/HYPERTENSIONAHA.111.173799. [DOI] [PubMed] [Google Scholar]
- 46.Morgan D.A., Rahmouni K. Differential effects of insulin on sympathetic nerve activity in agouti obese mice. Journal of Hypertension. 2010;28(9):1913–1919. doi: 10.1097/HJH.0b013e32833c2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandes-Santos C., Zhang Z., Morgan D.A., Guo D.F., Russo A.F., Rahmouni K. Amylin acts in the central nervous system to increase sympathetic nerve activity. Endocrinology. 2013;154(7):2481–2488. doi: 10.1210/en.2012-2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chhabra K.H., Xia H., Pedersen K.B., Speth R.C., Lazartigues E. Pancreatic angiotensin-converting enzyme 2 improves glycemia in angiotensin II-infused mice. American Journal of Physiology—Endocrinology and Metabolism. 2013;304(8):E874–E884. doi: 10.1152/ajpendo.00490.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.do Carmo J.M., Tallam L.S., Roberts J.V., Brandon E.L., Biglane J., da Silva A.A. Impact of obesity on renal structure and function in the presence and absence of hypertension: evidence from melanocortin-4 receptor-deficient mice. American Journal of Physiology—Regulatory, Integrative and Comparative Physiology. 2009;297(3):R803–R812. doi: 10.1152/ajpregu.00187.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Serra A., Romero R., Lopez D., Navarro M., Esteve A., Perez N. Renal injury in the extremely obese patients with normal renal function. Kidney International. 2008;73(8):947–955. doi: 10.1038/sj.ki.5002796. [DOI] [PubMed] [Google Scholar]
- 51.Simonds S.E., Pryor J.T., Ravussin E., Greenway F.L., Dileone R., Allen A.M. Leptin mediates the increase in blood pressure associated with obesity. Cell. 2014;159(6):1404–1416. doi: 10.1016/j.cell.2014.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan D.A., Despas F., Rahmouni K. Effects of leptin on sympathetic nerve activity in conscious mice. Physiological Reports. 2015;3(9) doi: 10.14814/phy2.12554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haynes W.G., Sivitz W.I., Morgan D.A., Walsh S.A., Mark A.L. Sympathetic and cardiorenal actions of leptin. Hypertension. 1997;30(3 Pt 2):619–623. doi: 10.1161/01.hyp.30.3.619. [DOI] [PubMed] [Google Scholar]
- 54.Aizawa-Abe M., Ogawa Y., Masuzaki H., Ebihara K., Satoh N., Iwai H. Pathophysiological role of leptin in obesity-related hypertension. Journal of Clinical Investigation. 2000;105(9):1243–1252. doi: 10.1172/JCI8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferrannini E., Buzzigoli G., Bonadonna R., Giorico M.A., Oleggini M., Graziadei L. Insulin resistance in essential hypertension. The New England Journal of Medicine. 1987;317(6):350–357. doi: 10.1056/NEJM198708063170605. [DOI] [PubMed] [Google Scholar]
- 56.Reaven G.M. Relationship between insulin resistance and hypertension. Diabetes Care. 1991;14(Suppl 4):33–38. doi: 10.2337/diacare.14.4.33. [DOI] [PubMed] [Google Scholar]
- 57.Scherrer U., Sartori C. Insulin as a vascular and sympathoexcitatory hormone: implications for blood pressure regulation, insulin sensitivity, and cardiovascular morbidity. Circulation. 1997;96(11):4104–4113. doi: 10.1161/01.cir.96.11.4104. [DOI] [PubMed] [Google Scholar]
- 58.Kuhnen P., Clement K., Wiegand S., Blankenstein O., Gottesdiener K., Martini L.L. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. The New England Journal of Medicine. 2016;375(3):240–246. doi: 10.1056/NEJMoa1512693. [DOI] [PubMed] [Google Scholar]
- 59.Fan W., Dinulescu D.M., Butler A.A., Zhou J., Marks D.L., Cone R.D. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141(9):3072–3079. doi: 10.1210/endo.141.9.7665. [DOI] [PubMed] [Google Scholar]
- 60.Arakawa K., Ishihara T., Oku A., Nawano M., Ueta K., Kitamura K. Improved diabetic syndrome in C57BL/KsJ-db/db mice by oral administration of the Na(+)-glucose cotransporter inhibitor T-1095. British Journal of Pharmacology. 2001;132(2):578–586. doi: 10.1038/sj.bjp.0703829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asirvatham-Jeyaraj N., Fiege J.K., Han R., Foss J., Banek C.T., Burbach B.J. Renal denervation normalizes arterial pressure with no effect on glucose metabolism or renal inflammation in obese hypertensive mice. Hypertension. 2016;68(4):929–936. doi: 10.1161/HYPERTENSIONAHA.116.07993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rafiq K., Fujisawa Y., Sherajee S.J., Rahman A., Sufiun A., Kobori H. Role of the renal sympathetic nerve in renal glucose metabolism during the development of type 2 diabetes in rats. Diabetologia. 2015;58(12):2885–2898. doi: 10.1007/s00125-015-3771-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dicker S.E., Shirley D.G. Compensatory renal growth after unilateral nephrectomy in the new-born rat. The Journal of Physiology. 1973;228(1):193–202. doi: 10.1113/jphysiol.1973.sp010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kellett G.L., Helliwell P.A. The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochemical Journal. 2000;350(Pt 1):155–162. [PMC free article] [PubMed] [Google Scholar]
- 65.Gouyon F., Caillaud L., Carriere V., Klein C., Dalet V., Citadelle D. Simple-sugar meals target GLUT2 at enterocyte apical membranes to improve sugar absorption: a study in GLUT2-null mice. The Journal of Physiology. 2003;552(Pt 3):823–832. doi: 10.1113/jphysiol.2003.049247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Komoroski B., Vachharajani N., Feng Y., Li L., Kornhauser D., Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clinical Pharmacology & Therapeutics. 2009;85(5):513–519. doi: 10.1038/clpt.2008.250. [DOI] [PubMed] [Google Scholar]
- 67.Vallon V., Platt K.A., Cunard R., Schroth J., Whaley J., Thomson S.C. SGLT2 mediates glucose reabsorption in the early proximal tubule. Journal of the American Society of Nephrology. 2011;22(1):104–112. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thorens B., Guillam M.T., Beermann F., Burcelin R., Jaquet M. Transgenic reexpression of GLUT1 or GLUT2 in pancreatic beta cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. Journal of Biological Chemistry. 2000;275(31):23751–23758. doi: 10.1074/jbc.M002908200. [DOI] [PubMed] [Google Scholar]
- 69.Grunert S.C., Schwab K.O., Pohl M., Sass J.O., Santer R. Fanconi-Bickel syndrome: GLUT2 mutations associated with a mild phenotype. Molecular Genetics and Metabolism. 2012;105(3):433–437. doi: 10.1016/j.ymgme.2011.11.200. [DOI] [PubMed] [Google Scholar]
- 70.Jurczak M.J., Lee H.Y., Birkenfeld A.L., Jornayvaz F.R., Frederick D.W., Pongratz R.L. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes. 2011;60(3):890–898. doi: 10.2337/db10-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bell-Reuss E., Trevino D.L., Gottschalk C.W. Effect of renal sympathetic nerve stimulation on proximal water and sodium reabsorption. Journal of Clinical Investigation. 1976;57(4):1104–1107. doi: 10.1172/JCI108355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jacobson W.E., Hammarsten J.F., Heller B.I. The effects of adrenaline upon renal function and electrolyte excretion. Journal of Clinical Investigation. 1951;30(12:2):1503–1506. doi: 10.1172/JCI102560. [DOI] [PMC free article] [PubMed] [Google Scholar]