

Abstract
Exosomes, nano‐sized secreted extracellular vesicles (EVs), are actively studied for their diagnostic and therapeutic potential. In particular, exosomes secreted by dendritic cells (DCs) have been shown to carry MHC‐peptide complexes allowing efficient activation of T lymphocytes, thus displaying potential as promoters of adaptive immune responses. DCs also secrete other types of EVs of different size, subcellular origin and protein composition, whose immune capacities have not been yet compared to those of exosomes. Here, we show that large EVs (lEVs) released by human DCs are as efficient as small EVs (sEVs), including exosomes, to induce CD4+ T‐cell activation in vitro. When released by immature DCs, however, lEVs and sEVs differ in their capacity to orient T helper (Th) cell responses, the former favouring secretion of Th2 cytokines, whereas the latter promote Th1 cytokine secretion (IFN‐γ). Upon DC maturation, however, these functional differences are abolished, and all EVs become able to induce IFN‐γ. Our results highlight the need to comprehensively compare the functionalities of EV subtypes in all patho/physiological systems where exosomes are claimed to perform critical roles.
Keywords: CD4+ T cells, dendritic cells, exosomes, extracellular vesicles, microvesicles
Subject Categories: Immunology, Membrane & Intracellular Transport
Introduction
Cells secrete into their environment different types of membrane‐enclosed structures, collectively called extracellular vesicles (EVs), which play an important role in cell‐to‐cell communication (Raposo & Stoorvogel, 2013; Colombo et al, 2014; Yanez‐Mo et al, 2015). EVs have been shown to contain proteins and nucleic acids that can be transferred to recipient cells, modifying their physiological state (Kosaka et al, 2016; Tkach & Théry, 2016).
Live cells release very heterogeneous EV populations that have distinct structural and biochemical properties depending on their subcellular site of origin. Exosomes are a subtype of EVs that are formed as intraluminal vesicles of multivesicular compartments of the endocytic pathway, and they therefore present a mean diameter of 50–150 nm, similar to the diameter of intraluminal vesicles. On the other hand, microvesicles display a diverse range of sizes (100–1,000 nm in diameter) and are formed and released by budding from the cells’ plasma membrane. Cells undergoing apoptosis also release EVs, generally called “apoptotic bodies”, with a wide range of sizes (from 50 nm to bigger than 1,000 nm) (Crescitelli et al, 2013; Colombo et al, 2014). EVs are commonly isolated and concentrated by differential ultracentrifugation (Thery et al, 2006). At each step of centrifugation, enrichment of different EVs is obtained. Large vesicles (lEVs) are pelleted at low speed centrifugation (2,000 × g), while vesicles smaller than 150 nm constitute the majority of EVs recovered at the last step of high‐speed ultracentrifugation (100,000 × g) (Kowal et al, 2016). In the literature, the latter EVs are generally called exosomes (Gould & Raposo, 2013). However, we now know that this small EV pellet is composed not only of exosomes but also of other non‐exosomal vesicles (Kowal et al, 2016). We will thus use the term “small EVs (sEVs)” instead of “exosomes” in this article.
In the past years, numerous studies have described functions of sEVs, in all pathological and physiological systems, from cancer to cardiovascular function, as well as development or neurobiology (Yanez‐Mo et al, 2015). Induction of T lymphocyte‐dependent immune responses by sEVs secreted by professional antigen presenting cells like B lymphocytes (Raposo et al, 1996) or dendritic cells (DCs) (Zitvogel et al, 1998; Thery et al, 2002; Segura et al, 2005) provided the first evidence that secretion of EVs could play a communication role between cells (Thery et al, 2009; Robbins & Morelli, 2014). DCs play a fundamental role in the initiation of immune responses (Merad et al, 2013) by presenting exogenous antigens to CD8+ T lymphocytes as peptides loaded onto MHC class I molecules (a process called cross‐presentation), or to CD4+ T lymphocytes as peptides bound to MHC class II molecules. Consistently, human DC‐derived sEVs have been shown to bear functional MHC class I and class II molecules that can be loaded with specific peptides to activate cognate T cells (Hsu et al, 2003; Admyre et al, 2006), demonstrating the capacity of DCs to spread immune responses through sEVs. These findings motivated the use of DC‐derived sEVs to boost anti‐tumour immune responses in cancer clinical trials (Escudier et al, 2005; Morse et al, 2005; Besse et al, 2016), although with limited clinical effects.
We have recently demonstrated that human DCs secrete simultaneously abundant and heterogeneous EVs of different sizes and from different intracellular origins, which all bear MHC class I and class II molecules (Kowal et al, 2016). These other types of EVs may thus represent alternative immunotherapy tools. However, side‐by‐side comparison of the immune effects of sEVs and other EVs has never been comprehensively performed. Therefore, it is unclear whether exosomes, despite the numerous studies describing their functions, are the best or only EVs with interesting therapeutic potential.
Here, we have isolated different subtypes of EVs simultaneously released by live human primary DCs and characterized their effects on primary CD4+ T lymphocytes in vitro. Our results show that both lEVs and sEVs secreted by immature DCs efficiently induce activation of T cells, but these activated T cells release different cytokines. Particularly, lEVs promote the secretion of Th2 cytokines like IL‐4, IL‐5 and IL‐13 while sEVs induce the secretion of the Th1 cytokine IFN‐γ. sEVs bearing the IFN‐γ‐inducing activity were recovered in intermediate floating fractions of a density gradient, with the most efficient sEVs in a subfraction not enriched in bona fide exosomes (Kowal et al, 2016). In addition, the most efficient IL‐13‐inducing EVs were recovered in floating fractions of the gradient displaying a higher density than sEVs. We could assign part of the activity of EVs to some transmembrane receptors involved in T‐cell activation: CD40 and DC‐SIGN for sEVs, and CD80 for lEVs. Upon DC maturation; however, all types of EVs became equally efficient to induce IFN‐γ secretion by Th lymphocytes, that is the kind of immune response expected to be efficient in anti‐tumour immunotherapy.
Results
Characterization of different types of EVs secreted by human DCs
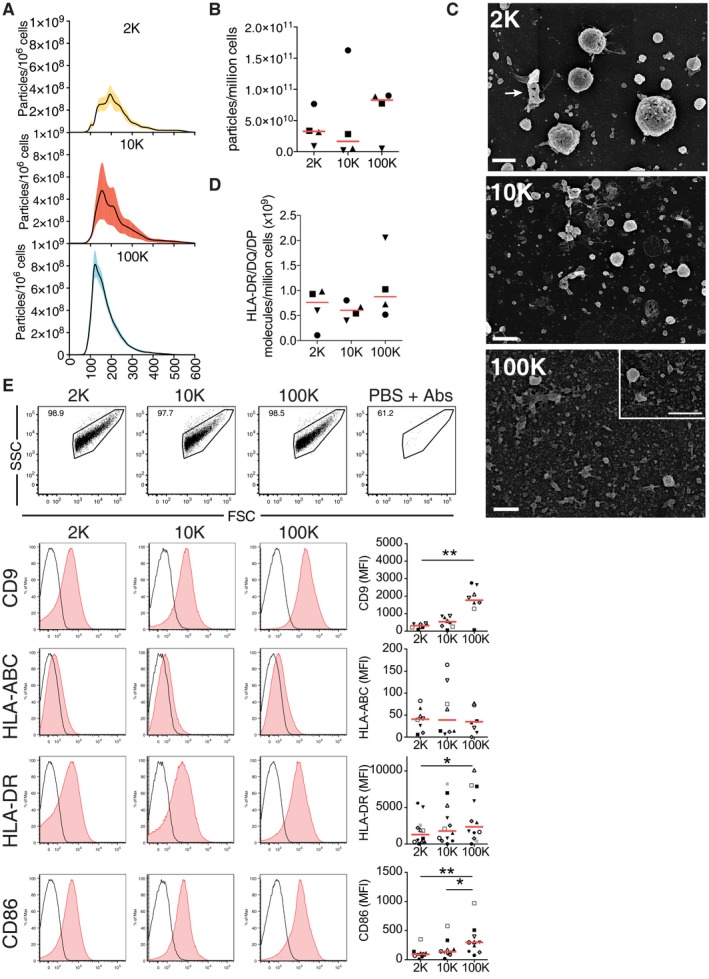
We have shown that human primary DCs differentiated in vitro from monocytes secrete a heterogeneous range of EVs, which can be in part separated by their pelleting properties (Thery et al, 2006; Kowal et al, 2016). In this previous work, we extensively characterized by transmission electron microscopy (TEM) and Western blot (WB) the lEVs recovered by low speed centrifugation (2,000 × g = 2K), intermediate speed (10,000 × g = 10K) centrifugation and sEVs pelleted by high‐speed (100,000 × g = 100K) ultracentrifugation. Our previous measuring of EV sizes on TEM pictures had shown that 95% of EVs were smaller than 200 nm in the 100K pellet, 85% in the 10K pellet and less than 50% in the 2K pellet (Kowal et al, 2016). Nanoparticle Tracking Analysis (NTA, Fig 1A) showed similar distributions, with 74.4% particles smaller than 200 nm in the 100K pellet (median size of 155.6 nm), less than 45.1% in the 2K pellet, which contained vesicles as big as 500 nm (median size of 199.7 nm), and a combination of both in the intermediate 10K pellet (median size 168.2 nm). The 2K pellet contained generally lower numbers of particles than the 100K, as quantified by NTA (Fig 1B), but more total proteins (Kowal et al, 2016), suggesting its enrichment in large, protein‐containing EVs. This was confirmed by scanning electron microscopy (SEM) imaging of the pellets loaded on slides coated with poly‐l‐lysine, which ensures efficient capture of all vesicles (Fig 1C). We observed numerous spherically shaped vesicles larger than 500 nm in diameter in the 2K pellet: these lEVs are clearly distinct from irregular‐shape debris (arrow, Fig 1C, 2K panel). The 2K pellet also contains EVs of about 200–500 nm in diameter, and a few even smaller EVs. While analysing the morphology of the whole DC by SEM, we observed various protrusions on the plasma membrane, including spherical structures of 100–250 nm, and larger tubules and ruffles that we speculate can lead to the formation of EVs (Fig EV1A). The 100K pellet contains a vast majority of the smallest EVs (< 100 nm, Fig 1C, 100K panel, enlargement), but also some rare intermediate size EVs. The 10K pellet contains a mixture of all these EVs. SEM therefore represents a complementary imaging technique providing a comprehensive view of the size and shape of EVs, and demonstrating the actual vesicular nature of the lEVs.
Figure 1. Characterization of DC‐derived EVs recovered in successive differential ultracentrifugation pellets.
-
A, BThe different pellets of EVs isolated from DCs (2K = 2,000 × g; 10K = 10,000 × g; 100K = 100,000 × g) were analysed by Nanoparticle Tracking Analysis (NTA). Each line corresponds to the mean ± SEM of four acquired donor samples for each pellet. A wide range of particle size is observed in the 2K and 10K pellet, whereas the size range of particles in the 100K pellets is more restricted (A). The number of purified particles secreted per million cells tracked by NTA for each pellet (2K, 10K and 100K) was calculated using the NTA3.2 software (B) (n = 4, one symbol per donor). Red line indicates median.
-
CThe 2K, 10K and 100K pellets were loaded on poly‐lysine‐coated slides, fixed and analysed by scanning EM (SEM). Inset in the 100K pellet image shows another field with higher magnification. Arrow in 2K panel indicates a cell debris. Scale bars = 1 μm.
-
DThe whole number of MHC II molecules on the EVs was quantified coupling the different pellets to beads and incubating them with mouse antibodies against HLA‐DR/DQ/DP. The absolute number of MHC II molecules per pellet was determined using calibration beads (see Materials and Methods for assay description) (n = 4, one symbol per donor). Red line indicates median.
-
EThe successive pellets were analysed by flow cytometry, to measure the overall level of surface expression of various markers. EVs were detected in a FSC/SSC gate, which did not contain any events when dilutions of antibodies in filtered PBS in the absence of EV pellets were analysed (upper panel). EVs were stained for the CD9 tetraspanin and immune molecules (HLA‐ABC, HLA‐DR and CD86) (red histogram). Isotype antibodies were used as control (black line). The specific mean fluorescence intensity (MFI with antibody–MFI with isotype control) was calculated as value of global molecule exposure on the bulk EV pellets. Representative histograms are shown in the lower left panel, and the quantification of the specific MFI are shown in the lower right panel (n = 8–12, one symbol per donor). Red line indicates median. *P < 0.05; **P < 0.01; (Friedman test).
Figure EV1. SEM of immature DCs and whole‐mount TEM analysis of each pellet.
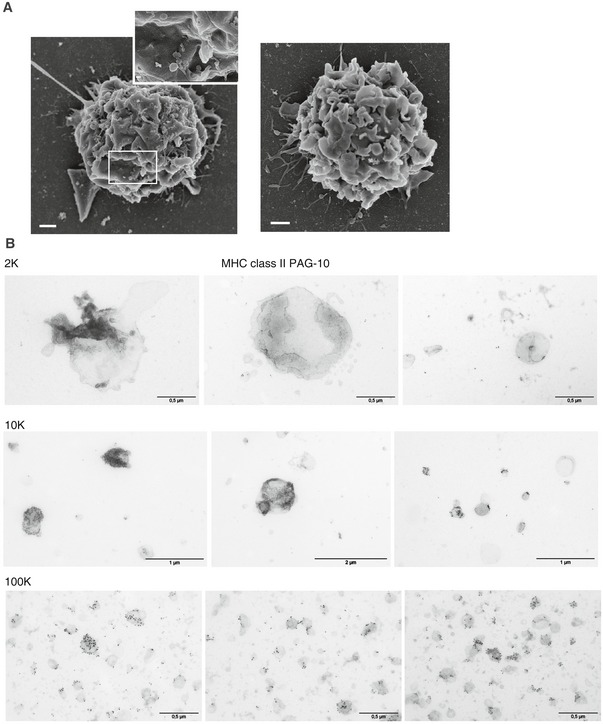
- DCs were loaded on poly‐lysine‐coated slides, fixed and analysed by SEM. Two DCs are shown as representative images. Scale bars = 1 μm for the whole cell and 100 nm for the inset.
- Whole‐mount EVs were stained for MHC II (PAG10) and analysed by TEM. Images are shown for the different pellets.
By WB and quantitative proteomic analyses, we had observed that the tetraspanin CD9 was present in all pellets but enriched in the sEVs, whereas equivalent levels of the immunologically relevant MHC class I and class II molecules were recovered in all EV pellets (Kowal et al, 2016). Thus, we next analysed whether the relevant immune molecules (i.e. MHC molecules) were equally displayed at the surface of all EVs, information that could not be retrieved from the analysis by WB of intracellular epitopes in lysates of EVs. We thereby used a FACS‐based calibration assay previously developed (Viaud et al, 2011) to quantify the absolute number of MHC class II (HLA‐DR, DQ and DP) molecules and observed not significantly different levels in all pellets (mean ± SEM: 2K = 7.6 × 1008 ± 1.9 × 1008, 10K = 6.7 × 1008 ± 9.5 × 1007 and 100K = 8.8 × 1008 ± 3.2 × 1008 molecules secreted on EVs per million cells) (Fig 1D). We have also analysed the presence of CD9, MHC class I (HLA‐A/B/C), MHC class II (HLA‐DR) and the co‐stimulatory molecule CD86 by direct flow cytometry with antibodies recognizing extracellular domains from each protein (Fig 1E). Forward scatter (FSC) and side scatter (SSC) parameters of the flow cytometer were set to log scale and low threshold, to allow detection of antibody‐dependent fluorescent signal in EV preparations above the background signal given by antibodies in the absence of EVs (Fig 1E, upper right panel), and by isotype control antibodies on EVs. Note that the flow cytometer used here cannot differentiate FSC/SSC properties of EVs or polystyrene beads of 100 nm, 400 nm or 1 μm in diameter (data not shown). Thus, it provides information about EVs as a bulk population and not single EVs. By using this assay, we could corroborate the presence of all the molecules in all EV pellets, although a clear enrichment of CD9 and CD86 was detected in sEVs. Besides, all different vesicles showed variable contents of MHC class I and II, with higher levels of HLA‐DR on the 100K compared to the 2K pellet. Finally, by TEM, we observed MHC class II staining on the surface of several (but not all) EVs in the 10K and 100K pellet, and a particular pattern of MHC class II distribution on lEVs (> 500 nm), which often displayed high content of MHC class II on membrane patches (Fig EV1B).
CD4+ T cells are activated by all DC‐derived EV subtypes
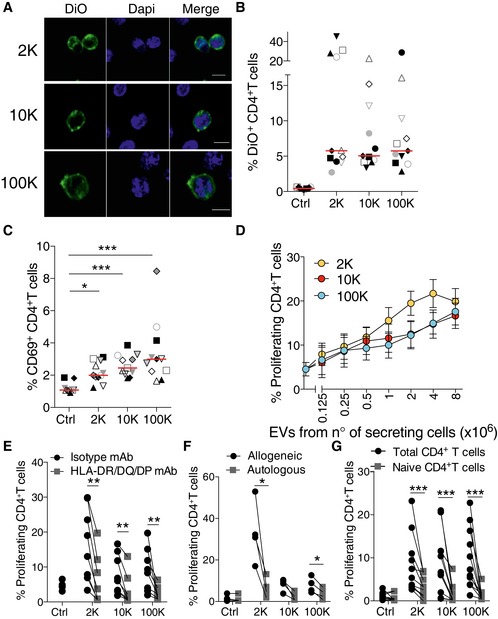
Since the 2K, 10K and 100K pellets all contain MHC class II, we decided to analyse whether the EVs could exert any immunomodulatory effect on CD4+ T cells. Given the biochemical complexity of EVs (proteins, lipids, nucleic acids), it is impossible to quantify their concentration in a rigorous manner in terms of molarity, as is done for single molecules. Furthermore, we observed that while the 2K pellet contained, in general, more proteins and fewer particles than the other two pellets, the 100K pellet contained, conversely, more particles and less proteins (Fig 1B and Kowal et al, 2016). Therefore, choosing either protein amount or particle number to equalize the amount of EVs used in functional assays would have biased the results in favour of either the 100K or the 2K pellet, respectively, leading to, potentially, opposite conclusions. We thus chose to expose T cells to EV pellets recovered from a given number of secreting DCs, that is a given volume of conditioned medium. Table 1 indicates the average particle number and protein content of each pellet used in such experiments. We first evaluated how the different EVs interacted with T cells, as compared to DC‐derived sEVs, which are known to be recruited by these cells (Nolte‐’t Hoen et al, 2009). We incubated freshly isolated total CD4+ T cells for 18 h with the different EVs labelled with DiO, and analysed them by confocal microscopy (Fig 2A) and flow cytometry (Figs 2B and EV2). Some CD4+ T cells became DiO+ upon treatment with each of the different EV pellets, with similar percentages of DiO+ cells observed in all conditions (Fig 2B), suggesting that CD4+ T cells bind all of the EV subtypes similarly. We did not observe any DiO‐labelled CD4+ T cell when we used as control the DiO‐labelled pellets of non‐conditioned medium (Fig EV2). We then assessed T‐cell activation by measuring the up‐regulation of the early T‐cell activation marker CD69. The three pellets induced surface expression of CD69 on allogeneic CD4+ T cells when compared to untreated cells (control), and the 100K was slightly more efficient than the 2K and the 10K (Fig 2C). Upon prolonged culture, CD4+ T cells proliferated in the presence of all EV pellets in a dose‐dependent manner and at similar rates (Fig 2D). At the highest dose (EVs from 8 × 106 DCs), 2K, 10K and 100K pellets induced similar proliferations of CD4+ T cells, 19.8% (±2.9%), 16.6% (±2.8%) and 17.5% (±3.0%), respectively. T‐cell proliferation was due to specific recognition of allogeneic MHC class II molecules in all EV subtypes, as it was decreased in the presence of antibodies against HLA‐DP/DR/DQ (Fig 2E), and not observed when T cells were exposed to autologous EVs secreted by DCs of the same donor (Fig 2F). Note that to perform the latter experiment, we had to keep both autologous and allogeneic CD4+ T cells frozen during the 6 days of DC culture required for their differentiation and subsequent EV isolation, which resulted in a weaker responsiveness of T cells to the 10K and 100K pellets (compare Fig 2F and E). Finally, activation by allogeneic EVs was only observed on total CD4+ T cells: none of the EV pellets were able to induce proliferation of isolated naïve T cells (Fig 2G), as it was previously shown for mouse DC‐derived sEVs (Thery et al, 2002). These observations indicate that all types of EVs, not only sEVs, induce direct activation of allogeneic CD4+ T cells in an MHC‐dependent manner, and that presence of activated/memory cells is necessary for an efficient response.
Table 1.
Calculation of the average number of particles and total proteins present in the EV pellets from 1, 2, 4 and 8 million DCs
| Million of secreting cells | Total number of particles | Total protein (μg) | ||||
|---|---|---|---|---|---|---|
| 2K | 10K | 100K | 2K | 10K | 100K | |
| 8 | 4.11 × 1008 ± 2.27 × 1007 | 5.18 × 1008 ± 3.83 × 1007 | 6.82 × 1008 ± 4.97 × 1007 | 23.2 ± 2.4 | 13.6 ± 2.4 | 8.8 ± 1.6 |
| 4 | 2.06 × 1008 ± 1.14 × 1007 | 2.59 × 1008 ± 1.92 × 1007 | 3.41 × 1008 ± 2.48 × 1007 | 11.6 ± 1.2 | 6.8 ± 1.2 | 4.4 ± 0.8 |
| 2 | 1.03 × 1008 ± 5.69 × 1006 | 1.29 × 1008 ± 9.58 × 1006 | 1.70 × 1008 ± 1.24 × 1007 | 5.8 ± 0.6 | 3.4 ± 0.6 | 2.2 ± 0.4 |
| 1 | 5.14 × 1007 ± 2.84 × 1006 | 6.47 × 1007 ± 4.79 × 1006 | 8.52 × 1007 ± 6.21 × 1006 | 2.9 ± 0.3 | 1.7 ± 0.3 | 1.1 ± 0.2 |
Figure 2. All EV subtypes from DCs interact with and activate T cells.
-
ADiO‐labelled DC‐derived EVs were cultured for 18 h with allogeneic primary total CD4+ T cells and analysed by confocal microscopy. Nuclei are stained with DAPI (DiO = green; DAPI = blue) (scale bars = 5 μm).
-
BThe percentage of DiO+ T cells was analysed by flow cytometry. Total CD4+ T cells incubated for 18 h with DiO‐labelled EVs coming from 4 × 106 DCs or cultured alone as control (n = 11, one symbol per donor). Red line indicates median.
-
CCD69 expression on CD4+ T cells cultured for 18 h with the subtypes of EVs released by DCs. The percentage of CD69+ CD4+ T cells for each pellet treatment is shown (n = 11 donors, one symbol per donor). Red line indicates median. *P < 0.05; ***P < 0.001 (Friedman test).
-
DTotal CD4+ T‐cell proliferation was evaluated after 6 days of culture with different amounts of DC‐derived EVs. Proliferation was calculated by dilution of Cell Trace Violet dye (n = 13 donors, mean ± SEM is shown).
-
EProliferation of total CD4+ T cells after culture for 6 days with EVs from 8 × 106 DCs in the presence of blocking antibodies against HLA‐DR/DQ/DP or control isotype antibodies (n = 8 donors).
-
F, GEVs from 8 × 106 DCs were cultured with autologous or allogeneic total CD4+ T cells (F) or with total versus naïve CD4+ T (G) for 6 days to evaluate T‐cell proliferation (n = 4 donors (F) and n = 10 donors (G)).
Figure EV2. Flow Cytometry analysis of fluorescently labelled‐EV transfer to CD4+ T cells.
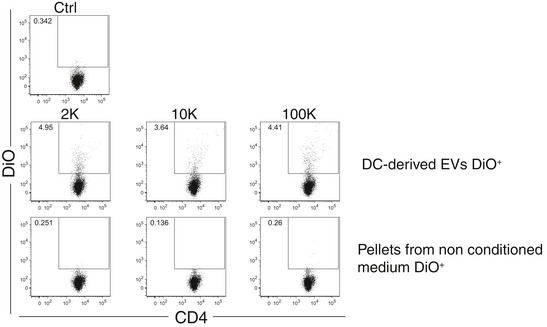
EV pellets (2K, 10K and 100K) coming from conditioned medium of DCs cultures or the pellets from non‐conditioned medium (depleted of serum EVs) were labelled with DiO lipophilic dye, washed in PBS and re‐ultracentrifuged three times and cultured for 18 h with allogeneic primary total CD4+ T cells. Transfer of the dye was analysed by flow cytometry. Dot‐plot graphs show the percentage of CD4+ DiO+ cells for both EV‐treated and control‐treated CD4+ T cells.
Distinct DC‐derived EVs promote the release of a specific cytokine signature by CD4+ T cells
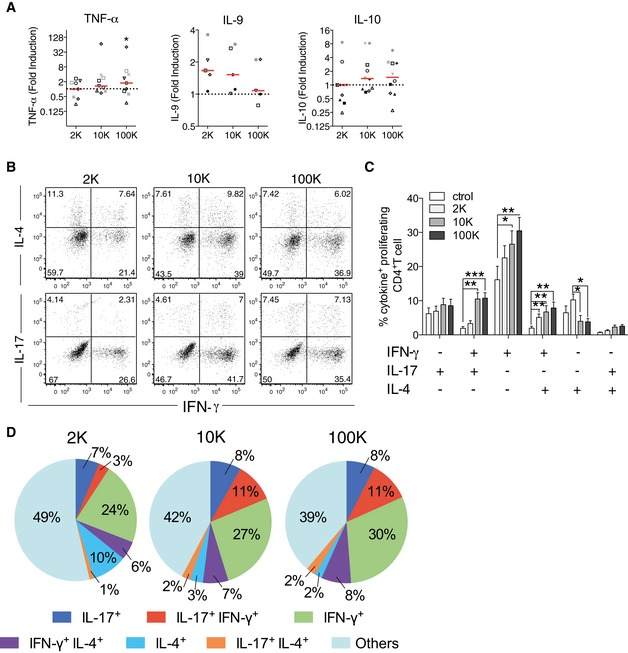
Since activation of CD4+ T cells can result in the release of different cytokines, which reflect the polarization state of the T cells, we examined CD4+ T‐cell cytokine profile after 6 days of culture with allogeneic EVs. IFN‐γ is the main cytokine secreted by Th1 effector cells involved in cell‐mediated immunity, and IL‐13, as well as IL‐5 and IL‐4, is preferentially produced by differentiated Th2 cells that regulate antibody‐mediated humoral immune responses (Chaudhry & Rudensky, 2013). T cells activated by the 2K pellet from 1 to 8 × 106 DCs secreted higher levels of IL‐13 as compared to untreated cells, but little or no IFN‐γ nor IL‐17 (Th17‐secreted cytokine) (Fig 3A and B). By contrast, T cells activated by the 10K and 100K pellet secreted IFN‐γ and IL‐17, but only low amounts of IL‐13 (Fig 3A and B), even at the highest dose of EV used. The highest dose of EVs (i.e. secreted by 8 × 106 DCs) was used to analyse secretion of other cytokines by T cells exposed to it: the other Th2 cytokines (IL‐4 and IL‐5) were also significantly secreted by T cells exposed to the 2K pellet, whereas the 10K and 100K pellets induced secretion of IL‐6 (Fig 3B) and of TNF‐α (100K pellet, Fig EV3A). The 10K and 100K pellets were also able to promote production of IL‐5 and IL‐13 in some cases, but not in a significantly different manner from spontaneous secretion by untreated CD4+ T cells. We also measured other cytokines in the supernatants, such as IL‐9 (Th9‐secreted cytokine) and IL‐10 (secreted by Treg) (Chaudhry & Rudensky, 2013), but we did not observe any significant differences compared to their basal levels secreted by non‐activated T cells (Fig EV3A). IFN‐γ and IL‐13 were the two cytokines produced in the greatest amounts in our cultures upon EV treatment (ranges of 0.2–2 ng/ml). We thus used the ratio of secreted IFN‐γ versus IL‐13 as an indication of the relative proportions of Th1 versus Th2 lymphocytes. The median IFN‐γ/IL‐13 ratio was below 1 (0.45) for the 2K‐stimulated CD4+ T cells, whereas it was above 1 (3.1 and 3.8) for the 10K‐ and 100K‐stimulated T cells, respectively (Fig 3C). This suggests that lEVs can tip the balance towards Th2, whereas medium and sEVs tip it towards Th1 lymphocytes. To study cytokine production by individual CD4+ T cells that proliferated upon EV stimulation (6 days culture), we performed intracellular staining for IFN‐γ, IL‐4 and IL‐17 after 4 h stimulation with PMA and ionomycin. The percentage of different cytokine‐producing subpopulations (Fig EV3B and C) and their relative proportions (Fig EV3D) were calculated for the proliferating CD4+ T cells. We observed that in all activation conditions, cytokine‐expressing cells were in majority single IFN‐γ producers (Fig EV3C). Cells activated by the 2K pellet, however, were more frequently IL‐4 single‐positive (10.2%) than cells activated by the 10K and 100K pellets (2–4%, Fig EV3C and D), whereas the latter were more frequently multifunctional, especially by producing simultaneously IFN‐γ and IL‐17A (10.7%), than cells activated by the 2K pellet (3.2%) (Fig EV3C and D). In addition, among the proliferating CD4+ T cells, the 10K and 100K pellets were more efficient at inducing cytokine‐producing cells (58–61%) than the 2K pellets (51%) (Fig EV3D). Therefore, even if all types of EVs efficiently induce CD4+ T‐cell activation, the resulting functionality of Th cells is different.
Figure 3. CD4+ T cells release different cytokines upon stimulation by DC‐derived EV subtypes.
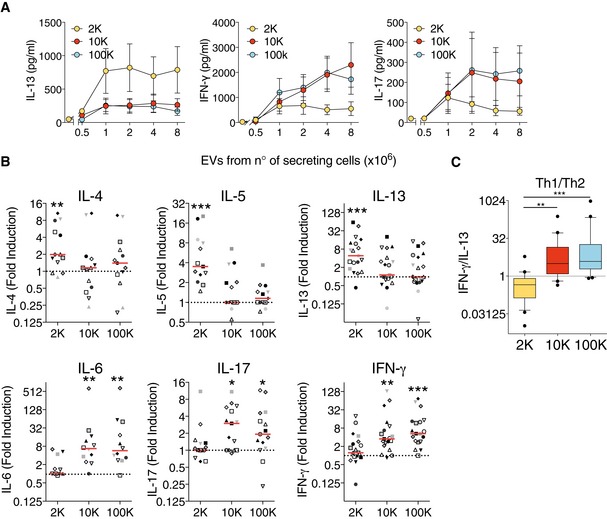
- The concentration of IL‐13, IFN‐γ and IL‐17 was measured by cytometric bead array (CBA) in supernatants after 6 days of culture of total CD4+ T cells with EVs released by 1 to 8 × 106 DCs (n = 9, mean ± SEM is shown).
- The concentration of IL‐4, IL‐5, IL‐6, IL‐13, IFN‐γ and IL‐17 was measured by CBA in supernatants after 6 days of culture of total CD4+ T cells with the highest dose of EVs (the released by 8 × 106 DCs). Each individual DC‐EV donor was used on T cells from two different donors, providing two biological replicates. Results are expressed as fold induction compared to untreated CD4+ T cells (n = 11, one symbol per individual DC‐EV:T‐cell combination). Red line indicates median. The range of cytokines secreted by untreated CD4+ T cells was 8.3–15 pg/ml of IL‐4; 18–109.1 pg/ml of IL‐5; 12.4–961.1 pg/ml of IL‐13; 18–115.2 pg/ml of IL‐6; 23–894.4 pg/ml of IFN‐γ; 20–152.5 pg/ml of IL17.
- Th1 to Th2 ratio was calculated by dividing the absolute amount of IFN‐γ (pg/ml) to the absolute amount of IL‐13 (pg/ml) secreted by a given T‐cell donor exposed to the different pellets obtained from an individual DC donor (n = 20 individual DC‐EV:T‐cell combination). The ratio is represented as a box plot (25th to 75th percentiles, the line represents the median; whiskers, min to max, points represent outliers as calculated by Tukey's test).
Figure EV3. Release of IL‐9, IL‐10 and TNF‐α and intracellular production of IL‐4, IFN‐γ and IL‐17 in CD4+ T cells upon DC‐derived EV subtypes stimulation.
-
AIL‐9, IL‐10 and TNF‐α cytokine concentration was measured in culture supernatant after 6 days of culture of total CD4+ T cell with DC‐derived EVs by cytometric bead array (n = 11, one symbol per donor). Red line indicates median.
-
B–DCultured T cells were re‐stimulated for 4 h with PMA and ionomycin in the presence of brefeldin A and permeabilized and stained for IFN‐γ, IL‐4 and IL‐17A. Proliferating cells were gated and evaluated for their single or double intracellular cytokine staining. Representative plots (B), quantification of the percentage (C) and relative proportions (D) of single or double positive proliferating CD4+ T cells for each cytokine (IL‐4+, IL‐17+, IFN‐γ+, IFN‐γ+IL‐17+, IL‐4+IL‐17+ and IFN‐γ+IL‐4+) are shown (four independent experiments, n = 12 donors). In (C), the mean of 12 donors ± SEM is shown. *P < 0.05; **P < 0.01; ***P < 0.001 (Friedman test).
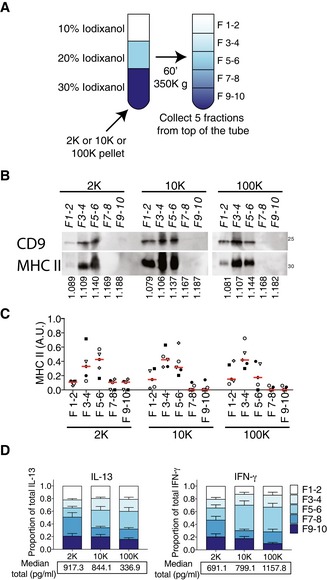
To determine whether each EV pellet contained functionally different subpopulations of vesicles, we used floatation into iodixanol gradients, which allowed us previously to separate two distinct EV populations from 10K and 100K pellets (Kowal et al, 2016). Here, we collected 5 × 1 ml fractions (instead of 10 × 0.5 ml in Kowal et al, 2016), which were numbered [1–2] to [9–10] from top to bottom (Fig 4A), to match Kowal's numbering of fractions. We observed that the majority of the MHC II‐ and CD9‐containing vesicles floated in the first three fractions ([1–2, density 1.08–1.09], [3–4, density 1.10–1.11] and [5–6, density 1.13–1.14]) (Fig 4B and C), consistent with our previous observation. However, for the 2K pellet of some donors, there were also MHC II‐containing vesicles in the dense fractions [7–8, density 1.17] and [9–10, density 1.18–1.19] (see quantification of individual Western blots: Fig 4C). We then analysed the effect of all fractions on CD4+ T cells and found that the IFN‐γ‐inducing activity was recovered in several fractions (Fig 4D, right panel) with a predominance in the fraction [5–6]. Therefore, all sEVs are able to polarize Th1 cells, but the exosome‐enriched fraction (fraction 3 in Kowal et al, 2016) is less efficient than the exosome‐depleted one (fraction 5 in Kowal et al, 2016). Fractions [5–6] of the 10K and 100K pellets also showed an ability to promote IL‐13‐secretion. Strikingly, the IL‐13 inducing activity of the 2K was significantly recovered in the first and lightest fraction [1–2] and in the denser fractions [7–8] and [9–10], with a predominance in the dense floating fraction [7–8] (Fig 4D, left panel). Therefore, the IL‐13‐inducing activity of the 2K pellet is present throughout the gradient, but the strongest in floating fractions that contain low levels of MHC II, suggesting that other EVs (with a low content of CD9 and MHC II) participate in the promotion of Th2 cells.
Figure 4. Analysis of EV function after floatation on iodixanol gradient.
-
A–CPellets obtained after 2K, 10K and 100K centrifugations from 80–120 × 106 cells were floated into an overlayed iodixanol gradient (A). Five fractions were collected and analysed by WB (representative of five experiments) (B), showing the floatation of MHC II‐ and CD9‐enriched EVs in fraction F3–4 and fraction F5–6. Densities of recovered fractions, as measured by refractometry, are shown below the WB figure (mean density of five independent gradients). Quantification of the distribution of MHC II relative abundance in all fractions of each centrifugation pellet analysed by WB of five independent experiments (C). For each pellet, arbitrary units (AU) = (SIfraction)/sum(SIF1–2 + SIF3–4 + SIF5–6 + SIF7–8 + SIF9–10) where SI = signal intensity (i.e. ratio of signal intensity in the given pellet to the total secreted protein) (n = 5, one symbol per donor). Red line indicates median.
-
DIL‐13 and IFN‐γ secretion was measured in supernatants after 6 days of total CD4+ T‐cell culture with the different fractions of the iodixanol gradients of the 2K, 10K and 100K pellets. The graph indicates the relative contribution of each fraction to the total cytokine secretion induced by each pellet. The relative contribution for each donor was calculated as CCfraction/sum(CCF1–2 + CCF3–4 + CCF5–6 + CCF7–8 + CCF9–10) for each pellet, where CC is cytokine concentration. Mean + SEM is shown. Below each graph, the sum of the cytokine concentration in all the fractions for each pellet is shown (median of 14 individual DC‐EV:T‐cell combinations).
Source data are available online for this figure.
Since all these EVs are simultaneously released by the same DC population, we next asked whether their distinct CD4+ T‐cell polarization abilities were determined by the secreting DCs themselves. A fixed number of CD4+ T cells were cultured with increasing amounts of DCs, side‐by‐side with the highest amount of vesicles used in Figs 2 and 3. We observed an equivalent proliferation rate for T cells cultured with 125–250 intact DCs as for the EVs of 8 × 106 DCs (Fig 5A). The CD4+ T cells stimulated by this amount of DCs (125 and 250) secreted both IL‐13 and IFN‐γ at similar levels as observed with the EV pellets (Fig 5B). When we calculated the Th1/Th2 ratio, we observed that intact DCs promote both Th1 and Th2 responses, with median values of the IFN‐γ/IL‐13 ratio not significantly different from 1, which is in contrast with the selective Th1 induction by both 10K and 100K pellets (ratio > 1) (Fig 5C). Finally, to determine whether DCs could activate T cells at a distance by releasing EVs, without the “artefactual” step of EV centrifugation and concentration, we co‐cultured DCs and T cells separated through either a 0.4‐μm or a 1‐μm porous membrane (Fig 5D). At a 1:1 DC:T‐cell ratio, we observed that T cells produced both IL‐13 and IFN‐γ at higher levels than T cells cultured alone (Fig 5D). Interestingly, at the 1:8 ratio, no IL‐13‐production was observed in the co‐culture through the 0.4‐μm filter, which retains EVs larger than 400 nm. Altogether, these results confirm that the DCs spread their capacity to induce Th1 and Th2 responses into distinct subtypes of EVs, carrying separately each activity.
Figure 5. Comparison of the effect of DC‐derived EVs versus the whole DC on T cells.
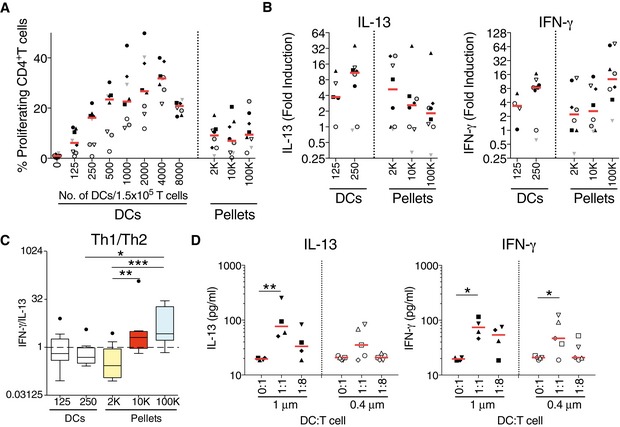
- Total CD4+ T cells were isolated, stained with Cell Trace Violet dye and cultured for 6 days with increasing numbers of in vitro differentiated DCs or with EVs purified from these DCs (2K, 10K and 100K). Proliferation was calculated by dilution of the fluorescent dye on CD3+CD4+ cells (n = 8, one symbol per individual DC‐EV:T‐cell combination). Red line indicates median.
- The concentration of IL‐13 and IFN‐γ was measured in culture supernatant after 6 days of culture of total CD4+ T cell with 125 and 250 whole DCs or with their secreted EVs. Results are expressed as fold induction compared to untreated CD4+ T cells (n = 8, one symbol per individual DC‐EV:T‐cell combination). Red line indicates median. The range of cytokines secreted by untreated CD4+ T cells was 20.8–402.7 pg/ml of IL‐13; 24.8–143.6 pg/ml of IFN‐γ.
- Th1 to Th2 ratio was calculated by dividing the concentration of IFN‐γ to the concentration of IL‐13 for each donor, for the whole DC‐T‐cell co‐culture (125 and 250 DCs) and for the EV‐T‐cell culture (n = 8). Results are shown as a box plot (25th to 75th percentiles, the line represents the median; whiskers, min to max, points represent outliers as calculated by Tukey's test). *P < 0.05; **P < 0.01; ***P < 0.001 (Friedman test).
- DC and T cells were co‐cultured in a Transwell system, separated through either a 0.4‐μm or a 1‐μm porous membrane at a 1:1 or 1:8 DC:T‐cell ratio. After 6 days, the supernatants were analysed for the presence of IL‐13 and IFN‐γ by cytometric bead array. Experiments done with 0.4‐μm or with 1‐μm pore are completely independent (not paired donors) (n = 4–5, one symbol per individual DC:T‐cell combination). Red line indicates median. *P < 0.05, **P < 0.01; one‐way ANOVA followed by Bonferroni's multiple comparison test.
The molecular composition of the EV surface is involved in the induction of T‐cell responses
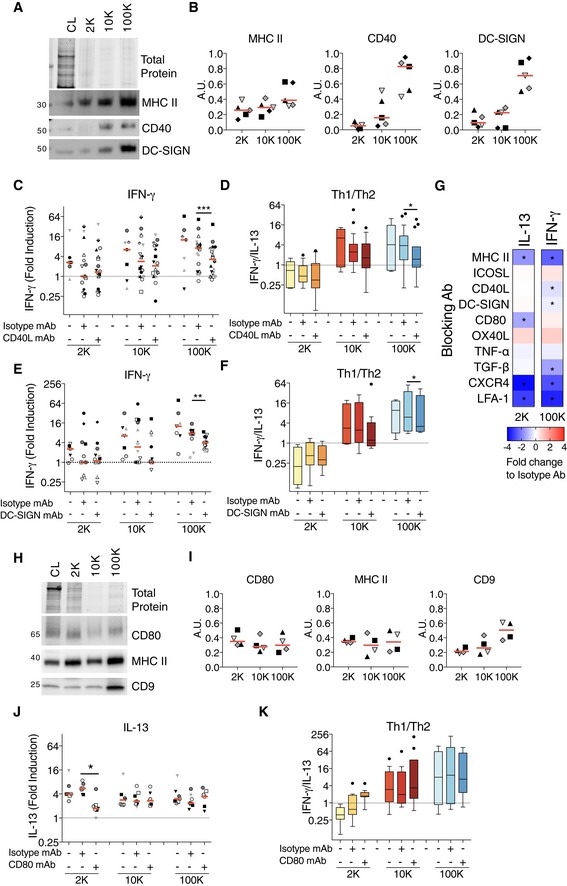
We next searched for molecular determinants that could explain differential functionalities of the 100K and 2K pellets. We thus analysed our extensive proteomic comparison of the medium (10K) and small DC‐derived EVs (100K) (Kowal et al, 2016), to identify differentially expressed proteins with immune functions. In this proteomic approach, we had not analysed the 2K pellets, but since proteins enriched in the 10K fractions were always even more abundant in the 2K pellet, as measured by WB, we used the proteomic analysis of 10K as an indication of the trend we could expect in the 2K pellet. We observed enrichments of some known immune regulators in the 100K fractions in our proteomic data: CD40 (Uniprot: TNR5, gene: TNFRSF5) was significantly detected with 3–5 peptides in each biological replicate of the 100K pellet and barely detected in the 10K (< 3 peptides), DC‐SIGN (Uniprot and gene: CD209), an adhesion molecule with immunomodulatory properties (den Dunnen et al, 2009), was very abundant in both 10K and 100K pellets (around 20 different peptides), but was significantly (P = 0.0006) 2.5 times enriched in the 100K as compared to 10K fractions of our proteomic data. We confirmed enrichment of DC‐SIGN and CD40 in the 100K pellets by WB (Fig 6A and B). We then asked whether these molecules were involved in the response induced by the medium and small EVs. We blocked the interaction between CD40 and CD40L present in T cells using blocking antibodies against CD40L and evaluated T‐cell proliferation (Fig EV4A) and cytokine secretion (Figs 6C and D, and EV4B). Note that the levels of cytokine secretion were decreased in the presence of irrelevant isotype‐control antibodies, although not significantly, as compared to those observed in the absence of antibodies (Figs 6C and E, and EV4B and D). Blocking CD40–CD40L interaction did not significantly affect EV‐induced proliferation of CD4+ T cells (Fig EV4A), nor IL‐13 secretion by CD4+ T cells exposed to the 2K pellet (Fig EV4B, 2K), nor IFN‐γ secretion induced by the 10K pellet (Fig 6C, 10K). By contrast, in cells exposed to the 100K pellet, IFN‐γ secretion was significantly decreased (P = 0.0004) by anti‐CD40L (Fig 6C, 100K). Moreover, in these cells, the ratio between IFN‐γ and IL‐13 levels was also decreased (Fig 6D, 100K). Similarly, blocking DC‐SIGN using an antibody in the EV‐CD4+ T‐cell cultures also resulted in minor, but significant decrease of 100K pellet‐induced IFN‐γ secretion (P = 0.0024), without altering T‐cell proliferation or IL‐13 secretion (Figs 6E and F, and EV4C and D). Again, the Th1 to Th2 ratio was also decreased upon blocking of DC‐SIGN (Fig 6F). Thus, CD40 and DC‐SIGN expression on sEVs both partially explain their ability to favour Th1 polarized CD4+ T cells.
Figure 6. Blockade of CD40–CD40L interaction or of DC‐SIGN decreases the response induced by sEVs, whereas blockade of CD80 decreases the response induced by lEVs.
-
A, BThe successive pellets (2K, 10K, 100K) of EVs isolated from DCs were analysed by WB side‐by‐side with the lysate of producing cells (CL, 0.4 × 106 cells), using antibodies to MHC II, CD40 and DC‐SIGN. Equivalent volume of each pellet was loaded on the gels (10 × 106 of secreting cells). Representative images (A) and quantifications (B) are shown (n = 5 donors, one symbol per donor). Red line indicates median.
-
C, DDC‐derived EVs (from 8 × 106 secreting cells) were cultured for 6 days with total CD4+ T cells pre‐incubated with blocking antibodies against CD40L. IFN‐γ secretion for the cells stimulated with the different pellets is shown (C). Th1 to Th2 ratio was calculated by dividing the concentration of IFN‐γ to the concentration of IL‐13 for each DC‐EV:T‐cell donor combination (D) (n = 7–18). The range of cytokines secreted by untreated CD4+ T cells was 20.3–483.1 pg/ml of IL‐13; 30–319.2 pg/ml of IFN‐γ.
-
E, FDC‐derived EVs (from 8 × 106 secreting cells) were pre‐incubated with blocking antibodies against DC‐SIGN for 30 min and then cultured with total CD4+ T cells for 6 days. Secretion of IFN‐γ for CD4+ T cells at the end of the culture with the 2K, 10K and 100K pellets is shown (E). Th1 to Th2 ratio was calculated as previously described for each DC‐EV: T‐cell donor combination (F) (n = 7–14). The range of cytokines secreted by untreated CD4+ T cells was 18–31 pg/ml of IL‐13; 21.5–194.8 pg/ml of IFN‐γ.
-
GMini‐screen using blocking antibodies against several molecules known to be relevant for DC:T‐cell interaction or to T‐cell activation by DCs (MHC II, ICOSL, CD40L, DC‐SIGN, CD80, OX40L, TNF‐α, TGF‐β, CXCR4, LFA‐1). DC‐derived 2K and 100K pellets (from 2 × 106 secreting cells) were pre‐incubated with the indicated blocking antibodies or its corresponding isotype control for 30 min before the addition of the CD4+ T cells. In some cases, CD4+ T cells were incubated with the blocking antibodies prior the addition of the DC‐derived EVs (see Materials and Methods for details). The CD4+ T cells were cultured for 6 days, and the IL‐13 and IFN‐γ were measured in the culture supernatants. Heat map representing the median of the log2‐transformed fold change respect to each isotype control is shown (n = 4–6 individual DC‐EV:T‐cell combinations). Asterisks indicate significant difference of the blocking antibody with respect to the isotype antibody treatment.
-
H, IThe successive pellets (2K, 10K, 100K) of EVs isolated from DCs were analysed by WB side‐by‐side with the lysate of producing cells (CL, 0.4 × 106 cells), using antibodies to MHC II, CD80 and CD9. Equivalent volume of each pellet was loaded on the gels (10 × 106 of secreting cells). Representative images (H) and quantifications (I) are shown (n = 4, one symbol per donor). Red line indicates median.
-
J, KDC‐derived EVs (from 2 × 106 of secreting cells) were pre‐incubated with blocking antibodies against CD80 for 30 min and then cultured with total CD4+ T cells for 6 days. Secretion of IL‐13 at the end of the culture with the 2K, 10K and 100K pellets is shown (J). Th1 to Th2 ratio was calculated as already described for each donor (K) (n = 7). The range of cytokines secreted by untreated CD4+ T cells was 200.3–730.5 pg/ml of IL‐13; 18–151.3 pg/ml of IFN‐γ.
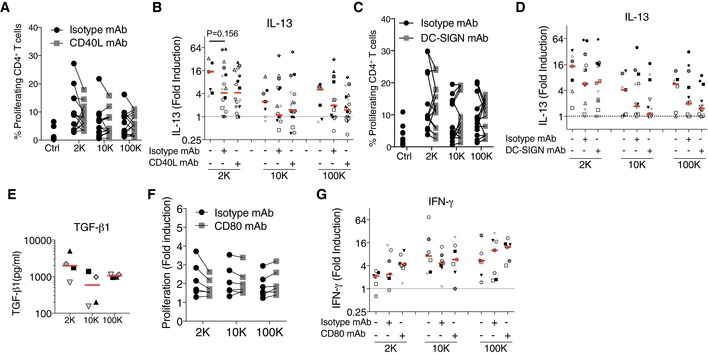
Figure EV4. Blockade of CD40–CD40L interaction or of DC‐SIGN decreases the response induced by sEVs, whereas blockade of CD80 decreases the response induced by lEVs.
-
A, BDC‐derived EVs (from 8 × 106 secreting cells) were cultured with total CD4+ T cells pre‐incubated with blocking antibodies against CD40L. Proliferation (A), secretion of IL‐13 for the 2K, 10K and 100K (B) were evaluated after 6 days (n = 7–18 , each symbol represents a DC‐EV:T cell donor combination). P‐value was calculated using a Wilcoxon signed‐rank test.
-
C, DDC‐derived EVs (from 8 × 106 secreting cells) were pre‐incubated with blocking antibodies against DC‐SIGN for 30 min and then cultured with total CD4+ T cells. Proliferation (C), and secretion of IL‐13 for the 2K, 10K and 100K (D) were evaluated after 6 days (n = 7–14 , each symbol represents a DC‐EV:T cell donor combination).
-
EThe presence of TGF‐β1 in the 2K, 10K and 100K derived from 10 × 106 cells was quantified by a high sensitivity ELISA (n = 4, one symbol per donor).
-
F, GDC‐derived EVs (from 2 × 106 secreting cells) were pre‐incubated with blocking antibodies against CD80 for 30 min and then cultured with total CD4+ T cells. Proliferation of CD4+ T cells was measured as the fold induction of the absolute cell number of each treatment to the absolute number of unstimulated CD4+ T cells at the end of the culture (F). Secretion of IFN‐γ for the CD4+ T cells stimulated with the 2K, 10K and 100K is shown (G) (n = 7 DC‐EV:T cell combinations, one symbol per each).
The same approach was not sufficient to identify molecular mechanisms underlying the specific T‐cell responses promoted by the 2K pellet, since we did not spot, in our previous proteomic analysis, an obvious protein candidate with immune regulatory properties that would be enriched in the 10K compared to the 100K. Thus, we performed a mini‐screen with blocking antibodies against various DC molecules involved in DC–T‐cell interaction or in T‐cell activity. We searched for those who blocked IL‐13 induction by the 2K pellet, without affecting IFN‐γ secretion induced by the 100K pellet, that is the opposite pattern as that observed with anti‐CD40L and DC‐SIGN antibodies (Fig 6G). As compared to cytokine secretion observed in the presence of isotype control antibodies, antibodies blocking ICOSL (ICOSLG), TNF‐α (TNFA) or TGF‐β (TGFB1) did not decrease IL‐13 secretion by the 2K pellet‐exposed T cells, and anti‐TGF‐β decreased IFN‐γ secretion induced by the 100K, even though TGF‐β was slightly more abundant in the 2K than 10K or 100K pellets (Fig EV4E). Anti‐OX40L (TNFSF4) slightly increased secretion of IL‐13, as well as of IFN‐γ. By contrast, antibodies blocking MHC class II, LFA‐1 (ITGAL), CD80 (CD80) or CXCR4 (CXCR4) induced a two‐ to fourfold decrease of IL‐13 secretion in the 2K‐exposed cultures (Fig 6G). However, blocking MHC Class II, LFA‐1 or CXCR4 also decreased more than twofold IFN‐γ secretion induced by the 100K pellets, suggesting, as expected, that these molecules are involved in EV–T‐cell interaction in general. By contrast, when blocking CD80, we observed that IFN‐γ secretion promoted by the 100K was not affected (Fig 6G). The potential involvement of CD80 in the specific activity of 2K pellets was therefore further investigated. Western blotting revealed similar levels of CD80 as well as MHC II in all pellets, whereas other molecules like the tetraspanin CD9 tended to be more abundant in the 100K pellets (Fig 6H and I), as shown before (Fig 1E, Kowal et al, 2016). By quantifying the secretion of the full‐range of cytokine induced by all three pellets, we confirmed that anti‐CD80 blocked IL‐13 induction exclusively in T cells cultured with the 2K (Fig 6J) without affecting significantly their proliferation (Fig EV4F), while IFN‐γ secretion induced by any pellet was unaffected (Fig EV4G). Consequently, the IFN‐γ/IL‐13 ratio became higher than 1, thus switched in favour of Th1 rather than Th2 cells, in cultures exposed to the 2K pellets and CD80‐blocking antibodies (Fig 6K). These results indicate that CD80 is partly responsible for the specific Th2‐promoting activity of lEVs.
All EV subtypes from IFN‐γ‐matured DCs promote secretion of the Th1 cytokine
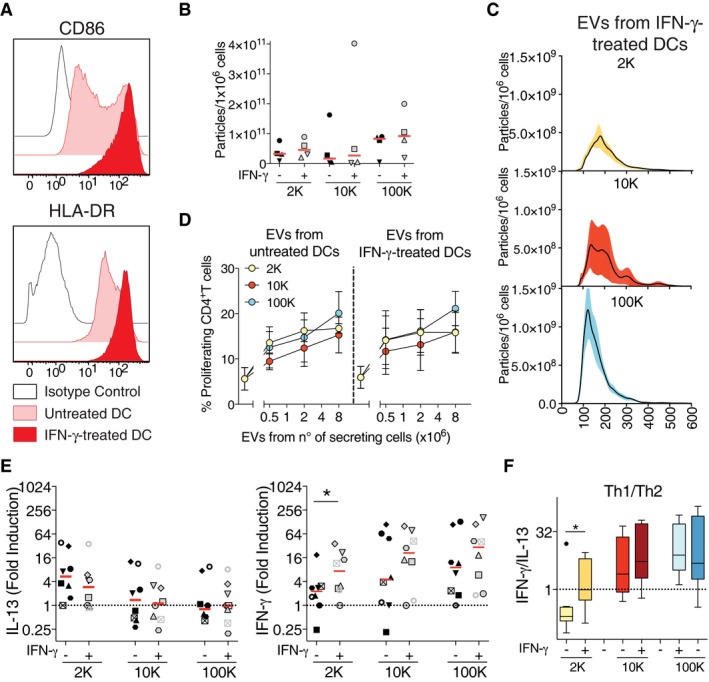
Lastly, we asked whether maturation of DCs alters the characteristic of the different subtypes of EVs and their capacity to promote differential responses. As done for the clinical trial using DC‐derived sEVs in lung cancer patients (Viaud et al, 2011; Besse et al, 2016), we treated DCs with IFN‐γ during the EV‐secretion time. This treatment induced maturation of the DCs, with a majority of cells expressing high levels of MHC class II and CD86 maturation markers, whereas the non‐treated DCs were composed of a mixed population of immature (low MHC class I and CD86) and mature DCs (Fig 7A). We observed by NTA analysis that the IFN‐γ treatment of DCs did not alter the number of particles present in the 2K, 10K and 100K pellets, as compared to those of untreated DCs (Fig 7B). However, we noticed that more sEVs were now present in the 2K pellet, since 65% of these particles were smaller than 200 nm (Fig 7C), as compared to 45% for 2K‐EVs from untreated DCs (Fig 1A). This observation is consistent with SEM images showing that IFN‐γ‐matured DCs displayed a smooth surface, devoid of the plasma membrane ruffles observed in immature DCs (compare Figs EV5A and EV1A). We then treated CD4+ T cells with vesicles from control or from IFN‐γ‐treated DCs and evaluated their proliferation (Fig 7D) and cytokine secretion (Fig 7E). As for immature DCs, the three EV pellets from IFN‐γ‐treated DCs induced similar T‐cell proliferation. Strikingly, however, the differential cytokine secretion induced by the distinct EVs was abolished, with the 2K pellets showing similar levels of IFN‐γ induction as the 10K and 100K pellets (Fig 7E, right panel, and F). Similar levels of IFN‐γ were measured in 2K pellets of immature and IFN‐γ‐treated DCs, ruling out artefactual presence of IFN‐γ coming from the DC treatment (Fig EV5B). Altogether, these results thus show that differential immune activities can be obtained from different subtypes of EVs released by human DCs, and that these activities can be modulated by treating the secreting DCs with a maturation‐inducing stimulus.
Figure 7. IFN‐γ treatment of DCs results in IFN‐γ secretion induced by lEVs.
-
ADCs treated for 24 h with IFN‐γ present a mature phenotype characterized by an up‐regulation of the co‐stimulatory molecule CD86 and of HLA‐DR measured by flow cytometry. One representative histogram profile is shown.
-
B, CThe different pellets of EVs isolated from untreated‐ or IFN‐γ‐treated DCs were analysed by NTA. The number of purified particles secreted per million DCs (B) or their size distribution (C) is shown (n = 4). In (C), each line corresponds to the mean ± SEM of four acquired donor samples for each pellet.
-
D, ETotal CD4+ T cells were cultured for 6 days with EVs derived from DCs treated or not with IFN‐γ. Proliferation (D) and IL‐13 (left panel) and IFN‐γ secretion (right panel) (E) were evaluated at the end of the culture. Individual donors from untreated‐ versus IFN‐γ‐treated DCs are shown. Results in (D) are expressed as fold induction compared to untreated CD4+ T cells, and the red line indicates median (n = 8, each symbol represents a DC‐EV:T cell donor combination). The range of cytokines secreted by untreated CD4+ T cells was 20.8–478.6 pg/ml of IL‐13; 24.8–688.1 pg/ml of IFN‐γ.
-
FTh1 to Th2 ratio was calculated by dividing the concentration of IFN‐γ to the concentration of IL‐13 for each donor, for the EV‐T‐cell culture (n = 8). Results are shown as a box plot (25th to 75th percentiles, the line represents the median; whiskers, min to max, points represent outliers as calculated by Tukey's test).
Figure EV5. Analysis of IFN‐γ incubated DCs and their EVs.
- IFN‐γ‐treated DCs were loaded on poly‐lysine‐coated slides, fixed and analysed by SEM. Four DCs are shown as representative images. Scale bar = 1 μm.
- The different pellets were analysed by direct flow cytometry, to measure the presence of IFN‐γ on EV surface. EVs were immunostained for IFN‐γ and analysed as for Fig 1D. Quantification of the MFI is shown (Gmean specific antibody–Gmean isotype antibody) (n = 4, one symbol per donor). Red line indicates median.
Discussion
Following our study showing that human monocyte‐derived DCs secrete a very wide and heterogeneous spectrum of vesicles (Kowal et al, 2016), we aimed to characterize and compare the effects of these different types of EVs on T cells. Our data show that: (i) all EV types efficiently induce T‐cell activation and expansion; (ii) distinct EV subtypes promote secretion of different cytokines by CD4+ T cells: both exosomal and non‐exosomal sEVs induce IFN‐γ secretion, suggesting Th1 polarization, whereas lEVs, which float at higher densities than sEVs in density gradients, preferentially favour IL‐13 secretion and Th2 orientation; (iii) DCs display both Th1 and Th2 polarizing capacities, which are released separately in their different EV subtypes; (iv) the molecules CD40 and DC‐SIGN on sEVs and CD80 on lEVs are involved in their specific Th activating abilities; (v) all EVs released by IFN‐γ‐activated DCs promote Th1 type of responses. One major outcome of this work is to provide, for the first time, an integrated view of the specific versus shared immune activities of DC‐derived EVs. Our results highlight the need for comprehensive deciphering of the functional diversity of EV subtypes, in the future, for any patho/physiological systems.
Activation of human CD4+ T clonal cell lines by exosomes from Epstein–Barr virus‐infected B lymphocytes was the first observation suggesting an immune effect of these vesicles (Raposo et al, 1996). The activation of primary CD4+ T cells by exosomes secreted by murine DCs was then demonstrated in vitro and in vivo (Zitvogel et al, 1998; Thery et al, 2002). However, our work is the first to disclose the effect of human DC‐derived sEVs on primary CD4+ T cells and to compare it to the effect induced by all the different EVs secreted by human DCs. The effect of exosomes derived from DCs loaded with tumour or non‐tumoral antigens on CD8+ T‐cell activation has also been extensively studied, both in humans (Andre et al, 2002; Admyre et al, 2006) and in mice (Zitvogel et al, 1998), but without comparing its efficiency with that of other EVs. The promising preclinical results showing that exosomes from DCs are capable of inhibiting tumour growth in mice in a T cell‐dependent manner motivated the use of these vesicles as a cell‐free tumour vaccine in the clinics. Three clinical trials have been done thus far, immunizing patients with exosomes released in vitro by their own immature (Escudier et al, 2005; Morse et al, 2005) or IFN‐γ‐matured DCs (Besse et al, 2016) loaded with tumour antigen‐derived peptides. In phase I and phase II trials done on melanoma and on non‐small cell lung cancer, clinical stabilization of some patients and NK cell stimulation were observed, but no T‐cell responses were detected. This could be due to several reasons, such as the advanced stage of cancer in the patients, possibly leading to compromised immune responses. Alternatively, the MHC I‐ and MHC II‐restricted tumour peptides may have been insufficient to generate an adequate T‐cell response, possibly due to the absence of intact antigen allowing simultaneous B‐cell stimulation on the injected exosomes. This feature has indeed been shown to be essential for T‐cell activation and for the induction of Th1 responses required for anti‐tumour effects (Qazi et al, 2009). The lack of T‐cell responses could also be due to an insufficient number of MHC molecules presenting the relevant peptides in the injected exosome preparation. In our work, we have observed that not only the sEVs (including exosomes) secreted by immature DCs can activate and promote T‐cell activation, expansion and Th1 differentiation, but also the medium and large EVs obtained from mature DCs (Fig 7). Furthermore, we also observed similar efficacy of CD8+ T‐cell activation by large, medium and sEVs secreted by DCs (Fig EV6A and B). Taken together, these results suggest that a mixture of all types of EVs released by IFN‐γ‐matured DCs, could be equally or even more efficient as cancer immunotherapy than exosomes or sEV preparation, which are much more difficult to obtain. Such immunotherapy would still qualify as “cell‐free” vaccine, as opposed to the use of membrane vesicles generated by sonication of whole DCs as previously suggested (Kovar et al, 2006). However, our proteomic comparative study of DC‐derived medium and sEVs revealed the presence of the immunoregulatory molecule PD‐L1 (PD1L1, CD274) which inhibits T‐cell activation, on both types of EVs (Kowal et al, 2016). Thus, simultaneous administration of neutralizing monoclonal antibodies to PD‐L1 together with the EVs, as proposed for exosome‐based therapy (Besse et al, 2016), could be also implemented in this setting to improve the therapy.
Figure EV6. CD8+ T‐cell activation induced by DC‐derived EVs.
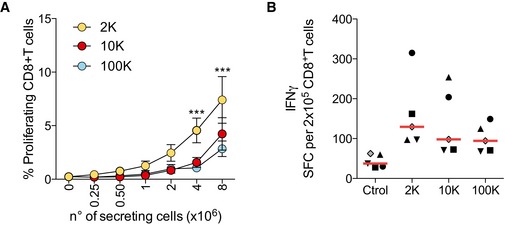
- Total CD8+ T‐cell proliferation was evaluated after 6 days of culture with different amounts of allogeneic DC‐derived EVs. Proliferation was calculated by dilution of Cell Trace Violet dye (n = 5 donors, mean ± SEM is shown). ***P < 0.001 2K compared to 10K and 100K (Friedman test).
- EVs purified from DCs incubated with an MHC class I‐restricted CEF viral peptides were cultured with total autologous CD8+ T cells on ELISPOT plates coated with IFN‐γ antibodies. After 48 h, the ELISPOT plates were developed and the number of spots per well was counted. The results are expressed as mean number of SFC/2 × 105 CD8+ T cells (n = 5 donors, one symbol per donor). Red line indicates median.
Concerning the molecular and cellular mechanisms of T‐cell activation by EVs, our work confirms previous results published on exosomes, expands them to the other EVs and describes surface molecules that mediate immune effects of EVs. In addition, we demonstrate that DC–T‐cell communication can occur in vitro at a distance, through secretion of EVs bearing functional MHC II molecules, without the need for artificial isolation and concentration of EVs (Fig 5D). MHC‐peptide complexes on exosomes can be presented to T cells either in a direct or an indirect manner (Thery et al, 2002; Montecalvo et al, 2008; Liu et al, 2016). In the former, MHC‐peptide complexes are directly engaged by antigen‐specific T cells leading to T‐cell activation, whereas in the latter recipient DCs acquire antigens or even the MHC‐peptide complexes carried by exosomes and then present them to T cells. This indirect mechanism is required for activation of naïve T cells by DC‐derived exosomes (Thery et al, 2002), whereas pre‐activated T cells can be directly activated by DC‐derived exosomes (Nolte‐’t Hoen et al, 2009). In our work, T‐cell expansion was dependent on MHC class II (Fig 2E) and was only achieved when EVs were cultured with allogeneic T cells (Fig 2F). Hence, T cells recognize allogeneic MHC class II molecules on EVs, in the absence of additional DCs. In the cultures used here, both naïve and memory T cells are present, and we observed that all EVs secreted by immature DCs were able to activate total T cells, and all were unable to activate in a direct manner naïve CD4+ T cells alone (Fig 2G). Therefore, memory T cells probably synergize with naïve T cells that have engaged antigens on the allogeneic MHC class II molecules present on the different EV types, to promote their activation, as described for presentation of alloantigens on intact cells (Morimoto et al, 1985; Akbar et al, 1988, 1991).
Despite the overall similarities in effects of the different EV subtypes, we also observed striking and clear differences in activities of large and small EVs. Another group has reported different quantitative effects of sEVs (called exosomes) and lEVs (called ectosomes) released by tumour cells (Keerthikumar et al, 2015), but here we also show a difference in qualitative effects. Large EVs that float at high densities in iodixanol gradient (Fig 4D, fractions [7–8]), and do not efficiently pass through 0.4‐μm pores (Fig 5D) promote secretion of Th2 cytokines. Note that separation of intracellular vesicles by iodixanol gradients, using the same conditions as used here for EVs, showed the presence of several transmembrane proteins in fractions [7–10] (TGN46, TfR, CD45), which therefore also contain membrane‐enclosed vesicles (Hivroz et al, 2017). We only detected MHC class II in these dense fractions by WB in some of the donors analysed. This added to the TEM observation that MHC Class II is not evenly distributed on lEVs but rather concentrated on patches (Fig EV1B) suggests that these few molecules on lEVs may fall below the detection limit of WB, but are sufficient to trigger T‐cell activation. By contrast, the sEVs, a majority of which bear MHC class II (Fig EV1B), float at lower densities in iodixanol (Fig 4C and D, fractions [3–6]) and promote secretion of Th1‐cytokines. Surprisingly, when analysing separately the two sEV subpopulations separated by iodixanol gradient, we observed (Fig 4D) that the sEVs floating around 1.14 g/ml (fractions [5–6]) were displaying stronger Th1 polarizing activity than those floating around 1.09 g/ml (fractions [3–4]). Indeed, our proteomic analysis had shown that the latter contain bona fide exosomes, enriched in late endosomal components, in addition to other sEVs, whereas the former contains a non‐exosomal subtype. Thus, our results suggest that some functional activities claimed to be exosomal features could be instead more prominent in other types of sEVs. We therefore now propose that, within the sEVs, exosomes and non‐exosomal EVs may not be functionally equivalent.
We could assign part of the differential activity of small and large EVs to the presence of CD40 and DC‐SIGN in the former, and their absence in the latter, and to CD80, which is present in all of them (Fig 6). DC functions have been extensively studied through allogeneic T‐cell stimulation assays, which led to fundamental understanding of DC biology and the finding of important DC immunoregulatory molecules (McDevitt, 2000): in our study, we used this molecular knowledge of DC functions to search for molecular players of T‐cell polarization in their EVs. Upon specific MHC‐peptide recognition by the T‐cell receptor (TCR) of a T cell, transient adhesive interactions between the T cell and DCs need to be strengthened in order to properly activate T cells, resulting in an immunological synapse. For instance, the co‐stimulatory molecules CD80 and CD86 interact with their counterparts in T cells, and the adhesion molecule LFA‐1 interacts with ICAM‐1, which all together provide a platform for sustained TCR engagement and signalling (Dustin & Chan, 2000; Montoya et al, 2002). It has long been documented in vitro and more recently in vivo that a strong TCR signal leads to a predominance of Th1 differentiation, whereas weak TCR signalling leads to Th2 differentiation (Constant et al, 1995; Hosken et al, 1995; van Panhuys et al, 2014; Besse et al, 2016). Consistently, DC‐SIGN has been described to have a transient role in immune synapse stabilization and to induce prolonged TCR signalling (Gijzen et al, 2007) when it binds to its counterpart ICAM‐3 (Geijtenbeek et al, 2000): this function may explain why its inhibition decreases sEV‐induced secretion of Th1 cytokines (Fig 6). In the case of CD40, it promotes Th1 polarization by inducing IL‐12 secretion by DCs upon interaction with CD40L on T cells (Ruedl et al, 2000). This mechanism cannot take place in our experimental system since DC‐derived EVs cannot be induced to secrete IL‐12. Thus, signalling induced by CD40L cross‐linking on T cells, together with signalling mediated by the TCR and other co‐stimulatory molecules also present on the EVs, are likely responsible for Th1 polarization. In addition to these molecules, we can hypothesize some other mechanisms linked to biophysical features of sEVs. For instance, the density of MHC class II molecules is higher on the 10K and 100K pellet EVs, compared to the lEVs found in the 2K pellet (Fig EV1B), which could thus explain their higher ability to promote the secretion of Th1 cytokines (van Panhuys et al, 2014). Furthermore, highly curved sEVs probably have different mechanical properties than lEVs, that is sEVs may be more difficult to deform. Indeed, the mechanical properties of antigen presenting substrates have recently been recognized as important modulators of T‐cell responses (Hivroz & Saitakis, 2016), and it would be worth exploring its role in EV‐mediated effects.
For the ability of lEVs to induce secretion of Th2‐cytokines, our work shows CD80 to be specifically involved in this process. Since CD80 is also detected on sEVs, we suspect that the ratio between CD80 and other immune synapse molecules in sEVs (CD40, DC‐SIGN, CD86 (Fig 1E) and possibly others) has an impact on the final EV function. Thus, quantitative differences in the protein composition of EVs may result in a completely different effect on the target cells, here Th polarization. Interestingly, previous works analysing the roles of CD80 in T‐cell polarization concluded that CD80 favours Th1 (Kuchroo et al, 1995; van Panhuys et al, 2014); however, others showed that CD80 is required for Th2‐responses in a model of lung inflammation (Harris et al, 2001). Therefore, the expression pattern of other co‐stimulatory ligands, together with CD80, should be further analysed to conclude on their respective polarization abilities.
Importantly, our experimental system uses mainly immature DCs, which are less efficient than mature DCs to induce T‐cell polarization, and is performed in vitro, which is a limitation of all human studies. Thus, our work cannot be interpreted in terms of the physiological roles of EVs in immune responses involving T‐cell polarization. As stated before, our goal was instead to provide a proof‐of‐principle of the importance to comprehensively compare the functionalities of EV subtypes. However, if we want to speculate, the observation that all EVs secreted by matured DCs favour Th1 responses, suggests that mature DCs could not only activate and polarize T cells by direct contact, but also via all their secreted EVs. On the contrary, different EVs secreted by immature DCs have different T‐cell orientation capacities, thus possibly immature DCs would send contradictory messages via their EVs, resulting in inefficient T‐cell polarization.
In conclusion, by simultaneously comparing side‐by‐side different types of EVs secreted by a given cell type, we here evidenced both common and different functional properties of EV subtypes, in antigen presentation to T lymphocytes. Performing similar comparative systematic studies in other physiological and pathological systems where exosomes or EVs are shown to perform interesting functions should be now considered. For instance, a myofibroblast‐inducing activity of tumour‐derived exosomes has been previously assigned to TGF‐β‐bearing exosomes (Webber et al, 2015): since we observed as much TGF‐β in lEVs than sEVs, (Fig EV4E), it would be important to determine if TGF‐β‐bearing lEVs do, or not, also carry this pro‐tumoral activity. Such studies will be crucial to determine which EVs should be used as therapeutic tools or targets in clinical approaches.
Materials and Methods
We have submitted all relevant data of our experiments to the EV‐TRACK knowledgebase for transparent reporting on EV‐related studies (EV‐TRACK ID: EV170008) (Van Deun et al, 2017).
Cells
Monocyte‐derived DCs were obtained from buffy coats from healthy human donors as previously described (Colombo et al, 2013). Briefly, peripheral blood mononuclear cells (PBMCs) were purified by density gradient centrifugation (LymphoPrep, Axis Shield). CD14+ cells were enriched by magnetic sorting (Miltenyi Biotec) and cultured at 2 million cells per ml for 5 days in RPMI 1640 (Gibco) supplemented with 10% FCS (Biowest or Lonza), 10 mM Hepes, 100 IU/ml penicillin and 100 μg/ml streptomycin (Gibco) in the presence of IL‐4 and GM‐CSF (50 and 100 ng/ml, respectively; Miltenyi Biotec). For IFN‐γ‐induced maturation, cells were treated 24 h with 1,000 IU/ml rIFN‐γ (Imukin, Boehringer Ingelheim, France) on day 5 in EV‐depleted FCS medium. Total or naïve CD4+ T cells were isolated from PBMCs or from CD14 negative fraction by negative selection using magnetic beads (CD4+ T Cell Isolation Kit or Naive CD4+ T Cell Isolation Kit, respectively, Miltenyi Biotec) following the manufacturer indications. Total CD8+ T cells were isolated from CD14 negative fraction by negative selection using magnetic beads (EasySep™ Human CD8+ T Cell Isolation Kit, Stem Cell Technologies) following the manufacturer indications. This study was conducted according to the Helsinki Declaration, with informed consent obtained from the blood donors, as requested by our Institutional Review Board.
EV isolation
Bovine EV‐depleted medium was obtained by overnight ultracentrifugation of RPMI 1640 medium (Gibco) supplemented with 20% of FCS at 100,000 × g in a 45Ti rotor (Beckman Coulter). On day 5 of DC‐differentiation, cells were washed in PBS (Gibco) and further cultured in EV‐depleted medium (10% EV‐depleted FCS final) for 24 h before collection of conditioned medium for EV isolation. EVs were isolated by differential ultracentrifugation as previously described (Kowal et al, 2016). Briefly, conditioned medium was centrifuged at 350 × g for 15 min at 4°C to pellet cells. Supernatant was centrifuged at 2,000 × g for 20 min at 4°C (2K pellet), transferred to new tubes and centrifuged in a 45Ti rotor for 40 min at 10,000 × g (9,000 rpm = 10K pellet), and finally for 90 min at 100,000 × g (30,000 rpm = 100K pellet). All pellets were washed in 50–60 ml of PBS and re‐centrifuged at the same speed before being resuspended in 50 μl of sterile PBS and stored at −20°C. Cells recovered from the first 350 × g pellet were pooled with cells detached from the plates by incubation at 4°C in PBS‐EDTA (DCs) and counted by Countess (Invitrogen). Viability was assessed by Trypan Blue stain 0.4% (Life Technologies) exclusion.
EV‐characterization by direct flow cytometry and Western Blotting
Vesicles secreted by 1 × 106 DCs (2K, 10K and 100K pellets) were stained with titrated amounts of mouse monoclonal anti‐human antibodies against CD9 (CD9‐FITC (clone M‐L13)), HLA‐DR (HLA‐DR‐APC (clone G46‐6)), HLA‐ABC (HLA‐ABC‐APC (clone G46‐2.6)) and CD86 (CD86‐FITC (clone 2331 (FUN‐1))) all from BD Biosciences for 30 min at 4°C in 50 μl of filtered (0.22 μm) PBS. As control, we acquired the dilution of the antibodies in filtered PBS without vesicles. Samples were acquired on a MACSQuant (Miltenyi Biotec) instrument. FSC and SSC parameters were set to log scale and low threshold. Data were analysed with FlowJo (Tree Star).
WB analysis was done as previously described (Kowal et al, 2016). Briefly, pellets recovered from a given number of cells (Fig 6A and H) or 10 μl of the optiprep gradient fractions (Fig 4B) were loaded for all samples from each preparation (2K, 10K and 100K pellet and cell) and analysed simultaneously on 4–15% Mini‐Protean® TGX Stain‐Free™ gels (Bio‐Rad), under non‐reducing conditions. Transferred membranes (Immun‐Blot PVDF Bio‐Rad) were developed using BM Chemiluminescence WB Substrate (POD) (Roche), and a ChemiDoc Touch imager (Bio‐Rad). Intensity of the bands was quantified using ImageLab Software (Bio‐Rad). Antibodies for WB were mouse monoclonal anti‐human CD9 (clone CBL162, Millipore), MHC class II (1B5 hybridoma anti‐DRa (Adams et al, 1983)), DC‐SIGN (clone 120507, R&D), CD40/TNFRSF5 (clone 82102, R&D) and CD80 (clone EPR1157(2), Abcam).
Nanoparticle Tracking Analysis
A LM10 NTA device (Malvern) was used following recommendations for EV acquisition by Gardiner et al (2013). Before each session, the size and concentration of standard silica beads (silica microspheres, 0.15 μm; Polysciences) was measured. All settings for the camera were determined using the bead solution and fixed for all measurements during the session [camera level 8, camera shutter 3.73–8.75 ms (depending on the set of measurements), camera gain 250.00]. For each sample, at least five videos of 30–60 s with more than 200 detected tracks per video, and in at least two dilutions, were taken and analysed using the Nanoparticle Tracking Analysis software 3.2 with default settings. Results represent the mean of the videos at different dilutions (multiplying by their correspondent dilution factor) of several donors.
EV floatation into iodixanol gradients
Iodixanol gradients were done as described previously (Kowal et al, 2016), with some modifications. Briefly, 2K, 10K and 100K pellets obtained by ultracentrifugation of conditioned medium from 80 to 120 million DCs were washed with PBS and resuspended in 1.2 ml of suspension buffer containing 0.25 M sucrose, 10 mM Tris pH 8.0 and 1 mM EDTA (pH 7.4). Vesicle suspension was mixed 1:1 with 60% stock solution of iodixanol/Optiprep (Sigma) and transferred to a SW55Ti rotor tube (Beckman). Then, 1.3 ml 20% iodixanol and 1.2 ml 10% iodixanol were successively layered on top of the vesicle suspension and tubes were centrifuged for 1 h at 4°C at 350,000 × g (54,000 rpm) in SW55Ti rotor, stopping without break. After centrifugation, five fractions of 1 ml were collected from the top of the tube. The density of each fraction was assessed with a refractometer (Carl Zeiss). Fractions were diluted with 1.5 ml PBS and centrifuged for 30 min at 100,000 × g (43,000 rpm) in a TLA 110 rotor (Beckman). The fractions were resuspended in 20 μl of PBS, and half of it was used for functional assays and the other half for WB analysis.
MHC class II quantification assay
The absolute number of molecules of MHC class II was calculated as described previously (Viaud et al, 2011). Briefly, a titrated mouse antibody (Ab) anti‐human HLA‐DP, DR, DQ (clone CR3/43, Dako) was incubated with increasing doses (from 0.5 to 8 million secreting cells) of EVs coupled with 5 μl beads for 30 min at 4°C. The quantification was determined using the Qifikit (Dako) containing calibration beads and the anti‐human HLA‐DP, DR, DQ in combination with a fluorescently labelled secondary antibody (anti mouse IgG1‐FITC, Dako). The absolute number of MHC II molecules per bead was determined as a function of the mean fluorescence intensity (MFI). Only doses in the linear range were used for calculation. Final quantification is expressed as numbers of MHC II molecules/million secreting cell.
Interaction of DC‐derived EVs with T cells
DiO‐labelled EVs were used to evaluate the interaction of vesicles released by DCs with T cells. Briefly, the different centrifugation pellets (2K, 10K and 100K) containing vesicles secreted by 24 × 106 DCs were stained with 1 μM fluorescent lipophilic carbocyanine dye 3,3′‐dioctadecyloxacarbocyanine perchlorate (DiO) (Vybrant® DiO Cell‐Labeling Solution, Molecular Probes) in 100 μl of PBS for 30 min at 37°C. To remove the excess of dye, vesicles were washed three times with 2 ml PBS and re‐centrifuged at the corresponding speed for each pellet using a TLA110 rotor (Beckman). The final pellet of DiO‐labelled EVs was resuspended in 30 μl of PBS. Pellets of non‐conditioned EV‐depleted FCS medium subjected to the differential ultracentrifugation protocol were stained as indicated for the DC‐derived EVs pellets and used as control.
DiO‐labelled EVs secreted by 4 × 106 DCs were cultured with 1.5 × 105 allogeneic total CD4+ T cells in T‐cell medium (RPMI 1640 medium supplemented with 10% FCS, 10 mM Hepes, 0.1 mM NEAA (Gibco), 1 mM sodium pyruvate (Gibco), 100 IU/ml penicillin and 100 μg/ml streptomycin (Gibco)) in a 96‐well plate for 18 h. For confocal microscopy analysis, T cells were adhered to poly‐l‐lysine‐coated cover slips and then fixed with 4% paraformaldehyde. Finally, nuclei were stained with DAPI. Confocal images were taken with LSM780 (Zeiss) with 40× (numerical apertures) and maximal pinhole closing. Single plane images or zstack images were acquired. For flow cytometry analysis of DiO+ T cells, data were acquired on a MACSQuant flow cytometer and analysed in FlowJo.
T‐cell proliferation and cytokine secretion assay
Freshly isolated total CD4+ T or CD8+ T cells were stained with CellTrace™ Violet Cell Proliferation (Thermo Fisher Scientific) according to the manufacturer protocol. DC‐derived EVs (secreted by increasing amount of DCs or fixed to those secreted by 8 million DCs) were cultured with total CD8+ or CD4+ or naïve CD4+ T cells (1.5 × 105 cells/well) for 6 days in 100 μl of T‐cell medium per well (96‐well plate). When intact DCs were used as stimulators, varying amounts of immature DCs were added to wells with 1.5 × 105 purified total CD4+ T cells stained with CellTrace™ Violet Cell Proliferation, and cultured for 6 days. For culture at a distance through porous membranes (0.4 and 1 μm), 2 × 105 T cells and either 2 × 105 (1:1 ratio) or 2.5 × 104 (1:8 ratio) allogeneic DCs were cultured on, respectively, the bottom and the upper part of Transwell inserts (Corning® HTS Transwell) for 6 days. In some experiments, where DC and CD4+ T cells from the same donor were used (Fig 2F, autologous), isolated CD4+ T cells were kept frozen at −80°C in 5% DMSO/95% FCS during the 6 days required for differentiation of monocytes into DCs and isolation of their EVs, until culture with the EV pellets. At the end of the T cell/EV or DC culture, cells were centrifuged and the supernatants were collected and frozen at −20°C before analysis of cytokine secretion by cytometric bead array (BD Biosciences) following manufacturer indications. For intracellular cytokine staining, cells were stimulated for 4 h with 1 μg/ml PMA (Sigma) and 1 μg/ml ionomycin (Calbiochem) in the presence of 10 μg/ml brefeldin A (Sigma). Cells were stained for the surface marker CD4 (CD4‐peCy5, clone RPA‐T4, BD Bioscience) and then with a live/dead dye (Fixable viability dye efluor 780, eBioscience). Cells were then fixed, permeabilized and labelled with intracellular staining reagents according to the manufacturer's instructions (BD Bioscience). Cells were stained with anti‐IL‐4‐APC (clone 8D4‐8, eBioscience), anti‐IFN‐γ‐PeCy7 (clone 45.B3, eBioscience), anti‐IL‐17A‐AF488 (clone BL168, Biolegend). Cells were analysed on MACSQuant instrument. Data were analysed with FlowJo software. In some experiments, EVs released by 2 or 8 × 106 DCs (as indicated in each figure legend) were incubated for 30 min prior to T cell addition with the following antibodies and their respective, provider matched, isotype controls: anti‐HLA‐DR/DQ/DP (10 μg/ml, clone CR3/43, isotype control: mouse IgG1, Dako), anti‐DC‐SIGN (5 μg/ml, clone 120507, isotype control: mouse IgG2, R&D), anti‐CD80 (10 μg/ml, clone 37711, isotype control: mouse IgG1, R&D), anti‐OX40L (10 μg/ml, clone 159403, isotype control: mouse IgG1, R&D), anti‐ICOSL (CD275 (B7‐H2) (10 μg/ml, clone MIH12, isotype control: mouse IgG1, eBioscience)), anti‐CXCR4 (10 μg/ml, clone 12G5, isotype control: mouse IgG2a, eBioscience), anti‐TNF‐α (10 μg/ml, clone clone 1825, isotype control: mouse IgG1, R&D), anti‐TGF‐β1 (5 μg/ml, clone 1D11, isotype control: mouse IgG1, R&D). In some experiments, T cells were pre‐incubated for 30 min with anti‐CD40L (1 μg/ml, clone MAB617, R&D) or its corresponding control isotype (mouse IgG2b, R&D), anti‐CD11a (LFA‐1α) (1 μg/ml, clone HI111, isotype control: mouse IgG1, eBioscience) before addition of the DC‐derived EVs.
TGF‐β1 quantification assay
Quantitation by ELISA of the EV pellets content of TGF‐β1 was performed with single‐plex assays (MesoScale Discovery) according to the manufacturer indications. EVs from 10 × 106 cells were subjected to an acid treatment for activation prior to use. All samples were run as technical duplicates, and averages were used for data analysis.
Electron microscopy
EV pellets resuspended in PBS were deposited on formvar–carbon‐coated copper/palladium grids for whole‐mount TEM analysis as previously described (Thery et al, 2006). For MHC class II labelling, samples on grids incubated with mouse anti‐MHC II, 10 nm protein‐A–gold (PAG) (PAG10), 1% glutaraldehyde (Electron Microscopy Sciences), before being contrasted and embedded in a mixture of methylcellulose and uranyl acetate. Samples were observed with a CM120 Twin FEI electron microscope (FEI Company, Eindhoven, the Netherlands) at 80 kV, and digital images were acquired with a numeric camera (Keen View, Soft Imaging System, Germany).
Scanning electron microscopy
For scanning electron microscopy, cells or EVs were loaded on a poly‐l‐lysine‐coated coverslip and fixed with 2% glutaraldehyde in phosphate buffer at room temperature for 1 h. Samples were vacuum‐dried and dehydrated by critical‐point drying with CO2. The specimens were mounted on metallic supports with carbon tape and ion sputtered with cathodic gold. Analysis of samples was performed using a Zeiss Ultra55 with high‐resolution Schottky thermal field emission gun (Schottky SEM‐FEG).
Author contributions
MT, JK, AEZ, LE and LM‐J performed research. MJ performed transmission electron microscopy staining and imaging. MS and DL performed scanning electron microscopy. MT, LM‐J and CT analysed data. MT and CT designed research and wrote the article, with additional input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 4B
Source Data for Figure 6A
Source Data for Figure 6H
Acknowledgements
This work was supported by the French National Research Agency through the “Investments for the Future” programme (France‐BioImaging, ANR‐10‐INSB‐04), the IDEX and Labex programmes (ANR‐10‐IDEX‐0001‐02 PSL* and ANR‐11‐LABX‐0043), Fondation ARC (SL220120605293 grant to C.T. and post‐doc grant to M.T. and to A.Z), ANRS (2015‐1, post‐doc grant to L. M‐J. and grant to C.T.), NIDA (DA040385, grant under subaward from Johns Hopkins Medical University to C.T.), CIC IGR‐Curie 1428, INSERM and Institut Curie (including PhD grant to J.K.). We thank V. Bazin (IBPS, Paris) for SEM analyses, Dr. Christel Goudot (INSERM U932) for help in analysing data, Drs. Elodie Segura, Claire Hivroz, Olivier Lantz, Andres Alloatti (INSERM U932), Dr. Nathalie Chaput (Institut Gustave, Roussy), Dr. Christian Neri (IBPS, Paris) and Dr Leandro Quadrana (ENS, Paris) for helpful discussions and comments on the manuscript.
The EMBO Journal (2017) 36: 3012–3028
References
- Adams TE, Bodmer JG, Bodmer WF (1983) Production and characterization of monoclonal antibodies recognizing the alpha‐chain subunits of human ia alloantigens. Immunology 50: 613–624 [PMC free article] [PubMed] [Google Scholar]
- Admyre C, Johansson SM, Paulie S, Gabrielsson S (2006) Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur J Immunol 36: 1772–1781 [DOI] [PubMed] [Google Scholar]
- Akbar AN, Terry L, Timms A, Beverley PC, Janossy G (1988) Loss of CD45R and gain of UCHL1 reactivity is a feature of primed T cells. J Immunol 140: 2171–2178 [PubMed] [Google Scholar]
- Akbar AN, Salmon M, Ivory K, Taki S, Pilling D, Janossy G (1991) Human CD4+CD45RO+ and CD4+CD45RA+ T cells synergize in response to alloantigens. Eur J Immunol 21: 2517–2522 [DOI] [PubMed] [Google Scholar]
- Andre F, Schartz NE, Chaput N, Flament C, Raposo G, Amigorena S, Angevin E, Zitvogel L (2002) Tumor‐derived exosomes: a new source of tumor rejection antigens. Vaccine 20(Suppl 4): A28–A31 [DOI] [PubMed] [Google Scholar]
- Besse B, Charrier M, Lapierre V, Dansin E, Lantz O, Planchard D, Le Chevalier T, Livartoski A, Barlesi F, Laplanche A, Ploix S, Vimond N, Peguillet I, Théry C, Lacroix L, Zoernig I, Dhodapkar K, Dhodapkar M, Viaud S, Soria J‐C et al (2016) Dendritic cell‐derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 5: e1071008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Rudensky AY (2013) Control of inflammation by integration of environmental cues by regulatory T cells. J Clin Invest 123: 939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Moita C, van Niel G, Kowal J, Vigneron J, Benaroch P, Manel N, Moita LF, Thery C, Raposo G (2013) Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J Cell Sci 126: 5553–5565 [DOI] [PubMed] [Google Scholar]
- Colombo M, Raposo G, Thery C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30: 255–289 [DOI] [PubMed] [Google Scholar]
- Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K (1995) Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J Exp Med 182: 1591–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I, Buzas EI, Lotvall J (2013) Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles 2: 20677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen J, Gringhuis SI, Geijtenbeek TBH (2009) Innate signaling by the C‐type lectin DC‐SIGN dictates immune responses. Cancer Immunol Immunother 58: 1149–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, Chan AC (2000) Signaling takes shape in the immune system. Cell 103: 283–294 [DOI] [PubMed] [Google Scholar]
- Escudier B, Dorval T, Chaput N, Andre F, Caby MP, Novault S, Flament C, Leboulaire C, Borg C, Amigorena S, Boccaccio C, Bonnerot C, Dhellin O, Movassagh M, Piperno S, Robert C, Serra V, Valente N, Le Pecq JB, Spatz A et al (2005) Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived‐exosomes: results of the first phase I clinical trial. J Transl Med 3: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner C, Ferreira YJ, Dragovic RA, Redman CW, Sargent IL (2013) Extracellular vesicle sizing and enumeration by nanoparticle tracking analysis. J Extracell Vesicles 2: 19671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TBH, Torensma R, van Vliet SJ, van Duijnhoven GCF, Adema GJ, van Kooyk Y, Figdor CG (2000) Identification of DC‐SIGN, a novel dendritic cell‐specific ICAM‐3 receptor that supports primary immune responses. Cell 100: 575–585 [DOI] [PubMed] [Google Scholar]
- Gijzen K, Tacken PJ, Zimmerman A, Joosten B, de Vries IJM, Figdor CG, Torensma R (2007) Relevance of DC‐SIGN in DC‐induced T cell proliferation. J Leukoc Biol 81: 729–740 [DOI] [PubMed] [Google Scholar]
- Gould SJ, Raposo G (2013) As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles 2: 20389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NL, Prout M, Peach RJ, Fazekas de St. Groth B, Ronchese F (2001) CD80 costimulation is required for Th2 cell cytokine production but not for antigen‐specific accumulation and migration into the lung. J Immunol 166: 4908 [DOI] [PubMed] [Google Scholar]
- Hivroz C, Saitakis M (2016) Biophysical aspects of T lymphocyte activation at the immune synapse. Front Immunol 7: 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hivroz C, Larghi P, Jouve M, Ardouin L (2017) Purification of LAT‐containing membranes from resting and activated T lymphocytes In The immune synapse: methods and protocols, Baldari CT, Dustin ML. (eds), pp 355–368. New York, NY: Springer New York; [DOI] [PubMed] [Google Scholar]
- Hosken NA, Shibuya K, Heath AW, Murphy KM, O'Garra A (1995) The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor‐alpha beta‐transgenic model. J Exp Med 182: 1579–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu DH, Paz P, Villaflor G, Rivas A, Mehta‐Damani A, Angevin E, Zitvogel L, Le Pecq JB (2003) Exosomes as a tumor vaccine: enhancing potency through direct loading of antigenic peptides. J Immunother 26: 440–450 [DOI] [PubMed] [Google Scholar]
- Keerthikumar S, Gangoda L, Liem M, Fonseka P, Atukorala I, Ozcitti C, Mechler A, Adda CG, Ang CS, Mathivanan S (2015) Proteogenomic analysis reveals exosomes are more oncogenic than ectosomes. Oncotarget 6: 15375–15396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N, Yoshioka Y, Fujita Y, Ochiya T (2016) Versatile roles of extracellular vesicles in cancer. J Clin Invest 126: 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar M, Boyman O, Shen X, Hwang I, Kohler R, Sprent J (2006) Direct stimulation of T cells by membrane vesicles from antigen‐presenting cells. Proc Natl Acad Sci USA 103: 11671–11676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal‐Bengtson B, Dingli F, Loew D, Tkach M, Théry C (2016) Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA 113: E968–E977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchroo VK, Prabhu Das M, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH (1995) B7‐1 and B7‐2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell 80: 707–718 [DOI] [PubMed] [Google Scholar]
- Liu Q, Rojas‐Canales DM, Divito SJ, Shufesky WJ, Stolz DB, Erdos G, Sullivan MLG, Gibson GA, Watkins SC, Larregina AT, Morelli AE (2016) Donor dendritic cell–derived exosomes promote allograft‐targeting immune response. J Clin Invest 126: 2805–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt HO (2000) Discovering the role of the major histocompatibility complex in the immune response. Annu Rev Immunol 18: 1–17 [DOI] [PubMed] [Google Scholar]
- Merad M, Sathe P, Helft J, Miller J, Mortha A (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31: 563–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecalvo A, Shufesky WJ, Stolz DB, Sullivan MG, Wang Z, Divito SJ, Papworth GD, Watkins SC, Robbins PD, Larregina AT, Morelli AE (2008) Exosomes as a short‐range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J Immunol 180: 3081–3090 [DOI] [PubMed] [Google Scholar]
- Montoya MC, Sancho D, Vicente‐Manzanares M, Sánchez‐Madrid F (2002) Cell adhesion and polarity during immune interactions. Immunol Rev 186: 68–82 [DOI] [PubMed] [Google Scholar]
- Morimoto C, Letvin NL, Distaso JA, Aldrich WR, Schlossman SF (1985) The isolation and characterization of the human suppressor inducer T cell subset. J Immunol 134: 1508–1515 [PubMed] [Google Scholar]
- Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, Valente N, Shreeniwas R, Sutton MA, Delcayre A, Hsu DH, Le Pecq JB, Lyerly HK (2005) A phase I study of dexosome immunotherapy in patients with advanced non‐small cell lung cancer. J Transl Med 3: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte‐’t Hoen EN, Buschow SI, Anderton SM, Stoorvogel W, Wauben MH (2009) Activated T‐cells recruit exosomes secreted by dendritic cells via LFA‐1. Blood 113: 1977–1981 [DOI] [PubMed] [Google Scholar]
- van Panhuys N, Klauschen F, Germain Ronald N (2014) T‐cell‐receptor‐dependent signal intensity dominantly controls CD4+ T cell polarization in vivo . Immunity 41: 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, Anckaert J, Martinez ZA, Baetens T, Beghein E, Bertier L, Berx G, Boere J, Boukouris S, Bremer M, Buschmann D, Byrd JB, Casert C, Cheng L, Cmoch A et al (2017) EV‐TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods 14: 228–232 [DOI] [PubMed] [Google Scholar]
- Qazi KR, Gehrmann U, Domange Jordo E, Karlsson MC, Gabrielsson S (2009) Antigen‐loaded exosomes alone induce Th1‐type memory through a B‐cell‐dependent mechanism. Blood 113: 2673–2683 [DOI] [PubMed] [Google Scholar]
- Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, Geuze HJ (1996) B lymphocytes secrete antigen‐presenting vesicles. J Exp Med 183: 1161–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200: 373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PD, Morelli AE (2014) Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 14: 195–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruedl C, Bachmann MF, Kopf M (2000) The antigen dose determines T helper subset development by regulation of CD40 ligand. Eur J Immunol 30: 2056–2064 [DOI] [PubMed] [Google Scholar]
- Segura E, Nicco C, Lombard B, Veron P, Raposo G, Batteux F, Amigorena S, Thery C (2005) ICAM‐1 on exosomes from mature dendritic cells is critical for efficient naive T‐cell priming. Blood 106: 216–223 [DOI] [PubMed] [Google Scholar]
- Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S (2002) Indirect activation of naive CD4+ T cells by dendritic cell‐derived exosomes. Nat Immunol 3: 1156–1162 [DOI] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3: Unit 3 22 [DOI] [PubMed] [Google Scholar]
- Thery C, Ostrowski M, Segura E (2009) Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9: 581–593 [DOI] [PubMed] [Google Scholar]
- Tkach M, Théry C (2016) Communication by extracellular vesicles: where we are and where we need to go. Cell 164: 1226–1232 [DOI] [PubMed] [Google Scholar]
- Viaud S, Ploix S, Lapierre V, Thery C, Commere PH, Tramalloni D, Gorrichon K, Virault‐Rocroy P, Tursz T, Lantz O, Zitvogel L, Chaput N (2011) Updated technology to produce highly immunogenic dendritic cell‐derived exosomes of clinical grade: a critical role of interferon‐gamma. J Immunother 34: 65–75 [DOI] [PubMed] [Google Scholar]
- Webber JP, Spary LK, Sanders AJ, Chowdhury R, Jiang WG, Steadman R, Wymant J, Jones AT, Kynaston H, Mason MD, Tabi Z, Clayton A (2015) Differentiation of tumour‐promoting stromal myofibroblasts by cancer exosomes. Oncogene 34: 290–302 [DOI] [PubMed] [Google Scholar]
- Yanez‐Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K , Casal E, Cappello F, Carvalho J, Colas E, Cordeiro‐da Silva A, Fais S , Falcon‐Perez JM, Ghobrial IM, Giebel B, Gimona M, Graner M, Gursel I, Gursel M et al (2015) Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4: 27066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, Ricciardi‐Castagnoli P, Raposo G, Amigorena S (1998) Eradication of established murine tumors using a novel cell‐free vaccine: dendritic cell‐derived exosomes. Nat Med 4: 594–600 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 4B
Source Data for Figure 6A
Source Data for Figure 6H