

Abstract
Lysyl oxidase–like-2 (LOXL2) is an enzyme secreted into the extracellular matrix that crosslinks collagens by mediating oxidative deamination of lysine residues. Our previous work demonstrated that this enzyme crosslinks the 7S domain, a structural domain that stabilizes collagen IV scaffolds in the basement membrane. Despite its relevant role in extracellular matrix biosynthesis, little is known about the structural requirements of LOXL2 that enable collagen IV crosslinking. In this study, we demonstrate that LOXL2 is processed extracellularly by serine proteases, generating a 65-kDa form lacking the first two scavenger receptor cysteine-rich domains. Site-specific mutagenesis to prevent proteolytic processing generated a full-length enzyme that is active in vitro toward a soluble substrate, but fails to crosslink insoluble collagen IV within the extracellular matrix. In contrast, the processed form of LOXL2 binds to collagen IV and crosslinks the 7S domain. Together, our data demonstrate that proteolytic processing is an important event that allows LOXL2-mediated crosslinking of basement membrane collagen IV.
Keywords: basement membrane, collagen, enzyme processing, extracellular matrix, lysyl oxidase, posttranslational modification (PTM), protein crosslinking, LOXL2, proteolytic processing
Introduction
Lysyl oxidase–like-2 (LOXL2) is a member of the lysyl oxidase family of copper-dependent amine oxidases (1). Like the rest of the family members, LOXL2 catalyzes the oxidative deamination of peptidyl lysine residues, which promotes the formation of lysyl-derived crosslinks in collagens and elastin (1–5). LOXL2-catalyzed crosslinks are essential posttranslational modifications in the biosynthesis and maintenance of the extracellular matrix (ECM),2 contributing to the tensile strength and stability of tissues (6). We have recently shown that LOXL2 is the main lysyl oxidase of the glomerular basement membrane, a specialized form of ECM that is an important functional component of the kidney glomerulus (7). Furthermore, together with peroxidasin-catalyzed sulfilimine crosslinking of the NC1 domain (8, 9), LOXL2-catalyzed crosslinking of the 7S domain contributes to stabilizing collagen IV networks, the main structural component of basement membranes (10–12). Other proposed biological functions of LOXL2 include regulation of tumor growth (13), metastasis (14), and angiogenesis (15). Because it is up-regulated in fibrotic diseases, LOXL2 has been proposed as an attractive pharmacological target to inhibit the characteristic increase in ECM deposition in fibrotic tissues (16–20).
The amino acid sequence of human LOXL2 consists of 774 residues organized by an N-terminal signal peptide contiguous to four N-terminal scavenger receptor cysteine-rich (SRCR) domains followed by a highly conserved lysyl oxidase catalytic domain. SRCR domains, commonly found on secreted and cell surface–bound proteins, have been proposed to mediate protein-protein interactions, ligand binding, and more recently deacetylation and deactylimination activity over peptidyl-acetyl lysines (21, 22). Although the predicted molecular mass derived from the amino acid sequence is about 84 kDa, LOXL2 is found as a ∼100-kDa protein in both the intracellular and the extracellular compartments, most likely because of posttranslational modifications such as N-glycosylation (23). Interestingly, several studies in cultured cells and tissues, including our own, have identified a smaller extracellular 65-kDa form (7, 24–26). The addition of a protease inhibitor mixture in the conditioned media of MCF-7 cells prevented the formation of the 65-kDa form, suggesting that it results from proteolytic processing of the large 100-kDa form (27). This observation is particularly relevant as other members of the LOX family such as LOX and LOXL1 become active only after they are proteolytically processed by bone morphogenetic protein-1 (28, 29). Although the crosslinking function of LOXL2 has been characterized in many biological systems, it is not known whether proteolytic processing regulates its enzymatic activity (30).
In this study, we investigated the role of LOXL2 processing on the crosslinking of the 7S domain of collagen IV, a key structural component of basement membranes. We identified the cleavage site in LOXL2 sequence by classical Edman microsequencing and site-directed mutagenesis. Furthermore, using CRISPR-Cas9 system, we developed a LOXL2-null cell line that allowed us to explore the effect of proteolytic processing on LOXL2 enzymatic activity. Our results suggest that LOXL2 proteolytic processing is a regulatory step on collagen IV and basement membranes biosynthesis.
Results
LOXL2 is proteolytically processed by serine proteases
To purify sufficient 65-kDa extracellular form of LOXL2 for the identification of the site of proteolytic cleavage by Edman microsequencing, we stably expressed the LOXL2 human ORF (NM_002318) in HEK293 cells (7). Immunoblotting characterization of HEK293 cells expressing this construct using a polyclonal antibody raised against the C-terminal region of the LOXL2 sequence showed that the intracellular fraction produced the full-length form of LOXL2 (∼100 kDa) without any sign of proteolytic cleavage (Fig. 1A). In contrast, the conditioned media of these cells showed the presence of both full-length and processed forms of LOXL2, similar to those observed in other cell types (24–26), and glomeruli (7). We purified LOXL2 forms from cell media by affinity chromatography and fractionated proteins by SDS-PAGE. The 65-kDa processed form was cut out of the gel and prepared for N-terminal Edman microsequencing. The results revealed the first 11 amino acid residues of processed form N-terminal end starting at Ala-318 of human LOXL2 sequence, indicating that proteolytic processing occurred at Lys-317. This site resides in the linker region between the SRCR2 and SRCR3 domains (Fig. 1B). Further inspection of the sequence showed that the P1 and P2 positions from the Lys-317 processing site are occupied by basic residues that are highly conserved in LOXL2 sequences of vertebrates (Fig. 1C).
Figure 1.

LOXL2 is processed at Lys-317 by a serine protease. A, immunoblot detection of LOXL2 via Myc-tag in intracellular (INT) and extracellular (EXT) fractions of HEK293 cells expressing the human LOXL2 sequence. Full-length (FL) and processed (P) forms of LOXL2 are indicated. B, illustration depicting the domain structure of full-length LOXL2 (top) primary sequence consisting of four scavenger receptor cysteine-rich (SRCR) domains, the catalytic lysyl oxidase domain (LOX) and three sites of N-glycosylation. Below is shown the proposed domain structure of the LOXL2 processed form observed in A. Edman microsequencing of the amino end of the 65 kDa processed LOXL2 yielded an 11-residue sequence (bold) that matched the human LOXL2 sequence starting from Ala-318, suggesting that proteolytic processing occurs at the Lys-317–Ala-318 peptide bond. The sequence motif (RFRK) recognized by serine proteases that precedes the cleavage site is also included. C, alignment of LOXL2 sequences showing the region encompassing the serine protease cleavage site in different organisms. Darker shadowing indicates higher degree of conservation. The conserved basic residues preceding the cleavage sites are denoted in a black rectangle. D, immunoblotting detection of LOXL2 in conditioned media from HEK293 cells expressing LOXL2 grown in culture media supplemented with different protease inhibitors as follows: aprotinin, 10 μg/ml; leupeptin, 10 μg/ml; pepstatin, 10 μm; PMSF, 1 mm; Dec-RVKR-CMK, 25 μm. The image shows a representative blot from three independent experiments. The graph shows the quantification of the amount of processed form with every treatment. Error bars represent S.D. from three independent experiments. Data were analyzed by Student's t test. Statistical significance is shown above the bars. *, p value ≤ 0.05; **, p value ≤ 0.01; ***, p value ≤ 0.001. Molecular mass standards in kilodaltons (kDa) are indicated on the left side of each gel.
The presence of these basic residues in P1 and P2 suggests that a serine protease cleaves LOXL2 at Lys-317. To test this hypothesis, HEK293 cells stably expressing human LOXL2 were grown in media containing serine protease inhibitors including leupeptin, aprotinin, and PMSF (Fig. 1D). Immunoblotting analyses of the culture media from these cells showed that the formation of the 65-kDa LOXL2 processed form was significantly reduced by aprotinin and PMSF, but not leupeptin, suggesting that serine proteases may be responsible for LOXL2 cleavage. HEK293 cells were also cultured in the presence of decanoyl-RVKR-chloromethylketone (Dec-RVKR-CMK), an inhibitor that blocks the activity of members of the subtilisin-like family of proprotein convertases such as furin (31). As shown in Fig. 1D, Dec-RVKR-CMK exhibited the highest inhibition of LOXL2 processing. Pepstatin, a potent inhibitor of aspartyl proteases, had no effect on LOXL2 cleavage. In summary, these data suggest that LOXL2 is proteolytically processed by a serine protease at Lys-317 in the extracellular compartment, removing the first two SRCR domains from LOXL2 sequence.
Processing is not required for LOXL2 to become enzymatically active with small, soluble substrates
To evaluate the effect of proteolytic processing on amine oxidase activity, we performed site-directed mutagenesis to generate either full-length or processed forms of LOXL2. To block proteolytic processing and generate a full-length LOXL2 form, the two basic residues preceding the cleavage site were mutated from RK to GE. The introduction of negatively charged glutamic acid at P1 was expected to block proteolytic processing by serine proteases (32–34). Transient expression of this construct in HEK293 cells predominantly generated the full-length form of LOXL2, indicating that proteolytic processing was abrogated (Fig. 2A). Initial studies in which only Lys-317 was mutated uncovered an anticipated atypical form of LOXL2 of about 70 kDa that is not observed in cells expressing wild-type LOXL2. A careful inspection of LOXL2 sequence identified Arg-256 as a potential cleavage site of serine proteases. Thus, to completely eliminate proteolytic processing and generate the full-length 100-kDa form, Arg-257 was mutated to glycine in addition to 316–317 residues RK (Fig. 2A). LOXL2BP was used to describe this “blocked processing LOXL2 mutant” construct. As most serine proteases have a preference for Arg at P1 (33, 34), we generated a second mutant by changing Lys-317 to Arg-317 to potentially enhance proteolytic cleavage. The resulting K317R processing LOXL2 mutant was named LOXL2K317R.
Figure 2.
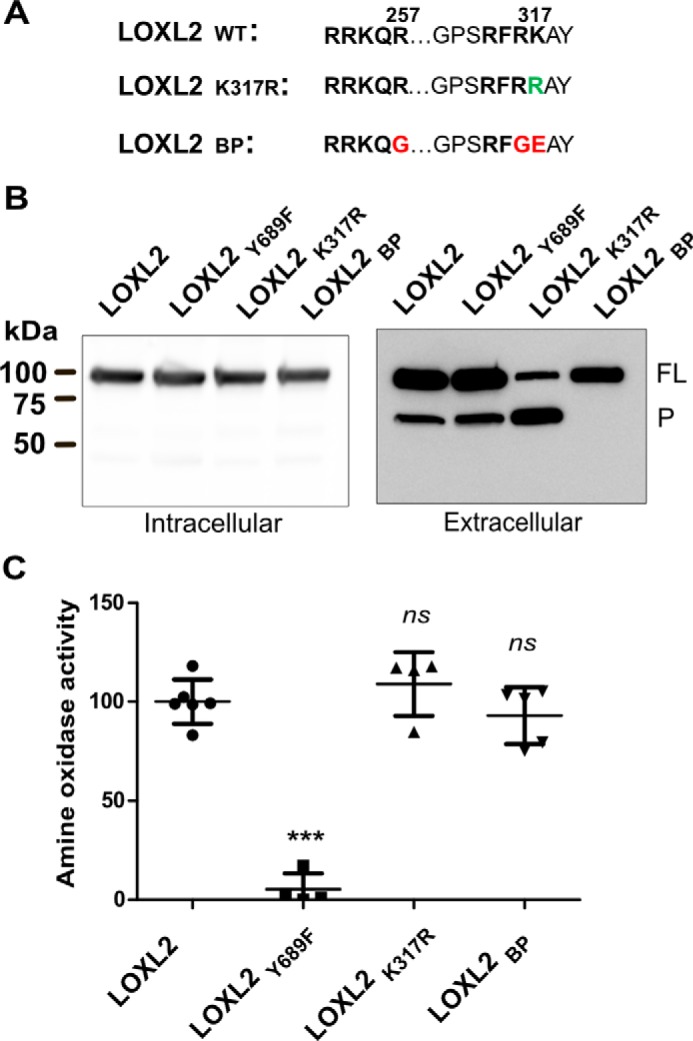
Proteolytic processing does not modulate amine oxidase activity of LOXL2. A, a scheme showing the mutation of specific residues in LOXL2 sequence. Green-colored residues represent changes expected to enhance LOXL2 processing (LOXL2K317R) and red-colored residues represent those predicted to block processing (LOXL2BP). B, immunoblotting analyses of intracellular and secreted proteins from HEK293 cells transfected with wild-type LOXL2 and processing (LOXL2K317R and LOXL2BP) and catalytically (Y689F) inactive LOXL2 mutants using an anti-LOXL2 antibody raised against a polypeptide encompassing the catalytic domain sequence. The image is representative of three independent experiments. The full-length (FL) and processed (P) forms of LOXL2 are indicated. C, amine oxidase activity measured by Amplex Red assay in media from HEK293 cells expressing the different LOXL2 constructs. Error bars represent S.D. from at least four independent experiments. Data were analyzed by Student's t test. Statistical significance is shown above the bars. ***, p value ≤ 0.001; ns, not significant.
To evaluate the effect of mutagenesis on proteolytic processing, these two new LOXL2BP and LOXL2K317R constructs, together with the catalytically inactive LOXL2Y689F mutant and wild-type LOXL2 as controls, were expressed in HEK293 cells. Immunoblot analyses for LOXL2 detection in whole cell extracts showed that all constructs produced LOXL2 proteins of 100 kDa. However, LOXL2 processing differences became evident when analyzing culture media containing the extracellular forms of LOXL2. As expected, the LOXLBP construct produced only the 100-kDa form. In contrast to the wild-type construct, the LOXLK317R construct showed a significant increase of the 65-kDa processed LOXL2 form (Fig. 2B).
To assess the effect of proteolytic processing on the amine oxidase activity, conditioned media of cells expressing LOXL2 wild type and mutants were assayed with Amplex Red assay using 1,5-diaminopentane as substrate (35). As shown in Fig. 2C, the amine oxidase activity showed no significant difference between processing mutants and wild-type LOXL2. These results show that cleavage at Lys-317 does not influence LOXL2 amine oxidase activity.
LOXL2 processing promotes collagen IV crosslinking
To evaluate the role of LOXL2 proteolytic processing on collagen IV crosslinking, we generated a LOXL2-null PFRH9 cell line by using CRISPR-Cas9 gene editing to target exon 2 of the mouse Loxl2 gene (Fig. 3A). Immunoblot analysis of the ECM-derived fraction, where both 100-kDa and 65-kDa forms are normally deposited in the wild-type cells, showed that both LOXL2 forms are absent in the knock-out cell line (Fig. 3B). In addition, no residual amine oxidase activity inhibited by BAPN was detected in LOXL2-null cell conditioned medium (Fig. 3C). Fig. 3D shows that 7S dodecamers extracted after collagenase digestion of the ECM derived from LOXL2-null PFHR9 cells are composed of only 7S monomeric subunits, confirming our previous crosslinking studies using pharmacological inhibitors of lysyl oxidase activity (7). Interestingly, the nonreduced 7S dodecamer of the LOXL2-null cells showed slightly faster electrophoretic mobility in comparison to the wild-type cells (Fig. 3E). These results show that LOXL2 is the sole source of lysyl oxidase activity for crosslinking the 7S dodecamer of collagen IV in PFHR9 cells. Furthermore, generation of a LOXL2-null PFHR9 cell line provides an excellent system to test the effect of LOXL2 proteolytic processing on collagen IV crosslinking.
Figure 3.

CRISPR/Cas9-mediated deletion of LOXL2 in PFHR9 cells yields 7S dodecamers devoid of lysyl-derived crosslinks. A, representation of the Loxl2 gene structure and the region of exon 2 targeted by the sgRNA sequences. B, immunoblotting analysis using an anti-LOXL2 antibody of ECM proteins extracted with urea from wild-type (WT) and LOXL2-null (KO) PFHR9 cells. A representative experiment from a total of three independent experiments is shown. Full-length (FL) and processed (P) forms of LOXL2 are indicated. C, the graph represents the fluorescence units generated by Amplex Red assay measuring the amine oxidase activity from urea-soluble matrix proteins from LOXL2-KO and WT PFHR9 cells in the presence or absence of BAPN. Error bars represent S.D. from five independent experiments. Data were analyzed by Student's t test. Statistical significance is shown above the bars. ***, p value ≤ 0.001; *, p value ≤ 0.05; ns, not significant. D, SDS-PAGE analyses of 7S dodecamer (7S) and NC1 hexamer (NC1) purified from LOXL2-KO and WT PFHR9 matrices. Proteins were reduced with 50 mm DTT. M, D, T, TT, P, and H indicate the electrophoretic mobility of monomer, dimer, trimer, tetramer, pentamer, and hexamer 7S subunits, respectively. E, SDS-PAGE analyses under nonreducing conditions of the same 7S dodecamers purified from WT and KO PFHR9 cell matrices.
To evaluate the 7S crosslinking activity of LOXL2 processing mutants, these constructs were expressed in the LOXL2-null PFHR9 cells. As expected, monomeric 7S subunits are predominant in the catalytically defective LOXL2Y689F mutant, indicating that the enzyme failed to reestablish the crosslinking profile observed in wild-type cells (Fig. 4). In contrast, the 7S dodecamer from wild-type LOXL2 and the enhanced processing mutant LOXL2K317R exhibited the typical crosslinking profile of oligomeric forms observed in wild-type cells. Surprisingly, the full-length, processing defective mutant LOXL2BP failed to crosslink the 7S dodecamer. These results demonstrate that, despite being catalytically active, the full-length processing mutant LOXL2BP was unable to crosslink the 7S dodecamer. These results suggest that removal of the first two SRCR domains by proteolytic processing is required for effective collagen IV crosslinking in the insoluble matrix.
Figure 4.
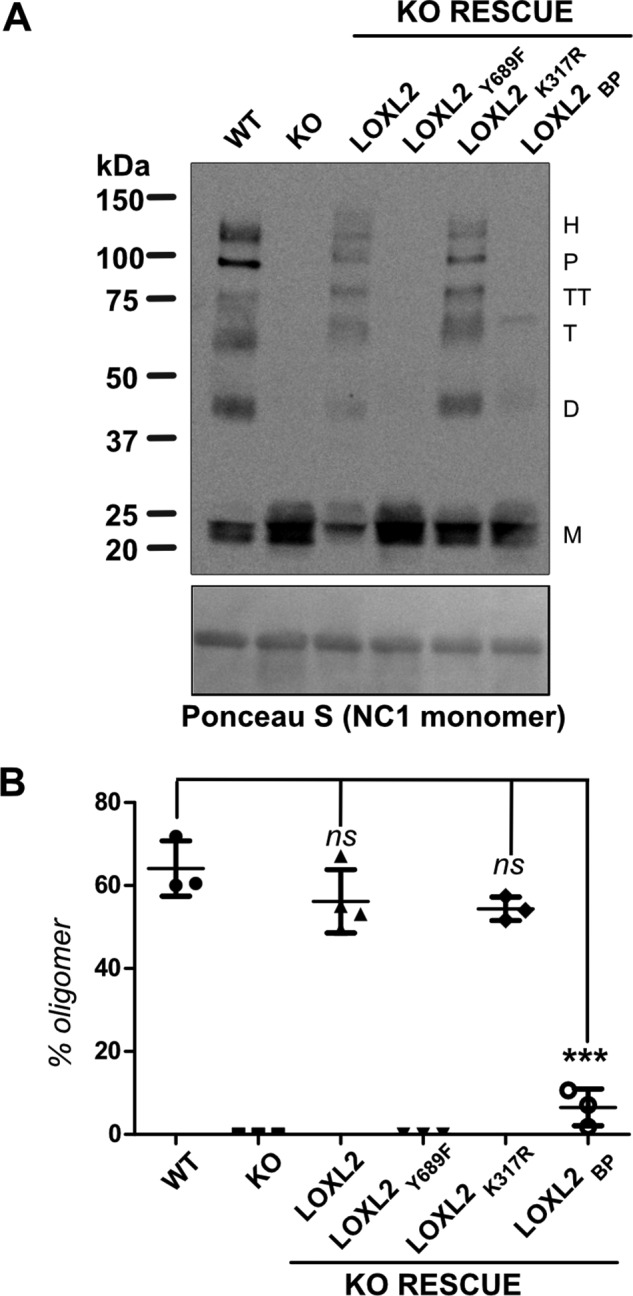
Proteolytic processing of LOXL2 promotes crosslinking of 7S dodecamer. A, immunoblotting detection of 7S subunits in collagenase digested samples of ECM produced by WT LOXL2, LOXL2-null PFHR9 cells (KO), and LOXL2-null PFHR9 overexpressing different LOXL2 mutants described in Fig. 2. Protein samples were fractionated under reducing conditions (50 mm DTT). The nitrocellulose membrane stained with Ponceau S showing the NC1 monomers is presented as a surrogate for total protein loading control. M, D, T, TT, P, and H indicate the electrophoretic mobility of monomer, dimer, trimer, tetramer, pentamer, and hexamer 7S subunits, respectively. B, representation of the percentage of 7S oligomers relative to the total 7S subunits per lane generated in each set of experiments. Error bars represent S.D. from three independent experiments. Data were analyzed by Student's t test. Statistical significance is shown above the bars. ***, p value ≤ 0.001; ns, not significant.
Proteolytic processing enables the interaction of LOXL2 with insoluble collagen IV
Although full-length and processed forms of LOXL2 have been reported to localize in the ECM of different tissues (7, 15), our results showed that only the processed LOXL2 crosslinks the 7S dodecamer. Thus, we hypothesized that the processed, but not the full-length, form of LOXL2 interacts with collagen IV. To test this hypothesis, we performed a collagenase digestion to solubilize proteins interacting with the insoluble collagen IV scaffold. As we wanted to detect a potential LOXL2–7S dodecamer interaction, we first performed immunoblotting analyses of purified LOXL2 to characterize its electrophoretic mobility under reducing and nonreducing conditions. As shown in Fig. 5A, under nonreducing conditions the full-length and processed LOXL2 forms migrated with an apparent molecular mass of ∼75 and ∼45 kDa, respectively. The faster electrophoretic mobility of both LOXL2 forms is consistent with the presence of many disulfide bridges that restrain the polypeptide chain yielding a smaller apparent molecular mass in SDS-PAGE. Immunoblotting analyses of collagenase-soluble proteins from wild-type PFHR9 matrix show a 45-kDa band equivalent to that of processed LOXL2 (Fig. 5B, right panel). Surprisingly, the immunoblot also showed an unexpected high molecular mass form of LOXL2 with an electrophoretic mobility slightly slower than the 7S dodecamer shown in the SDS-PAGE (Fig. 5B). None of these two LOXL2 forms were observed in the collagenase digest of LOXL2-null PFHR9 matrix (Fig. 5B, right panel). Thus, the presence of the high molecular mass LOXL2 form suggests a stable interaction between processed LOXL2 and the 7S dodecamer that is resistant to the highly dissociative conditions of SDS-PAGE analysis.
Figure 5.

Processed LOXL2 is covalently bound to collagen fragments of the 7S dodecamer. A, SDS-PAGE fractionation of purified LOXL2 under reducing (+DTT) or nonreducing conditions (−DTT). Full-length and processed forms in both conditions are indicated as FL and P, respectively. B, SDS-PAGE (left panel) and immunoblot anti-LOXL2 (right panel) of collagenase digests of WT and LOXL2-null PFHR9 cells under nonreducing conditions. C, the left panel shows immunoblotting detection of LOXL2 in purified 7S dodecamer and collagenase digests (CD) of PFHR9 and bovine glomeruli matrices under reducing conditions. The right panel shows the immunoblotting detection of LOXL2 in urea-extracted and collagenase-soluble proteins from PFHR9 matrix. D, left panel shows the SDS-PAGE analysis of purified 7S dodecamer incubated with or without pepsin. The right panel shows the immunoblotting detection of LOXL2 in collagenase-soluble proteins from PFHR9 matrix with or without pepsin digestion. E, SDS-PAGE analyses of purified 7S dodecamers digested with collagenase after reduction/alkylation and denaturation (left panel). The right panel shows the immunoblotting detection of LOXL2 in reduced/alkylated and denatured collagenase-soluble proteins digested with or without collagenase. Collagenase band observed in the gel is indicated by an asterisk (*). The position of processed LOXL2 (P) is indicated. Molecular mass standards in kilodaltons (kDa) are indicated on the left side of each gel.
To further characterize the interaction between 7S dodecamer and processed LOXL2, we performed immunoblotting analyses of purified 7S dodecamer, urea-soluble, and collagenase-soluble PHFR9 matrix proteins under reducing conditions. As shown in Fig. 5C, some bands are similar, but there are striking differences in the number and size of LOXL2 forms. Consistent with a LOXL2–7S dodecamer interaction, the unbound 65-kDa processed LOXL2 form was not detected in purified 7S dodecamer (Fig. 5C, left panel), as it is likely removed during chromatographic purification. However, a ladder of three or more additional bands, larger than the 65-kDa processed LOXL2, was observed in purified 7S dodecamer samples and collagenase digests of PFHR9 and glomerular matrices (Fig. 5C). Of note, the bands detected in the collagenase digest of bovine glomerular matrix showed a more complex pattern, which is probably a consequence of the highly heterogeneous collagenous material isolated from this tissue.
The striking differences between the electrophoretic profiles of LOXL2 forms present in urea-soluble and collagenase-soluble protein samples suggest that the enzyme interacts with collagen IV in different ways (Fig. 5C, right panel). For instance, the 100-kDa full-length LOXL2 band is detected in urea-extracted proteins, but not in collagenase digests, suggesting that it does not interact with collagen. In contrast, the 65-kDa processed LOXL2 (free form, equivalent to the 45-kDa band observed under nonreducing conditions) is present in both preparations, suggesting that this form associates with collagen IV in a noncovalent fashion. Lastly, the ladder of LOXL2 bands is only present in the collagenase digest, suggesting that processed LOXL2 forms a covalent complex with peptide fragments derived from 7S dodecamer that were not digested by collagenase under native conditions. Taken together, these results suggest that processed, but not full-length, LOXL2 is bound both covalently and noncovalently to collagen IV.
To validate that LOXL2 is bound to 7S polypeptides, we subjected collagenase-soluble PFHR9 proteins to pepsin digestion under nondenaturing conditions. As expected, the 7S dodecamer was resistant to pepsin digestion (Fig. 5D, left panel). However, LOXL2 was completely degraded under the same conditions (Fig. 5D, right panel), indicating that the LOXL2 signal detected by the antibody is specific despite the presence of excessively abundant 7S oligomeric subunits. In contrast, because it is possible that processed LOXL2 may have protected the attached 7S peptide fragments from collagenase digestion under native conditions, collagenase-soluble ECM proteins were subjected to disulfide reduction and denaturation followed by a second collagenase digestion. Fig. 5E shows that the 7S dodecamer was almost completely degraded by collagenase after reduction/alkylation and denaturation. At the same time, the ladder above processed LOXL2 was also completely degraded by collagenase followed by an evident increase of the 65-kDa processed LOXL2 (Fig. 5E, right panel). Collectively, these results clearly show that the ladder of LOXL2 forms observed in collagenase digests results from the covalent interaction of processed LOXL2 with 7S collagenous peptides.
To further characterize these findings, we analyzed the presence of LOXL2 in purified 7S from different biological samples. Although the bacterial collagenase preparation used for purification of 7S dodecamers is of the highest commercially available quality, to rule out LOXL2 cleavage by nonspecific proteases, we incubated recombinant LOXL2 with collagenase and monitored the ratio of processed and full-length forms of LOXL2 by immunoblotting. As shown in Fig. 6A, bacterial collagenase did not alter the relative abundance of the two forms of LOXL2, indicating that collagenase does not process LOXL2. Subsequently, we performed LC-MS/MS analyses of purified 7S dodecamers from PFHR9, bovine glomerular and placental matrices. In all samples, only peptides derived from processed LOXL2 sequences were detected (Fig. 6B). As expected, 7S dodecamer purified from the LOXL2-null PFHR9 cell line did not yield any LOXL2 peptides in the LC-MS/MS analyses. All together, these results revealed that only processed LOXL2 is solubilized from ECM by collagen degradation, and that a fraction of the enzyme is covalently bound to 7S dodecamer.
Figure 6.

Proteomic identification of LOXL2 peptides in purified 7S dodecamers from different ECM is shown. A, immunoblot detection of LOXL2 of purified recombinant human LOXL2 incubated for 24 h at 37 °C with or without collagenase. Full-length (FL) and processed (P) forms of LOXL2 are indicated. B, identification of LOXL2 peptides by mass spectrometry analyses of 7S purified from PFHR9 matrix (underlined in purple), bovine glomerular basement membrane (GBM) (cyan), and placenta basement membrane (PBM) (blue). Signal peptides (blue), SRCR domains (yellow), and LOX catalytic domains (green) are highlighted in the alignment.
To gain insight about the type of covalent interaction between processed LOXL2 and collagen IV, we examined the distribution of the different LOXL2 forms in the PFHR9–LOXL2-null rescue system. Processed LOXL2 and ladder bands were present in the collagenase-soluble proteins from PFHR9 cells transfected with wild-type LOXL2 and enhanced processing LOXL2K317R constructs (Fig. 7). As expected, none of the LOXL2 forms were detected in the collagenase digest of cells transfected with the full-length LOXL2BP, further supporting that proteolytic processing is required for LOXL2 localization to the insoluble matrix and interaction with collagen IV. However, although the catalytically defective LOXL2Y689F mutant undergoes proteolytic processing and it interacts noncovalently with collagen IV, the absence of LOXL2 ladder bands indicated that lysyl oxidase catalytic activity is required for the covalent interaction between processed LOXL2 and collagen IV. These findings demonstrate that covalent interaction between LOXL2 and collagen IV is mediated by lysyl oxidase enzymatic activity.
Figure 7.
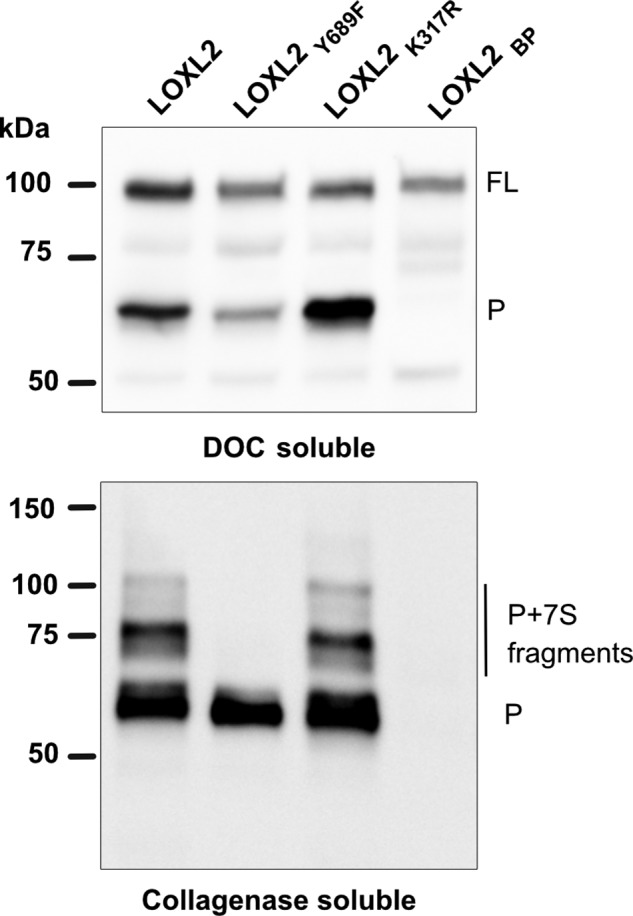
Amine oxidase activity is required for the covalent interaction between processed LOXL2 and collagen IV. Immunoblotting detection of LOXL2 in deoxycholate (DOC)–soluble fractions and collagenase-soluble proteins from the matrix of LOXL2-null PFHR9 cells overexpressing different LOXL2 constructs. Full-length (FL) and processed (P) forms of LOXL2 are indicated. The blots are representative of three independent experiments. Molecular mass standards in kilodaltons (kDa) are indicated on the left side of each gel.
Discussion
Our previous work identified LOXL2 as the main lysyl oxidase expressed in the glomerulus that crosslinks the 7S domain of collagen IV (7). This study provided the first direct evidence linking LOXL2 enzymatic activity and collagen IV crosslinking, an important step for basement membrane stabilization (10–12). Here, we extended those studies by investigating the role of LOXL2 proteolytic processing in 7S dodecamer crosslinking. Our results demonstrate that LOXL2 is cleaved by serine proteases at Lys-317, generating a processed form lacking the first two SRCR domains, without an apparent effect over the amine oxidase enzymatic activity. However, proteolytic processing enables the interaction of processed LOXL2 with collagen IV in the insoluble matrix promoting the formation of intermolecular crosslinks in the 7S dodecamer. Fig. 8 summarizes a proposed mechanism based on our findings.
Figure 8.
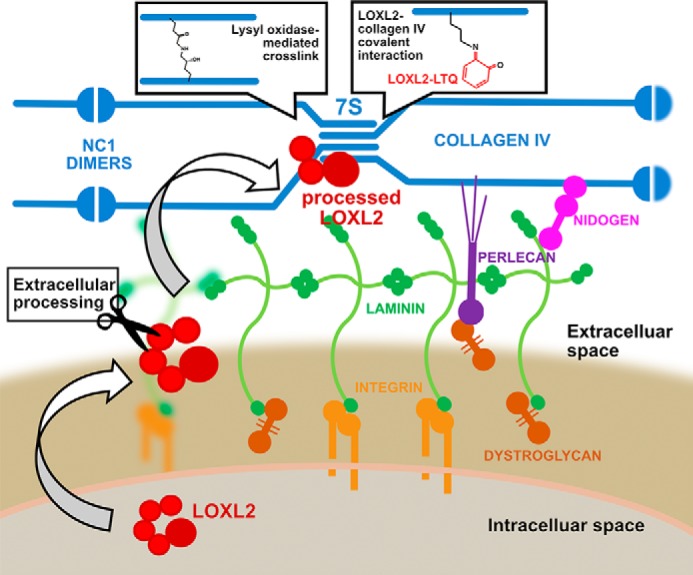
Proteolytic processing of LOXL2 enables binding to collagen IV and crosslinking of the 7S domain. The scheme depicts the secretion and proteolytic cleavage of LOXL2 by a serine protease in the ECM. The removal of SRCR1 and SRCR2 enables the interaction of processed LOXL2 with the 7S dodecamer and oxidative deamination of lysine residues forming reactive aldehydes, which may further condense into covalent lysyl-derived crosslinks. Alternatively, a small fraction of processed LOXL2 may also covalently bind to the 7S dodecamer, a process that depends on both proteolytic processing and amine oxidase activity. Covalent linkage may occur between the lysyl-tyrosyl quinone (LTQ) cofactor found in LOXL2 catalytic domain and a lysine residue from the 7S dodecamer, an event that may lead to enzyme inactivation.
Inhibition experiments with aprotinin, PMSF, and Dec-RVKR-CMK indicated that a serine protease is likely to be responsible for the proteolytic processing of LOXL2. Interestingly, it has recently been shown that a proprotein convertase (PC) activates peroxidasin, the enzyme responsible the formation of sulfilimine crosslinks in the C-terminal noncollagenous domain (NC1) of collagen IV (36). Although the blockade of LOXL2 proteolytic processing by Dec-RVKR-CMK inhibitor suggested the involvement of a PC, this family of proteases has a strong preference for Arg in P1 in the motif R-X-R/K-R. In contrast, the cleavage site of LOXL2 exhibits a Lys in P1, suggesting that other serine proteases may cleave LOXL2 (33). Interestingly, Dec-RVKR-CMK has been shown to also inhibit members of the trypsin-like (S1) family of membrane-anchored serine proteases. Unlike the PC family, trypsin-like proteases may cleave substrates encompassing either Arg or Lys at P1 (34, 37), suggesting that these proteases are more likely candidates to process LOXL2 at Lys-317. The importance of proteolytic cleavage as a regulatory event controlling LOXL2 crosslinking activity may be appreciated by the evolutionary conservation of Lys in the P1 site of LOXL2 sequences of many different organisms (Fig. 1C). Further studies are required to reveal the identity of the specific protease responsible for LOXL2 processing.
Our results using 1,5-diaminopentane as the substrate for in vitro activity assays demonstrated that both full-length and processed forms of LOXL2 are active. More importantly, proteolytic processing did not increase LOXL2 enzymatic activity toward a small substrate, indicating that LOXL2 crosslinking function is different to LOX and LOXL1, which requires proteolytic cleavage before becoming active. However, our results are in agreement with previous reports showing that LOXL4 exhibits amine oxidase activity in vitro regardless of the presence or absence of SRCR domains (38). The role of processing in collagen IV crosslinking became apparent in rescue experiments using LOXL2-null PFHR9 cells. The results indicated that removal of SRCR1 and SRCR2 domains by proteolytic processing was required before LOXL2 could effectively crosslink collagen IV deposited in the insoluble ECM. Indeed, our immunoblotting analyses of collagenase digests and purified 7S dodecamers from different tissues demonstrated that only the processed form of LOXL2 interacts with collagen IV. Conceivably, the presence of SRCR1 and SRCR2 domains blocks the proper interaction between LOXL2 and collagen IV by steric hindrance. These results suggest that, rather than an activation of enzymatic activity, LOXL2 proteolytic processing is a regulatory event that is required for an effective enzyme-substrate interaction to achieve oxidation of lysine residues and promote crosslinking of the 7S dodecamer.
Strikingly, we have found that a significant fraction of the processed LOXL2 is covalently attached to collagenase-sensitive 7S peptide fragments in a catalytically dependent manner, not only in the PFHR9-ECM but also in animal tissues, indicating that covalent crosslinking of LOXL2 to collagen IV can take place not only in matrix produced by cultured cells, but also in vivo. Although the nature of the collagenous sequences bound to LOXL2 were not identified, the evidence suggests that such fragments may be derived from the 7S dodecamer. These findings are the first report of a stable covalent interaction between a lysyl oxidase and its substrate. Although the biological implications of this finding are unclear at this time, one possibility is that the LOXL2-collagen IV interaction is the result of an arrested enzymatic intermediate in the enzyme's catalytic cycle. The chemical mechanism of lysyl oxidases proposed by Kagan (39) involves the formation of a Schiff base between the lysyl-tyrosyl quinone (LTQ) cofactor and the ϵ-amino group of Lys in the substrate. The covalent intermediate spontaneously breaks off from the enzyme's catalytic site and molecular oxygen regenerates the LTQ cofactor. Thus, it is possible that a Lys residue from the 7S dodecamer substrate is trapped in the active site of LOXL2 (Fig. 8). Obviously, covalent attachment of LOXL2 through its LTQ cofactor may render the enzyme inactive. It is not known whether this event is reversible, but perhaps this could be a mechanism to quench enzyme activity and avoid excessive lysine oxidation and collagen crosslinking. This new finding deserves further investigation to better understand the molecular mechanisms controlling LOXL2-catalyzed crosslinking of collagen.
LOXL2 proteolytic processing may also play a role as a regulatory mechanism for the crosslinking of other ECM substrates. Although our studies did not address whether proteolytic processing of LOXL2 is required for crosslinking of other ECM proteins that are also known substrates of LOXL2, such as type I and III collagens (40, 41), there is no reason to believe that this proteolytic event is a specific requirement for collagen IV crosslinking. Furthermore, because LOXL2 has been recognized as an important pharmacological target for fibrosis and cancer, it would be interesting to test if the proteolytic processing could regulate crosslinking and deposition of other ECM substrates.
Inhibition of the enzymatic activity of LOXL2 to suppress crosslinking of ECM proteins is a primary focus of anti-fibrotic therapeutic strategies (16–20). So far, a few LOXL2 inhibitors of different types have been developed for the treatment of several fibrotic and metastatic diseases (20, 41, 42). For instance, simtuzumab, a humanized monoclonal antibody described as an allosteric inhibitor of LOXL2, was shown to be an effective inhibitor in mouse models of lung and liver fibrosis (17, 20), but many of the clinical trials using this humanized antibody have failed to show improvement in the development of fibrosis (43). Although simtuzumab inhibitory characteristics were derived from preclinical studies using a mixture of full-length and processed LOXL2 (41), it is not clear whether processing may alter the antibody's inhibitory allosteric effect. LOXL2 has also been shown to be up-regulated in different nephropathies (44, 45) and, because of its role in collagen crosslinking, it has been suggested as a potential target to prevent increased deposition of collagen IV and other ECM proteins in kidney fibrosis (46). Although there are no published studies addressing this particular hypothesis, it would be interesting to see if any of the currently available LOXL2 inhibitors might indeed ameliorate kidney fibrosis. However, in light of our results, a better understanding of LOXL2 structural determinants mediating collagen crosslinking will better assist the development of more effective treatments targeting LOXL2 activity.
In summary, our study proposes proteolytic processing by serine protease(s) as an important regulatory mechanism of LOXL2-mediated crosslinking of the 7S domain of collagen IV. It is demonstrated that rather than increasing the enzyme catalytic activity, removal of SRCR1 and SRCR2 domains allows the interaction between LOXL2 and collagen IV, resulting in effective oxidation of lysine residues during basement membrane biosynthesis.
Experimental procedures
Cell culture
PFHR9 cells were grown in Dulbecco's modified Eagle's medium supplemented with 5% bovine growth serum from HyClone Laboratories (Logan, UT) and 1% penicillamine-streptomycin and incubated at 37 °C in 10% CO2. When ECM production was intended, PFHR9 were grown after confluency for 7 days and media were supplemented with 50 μg/ml ascorbic acid. HEK293 cells were cultured in Dulbecco's modified Eagle's medium and Ham's F-12 medium (1:1) with 10% bovine growth serum from HyClone Laboratories and 1% penicillamine-streptomycin and incubated at 37 °C in 5% CO2.
LOXL2 purification
LOXL2-His6 was generated and purified as previously reported (7). HEK293 cells stably expressing LOXL2-His6 were expanded, and secreted LOXL2-His6 was purified with a nickel nitrilotriacetic acid column to 95% purity as judged by SDS-PAGE. The lysyl oxidase activity of the purified recombinant protein was assayed by Amplex Red assay with 5 mm diaminopentane as substrate.
7S dodecamer purification
The collagen IV 7S dodecamer from ECM produced by PFHR9 was purified as previously published (7). Briefly, cells were homogenized in 1% (w/v) deoxycholate with sonication, and the insoluble material was isolated after centrifugation at 20,000 × g for 15 min. The pellet was then extracted with 1 m NaCl plus 50 mm Tris-Cl, pH 7.5, and 10 mm Tris-Cl, pH 7.5, and then digested in 50 mm Tris-Cl, pH 7.5, 5 mm CaCl2, 5 mm benzamidine, 25 mm 6-aminocaproic acid, 0.4 mm phenylmethylsulfonyl fluoride, and 0.1 mg/ml bacterial collagenase (Worthington Biochemical Corp.). Collagenase-solubilized material was dialyzed against 50 mm Tris-Cl, pH 7.5 and then subjected to DEAE-cellulose and gel filtration chromatography.
SDS-PAGE and immunoblotting
For immunoblotting assays, protein samples were loaded for 4–15% (w/v) SDS-PAGE and electrotransferred onto nitrocellulose membranes. Blots were incubated with polyclonal anti-7S collagen IV (Assay Biotech) or LOXL2 (Abcam) antibodies and visualized with horseradish peroxidase–conjugated donkey anti-rabbit as secondary antibody (Jackson ImmunoResearch Laboratories). Detection of HRP activity from secondary antibodies was revealed using Pierce enhanced chemiluminescence reagents (Thermo Scientific) following the instructions of the manufacturer. Quantification of immunoblots was performed using ImageJ software (47).
CRISPR-mediated LOXL2 knockout
The gRNA sequence for LOXL2 exon 2 (forward 5′-CACCGCGCGTGGAGGTCTACTACGA-3′; reverse 5′-AAACTCGTAGTAGACCTCCACGCGC-3′) was ligated into the BbsI sites of pSpCas9(BB)-2A-Puro (PX459) (gift from Dr. Feng Zhang, Addgene plasmid no. 48139) (48). PFRH9 cells were cotransfected with Lipofectamine 2000 (Invitrogen) with PX459 plasmid and a hygromycin resistance homologous recombination vector, with 500 base pairs' homology arms surrounding the targeted sequence of the exon 2. Transfection in absence of the homology recombination vector was performed to address the effectiveness of the designed sgRNA by Surveyor Assay (Integrated DNA Technologies (IDT)). Transfected PFHR9 cells were initially treated with 4 μg/ml puromycin to select cells containing the PX459 plasmid expressing the Cas9 protease and the specific sgRNA, after a first round of selection cells was selected with 250 μg/ml hygromycin B for the homologous recombination vector. Resistant clones were isolated and screened for the presence of the insertion cassette via homologous recombination with the following primers: forward/external 5′-CAGTCTGAGTCCTGCCAAGG-3′ and reverse/internal 5′-GCTTTGCTTCCGTACTGGAACTGAGGG-3′. Positive clones for the insertion were subjected to analysis by Western blot using two different anti-LOXL2 antibodies: rabbit polyclonal to LOXL2 (ab96233 and ab113960, Abcam).
Cloning, plasmid constructs, and transient transfection
TrueORF Gold cDNA for human LOXL2 (C-terminal Myc-DDK–tagged) construct cloned into the pCMV6-Entry vector (LOXL2) was purchased from Origene (Rockville, MD). The catalytically inactive mutant (LOXL2Y689F) was described in a previous publication (7). The following primers were designed to introduce the point mutations K317R, and K317E/R316G/R257G into the construct pCMV6-LOXL2-DDK-Myc: LOXL2-K317R forward 5′-GGACCCTCAAGATTCCGGAGAGCGTACAAGCCAGAGCAAC-3′; LOXL2-K317R reverse 5′-GTTGCTCTGGCTTGTACGCTCTCCGGAATCTTGAGGGTCC-3′; LOXL2-K317E forward 5′-GGACCCTCAAGATTCCGGGAAGCGTACAAGCCAGAGCAAC-3′; LOXL2-K317E reverse 5′-GTTGCTCTGGCTTGTACGCTTGCCGGAATCTTGAGGGTCC-3′; LOXL2-K316G forward 5′-GGACCCTCAAGATTCGGGGAAGCGTACAAGCCAGAGCAAC-3′; LOXL2-K316G reverse 5′-GTTGCTCTGGCTTGTACGCTTCCCCGAATCTTGAGGGTCC-3′; LOXL2-R257G forward 5′-CCTCACGGAGGAAGCAGGGCTACTGGCCATTCTC-3′; LOXL2-R257G reverse 5′-GAGAATGGCCAGTAGCGCTGCTTCCTCCGTGAGG-3′. Clones containing LOXL2-K317R (LOXL2K317R) and LOXL2-K317E/R316G/R257G (LOXL2BP) were verified by DNA sequencing. LOXL2, LOXL2Y689F, LOXL2K317R, and LOXL2BP constructs were transiently transfected in HEK293 cells using Lipofectamine 2000 reagent (Invitrogen).
Amine oxidase activity assay
LOXL2 amine oxidase enzymatic activity was measured in concentrated serum-free conditioned cell culture medium or 4 m urea-soluble PFHR9 matrix proteins using 1,5-diaminopentane as substrate and monitoring hydrogen peroxide production by an Amplex Red fluorescence assay (Invitrogen) (35). The enzymatic reaction was performed in 0.05 m sodium borate, pH 8.2, buffer containing 1 unit/ml of HRP, 10 mm Amplex Red, and 5 mm 1,5-diaminopentane in the presence or absence of 1 mm BAPN. Samples from 4 m urea-extracted ECM were diluted in a final 1 m urea concentration before measurement of amine oxidase activity. After 120-min incubation at 37 °C, fluorescence was measured on a SpectraMax M5 plate reader using excitation and emission wavelengths of 540 and 590 nm, respectively.
7S crosslinking by LOXL2 processing mutants
LOXL2-null PFHR9 cells were transfected with the previously described constructs LOXL2, LOXL2Y689F, LOXL2K317R, and LOXL2BP. Forty-eight h after transfection the cells were selected with 200 μg/ml G418 (Geneticin). Resistant clones were expanded and grown for 7 days after confluency in the presence of 50 μg/ml ascorbic acid. The cells and underlying ECM were scraped in deoxycholate buffer (1% sodium deoxycholate, 25 mm Tris-HCl pH 7.5, 1 mm EDTA-Na, 0.4 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 1 μg/ml pepstatin) and sonicated until homogeneous and centrifuged for 30 min, 14,000 × g. The insoluble pellet was subsequently washed with 1 m NaCl in 25 mm Tris-HCl pH 7.5. For some experiments urea-soluble proteins were extracted by incubating the insoluble pellet with 4 m urea in 25 mm Tris-HCl pH 7.5 for 1 h at room temperature. The remaining pellet was digested overnight with 50 μg/ml collagenase (Worthington Biochemical Corp.) in collagenase buffer (50 mm HEPES, pH 7.5, 5 mm CaCl2, 5 mm benzamidine, 25 mm 6-aminocaproic acid, 0.4 mm phenylmethylsulfonyl fluoride). The supernatant fraction was redigested overnight with 10 μg/ml collagenase. Collagenase-soluble proteins were analyzed by immunoblot using anti-7S polyclonal antibody. The ratio between crosslinked 7S subunits and uncrosslinked 7S monomers was calculated as percentage of the total protein per lane as quantified with Image J (47) gel analysis plugin.
Enzymatic digestion of 7S dodecamers
Ten micrograms of purified 7S dodecamer from PFHR9 cells, bovine glomerulus or bovine placenta were each mixed with an equal volume of 0.1 m Tris-HCl, pH 7.5, buffer containing 8 m guanidine-HCl, and 50 mm dithiothreitol to denature proteins and reduce disulfide bonds. Samples were heated in a boiling water bath for 10 min. After reaching room temperature, proteins were alkylated with 100 mm iodoacetamide in the dark for 45 min. Following ethanol precipitation at −20 °C overnight, the pellets were resuspended in 0.1 m ammonium bicarbonate pH 7.8 and digested with LysC (1:20 enzyme to protein ratio) for 4 h at 37 °C. The samples were heated at 95 °C for 7 min to inactivate LysC. Samples were cooled down to room temperature before adding trypsin at an enzyme to substrate ratio of 1:20 (w/w) for overnight at 37 °C. After trypsin inactivation, peptide samples were deglycosylated with PNGase F at 37 °C overnight. Peptide samples were cleaned using OMIX C18 tips (Agilent) before mass spectrometry analyses.
LC-MS/MS analyses and database searching
7S peptides were analyzed on a Q Exactive mass spectrometer equipped with an Easy nLC-1000 system (Thermo Scientific) as described earlier (49). Approximately a total of 1 μg of peptide was reconstituted in water containing 0.1% formic acid (solvent A) and separated on a PicoFrit (New Objective, Woburn, MA) column (75 μm inner diameter × 110 mm, 10 μm inner diameter tip) packed with Reprosil-Pur C18-AQ resin (3 μm particle size and 120 Å pore size) at a flow rate of 300 nl/min. Peptide mixture was fractionated by a 70-min gradient consisting of the following steps: 0–5 min, increase to 2% B (acetonitrile containing 0.1% formic acid); 5–55 min, 5–35% B; 55–60 min, 90% B, and held at 90% B for 10 min before returning to the initial conditions of 2% B. Data were acquired using a data-dependent method with a scanning window of 300–1800 m/z and dynamic exclusion enabled. Full scans were acquired at a resolution of 70,000, an automatic gain control (AGC) target of 3 × 106, and a 64 ms maximum injection time. The top 20 most abundant ions were selected for fragmentation with higher energy C-trap dissociation (HCD) at a resolution of 17,500, an automatic gain control target of 2 × 105, 100 ms maximum injection time, 2 m/z isolation width, and 27% normalized collision energy. The data were collected with unassigned and +1 charge excluded in the charge state settings. Dynamic exclusion was set to 20 s.
Thermo (.raw) files were independently searched by MyriMatch algorithm run on Windows server maintained by the Vanderbilt University Center for Structural Biology (50). Files were searched against full proteome-level mouse and bovine FASTA databases (NCBI), which also included the reverse sequences for each protein to compute the false discovery rate with a maximum of two missed cleavages and one missed terminus cleavage (semitryptic peptides), with a minimum peptide length of 5 residues and a maximum length of 50 residues. The search was configured to identify peptides with the following amino acid modifications: variable oxidation of methionine (+15.9949) as well as fixed carbamidomethylation of cysteine (+57.021). Precursor and fragment mass tolerance were set to 10 and 20 ppm, respectively. The pepXML files obtained from the search were imported into IDPicker (version 3.1) for the parsimonious grouping of identified proteins with a maximum Q value of 2% for peptide spectrum match (51, 52).
Author contributions
R. M. V. and A .J. L.-J. conceived and coordinated the study. A. J. L.-J., T. B., and R. M. V. designed experiments and acquired and analyzed data. R. M. V. and A. J. L.-J. interpreted data. A. J. L.-J. and R. M. V. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Mohamed Rafi for his skillful technical assistance. We also thank Dr. Gautam Bhave and Dr. Aaron Fidler for critical reading of the manuscript. The Dec-RVKR-CMK inhibitor was a gift from Dr Bhave.
This work was supported by National Institutes of Health Grant R01 DK099467. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ECM
- extracellular matrix
- SRCR
- scavenger receptor cysteine-rich
- Dec
- decanoyl
- CMK
- chloromethylketone
- BAPN
- β-aminopropionitrile
- PC
- proprotein convertase
- LTQ
- lysyl-tyrosyl quinone
- sgRNA
- single guide RNA.
References
- 1. Kagan H. M., and Ryvkin F. (2011) Lysyl oxidase and lysyl oxidase-like enzymes. In The Extracellular Matrix: An Overview (Mecham R. P., ed.), pp. 303–335, Springer-Verlag Berlin Heidelberg, Berlin, Germany [Google Scholar]
- 2. Eyre D. R., Paz M. A., and Gallop P. M. (1984) Cross-linking in collagen and elastin. Annu. Rev. Biochem. 53, 717–748 [DOI] [PubMed] [Google Scholar]
- 3. Reiser K., McCormick R. J., and Rucker R. B. (1992) Enzymatic and nonenzymatic cross-linking of collagen and elastin. FASEB J. 6, 2439–2449 [DOI] [PubMed] [Google Scholar]
- 4. Wang S. X., Mure M., Medzihradszky K. F., Burlingame A. L., Brown D. E., Dooley D. M., Smith A. J., Kagan H. M., and Klinman J. P. (1996) A crosslinked cofactor in lysyl oxidase: Redox function for amino acid side chains. Science 273, 1078–1084 [DOI] [PubMed] [Google Scholar]
- 5. Csiszar K. (2001) Lysyl oxidases: A novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol. 70, 1–32 [DOI] [PubMed] [Google Scholar]
- 6. Lucero H. A., and Kagan H. M. (2006) Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 63, 2304–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Añazco C., López-Jiménez A. J., Rafi M., Vega-Montoto L., Zhang M. Z., Hudson B. G., and Vanacore R. M. (2016) Lysyl oxidase-like-2 cross-links collagen IV of glomerular basement membrane. J. Biol. Chem. 291, 25999–26012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhave G., Cummings C. F., Vanacore R. M., Kumagai-Cresse C., Ero-Tolliver I. A., Rafi M., Kang J. S., Pedchenko V., Fessler L. I., Fessler J. H., and Hudson B. G. (2012) Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat. Chem. Biol. 8, 784–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vanacore R., Ham A. J., Voehler M., Sanders C. R., Conrads T. P., Veenstra T. D., Sharpless K. B., Dawson P. E., and Hudson B. G. (2009) A sulfilimine bond identified in collagen IV. Science 325, 1230–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Timpl R., Wiedemann H., van Delden V., Furthmayr H., and Kühn K. (1981) A network model for the organization of type IV collagen molecules in basement membranes. Eur. J. Biochem. 120, 203–211 [DOI] [PubMed] [Google Scholar]
- 11. Risteli J., Bächinger H. P., Engel J., Furthmayr H., and Timpl R. (1980) 7-S collagen: Characterization of an unusual basement membrane structure. Eur. J. Biochem. 108, 239–250 [DOI] [PubMed] [Google Scholar]
- 12. Yurchenco P. D. (2011) Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 3, a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cano A., Santamaría P. G., and Moreno-Bueno G. (2012) LOXL2 in epithelial cell plasticity and tumor progression. Future Oncol. 8, 1095–1108 [DOI] [PubMed] [Google Scholar]
- 14. Barker H. E., Cox T. R., and Erler J. T. (2012) The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 12, 540–552 [DOI] [PubMed] [Google Scholar]
- 15. Bignon M., Pichol-Thievend C., Hardouin J., Malbouyres M., Bréchot N., Nasciutti L., Barret A., Teillon J., Guillon E., Etienne E., Caron M., Joubert-Caron R., Monnot C., Ruggiero F., Muller L., and Germain S. (2011) Lysyl oxidase-like protein-2 regulates sprouting angiogenesis and type IV collagen assembly in the endothelial basement membrane. Blood 118, 3979–3989 [DOI] [PubMed] [Google Scholar]
- 16. Yang J., Savvatis K., Kang J. S., Fan P., Zhong H., Schwartz K., Barry V., Mikels-Vigdal A., Karpinski S., Kornyeyev D., Adamkewicz J., Feng X., Zhou Q., Shang C., Kumar P., Phan D., Kasner M., López B., Diez J., Wright K. C., Kovacs R. L., Chen P. S., Quertermous T., Smith V., Yao L., Tschöpe C., and Chang C. P. (2016) Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 7, 13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ikenaga N., Peng Z. W., Vaid K. A., Liu S. B., Yoshida S., Sverdlov D. Y., Mikels-Vigdal A., Smith V., Schuppan D., and Popov Y. V. (2017) Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut, 66, 1697–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aumiller V., Strobel B., Romeike M., Schuler M., Stierstorfer B. E., and Kreuz S. (2017) Comparative analysis of lysyl oxidase (like) family members in pulmonary fibrosis. Sci. Rep. 7, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chien J. W., Richards T. J., Gibson K. F., Zhang Y., Lindell K. O., Shao L., Lyman S. K., Adamkewicz J. I., Smith V., Kaminski N., and O'Riordan T. (2014) Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur. Respir. J. 43, 1430–1438 [DOI] [PubMed] [Google Scholar]
- 20. Barry-Hamilton V., Spangler R., Marshall D., McCauley S., Rodriguez H. M., Oyasu M., Mikels A., Vaysberg M., Ghermazien H., Wai C., Garcia C. A., Velayo A. C., Jorgensen B., Biermann D., Tsai D., et al. (2010) Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 16, 1009–1017 [DOI] [PubMed] [Google Scholar]
- 21. Ma L., Huang C., Wang X. J., Xin D. E., Wang L. S., Zou Q. C., Zhang Y. S., Tan M. D., Wang Y. M., Zhao T. C., Chatterjee D., Altura R. A., Wang C., Xu Y. S., Yang J. H., et al. (2017) Lysyl oxidase 3 is a dual-specificity enzyme involved in STAT3 deacetylation and deacetylimination modulation. Mol. Cell 65, 296–309 [DOI] [PubMed] [Google Scholar]
- 22. Resnick D., Pearson A., and Krieger M. (1994) The SRCR superfamily: A family reminiscent of the Ig superfamily. Trends Biochem. Sci. 19, 5–8 [DOI] [PubMed] [Google Scholar]
- 23. Xu L., Go E. P., Finney J., Moon H., Lantz M., Rebecchi K., Desaire H., and Mure M. (2013) Post-translational modifications of recombinant human lysyl oxidase-like 2 (rhLOXL2) secreted from Drosophila S2 cells. J. Biol. Chem. 288, 5357–5363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akiri G., Sabo E., Dafni H., Vadasz Z., Kartvelishvily Y., Gan N., Kessler O., Cohen T., Resnick M., Neeman M., and Neufeld G. (2003) Lysyl oxidase-related protein-1 promotes tumor fibrosis and tumor progression in vivo. Cancer Res. 63, 1657–1666 [PubMed] [Google Scholar]
- 25. Vadasz Z., Kessler O., Akiri G., Gengrinovitch S., Kagan H. M., Baruch Y., Izhak O. B., and Neufeld G. (2005) Abnormal deposition of collagen around hepatocytes in Wilson's disease is associated with hepatocyte specific expression of lysyl oxidase and lysyl oxidase like protein-2. J. Hepatol. 43, 499–507 [DOI] [PubMed] [Google Scholar]
- 26. Fong S. F., Dietzsch E., Fong K. S., Hollosi P., Asuncion L., He Q., Parker M. I., and Csiszar K. (2007) Lysyl oxidase-like 2 expression is increased in colon and esophageal tumors and associated with less differentiated colon tumors. Genes Chromosomes Cancer 46, 644–655 [DOI] [PubMed] [Google Scholar]
- 27. Hollosi P., Yakushiji J. K., Fong K. S., Csiszar K., and Fong S. F. (2009) Lysyl oxidase-like 2 promotes migration in noninvasive breast cancer cells but not in normal breast epithelial cells. Int. J. Cancer 125, 318–327 [DOI] [PubMed] [Google Scholar]
- 28. Uzel M. I., Scott I. C., Babakhanlou-Chase H., Palamakumbura A. H., Pappano W. N., Hong H. H., Greenspan D. S., and Trackman P. C. (2001) Multiple bone morphogenetic protein 1-related mammalian metalloproteinases process pro-lysyl oxidase at the correct physiological site and control lysyl oxidase activation in mouse embryo fibroblast cultures. J. Biol. Chem. 276, 22537–22543 [DOI] [PubMed] [Google Scholar]
- 29. Borel A., Eichenberger D., Farjanel J., Kessler E., Gleyzal C., Hulmes D. J., Sommer P., and Font B. (2001) Lysyl oxidase-like protein from bovine aorta. Isolation and maturation to an active form by bone morphogenetic protein-1. J. Biol. Chem. 276, 48944–48949 [DOI] [PubMed] [Google Scholar]
- 30. Moon H. J., Finney J., Ronnebaum T., and Mure M. (2014) Human lysyl oxidase-like 2. Bioorg. Chem. 57, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Denault J. B., D'Orleans-Juste P., Masaki T., and Leduc R. (1995) Inhibition of convertase-related processing of proendothelin-1. J. Cardiovasc. Pharmacol. 26, Suppl. 3, S47–S50 [PubMed] [Google Scholar]
- 32. Remacle A. G., Shiryaev S. A., Oh E. S., Cieplak P., Srinivasan A., Wei G., Liddington R. C., Ratnikov B. I., Parent A., Desjardins R., Day R., Smith J. W., Lebl M., and Strongin A. Y. (2008) Substrate cleavage analysis of furin and related proprotein convertases. A comparative study. J. Biol. Chem. 283, 20897–20906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duckert P., Brunak S., and Blom N. (2004) Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 17, 107–112 [DOI] [PubMed] [Google Scholar]
- 34. Barré O., Dufour A., Eckhard U., Kappelhoff R., Béliveau F., Leduc R., and Overall C. M. (2014) Cleavage specificity analysis of six type II transmembrane serine proteases (TTSPs) using PICS with proteome-derived peptide libraries. PLoS One 9, e105984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Palamakumbura A. H., and Trackman P. C. (2002) A fluorometric assay for detection of lysyl oxidase enzyme activity in biological samples. Anal. Biochem. 300, 245–251 [DOI] [PubMed] [Google Scholar]
- 36. Colon S., and Bhave G. (2016) Proprotein convertase processing enhances peroxidasin activity to reinforce collagen IV. J. Biol. Chem. 291, 24009–24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferguson T. E., Reihill J. A., Walker B., Hamilton R. A., and Martin S. L. (2016) A selective irreversible inhibitor of furin does not prevent pseudomonas aeruginosa exotoxin A-induced airway epithelial cytotoxicity. PLoS One 11, e0159868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim M. S., Kim S. S., Jung S. T., Park J. Y., Yoo H. W., Ko J., Csiszar K., Choi S. Y., and Kim Y. (2003) Expression and purification of enzymatically active forms of the human lysyl oxidase-like protein 4. J. Biol. Chem. 278, 52071–52074 [DOI] [PubMed] [Google Scholar]
- 39. Kagan H. M. (1994) Lysyl oxidase: Mechanism, regulation and relationship to liver fibrosis. Pathol. Res. Pract. 190, 910–919 [DOI] [PubMed] [Google Scholar]
- 40. Kim Y. M., Kim E. C., and Kim Y. (2011) The human lysyl oxidase-like 2 protein functions as an amine oxidase toward collagen and elastin. Mol. Biol. Rep. 38, 145–149 [DOI] [PubMed] [Google Scholar]
- 41. Rodriguez H. M., Vaysberg M., Mikels A., McCauley S., Velayo A. C., Garcia C., and Smith V. (2010) Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J. Biol. Chem. 285, 20964–20974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hutchinson J. H., Rowbottom M. W., Lonergan D., Darlington J., Prodanovich P., King C. D., Evans J. F., and Bain G. (2017) Small molecule lysyl oxidase-like 2 (LOXL2) inhibitors: The identification of an inhibitor selective for LOXL2 over LOX. ACS Med. Chem. Lett. 8, 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raghu G., Brown K. K., Collard H. R., Cottin V., Gibson K. F., Kaner R. J., Lederer D. J., Martinez F. J., Noble P. W., Song J. W., Wells A. U., Whelan T. P., Wuyts W., Moreau E., Patterson S. D., Smith V., Bayly S., Chien J. W., Gong Q., Zhang J. J., and O'Riordan T. G. (2017) Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: A randomised, double-blind, controlled, phase 2 trial. Lancet Respir. Med. 5, 22–32 [DOI] [PubMed] [Google Scholar]
- 44. Lorz C., Benito-Martín A., Boucherot A., Ucero A. C., Rastaldi M. P., Henger A., Armelloni S., Santamaría B., Berthier C. C., Kretzler M., Egido J., and Ortiz A. (2008) The death ligand TRAIL in diabetic nephropathy. J. Am. Soc. Nephrol. 19, 904–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neusser M. A., Lindenmeyer M. T., Moll A. G., Segerer S., Edenhofer I., Sen K., Stiehl D. P., Kretzler M., Gröne H. J., Schlöndorff D., and Cohen C. D. (2010) Human nephrosclerosis triggers a hypoxia-related glomerulopathy. Am. J. Pathol. 176, 594–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Breyer M. D., and Susztak K. (2016) The next generation of therapeutics for chronic kidney disease. Nat. Rev. Drug Discov. 15, 568–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: An open-source platform for biological-image analysis. Nat. Meth. 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Basak T., Vega-Montoto L., Zimmerman L. J., Tabb D. L., Hudson B. G., and Vanacore R. M. (2016) Comprehensive characterization of glycosylation and hydroxylation of basement membrane collagen IV by high-resolution mass spectrometry. J. Proteome Res. 15, 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tabb D. L., Fernando C. G., and Chambers M. C. (2007) MyriMatch: Highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Holman J. D., Ma Z. Q., and Tabb D. L. (2012) Unit 13.17 Identifying proteomic LC-MS/MS data sets with Bumbershoot and IDPicker. Curr. Protoc. Bioinformatics 37, 13.17: 13.17.1–13.17.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ma Z. Q., Dasari S., Chambers M. C., Litton M. D., Sobecki S. M., Zimmerman L. J., Halvey P. J., Schilling B., Drake P. M., Gibson B. W., and Tabb D. L. (2009) IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 8, 3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]