

Abstract
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, has developed multiple strategies to adapt to the human host. The five type VII secretion systems, ESX-1–5, direct the export of many virulence-promoting protein effectors across the complex mycobacterial cell wall. One class of ESX substrates is the PE–PPE family of proteins, which is unique to mycobacteria and essential for infection, antigenic variation, and host–pathogen interactions. The genome of Mtb encodes 168 PE–PPE proteins. Many of them are thought to be secreted through ESX-5 secretion system and to function in pairs. However, understanding of the specific pairing of PE–PPE proteins and their structure–function relationship is limited by the challenging purification of many PE–PPE proteins, and our knowledge of the PE–PPE interactions therefore has been restricted to the PE25–PPE41 pair and its complex with the ESX-5 secretion system chaperone EspG5. Here, we report the crystal structure of a new PE–PPE pair, PE8–PPE15, in complex with EspG5. Our structure revealed that the EspG5-binding sites on PPE15 are relatively conserved among Mtb PPE proteins, suggesting that EspG5–PPE15 represents a more typical model for EspG5–PPE interactions than EspG5–PPE41. A structural comparison with the PE25–PPE41 complex disclosed conformational changes in the four-helix bundle structure and a unique binding mode in the PE8–PPE15 pair. Moreover, homology-modeling and mutagenesis studies further delineated the molecular determinants of the specific PE–PPE interactions. These findings help develop an atomic algorithm of ESX-5 substrate recognition and PE–PPE pairing.
Keywords: bacterial pathogenesis, protein complex, protein secretion, protein structure, tuberculosis
Introduction
Tuberculosis (TB),2 which is primarily caused by Mycobacterium tuberculosis (Mtb) infection, causes ∼2 million deaths annually and therefore remains one of the most devastating diseases worldwide (1, 2). The recent emergence of multidrug-resistant TB and HIV co-infection has highlighted the urgent need for more effective new vaccines (3, 4). Therefore, it is critical to understand the virulent determinants and components of Mtb that are responsible for the host immune response and host–pathogen interactions during different stages of TB infection. The genomes of Mtb and other pathogenic mycobacteria have revealed the prominence of the pe and ppe gene families. For example, the Mtb H37Rv strain contains 99 pe genes and 69 ppe genes, thus highlighting the importance of this protein repertoire for mycobacterial survival and pathogenesis (5). Each PE or PPE protein contains a highly conserved N-terminal domain with a Pro-Glu or Pro-Pro-Glu motif, respectively. Most PE–PPE proteins also possess a variable C-terminal domain that contributes to structural and functional diversification within the protein family. Gene neighborhood and co-expression analyses suggest that PE and PPE proteins act in complexes (6, 7), and these interactions are well-exemplified by the PE25–PPE41 complex (8). Although the exact biological roles of most PE–PPE proteins remain unknown, some of them have been associated with antigenic variation (9–11), immune response modulation (12, 13), drug resistance (14–16), and Mtb virulence (17, 18).
PE–PPE proteins are commonly thought to be either secreted or presented on the cell surface, in line with their functional properties (19, 20). The secretion and surface translocation of PE–PPE proteins is associated with a unique, specialized set of type VII secretion systems (ESX-1 to ESX-5) (21, 22). Earlier studies have demonstrated that recognition of the ESX substrates by the cognate ESX machinery is mediated through a YXXXD/E secretion signal motif and a WXG motif, which is present in many ESX substrates, including PE–PPE proteins, WxG100 family proteins, and some Esp proteins. However, this signal motif does not define the specificity of secretion system (23, 24). Recently, the crystal structure of the ESX-5–encoded chaperone EspG5 in complex with PE25–PPE41 was solved to reveal the molecular determinants of PPE secretion through specific binding with EspGs (25, 26). Current predictions suggest that ∼95% of PPE proteins in Mtb interact with EspG5 and are secreted by the ESX-5 secretion system.
Phylogenetic analysis suggests that pe/ppe genes co-evolved with esx loci and underwent specific gene expansion (20, 27). For ESX-5, three duplicated gene clusters (ESX-5a, ESX-5b, and ESX-5c) are located distal to the ESX-5 region in the Mtb genome (28). Although little is known about the functions of these clusters, ESX-5a, which encodes two ESX proteins and PE8 and PPE15, is considered an accessory to the parental ESX-5 export apparatus and is responsible for the secretion of a subset of PE–PPE proteins (29). Up-to-date structural data of the PE–PPE protein complex are scarce, mainly because of difficulties associated with PE–PPE protein expression and purification (8). For example, individually expressed PEs and PPEs are highly insoluble. The only available relevant crystallographic structure of a PE–PPE pair was initially published more than 10 years ago to describe PE25–PPE41 (8), However, the atomic details of other PE–PPE pairs are essential to a better understanding of the distinct protein repertoire required for Mtb infection and pathogenesis. Here, we report the molecular interaction of a novel PE–PPE pair, PE8–PPE15, which is located within ESX-5a and is phylogenetically distinct from PE25–PPE41. A structural comparison with EspG5–PPE41 reveals that the EspG5-binding interface on PPE15 is relatively conserved than EspG5–PPE41, suggesting that the EspG5–PPE15 structure could represent a typical model for EspG5–PPE interactions. Our structure also highlights the structural flexibility induced by the highly conserved prolines and glycines present in the four helix bundle of the PE–PPE complex. Using a homology model of three other PE–PPE pairs and mutagenesis analysis, we identify the molecular determinants of specific PE–PPE recognition.
Results and discussion
Production of the EspG5–PE81–99–PPE151–194 complex for crystallographic studies
Previous bioinformatics analyses predicted the interaction of PE8 (Rv1040c) with PPE15 (Rv1039c) (6, 7). Here, our team used yeast two-hybrid and pulldown assays to validate the direct interaction of these proteins. Because full-length PE8 and PPE15 are highly insoluble, truncated fragments were constructed to improve solubility and define the minimum binding regions (supplemental Fig. S1a). Our results showed that the N-terminal domains of PE8 (residues 1–99) and PPE15 (residues 1–194) are necessary for PE8–PPE15 complex formation. Subsequently, PE81–99 and PPE151–194 were co-expressed and co-purified with the intent to obtain a sufficient sample for structural analysis. However, the protein complex was prone to aggregation at high concentrations, and crystallization trials using this recombinant material failed to yield well-diffracting crystals. To improve the solubility and stability of the protein complex, we included EspG5, which has been reported as a specific chaperone for PE–PPE proteins (30), in the co-purification experiment. Because pe8 and ppe15 are located within the ESX-5a duplicated gene cluster, we hypothesized that EspG5 might interact with the PE8–PPE15 pair. We confirmed the binding of EspG5 to PE81–99–PPE151–194 using a pulldown assay (supplemental Fig. S1b) and further purified this ternary complex to a high level of homogeneity (Fig. 1a). The results of sedimentation velocity and static light scattering experiments yielded a molecular mass of 65–66 kDa for the EspG5–PE81–99–PPE151–194 complex, indicating that these three proteins exist in a stoichiometric ratio of 1:1:1 (Fig. 1b and supplemental Fig. S2). The frictional ratio of 1.96, obtained through a sedimentation velocity analysis, also revealed that the protein complex forms an elongated shape in solution (Fig. 1b).
Figure 1.

Overview of the EspG5–PE81–99–PPE151–194 protein complex. a, elution profile of the EspG5–PE81–99–PPE151–194 complex from size-exclusion chromatography using Superdex 200. Peak fractions as indicated were analyzed by SDS-PAGE analysis. b, a sedimentation velocity ultracentrifugation analysis of the purified EspG5–PE81–99–PPE151–194 complex determined the following: molecular size of 65.0 kDa, frictional ratio of 1.9, suggested ratio of 1:1:1, and elongated shape. The calculated molecular masses of EspG5, PE81–99, and PPE151–194 are 35.0, 10.0, and 20.0 kDa, respectively. c, the crystal structure of the M. tuberculosis EspG5–PE81–99–PPE151–194 complex is depicted as a cartoon in two views with 180° rotation. EspG5 (warm pink) binds exclusively with PPE151–194 (cyan), whereas PE81–99 (yellow) interacts with PPE151–194 to form a four-helix bundle. d, the contact surfaces between EspG5 and PPE151–194 and between PPE151–194 and PE81–99. The molecular surfaces of EspG5 and PPE151–194 are colored according to the electrostatic potential. PPE151–194 (left, cyan) and PE81–99 (right, yellow) are depicted in cartoon mode.
Overall structure of EspG5–PE81–99–PPE151–194
The purified EspG5–PE81–99–PPE151–194 ternary complex was readily amenable to crystallization trials and yielded crystals that diffracted to a 2.9 Å resolution (Table 1). The structure was determined via molecular replacement as implemented in the program suite phenix.mr_rosetta, using the EspG5–PE25–PPE41 structure (PDB code 4W4L) as the search model. The electron density map was clearly defined throughout the structure, except for residues 85–99 in PE8 and residues 174–194 in PPE15, in line with a secondary structure prediction by Phyre2 (31), suggesting that these regions are highly disordered. This disordered region of PE8 includes the YXXXD/E secretion motif. The final structure contained residues 7–299 of EspG5, residues 7–84 of PE8, and residues 1–173 of PPE15. The overall structure of EspG5–PE81–99–PPE151–194 is similar to the previously reported structure of EspG5–PE25–PPE41 (Fig. 1c) (25, 26), with RMSD of 0.485 Å (EspG5), 2.475 Å (PE), and 2.037 Å (PPE). EspG5 interacts exclusively with helices α4 and α5 of PPE15 at the opposite end of the PE81–99–PPE151–194heterodimer, and no direct contact between EspG5 and PE81–99 was observed (Fig. 1, c and d). PE8 comprises two helices that interact with helices α1, α2, α3, and α5 of PPE15 to form a four-helix bundle. Structural comparisons between EspG5–PPE15 and EspG5–PPE41 and between PE8–PPE15 and PE25–PPE41 will be discussed in detail in later sections.
Table 1.
Data collection and refinement statistics
One crystal was used for each structure.
| EspG5–PE81–99–PPE151–194 (PDB code 5XFS) | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a (Å) | 54.74 |
| b (Å) | 69.96 |
| c (Å) | 203.55 |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 90 |
| Resolution (Å) | 29.60–2.90 (3.08–2.90)a |
| Rmerge (%) | 0.133 (0.533) |
| I/σ(I) | 6.7 (2.1) |
| Completeness | 99.5 (99.9) |
| Redundancy | 4.3 (4.4) |
| CC½ | 0.991 (0.775) |
| Refinement | |
| Resolution (Å)a | 29.599–2.900 (3.004–2.900) |
| No. reflections | 17,900 (1,762) |
| Rwork/Rfree (%) | 21.33/26.24 |
| No. of atoms | 3,880 |
| Protein | 3,853 |
| Water | 27 |
| B-factors | |
| Protein | 61.8 |
| Water | 39.4 |
| RMSD | |
| Bond lengths (Å) | 0.0086 |
| Bond angles (°) | 1.11 |
| Ramachandran (%) | |
| Favored | 95.93 |
| Allowed | 4.07 |
| Disallowed | 0 |
a The values given in parentheses are for the highest-resolution shell.
Recently, the atomic structure of a ESX-1 substrate EspB has been determined (32, 33), EspB adopts a PE–PPE like fold, and superimposition of EspB (PDB code 4WJ1) with PE8–PPE15 gives an RMSD of 2.114 Å (PE) and 1.764 Å (PPE) (supplemental Fig. S3a). Major structural differences lie on a short helix α1, extended α1–α2 loop and helix α2 in the PE domain of EspB, and a short α6–α7 loop for EspG binding in the PPE domain of EspB. We also compared the YXXXD/E secretion motif located in the C terminus of PE domain and the WXG motif in the helix-turn-helix region of PPE domain in PE8–PPE15, PE25–PPE41, and EspB (supplemental Fig. S3b). In the PE25–PPE41 and EspB structures, the YXXXD/E motif and the WXG motif are in close proximity, allowing van der Waals contact between Tyr87PE25 and Trp56PPE41, and a hydrogen bond formation between Tyr78EspB and Trp181EspB. Interestingly, the electron density for the 87YXXXE91 motif in PE8 cannot be seen, and the side chain of Trp57 in the WXG motif of PPE15 is flipped away from the PE–PPE–binding interface. Although our current structure only contains the PE–PPE domain of PE8–PPE15, it is difficult to predict the orientation of the 87YXXXE91 motif, which is located in the linker region before the C-terminal 184 residues of the full-length PE8. It is likely that Tyr87PE8 is in a flexible state, and its interaction with Trp57PPE15, if it exists, is distinct from that in PE25–PPE41 and EspB. However, the functional significance of these variations in ESX secretion needs further investigation.
EspG5–PPE151–194 provides a more typical model for EspG5–PPE interactions
A PDBsum (34) analysis of the crystal structure of EspG5–PE81–99–PPE151–194 showed that the contact surface between EspG5 and PPE15 measured ∼2654 Å, with an interface comprising 23 residues from EspG5 and 21 residues from PPE15. The interaction mainly involves helix α1′, the central β-sheet, the α1–α2 loop, and the β2–β3 loop of EspG5 and helices α4 and α5 of PPE15 (Fig. 2a). On PPE15, the main EspG5 contact regions are localized in helices α4 and α5, which contain residues 121–152. We further divided the binding interface of EspG5–PPE15 into three patches for comparison with the EspG5–PPE41 complex (Fig. 2, b and c). The first patch included residues Val121, Asn124, Thr130, and Trp144 from PPE15, which interact with the β2–β3 loop in EspG5. The molecular interactions in this patch are mediated by a hydrogen bond formation between Asn124PPE15 and Tyr96EspG5 and by hydrophobic contacts of Val121PPE15 with Val98EspG5 and Trp144PPE15 with Arg109EspG5. An identified intramolecular hydrogen bond between Thr130 and Asn124 in PPE15 likely stabilizes the helix-turn-helix tip of PPE15 (supplemental Fig. S4a). Residues in this interaction patch are highly conserved among the Mtb PPE proteins, including PPE41.
Figure 2.

The binding interface between EspG5 and PPE15. a, key structural elements involved in the EspG5–PPE15 interaction are highlighted in warm pink and cyan, respectively. b, an “open book” view of the EspG5–PPE15 interacting surface. EspG5 and the helix-turn-helix of PPE15 are shown with electrostatic surfaces. Residues that interact with the α1–α2 loop, with the helix α1′ and β sheet face, and with the β2–β3 loop of EspG5 are labeled in blue, black, and purple, respectively. c, sequence conservation of the EspG5 binding sites was presented by WebLogo using multiple sequence alignment of all PPE proteins in Mtb. Secondary structure elements of α4–α5 in PPE15 are indicated. A corresponding sequence alignment of PPE15 and PPE41 is shown underneath, and residues involved in EspG5 binding are labeled with red and blue triangles, respectively. d, microscale thermophoresis analysis of EspG5/PE81–99–PPE151–194 and EspG5/PE25–PPE41 interactions. The calculated values for the dissociation constant Kd are indicated.
The second interface patch is generated by the insertion of the helix-turn-helix tip of PPE15 into a hydrophobic pocket formed by the α1′-helix and central β-sheet of EspG5. Specifically, this patch comprises Val125, Leu126, Ile128, and Pro131 of PPE15 and Leu180, Leu216, Leu237, and Val241 of EspG5 (supplemental Fig. S4, b and c). Although the majority of PPE proteins adopt hydrophobic residues at residues equivalent to 125, 126, and 131 in PPE15, PPE41 contains a glutamine residue in the position equivalent to residue 128 in PPE15. Gln127 in PPE41 forms hydrogen bonds with the side chain of Gln256 and the main chain atoms of Val241 in EspG5, suggesting a relatively stronger interaction between PPE41 and EspG5. Nevertheless, interface patches 1 and 2 appear to be common among EspG5–PPE complexes.
The third binding interface patch includes interactions between the α5 helix of PPE15 and residues from the α1–α2 loop of EspG5. Interestingly, a comparison of the interactions in EspG5–PPE15 and EspG5–PPE41 revealed different binding modes in this patch. Specifically, in PPE15, this patch is rich in hydrophobic residues such as Met134, Ala138, Ala141, Ala148, Leu149, and Tyr152 and contains only one salt bridge, Glu137PPE15::Arg34EspG5 (supplemental Fig. S4d). By contrast, PPE41 in this patch contains a stretch of charged residues, as well as four salt bridges: Asp140PPE41::Arg109EspG5, Asp144PPE41::Arg27EspG5, Glu148PPE41::Arg27EspG5, and Arg154PPE41::Asp42EspG5. A sequence conservation analysis (Fig. 2c) reveals that apart from residue 141, many PPE proteins, including PPE15, carry hydrophobic residues in patch 3. This strongly suggests that the atomic structure of EspG5–PPE15 determined herein represents a typical model for EspG5–PPE interactions. Because patch 3 accounts for almost 50% of the total interface area, the binding affinity of EspG5–PPE41 is likely stronger relative to that of other EspG5–PPE proteins. We further analyzed the binding kinetics of EspG5 to PE81–99–PPE151–194 and EspG5 to PE25–PPE41 using microscale thermophoresis (Fig. 2d). The calculated dissociation constants of EspG5/PE81–99–PPE151–194 is 132 nm, whereas that of EspG5/PE25–PPE41 is 51 nm, indicating that EspG5–PPE15 or most EspG5–PPE proteins have a slightly weaker binding affinity than EspG5–PPE41.
Comparison of PE81–99–PPE151–194 with PE25–PPE41 reveals structural plasticity and a unique binding mode
Although PE81–99–PPE151–194 and PE25–PPE41 exhibit very similar folding characteristics, pronounced bending was observed in the four-helix bundle distal from the EspG5-binding area (Fig. 3a and supplemental Fig. S5). Specifically, the helical pairs α1 and α2 in PE8 and α2 and α3 in PPE15 are tilted by ∼26–29 and 20–23°, respectively, leading to dramatic shifts in the helical directions (Fig. 3b). In these four tilted helices, the kinks start at similar longitudinal positions and are facilitated by either a proline (Pro35PE8, Pro71PPE15) or a glycine residue (Gly59PE8, Gly39PPE15). In PE8 and PPE15, various highly conserved alanine residues are found proximal to the kinks, thus further promoting helical bending (Fig. 3c). It is noted that the α2 helix of PPE15 also contains two other highly conserved glycine residues (Gly22 and Gly33) that might also contribute to conformational changes. It is likely that the co-existing kinks in the helical pairs of PE8 and PPE15 is a cooperative effect that allows these two PE and PPE proteins to carry the same extent of helical bending for interaction. The presence of numerous highly conserved proline, glycine, and alanine residues in helices α1–α2 of PE and in helices α2–α3 of PPE proteins suggest that these helices may display different degrees of helical bending required for specific PE–PPE pair formation.
Figure 3.

Structural comparison of the PE8–PPE15 and PE25–PPE41 complexes. a, superposition of PE81–99–PPE151–194 (yellow and cyan) and PE25–PPE41 (orange and blue), showing different degrees of helical bending in PE and PPE (boxed in red). b, arrangement of the four-helix bundles of PE–PPE complexes viewed from the longitudinal axis (bottom to top view). The maximum distances between the two corresponding bent helices in PE8–PPE15 and PE25–PPE41 are indicated. c, helical kinks in α1 and α2 of PE8 and PE25 and α2 and α3 of PPE15 and PPE41. The prolines and glycines positioned at the kinks are highlighted as spheres, and proximal alanines are indicated by sticks. Sequence alignments of these residues between PE8 and PE25 and between PPE15 and PPE41 are shown. The sequence conservation of these residues among all Mtb PE–PPE proteins is presented by WebLogo.
As in PE25–PPE41, PE81–99–PPE151–194 complex formation is mediated by both electrostatic and hydrophobic interactions. Both complexes contain a hydrogen bond (Ser48 in the α2 helix of PE8 interacts with Tyr154 in the α5 helix of PPE15), and the interior of the four-helix bundle is lined with multiple hydrophobic contacts. However, distinct salt bridges and hydrogen bonds are found at the upper and lower areas of the PE81–99–PPE151–194 complex. We identified four sites in PE8–PPE15 interactions and further validated their importance using mutagenesis and pulldown assays (Fig. 4, a and b). These four sites include a salt bridge (Glu46PE8::Arg14PPE15) and three hydrogen bonds (Gln51PE8–Ser93PPE15, His73PE8–Tyr45PPE15, and Gln70PE8–Tyr72PPE15). Single alanine substitutions of the Arg14, Ser93, Tyr45, and Tyr72 residues in full-length PPE15 significantly reduced the interaction of this protein with PE81–99 (Fig. 4b), suggesting that these residues are essential for the PE8–PPE15 interaction. Interestingly, the PE8–PPE15 interaction was not completely abolished in PPE15 R14A/S93A and Y45A/Y72A double mutants or even quadruple mutant R14A/S93A/Y45A/Y72A. It is possible that the residual binding with PE8 observed in these PPE15 mutants was attributed by the conserved hydrogen bond Ser48PE8–Tyr154PPE15. Therefore, the PPE15 Y154A single mutant and R14A/S93A/Y45A/Y72A/Y154A quintuple mutant were created to examine the importance of this hydrogen contact in PE8–PPE15 interaction. Similar to other single mutants described above, mutation of residue Tyr154 reduced the binding of PE8. It is noteworthy that PE8–PPE15 interaction was totally impaired in the PPE15 quintuple mutant (Fig. 4b). These results indicate that the conserved hydrogen bond (Ser48PE8–Tyr154PPE15) is critical for minimal binding of PE and PPE proteins but strong and specific PE–PPE complex formation involves multiple binding sites along the helix bundles. To confirm that these PPE15 mutants were properly folded, their expression and solubility were examined by immunoblotting (supplemental Fig. S6). All PPE15 mutants exhibited solubility similar to that of the wild-type protein. When we analyzed the PE25–PPE41 structure and sequence alignment, the equivalent residues at these four sites were found to be mainly non-polar (Fig. 4c). The PE25–PPE41 complex binding interface contains one salt bridge (Arg24PE25::Glu37PPE41) and one hydrogen bond (Glu17PE25–Thr48PPE41). These two interactions are not seen in the PE8–PPE15 complex, which contains the equivalent residues of Ala24PE8 and Glu37PPE15 and Gln17PE8 and Val48PPE15 (supplemental Fig. S7). These findings suggest that apart from the conserved hydrogen bond (Ser48PE8–Tyr154PPE15) and hydrophobic contacts buried in the helix bundle, the two PE and PPE complexes have adopted unique sets of complementary residues that are essential for binding affinity and specificity.
Figure 4.

Specific recognition between PE8 and PPE15. a, detailed view of the interaction sites revealed from the PE8–PPE15 structure. b, validation of the PE8 and PPE15 interaction by pulldown assays. Lysates containing co-expressed GST-tagged PE81–99 and His-tagged PPE15 or PPE15 mutants were subjected to pulldown assays using glutathione-Sepharose. Pulldown products were examined by SDS-PAGE and immunoblotting using anti-His antibody. The result shows that the residues in PPE15 indicated in a are essential for the binding of PE8. c, structure based sequence alignments between PE8 (residues 1–99) and PE25 and between PPE15 (residues 1–104) and PPE41 are shown. Secondary structural elements of PE8 and PPE15 are shown above the sequences. Unique interacting sites in the PE8–PPE15 complex, as shown in a, are indicated by corresponding red numbers. The conserved hydrogen bond formed between Ser48PE8 and Tyr154PPE15 is not indicated.
PE–PPE interaction requires specific set of complementary residues and helical bending
We further extended our understanding to other PE–PPE complexes according to our obtained PE8–PPE15 structure. A homology detection by HHpred (35) identified 8 PE proteins and 29 PPE proteins in Mtb that share more than 45% sequence identities with PE8 and PPE15, respectively (supplemental Table S1). Of these, we selected three PE–PPE pairs that were previously predicted by a bioinformatics analysis (6): PE27–PPE43, PE13–PPE18, and PE32–PPE65, and examined their interactions using yeast two-hybrid assays (Fig. 5a). These three PE–PPE pairs and PE8–PPE15 are classified in the same phylogenetic sublineage IV and are believed to have co-evolved and co-expanded (supplemental Fig. S8). PE8–PPE15, PE13–PPE18, and PE32–PPE65 also constitute the three ESX-5 duplicated clusters (ESX-5a, ESX-5b, and ESX-5c) (28) (Fig. 5b), whereas PE27–PPE43 is associated with the ESX-5 secretion system (27). Homology models of these three PE–PPE pairs were generated using Modeller (36), and their binding interfaces were analyzed by PDBsum (34) and compared with the five interacting sites identified in PE8–PPE15 (Figs. 4 and 5c). The conserved hydrogen bond observed in Ser48PE8–Tyr154PPE15 was also found in these three PE–PPE pairs. However, for the other four PE–PPE binding sites, variations were noted. For site 1, the salt bridge between Glu46PE8 and Arg14PPE15 was conserved in PE27–PPE43 and PE13–PPE18. However, this site was replaced by hydrophobic contacts in PE32 (Leu46) and PPE65 (Leu15). The alignment of all PE–PPE proteins in Mtb revealed that 60% of PE–PPE complexes proteins contain the equivalent residues Glu and Arg, suggesting that most complexes adopt a salt bridge to maintain contact between the α2 helix of PE and the α1 helix of PPE. At site 2, hydrogen bonding between Gln51PE8 and Ser93PPE15 was only conserved in PE27–PPE43. In PE13–PPE18 and PE32–PPE65, however, Thr51PE13/PE32 can form a hydrogen bond with Thr163PPE18/PPE65. Interestingly, the residues at sites 3 and 4 were more variable. The hydrogen bond network in PE32–PPE65 is mediated through Gln73PE32, with Tyr46PPE65 and Gln73PPE65. Although PE13–PPE18 and PE27–PPE43 lack hydrogen bonds at sites 3 and 4, helical packing in the lower parts of the helix bundles is facilitated respectively by an Arg68PE13::Glu171PPE18 salt bridge and a Lys17PE27–Gln51PPE43 hydrogen bond. An additional hydrogen bond (His58PE32–Gln83PPE65) appears in the middle of the helix bundle in PE32–PPE65. Taken together, although only some of the interactions are highly conserved, PE–PPE proteins adopt specific sets of complementary residues for complex formation.
Figure 5.

Molecular interactions of other PE–PPE protein complexes in sublineage IV. a, interactions of the PE8–PPE15, PE13–PPE18, PE27–PPE43, PE32–PPE65, and PE25–PPE15 pairs as revealed by yeast two-hybrid assays. PE proteins were fused with the transcriptional activation domain (AD), and PPE proteins were fused with the DNA-binding domain (BD) or vice versa in the yeast two-hybrid assays. Positive interacting pairs are indicated by blue colonies grown on QDO/X/A plates. Interaction between PE25 and PPE15 was not observed. b, genome organization of ESX-5 and the three duplicated esx gene clusters in Mtb, namely ESX-5a, ESX-5b, and ESX-5c. The PE8–PPE15, PE13–PPE18, and PE32–PPE65 pairs are respectively located in the three duplicated ESX-5 regions. c, homology modeling of the PE27–PPE43, PE13–PPE18, and PE32–PPE65 complexes, showing a specific set of hydrogen bonds and salt bridges in each protein complex. The binding sites of interest in the overall structure are indicated by colored dotted circles, and the interacting residues are indicated by sticks in enlarged boxes. d, interaction studies of PE25–PPE15 via pulldown assays. A lysate containing co-expressed GST-tagged PE25 or mutant versions and His-tagged PPE15 was subjected to a pulldown assay using glutathione-Sepharose. PE25 and its mutants, as indicated in the table at left, did not interact with PPE15. The expression level and solubility of PPE15 were confirmed by Western blotting. Positive controls used co-expressed GST-PE25 and His-tagged PPE41 or GST-PE81–99 and His-tagged PPE15.
To test whether the specific interacting residues identified in PE8–PPE15 are the major determinants of binding specificity, we created various PE25 mutants, including PE25 A51Q, PE25 A51Q/L46E, PE25 A51Q/L46E/A70Q, and PE25 A51Q/L46E/A70Q/L73H. We hypothesized that the substitution of these residues in PE25 with their equivalents from PE8 would allow an interaction with PPE15 (Fig. 4a). Results from a GST pulldown assay revealed that none of these mutants could interact with PPE15 (Fig. 5d). We therefore considered that multiple sites along the interface are required for stabilization of the whole PE–PPE complex, which would explain why the single-, double-, and triple-amino acid PE25 mutant failed to interact with PPE15. However, we expected that PE25 A51Q/L46E/A70Q/L73H, which contained all complementary residues (including the conserved Ser48 in PE25) for the PPE15 interaction, would bind to PPE15. Although we did not find any electrostatic repulsion in the structural model, other determinants might contribute to the PE–PPE binding specificity and may have been responsible for the failure of PE25 A51Q/L46E/A70Q/L73H to pull down PPE15. As described in Fig. 3, PE8–PPE15 and PE25–PPE41 exhibit various degrees of helical bending. The crystal structure of the PE8–PPE15 complex shows that Gln70 and His73 are positioned closed to the kink of helix α2 in PE8. Therefore, although the mutant PE25 A51Q/L46E/A70Q/L73H contains residues equivalent to those in PE8, residues 70 and 73 in the PE25 mutant are distal from Tyr72 and Tyr45 in PPE15 and result in no interaction. Likely, the binding specificity of the PE8–PPE15 complex is defined by the specific set of complementary residues in the binding interface, as well as the conformation of helices in the bundle.
PE–PPE family members contribute a sophisticated protein repertoire to mycobacteria and are strongly associated with the pathogenesis and virulence of these organisms. However, this set of proteins is poorly understood, particularly regarding the formation and functions of PE and PPE pairs. The first crystal structure of the PE25–PPE41 complex, which was published more than 10 years ago, highlighted a conserved hydrophobic interface within the PE–PPE complex. Here, the crystal structure of a new PE–PPE pair, PE8–PPE15, in complex with EspG5 has elucidated the molecular basis underlying the binding specificities of PE–PPE pairs. In conjunction with our biochemical analysis, we propose a model for PE–PPE recognition (Fig. 6). Extensive hydrophobic contacts along the heterodimeric interface comprise the basic criterion for PE–PPE complex formation. The hydrogen bond observed between a highly conserved Ser48 on PE and Tyr154 on PPE was found to stabilize the interactions between the α2 helix of PE and the α5 helix of PPE and is likely a common property of PE–PPE complexes. Although α5 helical conformation stability is essential for EspG5 binding, the critical determinants of PE–PPE binding specificity depend on the coupling of multiple complementary residues positioned along the helix bundle, as well as the helical conformations of α1 and α2 in PE and α2 and α3 in PPE. Helical bending will determine whether these complementary residues are brought together for salt bridge and hydrogen bond formation and consequent PE–PPE interaction. On the other hand, analysis of the molecular surfaces of PE81–99–PPE151–194 and PE25–PPE41 revealed differences in the electrostatic surfaces (supplemental Fig. S9). PE81–99–PPE151–194 displays a relatively more hydrophobic and negatively charged surface, whereas PE25–PPE41 contains positively charged patches on each face of the four-helix bundle. It appears that the structural and functional properties of each PE–PPE protein is shaped by its unique electrostatic surface and extent of helical bending. However, the importance of the C-terminal domains of PE–PPE proteins cannot be excluded. Currently, PE–PPE structures are available for complexes in sublineage III (PE25–PPE41) and sublineage IV (PE8–PPE15 in this study). Therefore, structural solutions of other PE–PPE complexes, particularly those of the most recently evolved sublineage V, will provide a more comprehensive understanding of the evolution of this distinct protein family.
Figure 6.
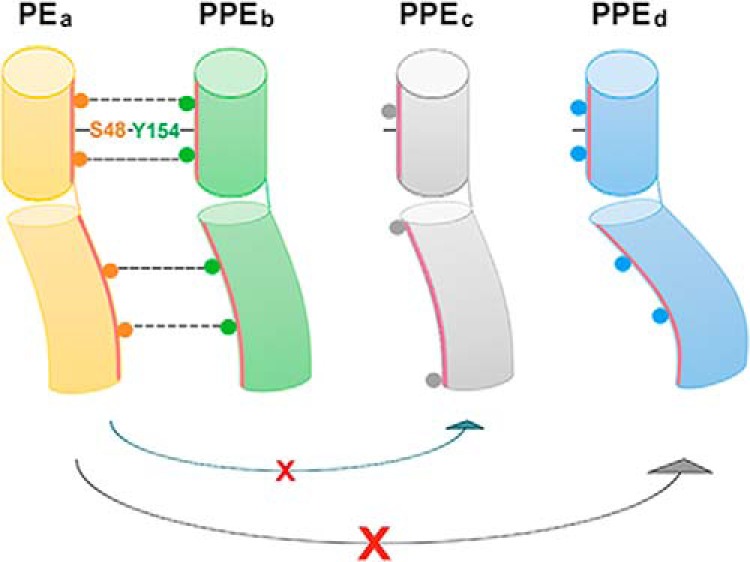
Proposed model for PE–PPE recognition. The basic principle is based on common properties of PE–PPE proteins, including the extensive non-polar binding interface and the highly conserved hydrogen bond between Ser48PE and Tyr154PPE to link the α2 helix of PE with the α5 helix of PPE, thus stabilizing the latter for EspG interaction. The specific PE–PPE interaction is determined by a specific set of multiple complementary residues along the helix bundle, as well as the helical conformation. A PE–PPE complex can form only when these two criteria are satisfied. For example, PEa cannot interact with PPEc and PPEd because PPEc does not contain complementary residues with PEa and because the helical bending of PPEd does not allow the complementary residues to interact with those in PEa.
Experimental procedures
Plasmid construction
Full-length and truncated versions of PE8 and PPE15, and EspG5 were amplified from M. tuberculosis strain H37Rv genomic DNA (ATCC). PE8 and PE81–99 were cloned into expression vector pGEX-6p-1 (GE Healthcare) via BamHI and SalI sites. EspG5 and PPE151–194 were cloned into vector pAC28 via NdeI and EcoRI and NdeI and BamHI sites, respectively (37). All mutations were introduced by using the QuikChange site-directed mutagenesis kit (Stratagene Corp., La Jolla, CA). All plasmid constructs obtained were confirmed by a DNA sequencing service (BGI) and then subjected to protein expression in Escherichia coli strain.
Protein expression and purification
The recombinant PE81–99–PPE151–194 were co-expressed in E. coli strain BL21 (DE3), whereas the pAC-EspG5 was expressed individually. Transformed bacteria were grown at 37 °C to an A600 of 0.4–0.6. Protein expression was then induced by 0.4 mm isopropyl-d-thiogalactopyranoside at 20 °C for 16–20 h. Cells expressing PE81–99–PPE151–194 and EspG5 were harvested and co-lysed with sonication in buffer of 20 mm HEPES, pH 7.5, 300 mm NaCl, 5 mm DTT, 5% glycerol. Lysate was cleared by centrifugation at 48,384 × g for 1 h, and the proteins were purified by affinity chromatography using glutathione-agarose 4B beads (Macherey Nagel). After cleavage of the GST tag from the fusion protein with PreScission protease overnight at 4 °C, the proteins were eluted in lysis buffer supplemented with 50 mm l-arginine and further purified using Mono Q 5/50 GL ion exchange column (GE Healthcare) and Superdex 200 (GE Healthcare) size-exclusion column. Purified protein complex containing EspG5–PE81–99–PPE151–194 were pooled and concentrated in buffer containing 20 mm HEPES, pH 7.5, 300 mm NaCl for crystallization trials.
Pulldown assay
For PE8–PPE15 interaction studies, GST-tagged PE81–99 and His-tagged PPE15 or PPE15 mutants were co-expressed and lysed in buffer containing 20 mm HEPES, pH 7.5, 300 mm NaCl, 5 mm DTT, and 5% glycerol. Clear lysate was mixed with glutathione agarose 4B beads (Macherey Nagel) and incubated for 2 h, followed by washing with lysis buffer for 8 times. Input material and the beads were boiled with SDS loading dye and analyzed by SDS-PAGE. For Western blotting detection, PPE15 or PPE15 mutants were probed with primary anti-His antibody (1:5000) (GE Healthcare). Same procedures were applied for PE25-PPE15 interaction analysis, but lysate containing co-expressed His-tagged PPE15 and GST-tagged PE25 or PE25 mutants were used. For nickel pull down, GST-tagged PE8 or -PE81–99 and His-tagged PPE15 or PPE151–194 were co-expressed and lysed in buffer containing 20 mm HEPES, pH 7.5, 300 mm NaCl, 20 mm imidazole, and 5% glycerol. Clear lysate was mixed with Ni-NTA agarose (Macherey Nagel) and incubated for 1h, followed by washing with lysis buffer eight times. Pulldown products were analyzed by SDS-PAGE. All experiments were performed in triplicate.
Size-exclusion chromatography/static light scattering (SEC/SLS)
The purified protein complex EspG5–PE81–99–PPE151–194 was injected into Superdex 200 analytical (GE Healthcare) size-exclusion column pre-equilibrated with buffer containing 20 mm HEPES, pH 7.5, 300 mm NaCl. The experiments were performed at a preadjusted temperature of 25 °C. Eluted protein from gel filtration was directed into a miniDawn light scattering detector and an Optilab DSP refractometer (Wyatt Technologies). The data were analyzed using the software ASTRA.
Analytical ultracentrifugation
Analytical ultracentrifugation experiments were performed using a Beckman proteomeLab XL-I analytical ultracentrifuge. The sample at a concentration of 1.6 AU absorbance at A280 was spun using at rotor 60-Ti at a speed of 42,000 rpm at 16 °C for 12 h. The data were collected at 280 nm in a continuous mode with a scan range from 6.05 to 7.20 cm. The data were processed according to continuous sedimentation coefficient distribution model using Sedfit (38) to determine the sedimentation coefficients.
Crystallization and structure determination
The EspG5–PE81–99–PPE151–194 crystals were grown by using the sitting drop vapor diffusion method. Crystals were obtained from optimized conditions containing 200 mm NaCl, 100 mm Tris, pH 8.5, 25% (w/v) PEG3350 after incubation at 16 °C for 4 days. For data collection, crystals were transferred to cryo protectant with 20% glycerol and immediately frozen under liquid nitrogen. X-ray diffraction data were collected at 100 K at Beamline 13B1 of the National Synchrotron Radiation Research Center in Taiwan. A 2.9 Å complete data set was processed by the imosflm (39). EspG5–PE81–99–PPE151–194 complex crystal belongs to the space group of P212121 with unit cell dimensions a = 54.74 Å, b = 69.96 Å, and c = 203.55 Å, and there is one complex per asymmetric unit. Phase determination was solved by molecular replacement using phenix.mr_rosetta (40, 41). Subsequent iterative refinement with the phenix.refine and manual model inspection and rebuilding with Coot (42) resulted in final Rwork/Rfree values of 21.33%/26.24%. A summary of X-ray data collection and model refinement statistics is shown in Table 1. The molecular graphics images were produced with PyMOL. The protein coordinates were submitted to Protein Data Bank with PDB code 5XFS.
Sequence analysis of PE and PPE proteins
All the sequence alignments were generated using Clustal Omega (43) and rendered by the ESPript server (44).
Microscale thermophoresis
EspG5, PE81–99–PPE151–194 and PE25–PPE41 were purified using affinity chromatography followed by gel filtration chromatography with final buffer containing 20 mm HEPES, pH 7.5, 300 mm NaCl. The microscale thermophoresis experiments were conducted using the Monolith NT.115 instrument (NanoTemper Technologies). In brief, 20 μm PE81–99–PPE151–194 or PE25–PPE41 was fluorescently labeled using the NanoTemper protein labeling kit RED-NHS (Amine Reactive). Labeled PE81–99–PPE151–194 or PE25–PPE41 were diluted to 0.4 μm and then mixed with 0.3–20 μm EspG5 in a final buffer containing 20 mm HEPES, pH 7.5, 300 mm NaCl. The reactions were set at 25 °C with 40% microscale thermophoresis power for 5-s/30-s/5-s laser off/on/off times. The microscale thermophoresis data were analyzed to obtain the dissociation constant Kd by using the software MO.Affinity Analysis.
Yeast two-hybrid screen
The yeast two-hybrid screen was performed twice according to the MatchmakerTM Gold yeast two-hybrid manual (Clontech). Briefly, recombinant pGBKT7 DNA-BD and pGADT7 DNA-AD plasmids were used to co-transform into Saccharomyces cerevisiae. Positive transformants having bait–prey interaction were selected on selective SD/−Leu/−Trp/(DDO) agar plates, the colonies on the DDO plates were patched onto higher stringency selective SD/−Ade/−His/−Leu/−Trp/X-α-Gal/aureobasidin A (QDO/X/A) agar plates (Clontech). The plate was incubated at 30 °C for 3 days. Those pairs detected both on double and quadruple selection plates were identified as potential interaction pairs. PE25–PPE41 was used as a positive control.
Comparative modeling of other PE–PPE complexes
The structures of PE27–PPE43, PE13–PPE18, and PE32–PPE65 complexes were predicted by homology modeling using our solved crystal structure of EspG5–PE81–99–PPE151–194 as the template by program MODELLERv9.18 (36). Sequences of individual PE and PPE proteins were aligned to PE8 and PPE15 by the program Clustal Omega (43). The sequence alignment was edited interactively using the program Chimera.
Author contributions
X. C., H.-F. C., K.-F. L., S. K.-W. T., and S. W.-N. A. designed research. X. C., H.-F. C., and J. Z. conducted the experiments. X. C. and S. W.-N. A. analyzed the data and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank the University Grants Council of Hong Kong Special Administrative Region One-Off Special Equipment Grant SEG UCHK08 for the equipment used in this work. We also thank the staff of Beamline 13B1 at National Synchrotron Radiation Research Center in Taiwan.
This work was supported by the Health and Medical Research Fund (HMRF/12110602). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5XFS) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains supplemental Table S1 and Figs. S1–S9.
- TB
- tuberculosis
- Mtb
- Mycobacterium tuberculosis
- PDB
- Protein Data Bank
- RMSD
- root-mean-square deviation.
References
- 1. Ejeta E., Legesse M., Ameni G., and Raghavendra H. L. (2013) Global epidemiology of tuberculosis: past, present and future. Sci. Technol. Arts Res. J. 2, 97–104 [Google Scholar]
- 2. World Health Organization (2016) Global tuberculosis report 2016, World Health Organization, Geneva, Switzerland [Google Scholar]
- 3. Corbett E. L., Watt C. J., Walker N., Maher D., Williams B. G., Raviglione M. C., and Dye C. (2003) The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch. Intern. Med. 163, 1009–1021 [DOI] [PubMed] [Google Scholar]
- 4. Kaufmann S. H. (2016) How can we improve the existing vaccine for tuberculosis to combat the growing number of multi-resistant strains?
- 5. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E. 3rd, Tekaia F, Badcock K., Basham D., Brown D., Chillingworth T., et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 6. Riley R., Pellegrini M., and Eisenberg D. (2008) Identifying cognate binding pairs among a large set of paralogs: the case of PE/PPE proteins of Mycobacterium tuberculosis. PLoS Comput. Biol. 4, e1000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tundup S., Akhter Y., Thiagarajan D., and Hasnain S. E. (2006) Clusters of PE and PPE genes of Mycobacterium tuberculosis are organized in operons: evidence that PE Rv2431c is co-transcribed with PPE Rv2430c and their gene products interact with each other. FEBS Lett. 580, 1285–1293 [DOI] [PubMed] [Google Scholar]
- 8. Strong M., Sawaya M. R., Wang S., Phillips M., Cascio D., and Eisenberg D. (2006) Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 8060–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Akhter Y., Ehebauer M. T., Mukhopadhyay S., and Hasnain S. E. (2012) The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie 94, 110–116 [DOI] [PubMed] [Google Scholar]
- 10. Banu S., Honoré N., Saint-Joanis B., Philpott D., Prévost M. C., and Cole S. T. (2002) Are the PE PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol. Microbiol. 44, 9–19 [DOI] [PubMed] [Google Scholar]
- 11. Karboul A., Mazza A., Gey van Pittius N. C., Ho J. L., Brousseau R., and Mardassi H. (2008) Frequent homologous recombination events in Mycobacterium tuberculosis PE/PPE multigene families: potential role in antigenic variability. J. Bacteriol. 190, 7838–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sayes F., Sun L., Di Luca M., Simeone R., Degaiffier N., Fiette L., Esin S., Brosch R., Bottai D., Leclerc C., and Majlessi L. (2012) Strong immunogenicity and cross-reactivity of Mycobacterium tuberculosis ESX-5 type VII secretion-encoded PE–PPE proteins predicts vaccine potential. Cell Host Microbe 11, 352–363 [DOI] [PubMed] [Google Scholar]
- 13. Khubaib M., Sheikh J. A., Pandey S., Srikanth B., Bhuwan M., Khan N., Hasnain S. E., and Ehtesham N. Z. (2016) Mycobacterium tuberculosis co-operonic PE32/PPE65 proteins alter host immune responses by hampering Th1 response. Front. Microbiol. 7, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Farhat M. R., Shapiro B. J., Kieser K. J., Sultana R., Jacobson K. R., Victor T. C., Warren R. M., Streicher E. M., Calver A., Sloutsky A., Kaur D., Posey J. E., Plikaytis B., Oggioni M. R., Gardy J. L., et al. (2013) Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 45, 1183–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Provvedi R., Boldrin F., Falciani F., Palù G., and Manganelli R. (2009) Global transcriptional response to vancomycin in Mycobacterium tuberculosis. Microbiology 155, 1093–1102 [DOI] [PubMed] [Google Scholar]
- 16. Fu L. M., and Tai S. C. (2009) The differential gene expression pattern of Mycobacterium tuberculosis in response to capreomycin and PA-824 versus first-line TB drugs reveals stress-and PE/PPE-related drug targets. Int. J. Microbiol. 2009, 879621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramakrishnan L., Federspiel N. A., and Falkow S. (2000) Granuloma-specific expression of Mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science 288, 1436–1439 [DOI] [PubMed] [Google Scholar]
- 18. Kohli S., Singh Y., Sharma K., Mittal A., Ehtesham N. Z., and Hasnain S. E. (2012) Comparative genomic and proteomic analyses of PE/PPE multigene family of Mycobacterium tuberculosis H37Rv and H37Ra reveal novel and interesting differences with implications in virulence. Nucleic Acids Res. 40, 7113–7122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sampson S. L. (2011) Mycobacterial PE/PPE proteins at the host–pathogen interface. Clin. Dev. Immunol. 2011, 497203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bottai D., and Brosch R. (2009) Mycobacterial PE, PPE and ESX clusters: novel insights into the secretion of these most unusual protein families. Mol. Microbiol. 73, 325–328 [DOI] [PubMed] [Google Scholar]
- 21. Abdallah A. M., Gey van Pittius N. C., Champion P. A., Cox J., Luirink J., Vandenbroucke-Grauls C. M., Appelmelk B. J., and Bitter W. (2007) Type VII secretion: mycobacteria show the way. Nat. Rev. Microbiol. 5, 883–891 [DOI] [PubMed] [Google Scholar]
- 22. Abdallah A. M., Bestebroer J., Savage N. D., de Punder K., van Zon M., Wilson L., Korbee C. J., van der Sar A. M., Ottenhoff T. H., van der Wel N. N., Bitter W., and Peters P. J. (2011) Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J. Immunol. 187, 4744–4753 [DOI] [PubMed] [Google Scholar]
- 23. Daleke M. H., Ummels R., Bawono P., Heringa J., Vandenbroucke-Grauls C. M., Luirink J., and Bitter W. (2012) General secretion signal for the mycobacterial type VII secretion pathway. Proc. Natl. Acad. Sci. U.S.A. 109, 11342–11347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pallen M. J. (2002) The ESAT-6/WXG100 superfamily–and a new Gram-positive secretion system? Trends Microbiol. 10, 209–212 [DOI] [PubMed] [Google Scholar]
- 25. Korotkova N., Freire D., Phan T. H., Ummels R., Creekmore C. C., Evans T. J., Wilmanns M., Bitter W., Parret A. H., Houben E. N., and Korotkov K. V. (2014) Structure of the Mycobacterium tuberculosis type VII secretion system chaperone EspG5 in complex with PE25–PPE41 dimer. Mol. Microbiol. 94, 367–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ekiert D. C., and Cox J. S. (2014) Structure of a PE–PPE-EspG complex from Mycobacterium tuberculosis reveals molecular specificity of ESX protein secretion. Proc. Natl. Acad. Sci. U.S.A. 111, 14758–14763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gey van Pittius N. C., Sampson S. L., Lee H., Kim Y., van Helden P. D., and Warren R. M. (2006) Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol. Biol. 6, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shah S., and Briken V. (2016) Modular organization of the ESX-5 secretion system in Mycobacterium tuberculosis. Front. Cell. Infect. Microbiol. 6, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shah S., Cannon J. R., Fenselau C., and Briken V. (2015) A duplicated ESAT-6 region of ESX-5 is involved in protein export and virulence of mycobacteria. Infect. Immun. 83, 4349–4361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Daleke M. H., van der Woude A. D., Parret A. H., Ummels R., de Groot A. M., Watson D., Piersma S. R., Jiménez C. R., Luirink J., Bitter W., and Houben E. N. (2012) Specific chaperones for the type VII protein secretion pathway. J. Biol. Chem. 287, 31939–31947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Solomonson M., Setiaputra D., Makepeace K. A., Lameignere E., Petrotchenko E. V., Conrady D. G., Bergeron J. R., Vuckovic M., DiMaio F., Borchers C. H., Yip C. K., and Strynadka N. C. (2015) Structure of EspB from the ESX-1 type VII secretion system and insights into its export mechanism. Structure 23, 571–583 [DOI] [PubMed] [Google Scholar]
- 33. Korotkova N., Piton J., Wagner J. M., Boy-Röttger S., Japaridze A., Evans T. J., Cole S. T., Pojer F., and Korotkov K. V. (2015) Structure of EspB, a secreted substrate of the ESX-1 secretion system of Mycobacterium tuberculosis. J. Struct. Biol. 191, 236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laskowski R. A. (2001) PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 29, 221–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Söding J., Biegert A., and Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Webb B., and Sali A. (2014) Protein structure modeling with MODELLER. Methods Mol. Biol. 1137, 1–15 [DOI] [PubMed] [Google Scholar]
- 37. Kholod N., and Mustelin T. (2001) Novel vectors for co-expression of two proteins in E. coli. BioTechniques 31, 322–328 [DOI] [PubMed] [Google Scholar]
- 38. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Terwilliger T. C., Dimaio F., Read R. J., Baker D., Bunkóczi G., Adams P. D., Grosse-Kunstleve R. W., Afonine P. V., and Echols N. (2012) phenix.mr_rosetta: molecular replacement and model rebuilding with Phenix and Rosetta. J. Struct. Funct. Genomics 13, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DiMaio F., Echols N., Headd J. J., Terwilliger T. C., Adams P. D., and Baker D. (2013) Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods 10, 1102–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sievers F., and Higgins D. G. (2014) Clustal Omega, accurate alignment of very large numbers of sequences. Methods. Mol. Biol. 1079, 105–116 [DOI] [PubMed] [Google Scholar]
- 44. Gouet P., Robert X., and Courcelle E. (2003) ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31, 3320–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.