

Abstract
The ability of the FdsABG formate dehydrogenase from Cupriavidus necator (formerly known as Ralstonia eutropha) to catalyze the reverse of the physiological reaction, the reduction of CO2 to formate utilizing NADH as electron donor, has been investigated. Contrary to previous studies of this enzyme, we demonstrate that it is in fact effective in catalyzing the reverse reaction with a kcat of 11 ± 0.4 s−1. We also quantify the stoichiometric accumulation of formic acid as the product of the reaction and demonstrate that the observed kinetic parameters for catalysis in the forward and reverse reactions are thermodynamically consistent, complying with the expected Haldane relationships. Finally, we demonstrate the reaction conditions necessary for gauging the ability of a given formate dehydrogenase or other CO2-utilizing enzyme to catalyze the reverse direction to avoid false negative results. In conjunction with our earlier studies on the reaction mechanism of this enzyme and on the basis of the present work, we conclude that all molybdenum- and tungsten-containing formate dehydrogenases and related enzymes likely operate via a simple hydride transfer mechanism and are effective in catalyzing the reversible interconversion of CO2 and formate under the appropriate experimental conditions.
Keywords: biofuel, carbon dioxide, enzyme catalysis, molybdenum, multifunctional enzyme
Introduction
Formate dehydrogenases are a heterogeneous group of enzymes that are widely distributed in nature, being found in anaerobic as well as aerobic bacteria, archaea, yeasts, fungi, plants, and vertebrates. These enzymes can be broadly classified as metal-independent/NAD(P)+-dependent, metal-containing/NAD(P)+-dependent, and metal-containing/NAD(P)+-independent (1). All these enzymes catalyze the oxidation of formic acid to carbon dioxide, generating two protons and two electrons (Equation 1).
| (Eq. 1) |
s
Some, but not all, metal-containing formate dehydrogenases have been reported to catalyze the reverse reaction, i.e. the reduction of carbon dioxide to formic acid, a reaction of considerable industrial interest given the general difficulty of activating CO2 for reduction (2–4). The main challenges in the use of these enzymes for the generation of formate are the low rates of CO2-reducing activity typically reported and the high O2 sensitivity exhibited by most of these enzymes (5). In the present study, we have examined the ability of an oxygen-tolerant formate dehydrogenase from Cupriavidus necator (FdsABG) to convert CO2 to formic acid.
C. necator (formerly known as Ralstonia eutropha) is an aerobic facultative chemoautotrophic bacterium capable of both heterotrophic and lithoautotrophic growth (6). The C. necator genome encodes four formate dehydrogenases, all of which are metal-dependent. Three are membrane-bound and expressed under various growth conditions, whereas one is cytoplasmic. This last enzyme, encoded by the fdsGBACD operon and consisting of the FdsABG gene products (7), is an O2-tolerant homodimer of trimers, (αβγ)2 (8, 9). The 105-kDa α-subunit (FdsA) contains the molybdenum-containing active site as well as four [4Fe–4S] clusters and a [2Fe–2S] cluster. Oxidation of formic acid occurs at the molybdenum center, which in the oxidized enzyme has an L2Mo(VI)S(S–Cys) structure (L represents the pyranopterin cofactor found in all molybdenum- and tungsten-containing enzymes other than nitrogenase; L is a guanine dinucleotide in the present case); reduction yields an L2Mo(IV)(SH)(S–Cys) center (9). The 55-kDa β-subunit (FdsB) contains FMN and a [4Fe–4S] cluster with the FMN being the site of NAD+ reduction. Finally, the 19-kDa γ-subunit (FdsG) contains one [2Fe–2S] cluster. The three subunits of the C. necator FdsABG formate dehydrogenase have strong sequence homology to the corresponding Nqo3, Nqo1, and Nqo2 subunits, respectively, of the crystallographically characterized Thermus thermophilus NADH dehydrogenase (10), and the enzyme thus belongs to the NADH dehydrogenase superfamily of enzymes. The enzyme is unusual among the metal-dependent formate dehydrogenases in being O2-tolerant and unusual among the NADH dehydrogenases in being cytosolic (and water-soluble) rather than membrane-integral. The large number of iron–sulfur clusters as well as the FMN is unique among the formate dehydrogenases and possibly affords a greater number of potential pathways for electrochemical reduction.
In previous work, we characterized the rapid reaction kinetics and spectroscopic properties of C. necator FdsABG and demonstrated that the enzyme operates via a ping–pong mechanism with a limiting rate constant, kred, for reduction of enzyme by formate of 140 s−1 at 10 °C (corresponding to 560 s−1 at 30 °C) and a Kd of 82 μm (11). We have also characterized the EPR signal of the molybdenum center in its Mo(V) state and demonstrated the direct transfer of the substrate Cα hydrogen to the molybdenum center in the course of the reaction, concluding that the reaction proceeds via direct hydride transfer from substrate to the Mo=S group of the active site molybdenum center with CO2 (rather than bicarbonate) as the immediate product of the overall reaction (Fig. 1). Formate is in fact known to be an effective hydride donor (as reflected in the reactivity of the metal-independent formate dehydrogenases that catalyze the direct reduction of NAD+ by formate via a ternary complex mechanism (involving an E-formate-NAD+ intermediate), and the Mo(VI)=S group of the molybdenum center is also known to be an effective hydride acceptor (as in the reaction mechanism of the molybdenum-containing xanthine oxidase (8)). Were the C. necator enzyme to be shown to effectively catalyze the reverse reaction, the implication would be that the Mo(IV)–SH center is an effective hydride donor to CO2 with the enzyme competent to activate CO2 for reduction.
Figure 1.

The proposed hydride transfer mechanism for the formate dehydrogenases. As the second carbon–oxygen double bond of the product forms, the displaced hydride attacks the Mo=S moiety, resulting in the formal two-electron reduction of the metal.
We report here that, contrary to an earlier report (6), the FdsABG formate dehydrogenase from C. necator is indeed able to effectively catalyze the reduction of CO2 using NADH as reductant, stoichiometrically generating formate (as confirmed by NMR) with a kcat of 11 ± 0.4 s−1. Importantly, we demonstrate that the enzyme's steady-state kinetic parameters in the forward and reverse reactions are consistent with the overall thermodynamics of the reaction. Finally, we establish the reaction conditions necessary to quantify the reverse reaction.
Results
Demonstration of CO2 as substrate for the reverse reaction
Carbon dioxide dissolved in aqueous solution exists in many forms: CO2(aq),3 carbonic acid, carbonate, and bicarbonate (12). In a hydride transfer mechanism for the oxidation of formate by FdsABG, the product of the reaction is CO2 rather than bicarbonate, with the implication that CO2 rather than bicarbonate is the substrate in the reverse direction. To ascertain whether this is in fact the case, we performed steady-state experiments whose results are summarized in Table 1.
Table 1.
Demonstration of CO2 as substrate for the reverse reaction
All reactions were measured by the change in NADH absorbance at 340 nm and performed in 100 mm potassium phosphate at pH 7.0 and 30 °C.
| Sample | kobs |
|---|---|
| s−1 | |
| +Bicarbonate, aerobic conditions | 8.1 ± 0.3 |
| −Bicarbonate, aerobic conditions | 7.6 ± 0.3 |
| +Bicarbonate, anaerobic conditions | 0.40 ± 0.04 |
| +CO2 gas, anaerobic conditions | 9.4 ± 0.6 |
First, we measured the ability of FdsABG to catalyze the reverse reaction using 100 mm potassium bicarbonate as the substrate at pH 7.0 and 30 °C under aerobic conditions, monitoring the reaction by the oxidation of NADH; the resulting rate was determined to be 8.1 ± 0.3 s−1. Significantly, a control experiment performed in the absence of bicarbonate yielded a similar rate, indicating that the observed oxidation of NADH by FdsABG was due to the diaphorase (O2:NADH oxidoreductase) activity of the enzyme (7.6 ± 0.3 s−1). Additionally, in the presence of bicarbonate, but in the absence of O2, NADH oxidation was virtually eliminated (0.40 ± 0.04 s−1). Finally, we performed an anaerobic experiment with a saturated solution of CO2 as the substrate; the resulting rate was determined to be 9.4 ± 0.6 s−1. Under our experimental conditions (30 °C, 1 atm, pH 7.0) the amount of CO2(aq) in solution under saturating conditions was calculated as 29.5 mm with a potassium bicarbonate concentration of ∼139 mm (at this pH there are only trace amounts of carbonic acid or carbonate) (12). The implication of the above experiments is thus that the substrate for the reverse reaction is CO2(aq) rather than carbonic acid, carbonate, or bicarbonate.
pH dependence of the reverse reaction
We next examined the pH dependence of the kinetics of the reverse reaction, with all experiments performed under anaerobic conditions. In these experiments, we performed duplicate assays at each pH using CO2-saturated solutions and solutions that were 50% CO2 to confirm that we were working at the high concentration limit of CO2 (control data not shown). As illustrated in Fig. 2 (data in black), the reaction of FdsABG with CO2 has a nominal pH optimum around 7.0. The values for kobs were fitted to the equation for a double-ionization mechanism, which yielded two apparent pKa values of 5.5 and 8.3. Both pKa values are in reasonable agreement with those measured for the reaction of FdsABG with formate (5.6 and 9.3, respectively). We note, however, that the value for pKa2eff may be subject to considerable error as a result of the increasing ionic strength of the CO2-saturated solutions at higher pH. Because the total dissolved carbon increases exponentially with pH due to the higher concentrations of bicarbonate (and eventually carbonate), the ionic strength of CO2-saturated solutions also increases very significantly as reflected by the increased conductivity at high pH (Fig. 2, data in blue). The conductivity reflects not only the amount of total carbon species but also significant concentrations of K+ as a consequence of adjusting pH with KOH. Contributions to total carbon from bicarbonate and carbonate can be calculated from well defined temperature-related equilibrium constants and partial pressures according to Equations 2 and 3 below.
| (Eq. 2) |
| (Eq. 3) |
where KH, Ka1, and Ka2 are the acid dissociation constants for CO2(aq), HCO3−, CO32−, respectively, and PCO2 is the partial pressure of CO2 (12). We tested the effect of increasing ionic strength on the ability of the enzyme to catalyze both the forward and reverse reactions. The kcatCO2 for the reaction of FdsABG with CO2 decreased to 13% in the presence of 3 m KCl (supplemental Fig. S1). The reaction of FdsABG with formate was similarly affected by increasing ionic strength; kcatformate decreased to 13% in the presence of 4 m KCl (supplemental Fig. S1). It is thus likely that the decrease in activity above pH 7.0 as described by pKa2eff was principally due to ionic strength effects rather than ionization phenomena. The identity of the ionizable group whose protonation results in loss of activity at low pH is not known, although a likely candidate is the Mo(VI)=S group of the molybdenum center, which is known to be protonated at pH 7.0 upon reduction of the molybdenum (11).
Figure 2.
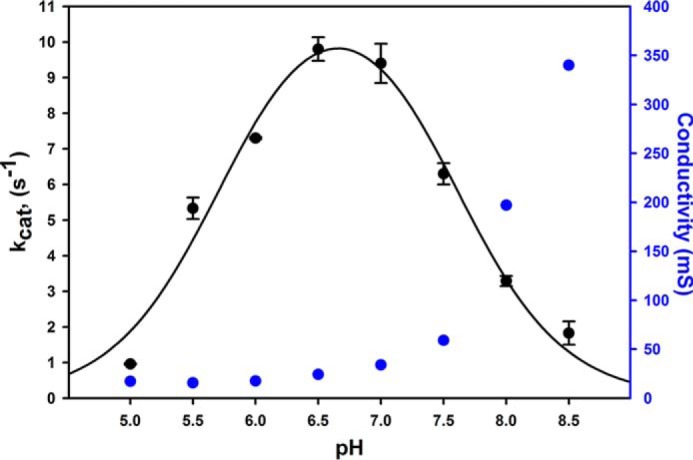
The dependence of kcat for FdsABG on pH. Kinetic experiments were performed using an overlapping buffer system (75 mm each of malate, potassium phosphate, and Tris base) containing 200 μm NADH and saturated with CO2(aq). The fit to the data (black solid line) yielded two pKa values of pKa1 = 5.5 and pKa2eff = 8.3. The plot in blue represents the corresponding conductivity, a consequence of increasing concentrations of bicarbonate/carbonate as well as KOH. All reactions were performed at 30 °C under anaerobic conditions. mS, millisiemens. Error bars represent standard deviations.
Further analysis of the reverse reaction
To demonstrate the ability of FdsABG to catalyze the reaction in both the forward and reverse directions, an experiment was performed in which the enzyme was sequentially supplemented with NAD+ or formate in a septum-sealed quartz cuvette under anaerobic conditions. Upon addition of enzyme to a cuvette containing 200 μm NADH in 100 mm potassium phosphate saturated with CO2 (final pH 6.3; Fig. 3, arrow 1), an absorbance decrease was observed as NADH was partially consumed upon catalysis of the reverse reaction, the reaction being driven by the high concentration of CO2 in the reaction mixture. The reaction was allowed to proceed until NADH was no longer consumed at which point the cuvette contained formate produced from CO2 reduction and an equivalent amount of NAD+. 400 μm NAD+ was injected to drive the reaction in the reverse direction (Fig. 3, arrow 2) with an absorbance increase as NAD+ was reduced back to NADH. The reaction was allowed to proceed until NADH was no longer produced at which point the solution was supplemented with 40 mm formate (Fig. 3, arrow 3), which resulted in a further absorbance increase as more NAD+ was reduced to NADH at the expense of the initially formed formate. These results confirm the thermodynamic and kinetic competence of the enzyme to catalyze both the forward and reverse reactions.
Figure 3.

Demonstration of the ability of FdsABG to catalyze both CO2 reduction and formate oxidation. The reaction was initiated by addition of FdsABG (arrow 1) via an argon-purged Gastight syringe to 200 μm NADH in 100 mm phosphate saturated with CO2(aq), final pH 6.3. At ∼200 s, 400 μm NAD+ was injected into the cuvette (arrow 2), and the reaction was allowed to proceed. At ∼400 s, 40 mm formate was further injected into the cuvette (arrow 3). The reaction was performed at 30 °C under anaerobic conditions.
Formic acid quantification and identification
Next, we confirmed by ion chromatography that the reaction product of the reduction of CO2 catalyzed by FdsABG was in fact formic acid using the protocol described under “Experimental procedures.” FdsABG was incubated with anaerobic saturated CO2(aq) and 300 μm NADH in 20 mm Bis-Tris propane (final pH 6.3, 30 °C), and the reaction was followed by the absorbance decrease at 340 nm (Fig. 4A). At completion of the reaction, an aliquot was withdrawn and submitted to ion chromatography with formate being detected with a characteristic retention of 5.9 min (Fig. 4B). Comparison against a calibration curve (supplemental Fig. S2) yielded a value of 120 μm formic acid generated in the experiment in very good agreement with the 130 μm NADH consumed on the basis of the absorbance change at 340 nm (retention times for bicarbonate/carbonate, NAD+/NADH, and buffer peaks are reported in supplemental Fig. S3). These results demonstrate the stoichiometric generation of formic acid at the expense of NADH for the enzyme-catalyzed reaction in the reverse direction. The identity of formic acid/formate generated enzymatically was further confirmed by comparison of the 13C NMR spectrum of the reaction product obtained with [13C]formate with an authentic natural abundance formic acid standard (Fig. 4, C and D).
Figure 4.

A, reaction of FdsABG with saturated CO2(aq) and 300 μm NADH in 20 mm Bis-Tris propane (final pH 6.3) performed at 30 °C under anaerobic conditions. The arrow indicates addition of enzyme. B, ion chromatography analysis of the product of the reaction in A as described under “Experimental procedures.” Retention times are indicated with arrows for formate (5.9 min), bicarbonate (7.4 min), and NADH/NAD+ (11.2 min). C, 13C NMR spectrum for [13C]formate generated enzymatically with dissolved [13C]bicarbonate at 161.08 ppm. D, 13C NMR standard spectrum with 100 mm natural abundance formic acid brought to pH ∼6.3 with sodium bicarbonate. μS, microsiemens.
Enzyme kinetics
Finally, with the reversibility of the reaction fully established, we sought to determine kcat, Km(CO2), and Km(NADH) for the reverse reaction. Having previously demonstrated that FdsABG operates via a ping–pong mechanism (11), the experiments simply involved determining the dependence of kobs as a function of [CO2] at a saturating concentration of NADH and as a function of [NADH] at a kinetically saturating concentration of CO2. As shown in Fig. 5, the results yielded hyperbolic plots from which a kcat of 11 ± 0.4 s−1, a Km(CO2) of 2.7 ± 0.3 mm (Fig. 5A), and a Km(NADH) of 46 ± 4.3 μm at pH 7.0 (Fig. 5B) were obtained. The kcat for CO2 reduction is ∼14 times slower than kcat for formate oxidation by FdsABG.
Figure 5.

A, hyperbolic plots for the reaction of FdsABG with CO2(aq) in the presence of 200 μm NADH. B, hyperbolic plots for the reaction of FdsABG with NADH in the presence of saturated CO2(aq). Plots in A and B yielded a kcat of 11 ± 0.37 s−1, a Km(CO2) of 2.7 ± 0.34 mm, and a Km(NADH) of 46 ± 4.3 μm, respectively. All reactions were performed in 100 mm potassium phosphate, pH 7.0, at 30 °C under anaerobic conditions. Error bars represent standard deviations.
Discussion
The FdsABG formate dehydrogenase from C. necator has been reported previously to be unable to catalyze the reverse reaction of formate oxidation (6, 7). The method used in this earlier work was similar to that used here, and we are unable to account for their (false) negative result. However, it was stated that a long (40-min) incubation period was used, and it is possible that the enzyme lost substantial activity over this period of time. In the present work, we find that the enzyme is in fact quite effective catalyzing the reduction of CO2, being able to stoichiometrically transfer reducing equivalents from NADH to CO2 with a steady-state kcat of 10 s−1. Given the previously observed false negative results with this (and possibly other) formate dehydrogenases, two specific points are important to bear in mind when assessing the ability of a given enzyme to catalyze the reverse reaction. First, the product of the reaction in the forward direction (and hence the substrate in the reverse) is CO2 rather than bicarbonate, and second, the reverse assay must be carried out under anaerobic conditions to avoid consumption of NADH by possible diaphorase activity of the enzyme. It must also be kept in mind that the pH optimum for the reverse reaction may be perturbed by ion strength effects given the higher concentration of bicarbonate/carbonate at higher pH.
Bicarbonate/carbonate is routinely used as a surrogate for CO2 in aqueous solutions, but it is important to recognize that it is an imperfect one. Although some equilibrium concentration of CO2 will always eventually be obtained, the uncatalyzed interconversion of CO2 and bicarbonate in aqueous solutions is in fact very sluggish on the time scales of catalysis for all formate dehydrogenases, even at low pH where CO2 is expected to predominate at equilibrium. Furthermore, for reactions performed in an experimental apparatus having a significant volume of headspace, partitioning into the gas phase will lower the concentration of dissolved CO2 for any given amount of bicarbonate/carbonate added. The final concentration of dissolved CO2 will in fact ultimately be dictated by the partial pressure of CO2 in the headspace according to Henry's law, not simply the total amount of bicarbonate initially added. It is likely that these considerations have all contributed to false negative results in assessing whether one or another formate dehydrogenase is capable of catalyzing the reduction of CO2 to formate. Our observation that, at least with the FdsABG formate dehydrogenase, there is significant diaphorase (O2:NADH oxidoreductase) activity that consumes NADH regardless of whether formate is formed by hydride transfer to CO2 further underscores the importance of performing assays under anaerobic conditions. It is a key aspect of the present work that we have not simply identified an(other) formate dehydrogenase capable of reducing CO2 under the appropriate conditions but provided an explanation for the disparate and confusing results of previous studies with this and other enzymes.
From a thermodynamic standpoint, E′0 for the NADH/NAD+ couple is −320 mV, and that for the formate/CO2 couple is −420 mV. From the Nernst equation, the overall Keq for the two-electron process is calculated to be ∼2100. To ensure compliance with the first law of thermodynamics, the following Haldane relationship relating the steady-state parameters for an enzyme operating via a ping–pong mechanism holds (13).
| (Eq. 4) |
where kcatforward = 201 ± 6.1 s−1, Km(formate) = 310 ± 6.5 μm, and Km(NAD+) = 130 ± 4.4 μm from our previous work (11) and kcatreverse = 11 ± 0.4 s−1, Km(CO2) = 2.7 ± 0.3 mm, and Km(NADH) = 46 ± 4.3 μm from the present work. Given the squared dependence of the calculation on kcatforward and kcatreverse, the agreement with the value of 2100 determined from the Nernst equation is considered to be quite good. This constitutes a stringent test of the validity of the steady-state parameters in both the forward and reverse reactions.
Moura and co-workers (14) have recently examined the reaction of the periplasmic formate dehydrogenase from Desulfovibrio desulfuricans and have concluded that this reaction also proceeds via a hydride transfer mechanism. Like the C. necator enzyme studied here, the D. desulfuricans enzyme possesses a terminal Mo(VI)=S sulfido group in the oxidized state but has a selenocysteine ligand to the molybdenum in place of the cysteine residue seen in FdsABG. The D. desulfuricans enzyme also differs in passing reducing equivalents into the intramembrane quinone pool (via subunits possessing four c-type cytochromes and two [4Fe–4S] clusters) rather than reducing NAD+, and thus these authors necessarily used reduced methyl viologen as a non-physiological reductant. It was not possible to assess the overall thermodynamics of the reverse reaction and the validity of the Haldane relationship as done here.
The active sites in the metal-dependent (both molybdenum- and tungsten-containing) formate dehydrogenases all possess a common active site structure as shown in Fig. 6. The present results strongly suggest that all operate via the same hydride transfer mechanism, and likely they all do so fully reversibly under the appropriate experimental conditions. The majority of these enzymes function physiologically in the direction of formate oxidation, but the molybdenum-dependent formate-hydrogen lyase complex of the acetogen Acetobacterium woodii has recently been shown to function physiologically in the direction of CO2 reduction using reducing equivalents obtained from H2 (15, 16). The hydride transfer mechanism is also likely to be relevant to both the molybdenum- and tungsten-containing formylmethanofuran dehydrogenases from methanogenic bacteria. These enzymes catalyze the first step in methanogenesis, the reductive condensation of CO2 and methanofuran to yield formylmethanofuran; they are bifunctional with a molybdenum or tungsten center involved in the oxidation–reduction reaction in one subunit and a di-zinc center that binds the methanofuran substrate in a second. The molybdenum- and tungsten-containing active sites of this second group of enzymes also strongly resemble those seen in the above mentioned formate dehydrogenases seen in Fig. 6. The tungsten-containing enzyme from Methanothermobacter wolfeii has recently been characterized crystallographically with the surprising observation that the tungsten and di-zinc active sites are separated by ∼43 Å with an intraprotein tunnel connecting them (17). The reaction had been thought to proceed via condensation of CO2 with methanofuran to form carboxymethanofuran followed by reduction of this intermediate to product formylmethanofuran. Given the structure of the enzyme, however, with the methanofuran-binding site so far removed from the tungsten center where reduction occurs, it is clear that the reaction must instead proceed via reduction of CO2 to formate at the tungsten center followed by diffusion of the formate thus formed through the intraprotein tunnel to the di-zinc site where it condenses with methanofuran to give the formylmethanofuran. The reaction at the tungsten sites of these enzymes is exactly equivalent to the reverse of the FdsABG formate dehydrogenase considered here, and it also likely proceeds via a hydride transfer mechanism (it is known that CO2 rather than bicarbonate is the substrate for the enzyme). Again, all these systems in their oxidized states have active sites with the structure shown in Fig. 6 where the oxidized active site can be formulated as L2M(VI)S(S/Se–Cys) where M is either molybdenum or tungsten; the reduced active site is formulated as L2M(IV)(SH)(S/Se–Cys). We suggest that all these systems function via the same basic hydride transfer mechanism regardless of the physiological direction of the reaction, with the oxidized M(VI)=S moiety constituting an effective hydride acceptor from formate and the reduced M(IV)–SH moiety an effective hydride donor to CO2. The mechanistic corollary is that just as formate is an effective hydride donor in the one direction, CO2 is an effective hydride acceptor in the reverse. With regard to the specific manner in which these enzymes activate CO2 for reduction, it is noteworthy that the active sites of these enzymes possess two strictly conserved Arg and His residues (illustrated in Fig. 7 in the case of the FdhF formate dehydrogenase from Escherichia coli (18)) that are known to be involved in formate binding in the forward direction and which we suggest are well positioned to accommodate the accumulating negative charge and molecular dipole on CO2 as the transition state in the reverse direction forms.
Figure 6.

Active site structures for formate dehydrogenases and formylmethanofuran dehydrogenases (oxidized and reduced forms, respectively).
Figure 7.
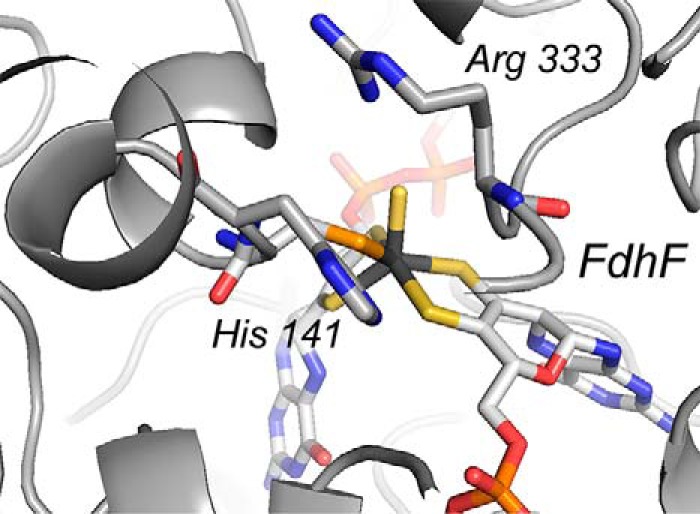
The active site of the FdhF formate dehydrogenase (Protein Data Bank code 1FDO) from E. coli. The highly conserved Arg and His residues implicated in formate binding are indicated.
The ability to reduce CO2 rapidly and efficiently has taken on great importance over the past decade, linked as it is to the emergence of inexpensive and sustainable sources of electricity (19). A variety of biohybrid systems have been developed utilizing metal-dependent formate dehydrogenases from a variety of organisms to bring about electrochemically driven reduction of CO2 to formate (for reviews, see Refs. 15, 20, and 21). In this context, the importance of the present work is twofold. First, the conditions have now been identified under which the ability of a given enzyme to catalyze the reduction of CO2 can be assessed, and second, the reaction mechanism utilized by all these enzymes involves hydride rather than H2 as reductant with formate rather than formic acid being generated. Even in the case of the A. woodii system (16, 17), H2 is oxidized to electrons and protons at the di-iron site of the hydrogenase component with the electrons subsequently transferred to the molybdenum center of the formate dehydrogenase component, which reduces CO2 via hydride transfer. The now demonstrated ability of biological systems to utilize a lower-barrier reaction for CO2 reduction involving the reaction of hydride to yield formate, rather than H2 to yield formic acid, has significant implications for the development of improved biohybrid and synthetic systems for CO2 fixation.
Conclusions
We have characterized here the kinetics of the reverse reaction for the molybdenum-containing FdsABG formate dehydrogenase of C. necator using CO2 as the reaction substrate and shown that the relevant Haldane relationship holds for the steady-state kinetic parameters in the forward and reverse reactions. We have further quantified for the first time the stoichiometric accumulation of formic acid as the product of the reaction and demonstrated the importance of performing enzymatic experiments with CO2 under anaerobic conditions as detailed under “Experimental procedures.” We submit that all molybdenum- and tungsten-containing enzymes of the formate dehydrogenase family, including the formylmethanofuran dehydrogenases, operate via the same hydride transfer mechanism and do so fully reversibly. The present work thus provides a framework for the development of increasingly efficient biocatalytic processes for CO2 fixation.
Experimental procedures
Chemicals
All chemicals were purchased from Fisher Scientific except where otherwise specified. NAD+, NADH, and [13C]CO2 were purchased from Sigma-Aldrich. Argon and CO2 were purchased from Airgas. Sodium [13C]bicarbonate was obtained from Cambridge Isotope Laboratories.
Protein preparation
C. necator (formerly known as R. eutropha) strain HF210 was grown as described previously (11). All protein purification steps were performed at 0–4 °C with an ÄKTA FPLC system (GE Healthcare) in a procedure modified from that of Niks et al. (11). The butyl-Sepharose 4 column was replaced with a 1.6 × 22-cm butyl-Sepharose HP (GE Healthcare), and the 5-ml butyl-Sepharose HP step at the end of the procedure was omitted.
Routine activity assays were performed at 30 °C in 75 mm potassium phosphate, pH 7.7, with 2 mm NAD+ and 40 mm sodium formate; formation of NADH was monitored at 340 nm (ϵ = 6,220 m−1 cm−1) with 1 unit of activity being defined as the amount of enzyme catalyzing the reduction of 1 μmol of NAD+/min. Typically 10 s were used to calculate the initial slope. Enzyme concentrations were determined using an estimated extinction coefficient at 410 nm of 51,500 m−1 cm−1, and activities were calculated with respect to one trimer with a molecular mass of 178 kDa (11).
Kinetic characterization
Kinetic assays were performed under anaerobic conditions at 30 °C. Dissolved carbon dioxide gas in reaction buffer was used as the source of CO2. For measuring the effect of pH on kobs, an overlapping buffer system consisting of 75 mm malate, 75 mm potassium phosphate, and 75 mm Tris base were used to cover the pH 4–9 range. All the reaction buffers were first bubbled with carbon dioxide continuously and titrated with potassium hydroxide or hydrochloric acid until the target pH stabilized. Upon addition of 200 μm NADH to each buffer, the solution was filtered and stored in a septum-sealed vial (Wheaton). Immediately before use each vial was resaturated with CO2, and the solution was transferred to an argon-purged quartz cuvette sealed with a rubber stopper using an argon-purged Gastight syringe (Hamilton). Reactions were started with addition of enzyme, which was injected via an argon-purged Gastight syringe. The CO2 reduction activities were measured by the change in NADH absorbance at 340 nm. The first 10 s after addition of the enzyme were used to calculate the initial slope. Values for kobs were fitted to an equation (Equation 5) for a double-ionization mechanism as described previously (11).
| (Eq. 5) |
To measure the effect of ion strength on kobs and the steady-state parameters for the reaction of FdsABG with NADH, the same experimental setup was used as above other than that 100 mm potassium phosphate pH 7.0 buffer was using instead of the overlapping buffer system. A range of 9–300 μm NADH was used in these assays, and the results were plotted according to the Michaelis-Menten equation for calculating the Km and kcat values using SigmaPlot.
Steady-state parameters were determined for the reaction of FdsABG with CO2 in 100 mm potassium phosphate pH 7.0 buffer. Three septum-sealed vials were used in these experiments. Vial A contained the buffer with 200 μm NADH and saturated with CO2, vial B contained the buffer with 200 μm NADH and saturated with argon, and vial C was flushed with argon and used for mixing reaction buffers from vial A and vial B for generating different concentrations of dissolved CO2. A range of 0.8–29.5 mm aqueous CO2 was used in these assays, and the results were plotted according to the Michaelis-Menten equation for calculating the Km and kcat values.
To demonstrate the ability of FdsABG to catalyze both the forward and reverse directions, a reaction was performed in a septum-sealed quartz cuvette containing 200 μm NADH in 100 mm potassium phosphate, final pH ∼6.3, saturated with CO2. FdsABG and formate were injected into the cuvette during the reaction via an argon-purged Gastight syringe.
Formic acid detection and identification
A solution containing 20 mm Bis-Tris propane buffer and 200 μm NADH was first saturated with CO2 in a septum-sealed quartz cuvette (final pH ∼6.3). The reaction was started with injection of FdsABG with an argon-purged Gastight syringe, and the reaction was allowed to proceed until NADH was no longer consumed. The amount of formic acid produced was estimated spectrophotometrically by the change in NADH absorbance at 340 nm. The formate-containing solution was subsequently separated from the enzyme via ultrafiltration with Amicon Ultra 4 (Millipore). The eluate was finally analyzed by ion chromatography (Dionex DX-120) using a Dionex Ionpac AS22 4 × 250-mm column and 4.5 mm sodium carbonate, 1.4 mm sodium bicarbonate as eluent with a flow rate of 0.49 ml/min. A range of 0–200 μm formic acid dissolved in 20 mm Bis-Tris propane buffer was used for standard calibration; the detection limit was estimated at 10 μm formic acid. At completion of the reaction, the formate was detected with a characteristic retention time of 5.9 min.
NMR spectroscopy was performed using a Bruker Avance III 700 instrument fitted with a 5-mm TCI 1H,2H,13C,15N quadruple resonance, z-gradient cryoprobe optimized for 1H and 13C. A solution containing 20 mm Bis-Tris propane buffer, 6 mm NADH, and 10% D2O (for NMR frequency lock) was first saturated with 13CO2 in a septum-sealed quartz cuvette with a 4-mm path length (final pH ∼6.3). The reaction was started by injection of FdsABG using an argon-purged Gastight syringe, and the reaction was allowed to proceed until NADH was no longer consumed (monitored at 388 nm). The formate-containing solution was subsequently separated from the enzyme via ultrafiltration using an Amicon Ultra 4 filter, and the eluate was submitted for NMR analysis. A separate NMR standard sample was prepared by bringing a solution of 20 mm Bis-Tris propane and 100 mm natural abundance formic acid to pH ∼6.3 with a mixture of 13C-labeled and natural abundance sodium bicarbonate (10% D2O was added prior to NMR analysis). The bicarbonate resonance was set to 161.08 ppm (calibrated relative to methanol (22)).
Author contributions
X. Y. performed kinetic characterization and formic acid detection experiments and assisted in preparing the manuscript. D. N. prepared the protein for this study, provided technical assistance, and assisted in preparing the manuscript. R. . and A. M. conceived the project, directed the research, and were principally responsible for preparation of the manuscript. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We acknowledge Dr. Haizhou Liu of the University of California, Riverside for use of the ion chromatography equipment and Dr. Dan Borchardt of the University of California, Riverside for NMR analysis.
This work was supported by National Science Foundation Grant NSF-CBET 1265044 (to A. M.) and United States Department of Energy Grant DE-SC0010666 (to R. H.) as well as a seed grant from the University of California, Riverside. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S3.
- aq
- aqueous
- Bis-Tris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
References
- 1. Maia L. B., Moura J. J., and Moura I. (2015) Molybdenum and tungsten-dependent formate dehydrogenases. J. Biol. Inorg. Chem. 20, 287–309 [DOI] [PubMed] [Google Scholar]
- 2. Sun Q., Jiang Y., Jiang Z., Zhang L., Sun X., and Li J. (2009) Green and efficient conversion of CO2 to methanol by biomimetic coimmobilization of three dehydrogenases in protamine-templated titania. Ind. Eng. Chem. Res. 48, 4210–4215 [Google Scholar]
- 3. Olah G. A. (2005) Beyond oil and gas: the methanol economy. Angew. Chem. Int. Ed. Engl. 44, 2636–2639 [DOI] [PubMed] [Google Scholar]
- 4. Qiao J., Liu Y., Hong F., and Zhang J. (2014) A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675 [DOI] [PubMed] [Google Scholar]
- 5. Maia L. B., Moura I., and Moura J. J. G. (2017) Molybdenum and tungsten-containing formate dehydrogenases: aiming to inspire a catalyst for carbon dioxide utilization. Inorg. Chim. Acta 455, 350–363 [Google Scholar]
- 6. Friedebold J., and Bowien B. (1993) Physiological and biochemical characterization of the soluble formate dehydrogenase, a molybdoenzyme from Alcaligenes eutrophus. J. Bacteriol. 175, 4719–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pohlmann A., Fricke W. F., Reinecke F., Kusian B., Liesegang H., Cramm R., Eitinger T., Ewering C., Pötter M., Schwartz E., Strittmatter A., Voss I., Gottschalk G., Steinbüchel A., Friedrich B., et al. (2006) Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 24, 1257–1262 [DOI] [PubMed] [Google Scholar]
- 8. Hille R., Hall J., and Basu P. (2014) The mononuclear molybdenum enzymes. Chem. Rev. 114, 3963–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oh J. I., and Bowien B. (1998) Structural analysis of the fds operon encoding the NAD+-linked formate dehydrogenase of Ralstonia eutropha. J. Biol. Chem. 273, 26349–26360 [DOI] [PubMed] [Google Scholar]
- 10. Sazanov L. A., and Hinchliffe P. (2006) Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436 [DOI] [PubMed] [Google Scholar]
- 11. Niks D., Duvvuru J., Escalona M., and Hille R. (2016) Spectroscopic and kinetic properties of the molybdenum-containing, NAD+-dependent formate dehydrogenase from Ralstonia eutropha. J. Biol. Chem. 291, 1162–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Butler J. (1991) Carbon Dioxide Equilibria and Their Applications, CRC Press, Boca Raton, FL [Google Scholar]
- 13. Segel H. I. (1993) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, Wiley-Interscience, New York [Google Scholar]
- 14. Maia L. B., Fonseca L., Moura I., and Moura J. J. (2016) Reduction of carbon dioxide by a molybdenum-containing formate dehydrogenase: a kinetic and mechanistic study. J. Am. Chem. Soc. 138, 8834–8846 [DOI] [PubMed] [Google Scholar]
- 15. Schuchmann K., and Müller V. (2013) Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science 342, 1382–1385 [DOI] [PubMed] [Google Scholar]
- 16. Schuchmann K., and Müller V. (2014) Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat. Rev. Microbiol. 12, 809–821 [DOI] [PubMed] [Google Scholar]
- 17. Wagner T., Ermler U., and Shima S. (2016) The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46 4Fe-4S clusters. Science 354, 114–117 [DOI] [PubMed] [Google Scholar]
- 18. Boyington J. C., Gladyshev V. N., Khangulov S. V., Stadtman T. C., and Sun P. D. (1997) Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 275, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 19. Appel A. M., Bercaw J. E., Bocarsly A. B., Dobbek H., DuBois D. L., Dupuis M., Ferry J. G., Fujita E., Hille R., Kenis P. J., Kerfeld C. A., Morris R. H., Peden C. H., Portis A. R., Ragsdale S. W., et al. (2013) Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 113, 6621–6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mondal B., Song J., Neese F., and Ye S. (2015) Bio-inspired mechanistic insights into CO2 reduction. Curr. Opin. Chem. Biol. 25, 103–109 [DOI] [PubMed] [Google Scholar]
- 21. Shi J., Jiang Y., Jiang Z., Wang X., Wang X., Zhang S., Han P., and Yang C. (2015) Enzymatic conversion of carbon dioxide. Chem. Soc. Rev. 44, 5981–6000 [DOI] [PubMed] [Google Scholar]
- 22. Gottlieb H. E., Kotlyar V., and Nudelman A. (1997) NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 62, 7512–7515 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.