

Abstract
Among primary brain cancers, gliomas are the most deadly and most refractory to current treatment modalities. Previous reports overwhelmingly support the role of the RNA-binding protein Hu antigen R (HuR) as a positive regulator of glioma disease progression. HuR expression is consistently elevated in tumor tissues, and a cytoplasmic localization appears essential for HuR-dependent oncogenic transformation. Here, we report HuR aggregation (multimerization) in glioma and the analysis of this tumor-specific HuR protein multimerization in clinical brain tumor samples. Using a split luciferase assay, a bioluminescence resonance energy transfer technique, and site-directed mutagenesis, we examined the domains involved in HuR multimerization. Results obtained with the combination of the split HuR luciferase assay with the bioluminescence resonance energy transfer technique suggested that multiple (at least three) HuR molecules come together during HuR multimerization in glioma cells. Using these data, we developed a model of HuR multimerization in glioma cells. We also demonstrate that exposing glioma cells to the HuR inhibitor tanshinone group compound 15,16-dihydrotanshinone-I or to the newly identified compound 5 disrupts HuR multimerization modules and reduces tumor cell survival and proliferation. In summary, our findings provide new insights into HuR multimerization in glioma and highlight possible pharmacological approaches for targeting HuR domains involved in cancer cell-specific multimerization.
Keywords: bioluminescence resonance energy transfer (BRET), cancer biology, cell proliferation, ELAV-like protein 1 (HuR (human antigen R)), RNA, glioma
Introduction
Hu antigen R (HuR)2 is an RNA-binding protein belonging to the ELAV gene family that binds to AU-enriched motifs and regulates mRNA splicing, stabilization, and translation (1). ELAV was initially discovered in Drosophila where it was found to be essential for development (2–4). In the adult or mature CNS, HuR is primarily localized in the nucleus of unstressed and non-proliferating cells. HuR actively moves to the cytoplasm during cellular proliferation and stress where it is a major regulator of selective protein expression through its capacity to control targeted mRNA and miRNA behavior (5, 6). HuR is overexpressed in several types of cancer including colorectal, brain, lung, and breast (7–9). HuR promotes tumor progression through regulation of mRNA and miRNA pools involved in proliferation, differentiation, and survival of the transformed cell (10–13). The cell-transforming capacity of HuR in cancer cells may be the result of a number of mechanisms including its overexpression, alterations in phosphorylation, subcellular localization, and generation of HuR aggregates or multimers that promote oncogenic transformation of mRNA and miRNA content (14–22).
The mechanisms of HuR phosphorylation, subcellular trafficking, and mRNA binding are well-established in cancer cells (23–27). However, despite the influence on HuR function (subcellular localization and mRNA binding), the mechanisms of HuR aggregation (referred to as multimerization) observed in numerous types of cancers remain unknown. HuR consists of three mRNA-binding domains (RRM1, RRM2, and RRM3) and a hinge region (between RRM2 and RRM3 domains), which has been implicated in the control of HuR subcellular trafficking and affinity for mRNA targets (28, 29). The tertiary structure of HuR has been identified for truncated HuR oligomers composed of RRM1-RRM2 domains; however, the structure of full-length HuR is only available by computational simulation (30–32). Currently, there are three distinct HuR regions that could be involved in the formation of multimers: the N-terminal domain containing the cysteine (Cys13) residue, the hinge region with several phosphorylation sites, and the N-terminal part of RRM3 containing Trp262 and Trp271 residues (30–33). The Cys13 residue has been implicated in disulfide bond formation in HuR RRM1-RRM2 oligomers, and the hinge and RRM3 domains have been implicated in homo- and heterogeneous helical hydrophobic interactions between ELAV family members.
HuR oligomerization has been reported in the cytoplasmic fractions of the U87 cell line; HeLa cell line derived from cervical cancer; and LNCaP, PC3, and DU-145 prostate cancer cell lines (18, 25, 34, 35). In our current study, we performed a detailed analysis of HuR multimerization in samples of primary brain tumor, primary patient-derived glioblastoma xenolines (PDGx), and the established glioma U251 cell line. We found a significant increase in HuR multimerization with advanced tumor grade. We observed HuR multimer formation in tumor samples predominantly in the cytoplasmic fraction. The multimers can be reduced to dimers or disrupted by exposure to the HuR-specific regulator MS-444, the tanshinone group compound 15,16-dihydrotanshinone-I (DHTS), or the newly identified compound 5. We developed and utilized several novel assays including split firefly luciferase assay (N- and C-terminal parts of firefly luciferase were attached to the C-terminal end of HuR) and bioluminescence energy transfer (BRET) assay (between HuR-Renilla luciferase and EGFP-HuR or HuR-EGFP constructs) to analyze HuR multimerization in U251 cells and to map HuR domains that may be involved in multimerization. By using the split luciferase assay and BRET technique in conjunction with the mutagenesis of HuR, we were able to confirm that the cysteine residue Cys13, HuR hinge, and RRM3 domains make significant contributions in HuR multimer formation. The removal of the hinge and/or RRM3 domains from the HuR molecule significantly decreases the chances of HuR multimer formation. The C13R mutation, in addition to removing the hinge and RRM3 domains, further impairs HuR multimerization. In summary, we provide new insights into HuR multimerization in glioma and highlight possible pharmaceutical pathways for its tumor cell-specific disruption based on the differentiation of targeted domains involved in HuR multimerization.
Results
HuR expression and subcellular localization in brain tumor samples
In a recent report, we observed HuR overexpression in brain tumor samples compared with control samples and noted a significant reduction of survival length for patients with higher HuR expression (16). In this report, we extend those results with an analysis of HuR expression and subcellular distribution in an array of brain tumor samples (World Health Organization (WHO) grades I–IV) and control brain tissue. Fig. 1A illustrates the low intensity of HuR staining in control brain samples and the striking enhancement in HuR intensity and cytoplasmic localization with increasing tumor grade. We observed weak HuR cytoplasmic staining in three of seven samples from control brain, weak to moderate HuR cytoplasmic staining in 14 of 27 samples from low-grade astrocytoma (WHO grades II and III), and moderate to strong HuR cytoplasmic staining in 13 of 15 high-grade astrocytoma (GBM, WHO grade IV) samples (Fig. 1A). Fig. 1B presents the normalized weight of HuR intensity detected in the tissue microarray from control, low-grade, and high-grade (GBM) samples. Fig. 1C illustrates examples of HuR protein distribution in nuclear and cytoplasmic fractions of control and GBM samples detected by a Western blotting technique. In the control samples, HuR was localized to the nuclear fraction and was almost below detection in the cytoplasmic fraction. In high-grade (GBM) samples, HuR resided in both nuclear and cytoplasmic fractions. On average, we observed 1.7 ± 0.26 (p = 0.04, n = 5)- and 1.6 ± 0.19 (p = 0.04, n = 5)-fold enhancement in the HuR protein in the nuclear and cytoplasmic fractions, respectively, in tumor samples compared with control brain (Fig. 1D). The HuR values were normalized to GAPDH in the cytoplasmic fraction and to lamin A/C in the nuclear fraction. In summary, these results demonstrate increased HuR expression and increased redistribution from the nuclear compartment to the cytoplasm with increasing tumor grade.
Figure 1.
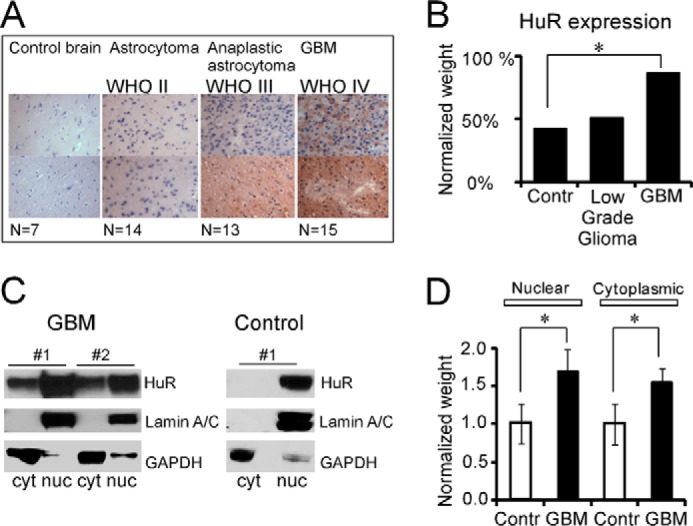
HuR protein expression in normal and brain tumor tissues. A, immunohistochemical detection of HuR in tissue microarray of normal and brain tumor samples. The images were taken at 40× magnification. B, graph representing the averaged HuR intensity detected in tissue microarray for normal, low- (WHO grades II and III), and high-grade GBM (WHO grade IV) brain tumor samples. The HuR intensity was normalized to the HuR averaged value in GBM samples. The difference is significant between normal and GBM samples with p = 0.05 (*). C, example of the assessment of HuR subcellular distribution (nuclear (nuc)/cytoplasmic (cyt) fractions) in control and brain tumor samples by Western blotting. Lamin A/C and GAPDH antibodies were used to confirm nuclear and cytoplasmic fractions, respectively. D, graph representing the averaged HuR protein expression detected by Western blotting (in nuclear and cytoplasmic fractions) in GBM samples compared with the control (Contr). The HuR data were normalized to the GAPDH expression for cytoplasmic fractions and to lamin A/C for nuclear fractions, then averaged for each tissue group, and finally normalized to the averaged values of nuclear control and cytoplasmic control, respectively. The difference between groups is significant with p = 0.04 (*) for both nuclear and cytoplasmic fractions. The error bars represent S.D. calculated for each group.
HuR multimerization in clinical brain tumor samples
HuR has been reported in the cytoplasmic fraction of several types of cancer, and dimer or multimer formation in vitro has been implicated in HuR function (18, 25, 34, 35). To evaluate the degree of HuR multimerization in brain tumor samples, we analyzed HuR content under non-reducing/non-denaturing conditions by Western blotting. Fig. 2A illustrates HuR content in the cytoplasmic fraction of control and high-grade GBM samples in non-reduced/non-denatured conditions versus reduced/denatured conditions. The presence of HuR multimers was observed in GBM samples compared with a predominantly monomeric HuR appearance in the control samples. Table 1 summarizes the analysis of HuR protein multimerization in the cytoplasmic fraction of control samples (n = 4) and brain tumors of different grades, low-grade astrocytoma (WHO grade II; n = 3) and high-grade (GBM WHO grade IV; n = 5). We conclude that there is an increase in HuR multimerization with an increase in tumor grade: four of five GBM samples exhibited HuR bands of high molecular weight (bands 3 and 4) with strong intensity compared with no high-molecular-weight HuR bands with strong intensity in control samples. The low-grade tumors exhibited high-molecular-weight HuR bands (bands 3 and 4) with low and medium intensity. HuR multimerization was observed predominantly in the cytoplasmic fraction and to a limited extent in the nuclear fraction of tumor samples (Fig. 2B). Similar results were detected in five of five tumor samples. To evaluate the nature of HuR multimerization in brain tumors, RNase treatment was used to exclude RNA as a cofactor promoting HuR multimerization, and treatment with β-mercaptoethanol was used to assess the role of disulfide bond formation in HuR multimerization. In Fig. 2C, HuR multimerization is illustrated in the cytoplasmic fraction of a GBM sample in non-reduced/non-denatured conditions before treatment (lane 1), after treatment with RNase A/T (lane 2), after treatment with β-mercaptoethanol (lane 3), and after treatment with RNase A/T plus β-mercaptoethanol (lane 4). Four of four analyzed GBM samples exhibited a similar pattern and reveal that HuR multimerization in GBM is not sensitive to RNase treatment but is partially reduced by β-mercaptoethanol. These results reveal an RNA-independent and tumor-specific HuR multimerization predominantly in the cytoplasmic fraction of transformed cells.
Figure 2.
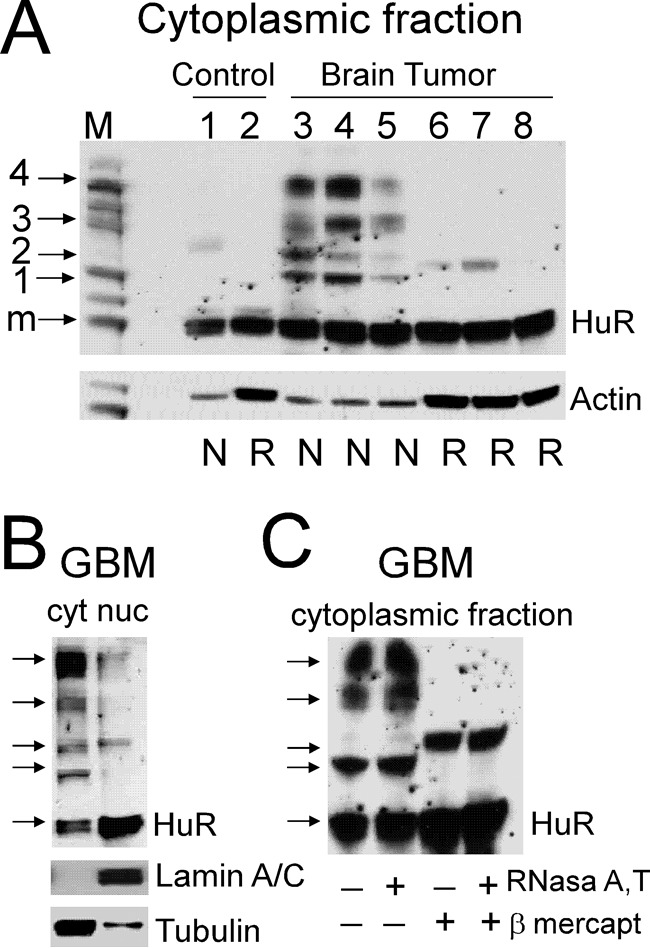
Assessment of HuR protein multimerization in control and brain tumor samples by Western blotting. A, examples of HuR protein dimerization/multimerization detected in non-reducing and non-denaturing conditions (N) versus reducing and denaturing conditions (R) in control and tumor samples. Note that lanes 1 and 2, 3 and 6, 4 and 7, and 5 and 8 represent proteins from the same samples at two different conditions. Each lane was loaded with an equal amount of protein. The actin level served to confirm equal loading. Note that the actin antibody has differing ability to recognize actin in the non-denatured, non-reduced condition compared with the denatured, reduced condition. The protein ladder is shown in lane M; the arrows correspond to 30 (monomeric HuR; m), 50 (1), 60 (2), 90 (3), and 120 kDa (4) molecular masses. B, examples of HuR protein multimerization in the non-reduced and non-denatured condition in cytoplasmic (cyt) and nuclear (nuc) fractions of the tumor sample. Lamin A/C and α-tubulin antibodies were used to confirm nuclear and cytoplasmic fractions, respectively. C, examples of HuR protein dimerization/multimerization from the same tumor sample in four different conditions: non-denatured and non-reduced (lane 1), pretreated with RNase A/T (lane 2), pretreated with β-mercaptoethanol (lane 3), and pretreated with both RNase A/T and β-mercaptoethanol (lane 4).
Table 1.
HuR multimerization
The numbers in parentheses in the second, third, and fourth columns represent the number of samples exhibiting the corresponding HuR band with the particular molecular mass and intensity. The band molecular masses are indicated in the first column: 30 kDa (monomer), 50 kDa (1), 60 kDa (2), 90 kDa (3), and 120 kDa (4). The band intensity is indicated as weak (w), medium (m), and strong (str).
| Band no. | Control (n = 4) | Low grade (n = 3) | High grade (n = 5) |
|---|---|---|---|
| Monomer | w (1) | w (0) | w (0) |
| Monomer | m (2) | m (3) | m (3) |
| Monomer | str (1) | str (0) | str (2) |
| 1 | w (1) | w (2) | w (1) |
| 1 | m (0) | m (0) | m (0) |
| 1 | str (0) | str (1) | str (4) |
| 2 | w (1) | w (1) | w (2) |
| 2 | m (0) | m (0) | m (1) |
| 2 | str (3) | str (0) | str (0) |
| 3 | w (2) | w (0) | w (1) |
| 3 | m (0) | m (2) | m (1) |
| 3 | str (0) | str (0) | str (3) |
| 4 | w (2) | w (3) | w (0) |
| 4 | m (0) | m (0) | m (1) |
| 4 | str (0) | str (0) | str (4) |
HuR multimerization in glioma cell lines
In agreement with the data generated in brain tumor tissue samples, we observed HuR multimerization in both established (U251) (Fig. 3A) and primary PDGx cell lines (supplemental Fig. 1) predominantly in the cytoplasmic fraction. We confirmed that a HuR/HuR interaction occurred in the cytoplasmic fraction by coimmunoprecipitation of endogenous HuR with a HuR-EGFP construct transiently transfected into the U251 cell line (Fig. 3B). We did not detect HuR coimmunoprecipitation with EGFP alone as a control. To identify HuR multimerization in living cells, we developed a split luciferase assay based on the reconstitution of firefly luciferase enzymatic activity during HuR protein multimerization resulting from the fusion of the N-terminal (amino acids 2–416) and C-terminal (amino acids 398–550) parts of firefly luciferase attached to the HuR protein. The system is detailed in Fig. 4A. As controls, the N-terminal piece of firefly luciferase (Nluc), C-terminal piece of firefly luciferase (Cluc), full-length firefly luciferase (Fluc), and HuR-Fluc constructs were created (Fig. 4B). We generated several doxycycline (dox)-inducible stable cell lines: (a) cell lines coexpressing HuR-Nluc and HuR-Cluc constructs to detect HuR multimerization, (b) cell lines coexpressing Nluc and Cluc constructs as a control, (c) cell lines expressing HuR-Fluc as a control, and (d) cell lines expressing Fluc as a control (Fig. 5A). Fig. 5B illustrates the averaged luminescence signal from the above cell lines with similar levels of construct induction. Note that the cell line coexpressing HuR-Nluc and HuR-Cluc constructs exhibits strong luminescence signal in the presence of the luciferase substrate luciferin, confirming HuR/HuR interaction in the U251 cell line. The averaged ratio of luminescence signal from HuR-Nluc+HuR-Cluc to background showed a 27 ± 6-fold (n = 8) increase. To reduce the chance of the reporter forming multimers with endogenous HuR, we transfected the HuR-Nluc+HuR-Cluc stable cell line with siRNA against the 3′-UTR of endogenous HuR to decrease endogenous HuR protein expression. In this setting, we measured an enhanced ratio of luminescence signal from HuR-Nluc+HuR-Cluc to background of 71 ± 7 (n = 11)-fold, whereas the opposite condition of transfection of the HuR-Nluc+HuR-Cluc stable cell line with recombinant HuR (without Nluc or Cluc tags) decreased the ratio of luminescence signal from HuR-Nluc+HuR-Cluc to 15 ± 2-fold relative to background (n = 4). Thus, our data confirm HuR multimerization in the U251 cell line and validate the use of the HuR-Nluc+HuR-Cluc system for the detection of this process.
Figure 3.
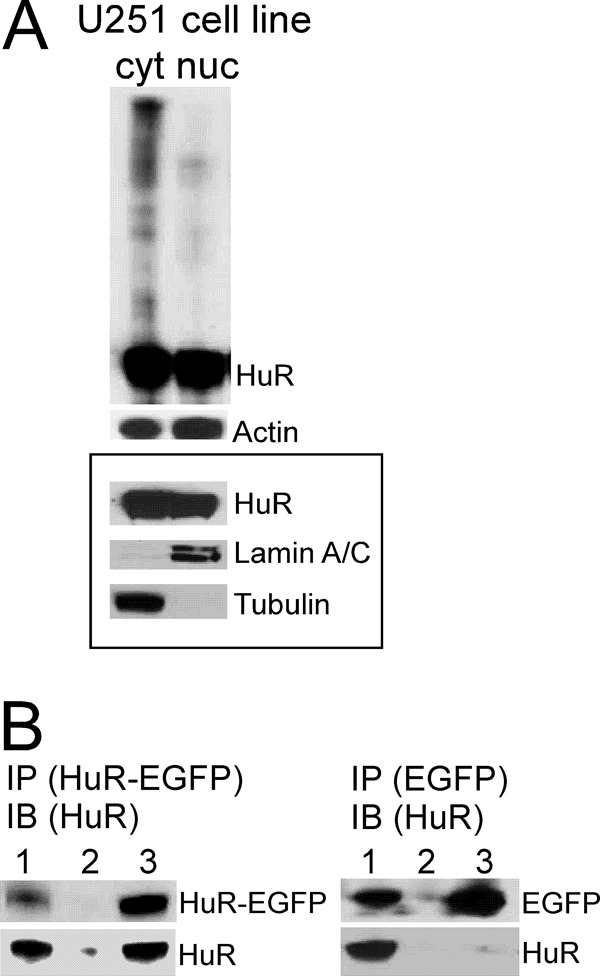
Assessment of HuR protein multimerization in protein samples from U251 glioma cell line. A, example of HuR protein multimerization in the non-reduced and non-denatured condition in cytoplasmic (cyt) and nuclear (nuc) fractions detected by Western blotting. Actin served to confirm equal protein loading. The framed inset represents HuR distribution in nuclear and cytoplasmic fractions in the reduced and denatured condition. Lamin A/C and α-tubulin antibodies were used to confirm nuclear and cytoplasmic fractions, respectively. B, example of coimmunoprecipitation of endogenous HuR protein (30 kDa) with HuR-EGFP protein (50 kDa) transfected in the U251 cell line and with EGFP (20 kDa) as a control. Immunoprecipitation (IP) of HuR-EGFP protein or EGFP was performed using EGFP antibody; immunoblotting (IB) was performed using HuR3A2 antibody. The lanes correspond to input (lane 1), immunoprecipitation using rabbit IgG antibody (lane 2), and immunoprecipitation using EGFP antibody (lane 3).
Figure 4.
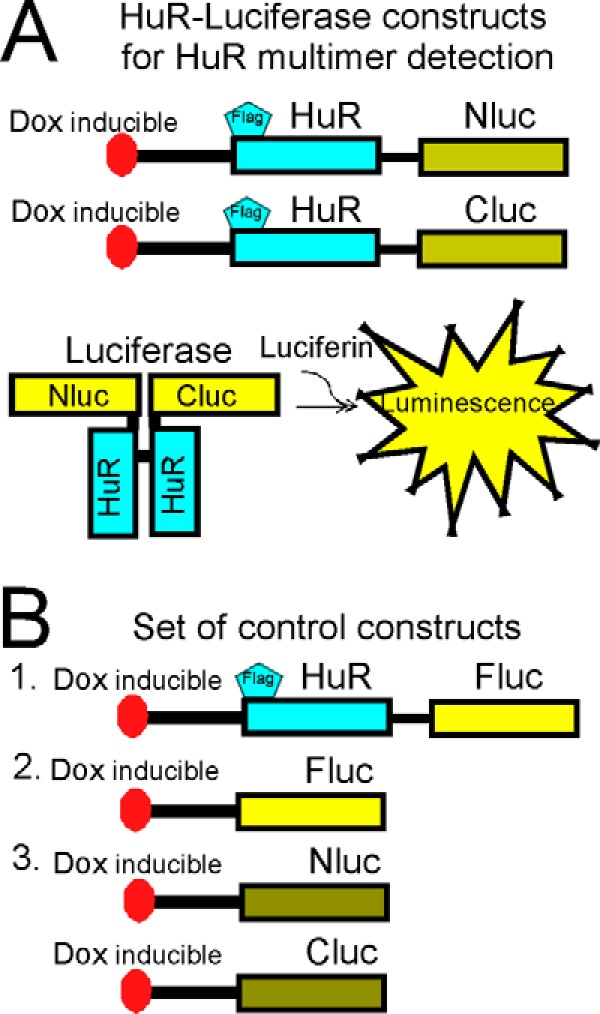
Schematic illustration of reporter constructs utilized in split firefly luciferase assay to detect HuR dimerization. A, the HuR constructs to detect HuR dimerization. Nluc corresponds to amino acids 1–398 of firefly luciferase, and Cluc corresponds to amino acids 394–550 of firefly luciferase. Both were fused with HuR (see “Experimental procedures” for details). B, sets of control constructs utilized in the split firefly luciferase assay (see “Experimental procedures” for details).
Figure 5.
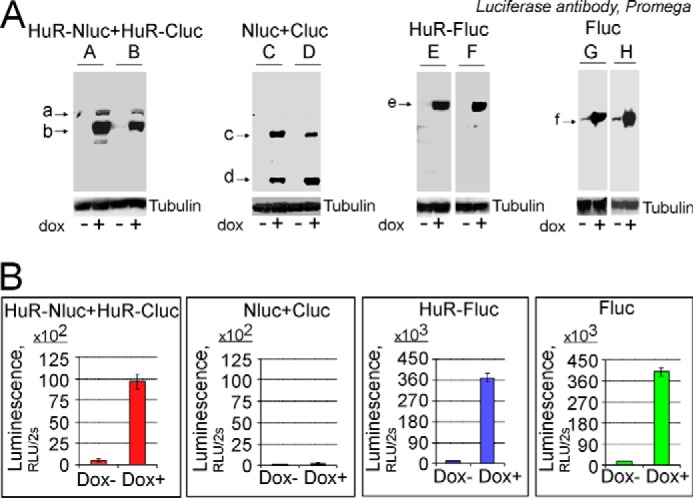
Confirmation of HuR protein dimerization in split firefly luciferase assay. A, illustration of the expression of reporter proteins encoded by dox-inducible reporter constructs utilizing denatured and reduced Western blotting technique. There are no oligomer and multimer protein complexes in these blots because of the denatured and reduced condition. Arrows a and b mark HuR-Nluc and HuR-Cluc denatured and reduced monomeric proteins, respectively, detected in the protein lysates from cells coexpressing HuR-Nluc and HuR-Cluc constructs; arrows c and d mark Nluc and Cluc, respectively, detected in the protein lysate from cells coexpressing Nluc and Cluc constructs; arrow e marks HuR-Fluc detected in the protein lysate from cells expressing the HuR-Fluc construct; and arrow f marks Fluc detected in the protein lysate from cells expressing the Fluc construct. α-Tubulin was used to confirm equal protein loading. The constructs were detected using luciferase antibody (Promega), which recognizes epitopes on the N and C termini of firefly luciferase (see “Experimental procedures”). At least two clones were generated for each condition (A–H). B, the averaged luminescence signals detected from the reporter stable cell lines with a similar level of construct induction in the presence of the firefly luciferase substrate d-luciferin (see “Experimental procedures” for details). The error bars represent S.D. for each group.
Cysteine 13, hinge, and RRM3 HuR domains regulate HuR multimerization
Several HuR domains including the N-terminal disulfide bond formed by cysteine 13, the hinge region between RRM2 and RRM3, and the RRM3 domain could be implicated in HuR multimerization. To determine whether the hinge and RRM3 domains of HuR are involved in the multimerization process, we generated dox-inducible stable cell lines expressing full-length HuR or truncated HuR versions fused with N- and C-terminal parts of firefly luciferase. The truncated constructs are truncated before the hinge region (residue 198) or after the hinge region (residue 244), and all are fused with the Nluc and Cluc parts of firefly luciferase (Fig. 6A). We were able to detect luminescence signal in the split luciferase assay from HuR-Nluc and HuR-Cluc constructs truncated before and after the hinge region but at significantly reduced signal intensity relative to the full-length constructs. The HuR construct without hinge, without RRM3, and with the C13R mutation failed to generate a luminescence signal above the background level in the split luciferase assay. Fig. 6B illustrates relative luminescence signal normalized to the protein expression of the above cell lines, confirming that the residue Cys13, hinge, and RRM3 domain of HuR are required for the HuR multimerization process.
Figure 6.
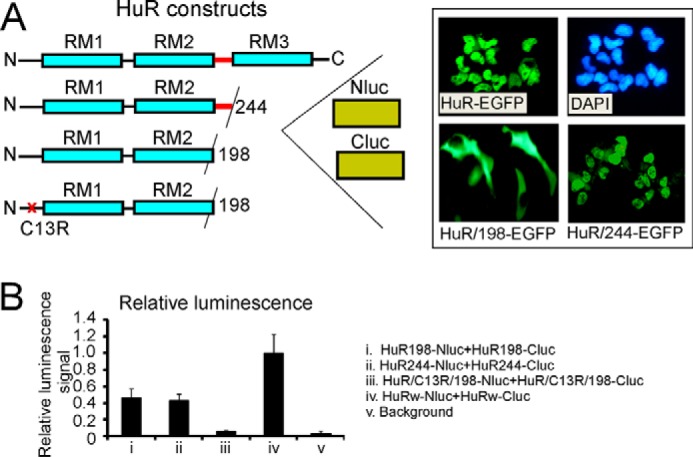
Assessment of HuR oligomerization domains in split firefly luciferase assay using reporter constructs with truncated and mutated HuR. A, schematic illustration of truncated and mutated HuR constructs used in split firefly luciferase assays (see “Experimental procedures”). The inset illustrates subcellular distribution of HuR-EGFP, HuR/244-EGFP (without RRM3 (RM3)), and HuR/198-EGFP (without hinge and without RRM3) constructs. Note that HuR proteins with deleted hinge regions lack the HuR nuclear localization signal and reside in the cytoplasmic fraction. The HuR proteins with only RRM3 domains deleted have nuclear/cytoplasmic distribution similar to that of full-length HuR. The subcellular distribution of the above constructs is in agreement with previous data (28). B, graph representing averaged relative luminescence signals from cells coexpressing dox-inducible HuR/198-Nluc and HuR/198-Cluc (i), HuR/244-Nluc and HuR/244-Cluc (ii), HuR/C13R/198-Nluc and HuR/C13R/198-Cluc (iii), and HuRw-Nluc and HuRw-Cluc (iv) constructs. Note that averaged luminescence signals from HuR/198-Nluc and HuR/198-Cluc (i), HuR/244-Nluc and HuR/244-Cluc (ii), and HuRw-Nluc and HuRw-Cluc (iv) cell lines significantly exceed background luminescence signal (v). The error bars represent S.D. for each group.
Analysis of HuR multimerization by BRET technique
To confirm and validate the properties of HuR multimerization, we used a BRET technique between HuR-Renilla luciferase and HuR-EGFP or EGFP-HuR constructs (Fig. 7A). By fusing the EGFP protein either to the N terminus or C terminus of HuR, we were able to evaluate the properties of HuR orientation in multimer formation. The HuR-Renilla luciferase alone or together with HuR-EGFP or EGFP-HuR constructs were transfected into U251 cells, and the BRET ratio was analyzed in the presence of coelenterazine h or native coelenterazine (see “Experimental procedures”). The PRKAR2A-EGFP construct was used as a control and did not alter the HuR-Renilla luciferase spectrum. The averaged BRET ratio detected at 510 nm was 0.23 ± 0.05 (n = 5) for HuR-Renilla luciferase plus HuR-EGFP constructs and 0.21 ± 0.06 (n = 5) for HuR-Renilla luciferase plus EGFP-HuR constructs (Fig. 7B). The difference was not significant, suggesting that HuR molecules have equal probability of converse and direct orientations in multimers. Fig. 7C illustrates examples of spectra detected from U251 cells transfected with HuR-Renilla luciferase alone or with HuR-Renilla luciferase plus HuR-EGFP.
Figure 7.
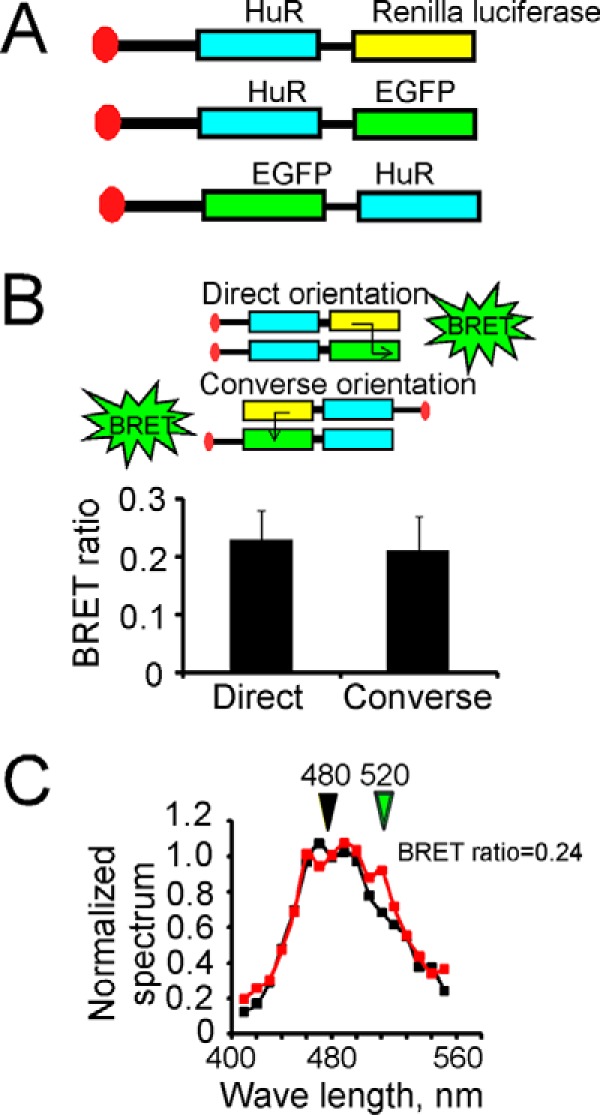
Confirmation of HuR dimerization by BRET technique. A, schematic representation of HuR constructs (HuR fused with Renilla luciferase and HuR fused with green fluorescent protein on N or C terminal end) utilized in BRET assay. B, graph representing averaged BRET ratios of 0.23 ± 0.05 (n = 5) for HuR-Renilla luciferase plus HuR-EGFP and 0.21 ± 0.06 (n = 5) for HuR-Renilla luciferase plus EGFP-HuR constructs, respectively. The error bars represent S.D. for each group. The difference between the two groups is not significant (p > 0.05). C, examples of superimposed emission spectra (from 410 to 570 nm; 10-nm individual measurement interval) for HuR-Renilla luciferase plus scrambled vector (marked with black squares) and HuR-Renilla luciferase plus HuR-EGFP constructs (marked with red squares) in the presence of Renilla luciferase substrate native coelenterazine. The luminescence signals at wavelengths 480 ± 10 and 520 ± 10 nm were used to calculate the BRET ratio (see “Experimental procedures” for details).
To confirm that at least three HuR molecules may come together for multimerization, we developed a BRET assay between the split luciferase assay constructs consisting of HuR-Nluc and HuR-Cluc (which when reconstituted would generate firefly luciferase activity) and a HuR-DsRed (red fluorescent protein fused with HuR) construct (Fig. 8A). The HuR-Nluc+HuR-Cluc stable cell line was transfected with the HuR-DsRed construct for BRET detection and cotransfected with siHuR against the 3′-UTR of endogenous HuR to improve the interaction among recombinant HuR-Nluc, HuR-Cluc, and HuR-DsRed proteins in these experiments. The BRET was initiated by the addition of the firefly luciferase substrate luciferin. The averaged BRET ratio between HuR-Nluc plus HuR-Cluc constructs and HuR-DsRed construct detected at 590 nm was 0.41 ± 0.1 (n = 4) (Fig. 8A), confirming that at least three HuR molecules may coexist in multimers. Based on the above results, we developed a model of HuR multimerization (presented in Fig. 8B). We propose that HuR multimerization is influenced by disulfide bond formation between Cys13 residues and oligomerization with the hinge and RRM3 domains of HuR (see “Discussion”). This model is consistent with the congruent BRET ratio between HuR-Renilla luciferase and HuR-EGFP or EGFP-HuR constructs observed in the experiments above.
Figure 8.
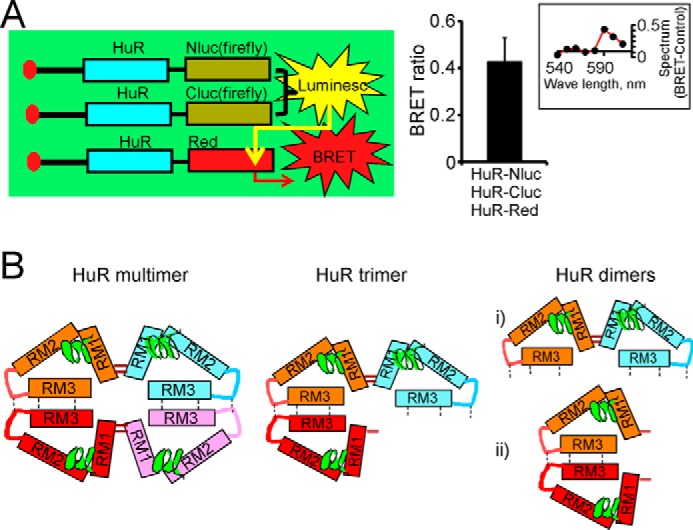
Confirmation of HuR multimerization by combined split firefly luciferase and BRET assays. A, schematic representation demonstrating detection of HuR multimerization in combined split firefly luciferase and BRET assays (left). The graph provides averaged BRET ratio detected between firefly luciferase reconstituted during HuR dimerization in split luciferase assay and HuR-DsRed fluorescence construct coexpressed in the same cells (see “Experimental procedures” for details). The error bars represent S.D. d-Luciferin was used as a firefly luciferase substrate. The inset illustrates the difference between the superimposed and normalized emission spectra of BRET condition (from cells coexpressing HuR-Nluc, HuR-Cluc, and HuR-DsRed constructs) and the control condition (cells coexpressing HuR-Nluc, HuR-Cluc, and DsRed constructs). The spectrum difference is presented from 540 to 610 nm with 10-nm individual measurement interval. The averaged ratio of the signal at 590 nm to the signal at 570 nm was 0.81 ± 0.1 (n = 4) for control and 1.22 ± 0.1 (n = 4) for BRET condition. The expression of endogenous HuR was decreased by using siHuR against HuR 3′-UTR (see “Experimental procedures” for details). B, potential models of HuR multimerization and dimerization. The HuR tetramer and trimer models (left and center, respectively) include concurrent disulfide bond formation between HuR N termini and hydrophobic oligomerization of hinge and RRM3 domains. HuR dimerization models (right) rely on formation of a disulfide bond between HuR N termini or oligomerization of hinge and RRM3 domains.
Disruption of HuR multimerization in tumor cells
Fig. 9 illustrates that HuR multimerization may be disrupted by (a) the tanshinone group compound DHTS, which has been implicated in HuR/RNA interaction (36), and (b) the new compound 5, which has been identified as a regulator of hinge region function (supplemental Fig. 2). We used the identified HuR-specific inhibitor MS-444 (37) as a control (supplemental Fig. 3). Note the specific and robust inhibition of HuR multimer formation after short (6–8-h) treatment with MS-444 that was specific for the luminescence signal from HuR-Nluc+HuR-Cluc-expressing cells compared with the luminescence signal from control cells expressing the Fluc construct. Fig. 9A, panel a, illustrates the inhibitory dose response of DHTS on HuR multimerization after 1.5 h of treatment. Fig. 9A, panel b, illustrates the inhibitory dose response of compound 5 on HuR multimerization (after 6–8 h of treatment). A specific and robust inhibition of HuR multimer formation is noted. In Fig. 9B, we show the cytotoxicity curves for DHTS (panel a) and compound 5 (panel b) on the U251 cell line; those for PDGx lines are shown in supplemental Fig. 4. We did not detect a significant alteration in total HuR cytoplasmic levels at the 1.5-h time point for DHTS (50 μm) or at the 6-h time point for MS-444 (up to 100 μm) or compound 5 (50 μm) (Fig. 9C, panel a). However, we did observe a dramatic decrease in the cytoplasmic HuR level after 24 h of treatment with compounds MS-444 (by 84%; lane 1), DHTS (by 95%; lane 2), and compound 5 (by 90%; lane 3) compared with the cytoplasmic HuR in untreated cells (lane 4) (Fig. 9C, panel b). Similar results were observed in three experiments. Thus, we conclude that the disruption of HuR multimerization could be achieved through the specific targeting of different HuR domains and leads to a decrease in tumor progression.
Figure 9.
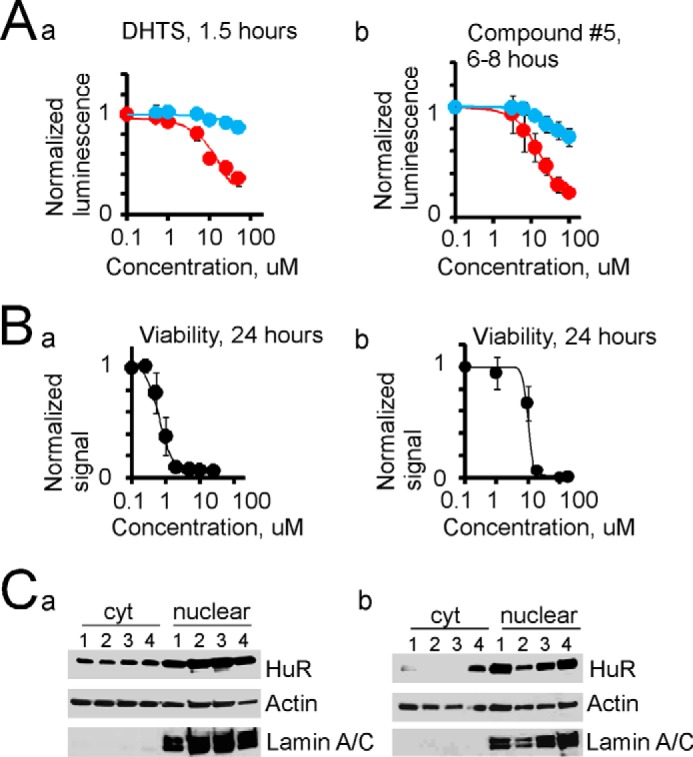
Disruption of HuR multimerization is toxic for tumor cells. A, dose-response curves of inhibition of HuR multimerization detected in reporter assay for compounds DHTS (1.5 h of treatment; panel a) and compound 5 (6–8 h of treatment; panel b). The red circles represent luminescence signal from U251 cells coexpressing HuR-Nluc and HuR-Cluc constructs, the blue circles represent luminescence signal from U251 cells expressing Fluc construct. For each experiment, data were normalized to the value of luminescence signal before treatment. The error bars represent S.D. IC50 = 20 ± 6 μm (n = 4) for DHTS, and IC50 = 21 ± 6 μm (n = 5) for compound 5. B, cytotoxicity dose responses for DHTS (24 h of treatment; panel a) and compound 5 (24 h of treatment; panel b) on the U251 cell line. The error bars represent S.D. C, cytoplasmic and nuclear HuR values detected by Western blotting after short (panel a) and long (panel b) cell treatments with compounds MS444 (50 μm; lane 1), DHTS (25 μm; lane 2), compound 5 (25 μm; lane 3), and DMSO control (lane 4). The short treatment corresponds to 6 h for MS444 and compound 5 and to 1.5 h for DHTS. The long treatment corresponds to 24 h for all compounds. The HuR values were normalized to the actin value in the cytoplasmic fraction and to lamin A/C in the nuclear fraction. Note the decrease of the cytoplasmic HuR level by 84, 95, and 90% after 24 h of treatment with compounds MS444, DHTS, and compound 5, respectively, compared with control (panel b). Lamin A/C antibody was used to confirm the separation of nuclear and cytoplasmic fractions.
Discussion
In our current work, we have developed several lines of evidence confirming the formation of HuR multimers in the cytoplasmic fraction of brain tumor cells. We confirmed HuR protein multimerization by Western blotting, split HuR-firefly luciferase assay, and BRET in living cells. The combination of split HuR-luciferase assay with BRET technique suggests that at least three HuR molecules come together in glioma cells during HuR multimerization. Based on our data, we have developed a model of HuR multimerization in cancer cells. Our data support the role of HuR protein multimerization as a promoter of the neoplastic process. By utilizing BRET and split luciferase assays with truncated and full-length HuR constructs, we demonstrated that the HuR hinge region and the RRM3 domain play a significant role in HuR multimerization in living cells. We found that a loss of the hinge region causes an accumulation of the recombinant HuR protein in the cytoplasmic fraction but with a significantly impaired ability for dimerization/multimerization. The HuR without RRM3 (but with hinge region) exhibits less protein in the cytoplasmic compartment but has a better chance for multimerization compared with the HuR that loses RRM3 and hinge together. Our data are in agreement with the RRM3 dimerization model proposed by using Brownian dynamics, molecular dynamics, and computational data analysis (29, 31, 32). The yeast two-hybrid system screening of HuR oligomerization domains also confirmed that the hinge and RRM3 regions of HuR are essential for HuR/HuR interaction (33). In addition, the analyses of truncated and full-length HuR protein binding with RNA indicate that the RRM3 domain is required for cooperative assembly of HuR oligomers on RNA (29). The comparison of primary sequences of ELAV family members confirmed that the HuR RRM3 domain has strong homology with HuC and HuD proteins; however, the hinge regions are very diversified. Interestingly, the oligomerization of full-length HuC and HuD proteins strongly relies on the hinge and RRM3 domains (38, 39). Thus, there is a substantial consensus that the RRM3 and hinge regions contribute to ELAV protein oligomerization and the unique sequence of the hinge region may serve as a novel molecular target for therapeutic development.
One of the intriguing and unique aspects of HuR multimerization noted in our work is the sensitivity of HuR multimers to redox condition. The HuR C13R mutant without the hinge and without RRM3 fails to generate a luminescence signal above the background level in the split luciferase assay. The sensitivity of HuR multimerization to redox changes could be explained by disulfide bond formation between cysteine (Cys13) residues, which have been implicated in the process of RRM1 and RRM1-RRM2 HuR dimer formation (33). The exclusively monomeric state has been reported for the HuR C13A (amino acids 2–189) construct, confirming the stringent dependence of the HuR RRM1-RRM2 dimerization on disulfide Cys13–Cys13 bond formation (30). The analysis of primary sequences of ELAV family members revealed that HuR is the only protein of the ELAV family members that has the Cys13 residue on the N terminus. The in situ S–S bond-dependent chemical cross-linking of HuD proteins in multimer modules suggests that the redox state-dependent multimerization of HuD proteins may exist at the cellular level in certain pathological conditions and may rely on cysteine residues probably located in the RRM3 domain of HuD (38). There are no reports of redox-sensitive HuD multimerization in normal physiological conditions. Although HuR multimerization has been observed in several cell lines, the functional role of HuR oligomerization and multimerization remains uncertain. HuR multimerization may occur prior to mRNA binding as well as allosterically during HuR-mRNA complex formation (24, 29, 36). We believe that HuR multimerization impacts HuR function such as affecting HuR-mRNA binding, HuR ability to participate in splicing, HuR subcellular localization, or HuR stability. The disruption of HuR dimerization by low-molecular-weight compounds through the interaction with RRM1-RRM2-hinge HuR core or hinge-RRM3 domains in vitro significantly diminishes HuR-mRNA binding and ultimately leads to the reduction in cancer cell proliferation and survival (36, 37, 40, 41). However, the modification of HuR function should be done with caution, using cell-specific targeting, because of potential side effects on normally proliferating cells.
In Fig. 8B, we propose a model of HuR multimerization in cancer cells that combines the interactions of several HuR-oligomerizing domains. This model includes disulfide bond formation (Cys13–Cys13) between N-terminal regions of HuR and at the same time oligomerizations of hinge and RRM3 HuR domains. Current HuR tertiary models are based on biochemical data with limited sets of physiological variables and computational analysis of truncated HuR constructs only designed to simulate HuR dimerization (28–33). Our HuR tertiary model is built on quantitative data from living cells with truncated and full-length HuR constructs. The cell-based environment and native full-length HuR molecule allowed us to reveal complex interactions between HuR-oligomerizing domains that may be different in nature compared with biochemical assays. Importantly, this model of HuR multimerization is supported by the congruent BRET ratio for HuR fused to either the N or C terminus with fluorescent protein that was observed in our BRET assay experiment between HuR-Renilla luciferase and HuR-EGFP or EGFP-HuR constructs.
The formation of HuR multimers is most likely a complex process, which could be influenced by intracellular conditions (like redox state, pH, and ionic strength), cofactors (like mRNA and protein partners), or post-translational modifications (like phosphorylation/dephosphorylation, methylation, and acetylation). Primary brain tumors are characterized by oxidative stress, abnormal patterns of protein phosphorylation, and abnormal protein localization (12–14, 16, 21, 27, 42, 43). Particularly, the oxidative stress in brain tumors is well-established and could be caused by known oncogenic or metabolic alterations such as isocitrate dehydrogenase-1 (IDH1) Arg132 mutations, mitochondrial dysfunction, ATP withdrawal, or inflammatory conditions related to viral infection (44–48). Supplemental Fig. 5 illustrates an induction of the HuR/HuR multimerization in stress conditions evoked by coexpression of endogenous IDH1 and recombinant IDH1 R132H mutant in U251 cells, suggesting that HuR multimerization may be regulated on the post-translational level. Indeed, cellular stress and ATP depletion are significant factors involved in HuR cytoplasmic shuttling and induction of the antiapoptotic function in normal and cancer cells (49–52). The influence of HuR phosphorylation/dephosphorylation on HuR oligomerization and subcellular localization has been reported in cancer cells (18, 34, 35, 52). Alterations of HuR phosphorylation in the hinge region affect HuR nuclear/cytoplasmic shuttling, assembly in stress granules, and attachment to centrosomes (1, 26, 27, 52). Thus, HuR multimerization influenced by the cellular environment could be considered as an adaptative survival mechanism of cancer cells under stress conditions. Overall, our work provides an understanding of HuR multimerization in cancer cells and highlights specific pathways for its disruption.
Experimental procedures
Cell culture and patient material
The XD459 and JX10 primary PDGx lines were previously established from human brain tumor tissue (10). The U251 cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA). The U251 Tet-On cell line was previously developed from the U251 cell line by using pTet-On plasmid (Clontech) (10). The stable cell lines U251 Tet-On-(HuR-Nluc+HuR-Cluc), U251 Tet-On-(Nluc+Cluc), U251 Tet-On-(HuR-Fluc), U251 Tet-On-(Fluc), U251 Tet-On-(HuR198-Nluc+HuR198Cluc), and U251 Tet-On-(HuR244-Nluc+HuR244-Cluc) were established by using antibiotic selection (hygromycin B and G418 sulfate, both purchased from Mediatech, Inc., Manassas, VA). Cell lines were expanded in DMEM/F-12 (Mediatech, Inc.) supplemented with 10% fetal bovine serum (Mediatech, Inc.), 100 units/ml penicillin/streptomycin (Mediatech, Inc.), and 400 mol/liter l-glutamine (Mediatech, Inc.). The identity of stable cell line clones was maintained by supplementing cell medium with hygromycin B (0.1 mg/ml final concentration) and G418 sulfate (0.4 mg/ml final concentration). Cells were grown at 37 °C and 5% CO2. The tissue microarray of control and tumor samples was a gift from Dr. Tom Mikkelsen of the Hermelin Brain Tumor Center, Henry Ford Hospital, Detroit, MI. The GBM biopsy tissues and control samples for protein extraction were obtained from Dr. Y. Gillespie, Brain Tumor Tissue Core, University of Alabama, Birmingham, AL. Patient materials were used with the informed consent of the patients and with the approval of the University of Alabama Institutional Review Board. Compound 5 (N-[4-(1H-benzimidazol-2-yl)phenyl]-2-chloro-5-nitrobenzamide) was purchased from ChemBridge, San Diego, CA. DHTS was purchased from Sigma-Aldrich. All compounds were solubilized with dimethyl sulfoxide (DMSO) (Sigma-Aldrich).
Construct design and cloning
The firefly luciferase constructs Fluc, Nluc (amino acids 1–398), and Cluc (amino acids 394–550) were a gift from Dr. Stuart J. Frank in pcDNA3.1+ vector. Fluc, Nluc, and Cluc were cut from pcDNA3.1+ vector using NotI and XhoI enzymes (New England Biolabs) and subcloned in pTre2Hygro dox-inducible vector using NotI and SalI sites. The full-length HuR and truncated HuR (amino acids 1–198, 1–244, and 1–282) constructs, all with FLAG tag at the N terminus and without a stop codon, were amplified using Forw (5′-TGGAGACGCCATCCACGCTG) and Rev-HuR-full-NotI (5′-AAAAAAAGCGGCCGCTTTGTGGGACTTGTTGGTTTTGAAGG), Rev-HuR-198-NotI (5′-AAAAAAAGCGGCCGCCTGCGAGAGGAGTGCCACGTTTTTGTTCTG), Rev-HuR-244-NotI (5′-AAAAAAAGCGGCCGCCCAGCCGGAGGAGGCGTTTCCTGGCACG), and Rev-HuR-282-NotI (5′-AAAAAAAGCGGCCGCGTTGGTGTTGAAGTCGCGGATCACTTTC) primers, respectively by PCR from the previously made FLAG-HuR construct, digested with NotI enzyme, and cloned in-frame in pTre2Hygro vectors with firefly luciferase constructs using the NotI site. All insert orientations, frame, and correct sequences were verified by sequencing in the University of Alabama Genomics core facility. The C13R mutation in HuR/198-Nluc, HuR/198-Cluc, and HuR/198-Fluc constructs was generated by site-directed mutagenesis with the following primers: Forw-C13R, 5′-CCACATGGCCGAAGACCGCAGGGGTGACATCGGGAG, and Rev-C13R, 5′-CTCCCGATGTCACCCCTGCGGTCTTCGGCCATGTGG. The Renilla luciferase was amplified by PCR with Rluc-Forw-NotI (5′-GTGCTAGCGCGGCCGCGGGTGGTGGTGCTTCCAAGGTGTACGACCCCGAG) and Rluc-Rev-SalI (5′-GTGCTAGCGTCGACTTACTGCTCGTTCTTCAGCACGCG) primers from pGL4.82 vector (Promega, Madison, WI) and cloned in pTre2Hygro vector using the NotI and SalI sites. The full-length HuR (with FLAG tag and without stop codon) was amplified by PCR with primers Forw (5′-TGGAGACGCCATCCACGCTG) and Rev-HuR-full (5′-AAAAAAAGCGGCCGCTTTGTGGGACTTGTTGGTTTTGAAGG), digested with NotI, and fused with Renilla luciferase using the NotI site. The EGFP-HuR construct was previously made. Briefly, to make the HuR-EGFP construct, EGFP (without ATG codon and with stop codon) was amplified by PCR with primers Forw-EGFP-NotI (5′-GTGCTAGCGCGGCCGCGGGTTCTGGTGGTgtgagcaagggcgaggagctgttcaccggggtg) and Rev-EGFP-SalI (5′-GTGCTAGCGTCGACTTACTTGTACAGCTCGTCCATGCCGAGAG), digested with NotI and SalI enzymes, and cloned in pTre2Hygro vector using the NotI and SalI sites. Full-length or truncated HuR (without stop codon) constructs were fused in-frame with EGFP using the NotI site with the same strategy as described above. To make the PRKAR2A-EGFP construct, PRKAR2A (without stop codon) was amplified by PCR with primers Rev-NotI-PRKAR2A (5′-GTGCTAAGGCGGCCGCCTGCCCGAGGTTGCCCAGATCCACGCT) and Forw-NotI-PRKAR2A (5′-GTGCTAGCGCGGCCGCATGAGCCACATCCAGATCCCGCCGGGGCTCACGG) from LgBiT-PRKAR2A vector (Promega), digested with NotI, and fused in-frame with EGFP using the NotI site. To make the DsRed-HuR construct, red fluorescent protein (without stop codon) was cut from the DsRed2-C1 vector using NheI and SalI enzymes and cloned in pTre2Hygro vector using the NheI and SalI sites. HuR was amplified by PCR with primers Forw-HuR-SalI (5′-GCGGCCGCGTCGACCAAGACTACAAGGACGACGATGACAAG) and Rev HuR-SalI (5′-GTGCTAGCGTCGACTTATTTGTGGGACTTGTTGG), digested with SalI enzyme, and fused with red fluorescent protein using the SalI site. The firefly luciferase HuR constructs were linearized with FspI restriction enzyme for stable cell line creation. The DNA constructs were transfected using TransIT-293 (Mirus) transfection reagent or by an electroporation technique using a 4D-NucleofectorTM core unit and X kit (Lonza, Koln, Germany). To make IDH1constructs, the FLAG epitope was attached to the IDH1 gene using the following primers: Forw1-ATG-FLAG-SalI-IDH1, 5′-ACATGGACTACAAGGACGACGATGACAAGTCCAAAAAAATCAGTGGCGGTTCTGTGGTAGAG, and Rev-stop-SalI-IDH1, 5′-AAGCAACAGTCGACTTAAAGTTTGGCCTGAGCTAGTTTGATC. The mutations were achieved by PCR with the following primers: Forw-IDH1-R132H, 5′-GTAAAACCTATCATCATAGGTCATCATGCTTATGGGGATC, and Rev-IDH1-R132H, 5′-GATCCCCATAAGCATGATGACCTATGATGATAGGTTTTAC; Forw-IDH1-R132C, 5′-GTAAAACCTATCATCATAGGTTGTCATGCTTATGGGGATC, and Rev-IDH1-R132C, 5′-GATCCCCATAAGCATGACAACCTATGATGATAGGTTTTAC; and Forw-IDH1-R132S, 5′-GTAAAACCTATCATCATAGGTAGTCATGCTTATGGGGATC, and Rev-IDH1-R132S, 5′-GATCCCCATAAGCATGACTACCTATGATGATAGGTTTTAC. The constructs were cloned in pTre2Hygro vector using the SalI restriction site. All constructs were verified by sequencing.
Luciferase assay
96-well assay plates (white, clear bottom polystyrene plates treated for tissue culture; Corning) were utilized for all luminescence detection assays. Cells from expanded cell lines were detached by trypsinization, counted using a TC10 automated cell counter (Bio-Rad), plated at 60,000/well, and induced for 48 h by dox (0.75 μg/ml) or non-induced (as control). After 48 h of construct induction, beetle luciferin (d-luciferin) (Promega) was used as a substrate of firefly luciferase constructs for bioluminescence imaging of luminescence signals in split luciferase assays for HuR dimer detection and controls. The plates were equilibrated at room temperature, fresh cell culture medium containing freshly reconstituted d-luciferin (0.5–1 mg/ml) was added at 1:1 ratio to the well medium using a multichannel pipette (XL 3000ITM, Denville Scientific), plates were gently rocked for 5–10 min, and the accumulative luminescence signals were detected using an InfinitiM200 plate reader (Tecan). To read luminescence spectra, a SynergyH1 microplate reader (BioTek, Winooski, VT) was used. To ensure a stable signal, the first-read well was reread after each series of reading. Each sample was at least triplicated in the plate, and four series of readings were performed for each plate. Native coelenterazine or coelenterazine h (1–10 μm final concentration) (both from NanoLight Technology, Pinetop, AZ) was used in experiments with Renilla luciferase constructs. The plates were equilibrated at room temperature, PBS medium containing freshly reconstituted coelenterazine was added to the wells using a multichannel pipette (XL 3000I), plates were gently rocked for 2–5 min, and the luminescence signals were detected using a SynergyH1 microplate reader. To ensure a stable signal, the first-read well was reread after each series of reading. Each sample was at least triplicated in the plate, and three series of readings were performed for each plate.
BRET technique
To measure the BRET signal between Renilla luciferase and EGFP constructs (HuR-Renilla luciferase and HuR-EGFP, EGFP-HuR, and PRKAR2A-EGFP), the corresponding plasmids at a 1:1 ratio were cotransfected using a 4D-Nucleofector core unit and X kit. The emission spectra were initiated by the Renilla luciferase substrate native coelenterazine or coelenterazine h (1–10 μm final concentration) and detected in a range from 400 to 550 nm using a SynergyH1 microplate reader. The detected signals at wavelengths 480 ± 10 and 520 ± 10 nm, corresponding to the maxima of the emission spectra for Rluc and EGFP, respectively, were chosen for BRET calculation. Note that the 480 ± 10 nm wavelength is the maximum of EGFP excitation as well. The BRET ratio was calculated by using the equation [(emission at 520 ± 10 nm) − (emission at 480 ± 10 nm × Cf)]/(emission at 480 ± 10 nm) where Cf is (emission at 520 ± 10 nm)/(emission at 480 ± 10 nm) measured in cells cotransfected with HuR-Renilla luciferase and scrambled control plasmid. The expression of EGFP constructs was confirmed by measuring fluorescence signals acquired by excitation at 480 ± 10 nm and emission at 520 ± 10 and 560 ± 10 nm (as a control) using a SynergyH1 microplate reader.
To measure BRET signal between reconstituted firefly luciferase (firefly luciferase activity was achieved during HuR protein multimerization in the split firefly luciferase assay) and the cotransfected HuR-DsRed fluorescence construct, d-luciferin (0.5–1 mg/ml) was used as the initiation signal and as a substrate for firefly luciferase. The luminescence signals, detected using a SynergyH1 microplate reader at wavelengths of 570 ± 10 and 590 ± 10 nm, corresponding to the maxima of the emission spectra for Fluc and red fluorescent protein, respectively, were chosen for BRET calculation. Note that the 570 ± 10 nm wavelength is the maximum of DsRed excitation. The BRET ratio was calculated by using the equation [(emission at 590 ± 10 nm) − (emission at 570 ± 10 nm × Cf)]/(emission at 570 ± 10 nm) where Cf is (emission at 590 ± 10 nm)/(emission at 570 ± 10 nm) measured in cells cotransfected with HuR-Nluc+HuR-Cluc and scrambled control plasmid. The expression of DsRed constructs was confirmed by measuring the fluorescence signal acquired by excitation at 570 ± 10 nm and emission at 590 ± 10 and 620 ± 10 nm (as a control) using a SynergyH1 microplate reader. Note, in this experiment, cells were cotransfected with siHuR against non-coding 3′-UTR of endogenous HuR protein (HuR siRNA 1027417, Qiagen GmbH, Hilden, Germany) to decrease expression of endogenous HuR protein (16) and improve interaction among recombinant HuR-Nluc, HuR-Cluc, and DsRed-HuR proteins or an equal amount of control siRNA (Cell Signaling Technology, Danvers, MA).
Immunohistochemistry
A standard immunohistochemistry protocol for paraffin-embedded tissue sections (University of Alabama core facility) was used for the array of brain tumor samples (WHO grades I–IV) and control brain tissue. Briefly, tissue sections underwent deparaffinization/rehydration by a series of incubations in xylene and 100 and 95% ethanol and a wash in distilled H2O followed by antigen unmasking in 10 mm sodium citrate buffer for 10 min at 95 °C. Tissue sections were then cooled at room temperature for 30 min and stained with 3% hydrogen peroxide for 10 min to quench endogenous peroxidase activity before the staining procedure. To prevent nonspecific antibody binding to the tissue, tissue sections were incubated with universal blocking buffer consisting of 1% BSA, 0.2% nonfat powdered skim milk, 0.3% Triton X-100, and 1× PBS for 1 h at room temperature in a humidified chamber. The staining procedure utilized the HuR3A2 (Santa Cruz Biotechnology, Inc., Dallas, TX) primary antibody at 1:200 dilution at 4 °C overnight. SignalStain Boost Detection Reagent (HRP; mouse) was used at room temperature for 30 min for signal detection, and for signal development SignalStain diaminobenzidine chromogen concentrate was mixed with diluent. Hematoxylin (Fisher Scientific) was used for nuclear counterstaining. Washing buffer was Tris-buffered Saline with Tween 20. 0.3% Triton was used for cell permeabilization, and VectaMount mounting medium (Vector Laboratories, Burlingame, CA) was used for coverslip mounting.
Immunoprecipitation and Western blotting
The immunoprecipitations were performed by using rabbit immunoprecipitation matrix (Santa Cruz BioTechnology, Inc.) coated with EGFP antibody (Santa Cruz Biotechnology, Inc.) or anti-rabbit IgG antibodies as a control (Santa Cruz Biotechnology, Inc.) overnight at 4 °C. The cells lysates were precleared for 2 h with immobilized A/G protein slurry (Life Technologies) before immunoprecipitation. The immunoprecipitation was achieved during a period of 6 h at 4 °C, the beads were washed three to five times in appropriate buffers at 4 °C, and contents were released using 5× SDS gel loading buffer and 95 °C heat for 9 min. The cell lysis buffer (Cell Signaling Technology) supplemented with protease inhibitor mixture (Thermo Fisher Scientific, Waltham, MA), and sodium orthovanadate was utilized for cells lysis and total protein extraction. For nuclear/cytoplasmic protein fractionation, the NE-PERTM nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific) were used according to accompanying protocol. The lamin A/C antibody (Cell Signaling Technology) and α-tubulin antibody (Sigma-Aldrich) were used to confirm correct nuclear/cytoplasmic fractionation, respectively. Samples were premixed with 5× gel loading buffer supplemented with β-mercaptoethanol and preheated at 95 °C for 9 min before gel loading in reduced, denatured conditions. For samples in non-reduced and non-denatured conditions, 5× gel loading buffer without reducing agents was used, and the preheating step was omitted. In some experiments, protein samples were pretreated with RNase T1 (Ambion, Waltham, MA) and RNase A (Qiagen GmbH). 4–5% blotting-grade blocker (Bio-Rad) in TBS with Triton X-100 was used as a blocking solution for Western blotting. The ImageJ program was used for Western blot data analysis. The following antibodies were used for protein detection in Western blotting: lamin A/C, GAPDH, cleaved poly(ADP-ribose) polymerase, anti-rabbit HRP, anti-mouse HRP antibodies (Cell Signaling Technology); HuR3A2, EGFP, actin, anti-rabbit, anti-mouse IgG1-HRP, anti-rabbit HRP, anti-goat IgG-HRP antibodies (Santa Cruz Biotechnology, Inc.); α-tubulin, anti-FLAG antibodies (Sigma-Aldrich); and anti-luciferase goat polyclonal antibody (Promega).
Viability assay
PrestoBlue cell viability reagent (Thermo Fisher Scientific) was used for the cytotoxicity assay according to the manufacturer's protocol. Briefly, cell were plated in 48-well plates (Costar®, Corning) at 15,000–20,000/well to evaluate the inhibitory dose response for the desired drug and from 2,000 to 60,000/well in the same plate to ensure linear working range of PrestoBlue reagent. The drugs were freshly reconstituted in cell medium and added to the wells using a multichannel pipette (XL 3000I). Each drug concentration was at least triplicated in the plate. After 24 or 48 h of cell exposure to various drug concentrations, the PrestoBlue reagent (15–20 μl) was added to each well using a multichannel pipette, and plates were incubated at 37 °C and 5% CO2 for 10–20 min. To avoid the influence of alterations of redox and alkylation states of the solution on PrestoBlue performance, the cell medium was exchanged for fresh medium before PrestoBlue addition. Following the incubation period, the PrestoBlue fluorescence signal (560-nm excitation/590-nm emission) was detected using an InfinitiM200 plate reader. At least three series of readings were performed for each plate. The background signal (detected from wells with medium without cells) was subtracted from the readings in each plate during data analysis. The dose-response curves were normalized to the corresponding cell signals without drug treatment. The IC50 value was determined as the concentration of an inhibitor at which the response is reduced by half.
d-2-Hydroxyglutarate evaluation
d-2-Hydroxyglutarate evaluation was performed using a d-2-hydroxyglutarate assay kit (K213-100, BioVision) according to the manufacturer's protocol.
Statistical analysis
Statistical analysis and graphing were performed using Excel and Origin Pro software. Statistical significance was determined by Student's t test (analysis of variance), and a p value ≤0.05 was considered significant. Values are expressed as mean ± S.D. Statistically significant data are labeled by an asterisk in the graphs.
Author contributions
N. F. designed experiments and constructs, performed experiments and cloning, analyzed data, and wrote the manuscript. X. Y. performed experiments and contributed to cloning and data analysis. S. A. contributed to manuscript discussion. A. S. contributed to cloning and manuscript preparation. J. R. H. analyzed and interpreted immunohistological data. Z. G. contributed to cell lines development. P. K. contributed to scientific planning, experiments pertaining to MS-444, and manuscript preparation. S. B. contributed to data analysis and manuscript discussion. L. B. N. contributed to experiment discussion, design, data analysis, and manuscript writing.
Supplementary Material
This work was supported by National Institutes of Health Grant R01 CA200624 (to L. B. N.) and the University of Alabama Comprehensive Cancer Center Neuro-oncology Research Acceleration Fund. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. 1–5.
- HuR
- Hu antigen R
- BRET
- bioluminescence resonance energy transfer
- miRNA
- microRNA
- PDGx
- patient-derived glioblastoma xenoline
- DHTS
- 15,16-dihydrotanshinone-I
- compound 5
- N-[4-(1H-benzimidazol-2-yl)phenyl]-2-chloro-5-nitrobenzamide
- EGFP
- enhanced GFP
- GBM
- glioblastoma multiforme
- WHO
- World Health Organization
- Fluc
- full-length firefly luciferase
- Nluc
- N-terminal piece of firefly luciferase
- Cluc
- C-terminal piece of firefly luciferase
- dox
- doxycycline
- IDH1
- isocitrate dehydrogenase-1
- Rluc
- Renilla luciferase
- ELAV
- embryonic lethal abnormal vision.
References
- 1. Brennan C. M., and Steitz J. A. (2001) HuR and mRNA stability. Cell. Mol. Life Sci. 58, 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campos A. R., Grossman D., and White K. (1985) Mutant alleles at the locus elav in Drosophila melanogaster lead to nervous system defects. A developmental-genetic analysis. J. Neurogenet. 2, 197–218 [DOI] [PubMed] [Google Scholar]
- 3. Robinow S., Campos A. R., Yao K. M., and White K. (1988) The elav gene product of Drosophila, required in neurons, has three RNP consensus motifs. Science 242, 1570–1572 [DOI] [PubMed] [Google Scholar]
- 4. Bronicki L. M., and Jasmin B. J. (2013) Emerging complexity of the HuD/ELAVl4 gene; implications for neuronal development, function, and dysfunction. RNA 19, 1019–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang W., Caldwell M. C., Lin S., Furneaux H., and Gorospe M. (2000) HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 19, 2340–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gorospe M. (2003) HuR in the mammalian genotoxic response: post-transcriptional multitasking. Cell Cycle 2, 412–414 [PubMed] [Google Scholar]
- 7. López de Silanes I., Fan J., Yang X., Zonderman A. B., Potapova O., Pizer E. S., and Gorospe M. (2003) Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene 22, 7146–7154 [DOI] [PubMed] [Google Scholar]
- 8. Nabors L. B., Gillespie G. Y., Harkins L., and King P. H. (2001) HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 61, 2154–2161 [PubMed] [Google Scholar]
- 9. Heinonen M., Fagerholm R., Aaltonen K., Kilpivaara O., Aittomäki K., Blomqvist C., Heikkilä P., Haglund C., Nevanlinna H., and Ristimäki A. (2007) Prognostic role of HuR in hereditary breast cancer. Clin. Cancer Res. 13, 6959–6963 [DOI] [PubMed] [Google Scholar]
- 10. Filippova N., Yang X., Wang Y., Gillespie G. Y., Langford C., King P. H., Wheeler C., and Nabors L. B. (2011) The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol. Cancer Res. 9, 648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poria D. K., Guha A., Nandi I., and Ray P. S. (2016) RNA-binding protein HuR sequesters microRNA-21 to prevent translation repression of proinflammatory tumor suppressor gene programmed cell death 4. Oncogene 35, 1703–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J., Wang B., Bi J., and Zhang C. (2011) Cytoplasmic HuR expression correlates with angiogenesis, lymphangiogenesis, and poor outcome in lung cancer. Med. Oncol. 28, Suppl. 1, S577–S585 [DOI] [PubMed] [Google Scholar]
- 13. Wang J., Guo Y., Chu H., Guan Y., Bi J., and Wang B. (2013) Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 14, 10015–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doller A., Akool el-S., Huwiler A., Müller R., Radeke H. H., Pfeilschifter J., and Eberhardt W. (2008) Posttranslational modification of the AU-rich element binding protein HuR by protein kinase Cδ elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol. Cell. Biol. 28, 2608–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chou S. D., Murshid A., Eguchi T., Gong J., and Calderwood S. K. (2015) HSF1 regulation of β-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene 34, 2178–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Filippova N., Yang X., and Nabors L. B. (2015) Growth factor dependent regulation of centrosome function and genomic instability by HuR. Biomolecules 5, 263–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galbán S., Kuwano Y., Pullmann R. Jr., Martindale J. L., Kim H. H., Lal A., Abdelmohsen K., Yang X., Dang Y., Liu J. O., Lewis S. M., Holcik M., and Gorospe M. (2008) RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1α. Mol. Cell. Biol. 28, 93–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurgis F. M., Yeung Y. T., Tang M. X., Heng B., Buckland M., Ammit A. J., Haapasalo J., Haapasalo H., Guillemin G. J., Grewal T., and Munoz L. (2015) The p38-MK2-HuR pathway potentiates EGFRvIII-IL-1β-driven IL-6 secretion in glioblastoma cells. Oncogene 34, 2934–2942 [DOI] [PubMed] [Google Scholar]
- 19. Lucchesi C., Sheikh M. S., and Huang Y. (2016) Negative regulation of RNA-binding protein HuR by tumor-suppressor ECRG2. Oncogene 35, 2565–2573 [DOI] [PubMed] [Google Scholar]
- 20. Sharma S., Verma S., Vasudevan M., Samanta S., Thakur J. K., and Kulshreshtha R. (2013) The interplay of HuR and miR-3134 in regulation of AU rich transcriptome. RNA Biol. 10, 1283–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vo D. T., Abdelmohsen K., Martindale J. L., Qiao M., Tominaga K., Burton T. L., Gelfond J. A., Brenner A. J., Patel V., Trageser D., Scheffler B., Gorospe M., and Penalva L. O. (2012) The oncogenic RNA-binding protein Musashi1 is regulated by HuR via mRNA translation and stability in glioblastoma cells. Mol. Cancer Res. 10, 143–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang L. F., Lou J. T., Lu M. H., Gao C., Zhao S., Li B., Liang S., Li Y., Li D., and Liu M. F. (2015) Suppression of miR-199a maturation by HuR is crucial for hypoxia-induced glycolytic switch in hepatocellular carcinoma. EMBO J. 34, 2671–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rebane A., Aab A., and Steitz J. A. (2004) Transportins 1 and 2 are redundant nuclear import factors for hnRNP A1 and HuR. RNA 10, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang H., Zeng F., Liu Q., Liu H., Liu Z., Niu L., Teng M., and Li X. (2013) The structure of the ARE-binding domains of Hu antigen R (HuR) undergoes conformational changes during RNA binding. Acta Crystallogr. D Biol. Crystallogr. 69, 373–380 [DOI] [PubMed] [Google Scholar]
- 25. Chu P. C., Chuang H. C., Kulp S. K., and Chen C. S. (2012) The mRNA-stabilizing factor HuR protein is targeted by β-TrCP protein for degradation in response to glycolysis inhibition. J. Biol. Chem. 287, 43639–43650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yoon J. H., Abdelmohsen K., Srikantan S., Guo R., Yang X., Martindale J. L., and Gorospe M. (2014) Tyrosine phosphorylation of HuR by JAK3 triggers dissociation and degradation of HuR target mRNAs. Nucleic Acids Res. 42, 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Filippova N., Yang X., King P., and Nabors L. B. (2012) Phosphoregulation of the RNA-binding protein Hu antigen R (HuR) by Cdk5 affects centrosome function. J. Biol. Chem. 287, 32277–32287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fan X. C., and Steitz J. A. (1998) HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc. Natl. Acad. Sci. U.S.A. 95, 15293–15298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fialcowitz-White E. J., Brewer B. Y., Ballin J. D., Willis C. D., Toth E. A., and Wilson G. M. (2007) Specific protein domains mediate cooperative assembly of HuR oligomers on AU-rich mRNA-destabilizing sequences. J. Biol. Chem. 282, 20948–20959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benoit R. M., Meisner N. C., Kallen J., Graff P., Hemmig R., Cèbe R., Ostermeier C., Widmer H., and Auer M. (2010) The x-ray crystal structure of the first RNA recognition motif and site-directed mutagenesis suggest a possible HuR redox sensing mechanism. J. Mol. Biol. 397, 1231–1244 [DOI] [PubMed] [Google Scholar]
- 31. Scheiba R. M., de Opakua A. I., Díaz-Quintana A., Cruz-Gallardo I., Martínez-Cruz L. A., Martínez-Chantar M. L., Blanco F. J., and Díaz-Moreno I. (2014) The C-terminal RNA binding motif of HuR is a multi-functional domain leading to HuR oligomerization and binding to U-rich RNA targets. RNA Biol. 11, 1250–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Díaz-Quintana A., García-Mauriño S. M., and Díaz-Moreno I. (2015) Dimerization model of the C-terminal RNA Recognition Motif of HuR. FEBS Lett. 589, 1059–1066 [DOI] [PubMed] [Google Scholar]
- 33. Toba G., and White K. (2008) The third RNA recognition motif of Drosophila ELAV protein has a role in multimerization. Nucleic Acids Res. 36, 1390–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. David P. S., Tanveer R., and Port J. D. (2007) FRET-detectable interactions between the ARE binding proteins, HuR and p37AUF1. RNA 13, 1453–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Durie D., Lewis S. M., Liwak U., Kisilewicz M., Gorospe M., and Holcik M. (2011) RNA-binding protein HuR mediates cytoprotection through stimulation of XIAP translation. Oncogene 30, 1460–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. D'Agostino V. G., Lal P., Mantelli B., Tiedje C., Zucal C., Thongon N., Gaestel M., Latorre E., Marinelli L., Seneci P., Amadio M., and Provenzani A. (2015) Dihydrotanshinone-I interferes with the RNA-binding activity of HuR affecting its post-transcriptional function. Sci. Rep. 5, 16478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meisner N. C., Hintersteiner M., Mueller K., Bauer R., Seifert J. M., Naegeli H. U., Ottl J., Oberer L., Guenat C., Moss S., Harrer N., Woisetschlaeger M., Buehler C., Uhl V., and Auer M. (2007) Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat. Chem. Biol. 3, 508–515 [DOI] [PubMed] [Google Scholar]
- 38. Kasashima K., Sakashita E., Saito K., and Sakamoto H. (2002) Complex formation of the neuron-specific ELAV-like Hu RNA-binding proteins. Nucleic Acids Res. 30, 4519–4526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hinman M. N., Zhou H. L., Sharma A., and Lou H. (2013) All three RNA recognition motifs and the hinge region of HuC play distinct roles in the regulation of alternative splicing. Nucleic Acids Res. 41, 5049–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu X., Lan L., Wilson D. M., Marquez R. T., Tsao W. C., Gao P., Roy A., Turner B. A., McDonald P., Tunge J. A., Rogers S. A., Dixon D. A., Aubé J., and Xu L. (2015) Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem. Biol. 10, 1476–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Z., Bhattacharya A., and Ivanov D. N. (2015) Identification of small-molecule inhibitors of the HuR/RNA interaction using a fluorescence polarization screening assay followed by NMR validation. PLoS One 10, e0138780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benoit R., and Auer M. (2011) A direct way of redox sensing. RNA Biol. 8, 18–23 [DOI] [PubMed] [Google Scholar]
- 43. Kodiha M., and Stochaj U. (2012) Nuclear transport: a switch for the oxidative stress-signaling circuit? J. Signal Transduct. 2012, 208650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cobbs C. S. (2013) Cytomegalovirus and brain tumor: epidemiology, biology and therapeutic aspects. Curr. Opin. Oncol. 25, 682–688 [DOI] [PubMed] [Google Scholar]
- 45. Ichimura K., Pearson D. M., Kocialkowski S., Bäcklund L. M., Chan R., Jones D. T., and Collins V. P. (2009) IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-oncology 11, 341–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Katsetos C. D., Anni H., and Dráber P. (2013) Mitochondrial dysfunction in gliomas. Semin. Pediatr. Neurol. 20, 216–227 [DOI] [PubMed] [Google Scholar]
- 47. Matlaf L. A., Harkins L. E., Bezrookove V., Cobbs C. S., and Soroceanu L. (2013) Cytomegalovirus pp71 protein is expressed in human glioblastoma and promotes pro-angiogenic signaling by activation of stem cell factor. PLoS One 8, e68176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thota B., Shukla S. K., Srividya M. R., Shwetha S. D., Arivazhagan A., Thennarasu K., Chickabasaviah Y. T., Hegde A. S., Chandramouli B. A., Somasundaram K., and Santosh V. (2012) IDH1 mutations in diffusely infiltrating astrocytomas: grade specificity, association with protein expression, and clinical relevance. Am. J. Clin. Pathol. 138, 177–184 [DOI] [PubMed] [Google Scholar]
- 49. Dickson A. M., Anderson J. R., Barnhart M. D., Sokoloski K. J., Oko L., Opyrchal M., Galanis E., Wilusz C. J., Morrison T. E., and Wilusz J. (2012) Dephosphorylation of HuR protein during alphavirus infection is associated with HuR relocalization to the cytoplasm. J. Biol. Chem. 287, 36229–36238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Burkhart R. A., Pineda D. M., Chand S. N., Romeo C., Londin E. R., Karoly E. D., Cozzitorto J. A., Rigoutsos I., Yeo C. J., Brody J. R., and Winter J. M. (2013) HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 10, 1312–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sokoloski K. J., Dickson A. M., Chaskey E. L., Garneau N. L., Wilusz C. J., and Wilusz J. (2010) Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 8, 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim H. H., Abdelmohsen K., Lal A., Pullmann R. Jr., Yang X., Galban S., Srikantan S., Martindale J. L., Blethrow J., Shokat K. M., and Gorospe M. (2008) Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev. 22, 1804–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.