

Abstract
Conotoxin GeXIVA inhibits the α9α10 nicotinic acetylcholine receptor (nAChR) and is analgesic in animal models of pain. α-Conotoxins have four cysteines that can have three possible disulfide connectivities: globular (CysI–CysIII and CysII–CysIV), ribbon (CysI–CysIV and CysII–CysIII), or bead (CysI–CysII and CysIII–CysIV). Native α-conotoxins preferably adopt the globular connectivity, and previous studies of α-conotoxins have focused on the globular isomers as the ribbon and bead isomers typically have lower potency at nAChRs than the globular form. A recent report showed that the bead and ribbon isomers of GeXIVA are more potent than the globular isomer, with low nanomolar half-maximal inhibitory concentrations (IC50). Despite this high potency, the therapeutic potential of GeXIVA is limited, because like most peptides, it is susceptible to proteolytic degradation and is challenging to synthesize in high yield. Here we used backbone cyclization as a strategy to improve the folding yield as well as increase the serum stability of ribbon GeXIVA while preserving activity at the α9α10 nAChR. Specifically, cyclization of ribbon GeXIVA with a two-residue linker maintained the biological activity at the human α9α10 nAChR and improved stability in human serum. Short linkers led to selective formation of the ribbon disulfide isomer without requiring orthogonal protection. Overall, this study highlights the value of backbone cyclization in directing folding, improving yields, and stabilizing conotoxins with therapeutic potential.
Keywords: drug design, nicotinic acetylcholine receptors (nAChR), peptide chemical synthesis, peptide conformation, protein folding, conotoxin, cyclization, oxidative folding, stability
Introduction
Acetylcholine receptors are present in the nervous system and at neuromuscular junctions and are defined as either muscarinic acetylcholine receptors or nicotinic acetylcholine receptors (nAChRs)2 (1, 2). Neuronal nAChR subtypes are homo- or heteropentamers formed by the combination of α (α2–α10) and β (β2–β4) subunits. These neuronal nAChR subtypes are involved in a range of conditions and diseases, including neuropathic pain, nicotine addiction, schizophrenia, Parkinson's disease, and Alzheimer's disease (3, 4).
Conotoxins are disulfide-rich peptides found in the venoms of marine cone snails that can selectively and potently alter the function of ion channels and receptors of the nervous system (5–7). Some have been used as pharmacological probes or developed as drug leads (8, 9). One notable example is ω-conotoxin MVIIA (also called ziconotide or Prialt®), which targets the calcium channel CaV2.2 and is an approved drug to treat neuropathic pain (10, 11). Likewise, Vc1.1 reached phase II clinical trials for the treatment of neuropathic pain, and RgIA also entered phase I trials. These latter two peptides have high potency at the α9α10 nAChR, which is considered a pain target, although the full mechanism(s) of analgesia are not understood (12, 13), and G protein-coupled receptors such as the GABAB receptor have also been suggested as targets (14–17).
The sequence of conotoxin GeXIVA was identified recently in a transcriptome analysis of the venom duct of Conus generalis (18). This 28-amino acid peptide has four cysteines (Cys I–IV) that can theoretically form three isomers depending on the disulfide connections: globular (CysI–CysIII and CysII–CysIV), ribbon (CysI–CysIV and CysII–CysIII), or bead (CysI–CysII and CysIII–CysIV) as shown in Fig. 1. Surprisingly, all three isomers of GeXIVA inhibit the rat α9α10 nAChR with similar potency, although the bead and ribbon isomers are slightly more potent than the globular (IC50 of the bead = 4.6 nm; ribbon = 7.0 nm; and globular = 22.7 nm) (18). NMR structure calculations have shown that ribbon GeXIVA is flexible while still having a more defined structure than the other two isomers (18). These isomers exhibit higher or similar potency at rat α9α10 nAChR compared with Vc1.1 (19) or RgIA (20). In addition, the ribbon and bead isomers of GeXIVA showed promise in relieving neuropathic pain in a chronic constriction injury model and also in a tail-flick model of acute pain (21).
Figure 1.

The three isomers of αΟ-conotoxin GeXIVA and the 3D NMR solution structure of ribbon GeXIVA (18). Cys residues are labeled with Roman numerals (I–IV), and the disulfide bonds are shown as yellow sticks. The structure shown for the ribbon GeXIVA is the lowest energy conformer from an ensemble of 10 NMR models.
Peptides, including conotoxins, typically have low oral bioavailability and are easily degraded by proteases in vivo, thus decreasing their therapeutic potential (22–24). Over the last decade, cyclic peptides have been discovered in animals, plants, fungi, and bacteria and offer clues toward improving the stability of peptides (25–27). Head-to-tail cyclic peptides are typically more resistant to thermal or enzymatic degradation than their linear counterparts (28, 29). Improving the stability of conotoxins in biological fluids using backbone cyclization was initially inspired by research into naturally occurring cyclic peptides (29, 30) and is a proven strategy to improve the stability of peptides, including conotoxins (31). For example, the stability of α-conotoxins MII, ImI, Vc1.1, RgIA, AuIB, and χ-conotoxin MrIA was significantly improved through backbone cyclization, and most of the cyclic peptides had similar or improved activity compared with their parent peptides (32). Most notably, Vc1.1 became orally active in animal models of pain after backbone cyclization (33). The length and amino acid composition of the linker used to join the N and C termini of the parent peptide are vital factors for backbone cyclization. For example, the length of the linker has been shown to modulate the yields of the three isomers of framework I conotoxins (corresponding to a CC-C-C pattern) and influence their biological activities (34–38).
Here we investigated whether a similar strategy could be used for α-conotoxin GeXIVA. We used backbone cyclization to control the selective formation of the ribbon isomer and investigated the effects of cyclization on structure, activity, and stability. The ribbon isomer is formed by the oxidation of two cysteine residues located near the N and C termini of the peptide, suggesting that backbone cyclization could be used to direct the selective formation of the resultant disulfide bond. The three isomers of linear GeXIVA and a suite of cyclic ribbon GeXIVA analogs were synthesized, and their serum stabilities and biological activities at the human α9α10 (hα9α10) nAChR were tested.
Results
Cyclic GeXIVA ribbon isomer retains native structure
NMR structure calculations for ribbon GeXIVA (18) showed that its N and C termini are maintained in close proximity (∼6.4 Å) by a disulfide bridge between the first and last Cys residues (Fig. 1). Five linkers were designed using molecular modeling to bridge the termini, and all were geometrically compatible with the linear peptide structure, with the cyclic backbones maintained within 1 Å root-mean-square deviation (RMSD) of the linear peptide. The data suggested that even a one-residue linker is sufficient to cyclize the backbone of ribbon GeXIVA without perturbing structure. Models of cyclic GeXIVA with minimum perturbation of the parent peptide backbone are shown in Fig. 2. For experimental validation, we focused on three short linkers (G, GG, and GAG) and the three corresponding cyclic variants: ribbon cGeXIVA_G, ribbon cGeXIVA_GG, and ribbon cGeXIVA_GAG. The three isomers of linear GeXIVA and the three cyclic ribbon analogs were synthesized using orthogonal protection of the cysteine side chains to orient the folding toward a unique disulfide isomer. The yields of three isomers of linear GeXIVA and cyclic ribbon GeXIVA were low (<1%) using a two-step oxidation method. Although we optimized the purification procedure, globular GeXIVA could not be completely purified. We therefore explored an alternative one-pot synthetic approach.
Figure 2.

Amino acid sequences and NMR structure of ribbon GeXIVA and cyclic ribbon isomers of GeXIVA. A, amino acid sequences of ribbon GeXIVA and cyclic analogs. The backbone cyclization of ribbon GeXIVA was carried out using different sized linkers (highlighted in red) spanning its N and C termini (indicated by the black bracket and line). The black curved lines below the sequence show the ribbon disulfide connectivity. The α-carbon (Cα) RMSD of the backbone for the parent peptide and molecular models of the cyclic peptides are shown in the last column. B, NMR solution structure of ribbon GeXIVA (green) and models of cyclic variants (backbones in blue and linkers in red). The disulfide bonds are represented by yellow sticks, and the Cα are shown as balls.
One-pot oxidative folding
Non-directed oxidative folding of GeXIVA and cGeXIVA_GG was carried out in NH4HCO3 (pH 8.3) at room temperature (Fig. 3A). For GeXIVA, three major products were formed within 20 h. The three major peaks were observed in the ultra-performance liquid chromatography trace, accounting for ∼25, 45, and 30% of the total peptide, respectively. Analysis of each peak using a TripleTOF 5600 mass spectrometer (AB SCIEX, Concord, Canada) with a nanoelectrospray ionization source revealed masses of 3450.62, 3450.62, and 3450.63 Da, respectively, each corresponding to fully oxidized GeXIVA. The three peaks were unambiguously identified by co-elution with the globular GeXIVA, bead GeXIVA, and ribbon GeXIVA isomers. The HPLC peaks of ribbon and bead GeXIVA partly overlapped and were difficult to separate (Fig. 3A, 24h). Interestingly, a single peak representing the majority of the oxidized forms transiently appeared from 1 to 4 h and then disappeared from 4 to 8 h (Fig. 3A, black asterisk). This transient peak partly overlaps with the major final product (Fig. 3A, 24h), which was identified as globular GeXIVA. We hypothesized that the globular isomer might form within 4 h and then gradually convert into other isomers. We incubated globular GeXIVA in NH4HCO3 (pH 8.3) at room temperature at a final concentration of 20 μm and the globular GeXIVA was found to be very stable in the folding buffer for 24 h (data not shown), suggesting that the first major peak at 4 h mainly comprises intermediate forms with similar retention time to the globular isomer.
Figure 3.

Time course of the oxidative folding of GeXIVA and cGeXIVA_GG. A, HPLC traces of samples taken at various times during the random folding process. For GeXIVA, the black asterisk indicates that the peak occurring in the folding process of GeXIVA comprises intermediate forms with similar retention time to the globular isomer, and the red asterisk indicates an impurity. The globular, bead, and ribbon GeXIVA from two-step oxidations are shown as red, green, and blue traces, respectively. For cGeXIVA_GG, the blue asterisk indicates the major intermediate occurring in the folding process of cGeXIVA_GG. The ribbon cGeXIVA_GG from two-step oxidation is shown as a blue trace. I1 represents the intermediate with one disulfide bond, and I2 represents the intermediates with two disulfide bonds. B, surface representations of ribbon GeXIVA and ribbon cGeXIVA_GG. Positively charged residues are shown in blue, negative residues are red, polar residues are white, and hydrophobic residues are green. Surface representations were created using the program PyMOL. Cyclization of ribbon GeXIVA resulted in an additional hydrophobic patch (circle) in cGeXIVA_GG.
The one-step oxidation of reduced cGeXIVA_GG was complete at 16 h, whereas some intermediates could still be seen at that time for the linear GeXIVA. Only three products were formed, all of mass 3546.66 Da, corresponding to that of the fully oxidized cGeXIVA_GG. The third major peak accounted for 86% of the total peptide content and was confirmed as being the ribbon isomer by co-elution. The two other peaks are presumably the globular and bead isomers. In contrast to linear GeXIVA, the retention time of globular, bead, and ribbon isomers of cGeXIVA_GG separate well (Fig. 3A) as the linker engenders ribbon cGeXIVA_GG with a larger hydrophobic patch than ribbon GeXIVA (Fig. 3B, green), which could explain its later retention time. Four intermediate forms were identified over the time course. The major intermediate (Fig. 3A, blue asterisk and labeled I1) was the most abundant of the oxidized forms from 1 to 4 h. The major intermediate I1 of cGeXIVA_GG was isolated, and its mass was 3548.66 Da, indicating that it has only one disulfide bond. The masses of the three minor intermediates were each 3546.65 Da, suggesting that they have two disulfide bonds (all labeled I2 in Fig. 3A).
NMR analyses of ribbon GeXIVA and ribbon cGeXIVA_GG
A comparison of the secondary αH chemical shifts of ribbon GeXIVA with those of ribbon cGeXIVA_GG suggested very little difference in structure between the linear and cyclic analogs (Fig. 4A). The different chemical environment created by the presence of the cyclizing linker can easily explain the minimal shift differences near the termini. Overlapping peaks in the 2D NMR spectra prevented the unambiguous assignment of the cGeXIVA_G and cGeXIVA_GAG spin systems.
Figure 4.

Structural characterization of cGeXIVA_GG. A, secondary αH chemical shifts of ribbon GeXIVA and ribbon cGeXIVA_GG (ΔδHα). The overall secondary structures of ribbon GeXIVA and ribbon cGeXIVA_GG are similar, and both peptides display a 310-helix from Pro12 to Arg16. The black lines below the sequence show the ribbon disulfide connectivity. B, ribbon diagram of the NMR solution structure of cGeXIVA_GG. Models were overlaid over the Cys9–Cys20 segment. Disulfide bonds are shown in yellow.
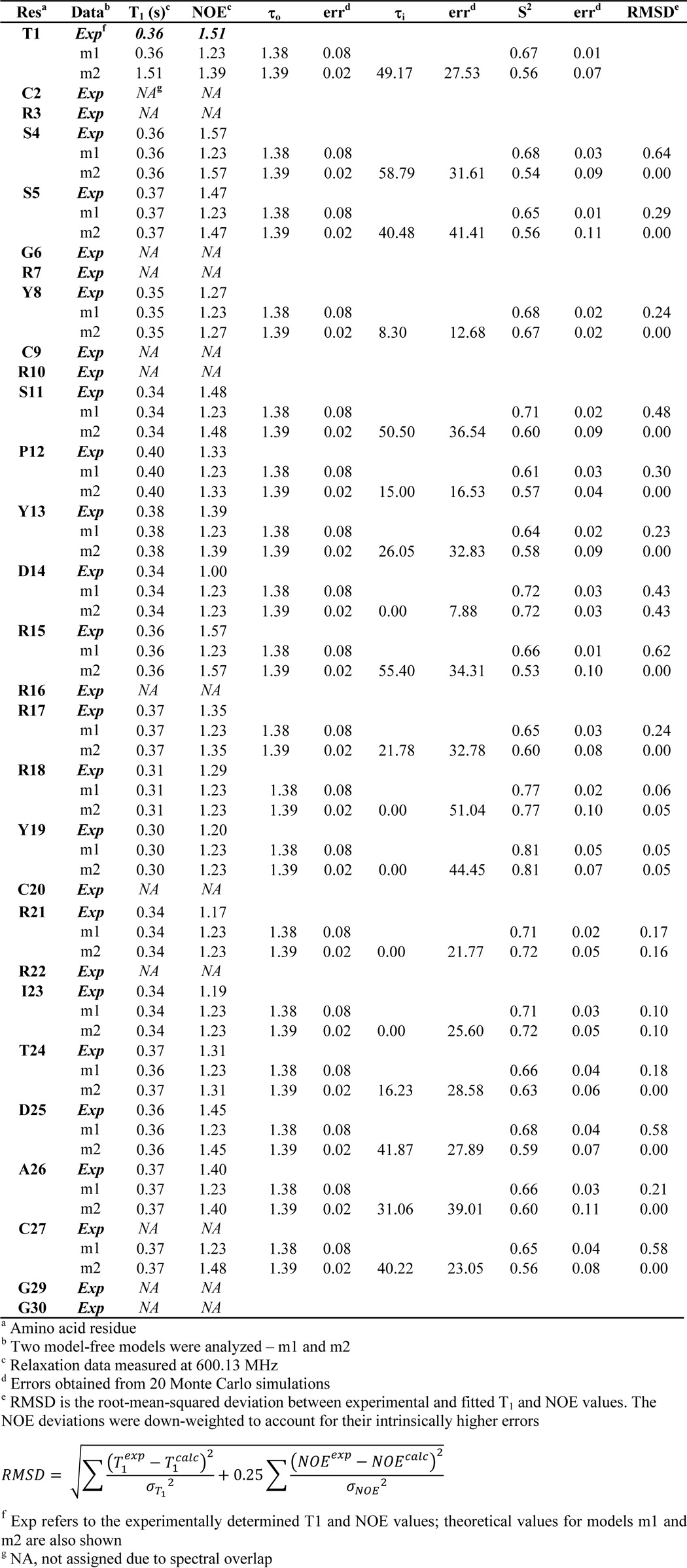
The 3D structure of cGeXIVA_GG was calculated with a total of 171 distance restraints and found to contain a short 310-helix (from Pro12 to Arg16; Fig. 4B). The Ser11–Pro12 peptide bond was determined to adopt a trans configuration, similar to the configuration of this bond in the ribbon and bead isomers of linear GeXIVA (18). As shown for ribbon GeXIVA (18), the loop formed between Cys9 and Cys20 overlays reasonably well between NMR models (RMSD of 1.38 ± 0.56 Å across backbone atoms of residues 9–20; Table 1), although the remainder of the peptide is somewhat disordered, suggesting a degree of flexibility. We thus used NMR spin relaxation measurements to characterize the backbone dynamics of cGeXIVA_GG (Fig. 5 and Table 2). Heteronuclear T1 and NOEs were measured and used to derive order parameters reflective of motion of the Cα-Hα bonds. Because of spectral overlap, only 18 of the 30 residues could be analyzed. Nevertheless, the order parameters (0.53–0.81; mean of 0.63) suggest that cGeXIVA_GG is moderately flexible, which agrees with the geometric analysis of the lowest energy structures. The least flexible region is at the C-terminal end of the Cys9 to Cys20 loop.
Table 1.
Statistical analysis of cGeXIVA_GG structures
All statistics are given as means ± S.D.
| Experimental restraints | |
| Total no. distance restraints | 171 |
| Intraresidue | 90 |
| Sequential | 73 |
| Medium range, i − j < 5 | 8 |
| Long range, i − j ≥ 5 | 0 |
| Hydrogen bond restraints | 2 |
| Dihedral angle restraints | 32 |
| NOE violations exceeding 0.3 Å | 0 |
| Dihedral violations exceeding 2.0 Å | 0 |
| RMSD from mean structure, Å | |
| Backbone atoms (residues 9–20) | 1.38 ± 0.56 |
| All heavy atoms (residues 9–20) | 3.33 ± 0.98 |
| Stereochemical qualitya | |
| Residues in most favored Ramachandran region, % | 77.9 ± 8.9 |
| Ramachandran outliers, % | 1.4 ± 1.8 |
| Unfavorable side-chain rotamers, % | 3.7 ± 2.6 |
| Clashscore, all atoms | 0.1 ± 0.4 |
| Overall MolProbity score | 1.6 ± 0.3 |
a According to MolProbity (76).
Figure 5.

Experimental relaxation data and derived order parameters for cGeXIVA_GG. T1 (top panel) and heteronuclear NOE (middle panel) values for the Cα nuclei were measured at 600 MHz. Errors are shown as standard deviations of triplicate measurements. Only 18 of the residues could be analyzed. A model-free analysis was used to derive order parameters (bottom panel). Models 1 and 2 of model-free were used in the analysis. Errors, shown as standard deviation, were estimated using a Monte Carlo algorithm.
Table 2.
Relaxation data, fitted parameters, and error estimates for cGeXIVA_GG
Ribbon cGeXIVA_GG and ribbon GeXIVA are equipotent inhibitors of the human α9α10 nAChR subtype
Linear GeXIVA isomers have been shown to selectively and potently inhibit the rat α9α10 nAChR subtype (18). Here, globular, bead, and ribbon isomers of linear GeXIVA, and cyclic constructs of ribbon GeXIVA (cGeXIVA_G, cGeXIVA_GG, and ribbon cGeXIVA_GAG) were tested for their functional activity at the hα9α10 nAChR subtype heterologously expressed in Xenopus laevis oocytes. All three cyclic ribbon constructs tested at 100 nm inhibited ACh-evoked currents mediated by hα9α10 nAChRs by ∼60–70% (n = 4–6). Similarly, the bead (n = 5) and ribbon isomers (n = 8) of GeXIVA inhibited the hα9α10 ACh-evoked current amplitude by ∼60–70%. In contrast, the globular GeXIVA was less potent, inhibiting hα9α10 ACh-evoked currents by ∼40% (n = 12) (Fig. 6A).
Figure 6.

Inhibition of human α9α10 nAChR by linear GeXIVA and ribbon cGeXIVA isomers. A, bar graph of globular GeXIVA, bead GeXIVA, ribbon GeXIVA, ribbon cGeXIVA_G, ribbon cGeXIVA_GG, and ribbon cGeXIVA_GAG (100 nm) inhibition of ACh-evoked peak current amplitude mediated by hα9α10 nAChRs. Whole-cell hα9α10 nAChR-mediated currents were activated by 6 μm ACh. B, concentration–response curves of globular and ribbon GeXIVA and ribbon cGeXIVA_GG inhibition of ACh-evoked currents mediated by hα9α10 nAChRs (means ± S.E., n = 4–12).
The concentration-dependent activity of globular and ribbon GeXIVA, and ribbon cGeXIVA_GG at hα9α10 nAChRs were determined (Fig. 6B). All peptides reversibly inhibited ACh-evoked currents in a concentration-dependent manner, giving IC50 values of 198.6 ± 18.9 nm (n = 4–12) for globular GeXIVA, 35.1 ± 2.7 nm (n = 4–8) for ribbon GeXIVA and 37.6 ± 4.9 nm (n = 4–11) for ribbon cGeXIVA_GG. Consistent with the previously reported potency of globular and ribbon GeXIVA at rat α9α10 nAChRs, the globular isomer of GeXIVA had lower potency at hα9α10 nAChRs than the ribbon analog. Ribbon GeXIVA and ribbon cGeXIVA_GG had similar IC50 values, suggesting that backbone cyclization had no impact on the potency of inhibition at hα9α10 nAChRs.
Serum stability of linear GeXIVA and ribbon cGeXIVA isomers
Stability assays for the ribbon isomer of GeXIVA and its three cyclic analogs were carried out in human serum. All four peptides degraded within 2 h, but the ribbon cGeXIVA_GG was slightly more stable than the other peptides (data for cGeXIVA_G and GeXIVA_GAG not shown). Subsequently, the stability of the three isomers of linear GeXIVA and of ribbon cGeXIVA_GG was compared with that of α-conotoxin cVc1.1 (33) in 25% human serum. The positive control, cVc1.1, was highly stable, whereas only ∼20% of the three isomers of GeXIVA remained after 4 h of incubation. Ribbon cGeXIVA_GG was more stable than the three linear isomers of GeXIVA (p < 0.001) at the 4-h time point, with 38% of cGeXIVA_GG remaining (Fig. 7).
Figure 7.
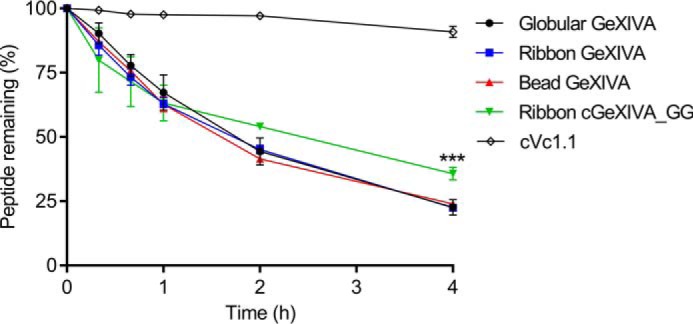
The stability of GeXIVA isomers, ribbon cGeXIVA_GG, and cVc1.1 (positive control) in 25% human serum. The one-way analysis of variance/Tukey test and was used for comparing the remaining peptides at the 4-h time point, and the data are expressed as the means ± S.E. ***, p < 0.001. All values (except cVc1.1) were compared with that of ribbon GeXIVA.
Discussion
The α9α10 nAChR mediates efferent olivocochlear innervations in outer hair cells and modulates immune responses in lymphocytes (39–41). It is also reported to be implicated in pain and cancer (42, 43). Only a handful of α-conotoxins is currently known to selectively inhibit the α9α10 nAChR (18, 44–46), so studies of new examples such as GeXIVA are important for defining receptor specificity. Furthermore, previous studies have highlighted the value of backbone cyclization to enhance the biopharmaceutical properties of α-conotoxins (33, 47), and so here we attempted to apply this approach to GeXIVA.
Molecular modeling was used to design linkers comprising only a small number of amino acids that would introduce minimal constraints into the peptide fold. A recent study highlighted the importance of the distance between the N and C termini in designing cyclizing linkers (48), but considering distances alone will not address limitations caused by accessible conformations of backbones nor the orientation of the termini. Structural perturbations may be introduced unintentionally into a cyclic peptide when a linker is designed on the basis of distance only, ultimately decreasing biological activity (47, 49). In the case of ribbon GeXIVA, modeling showed that the proximity of the termini would allow cyclization with a single residue linker without affecting the structure. Because non-neutral amino acids in the cyclizing linker sequence can have a negative effect on biological activity (50), only Gly and Ala amino acids were used in the linkers in the current study. These residues are small, flexible, non-charged, and likely to have minimal interference with biological activity. No more than two sequential Gly or Ala were used in any individual linker to avoid overlapping peaks in NMR spectra and thus avoid spectral assignment difficulties. Linkers composed of Gly and Ala residues have been successfully used to cyclize the α-conotoxins MII, RgIA, and Vc1.1, without adversely affecting biological activity or structure (33, 47, 49).
The linkers used in this study did not affect the activity and structure of ribbon GeXIVA. Indeed, all cyclic ribbon variants, i.e. cGeXIVA_G, cGeXIVA_GG, and cGeXIVA_GAG, had similar activity to the linear ribbon form at the hα9α10 nAChR. Although no structural data from peptides cyclized with a G or GAG linker were obtained because of NMR spectral overlap, the GG linker did yield a cyclized peptide with a very similar conformation to the linear ribbon GeXIVA. Alignment of the lowest energy model generated from NMR data and an analysis of the backbone relaxation measurements suggests that the cyclic variant is most rigid in the Cys9–Cys20 loop region but slightly more flexible in the remainder of the molecule. It has recently been shown that backbone cyclization can enhance or retain backbone rigidity for three different classes of peptides, including for the conotoxin Vc1.1 (51). Cyclization appears to have less of a rigidifying effect in GeXIVA.
The non-directed oxidation of GeXIVA resulted in the preferential formation of the bead isomer, followed by the ribbon and globular forms, which interestingly parallels their biological activity (18). By contrast, two previous studies employing the one-step oxidation of α-conotoxins showed preferential formation of the globular isomer under similar conditions (0.1 mNH4HCO3, pH 8, room temperature) (52, 53). A possible explanation for this difference in isomer formation is that GeXIVA displays a Cys framework XIV (C-C-C-C) with 6, 10, and 6 residues within the CysI–CysII, CysII–CysIII, and CysIII–CysIV backbone segments, respectively, rather than the Cys framework I (CC-C-C) of most α-conotoxins with 0, 4, and 3–7 residues between the cysteines.
The one-step oxidation of cGeXIVA_GG was notably different from that of the linear parent peptide because it resulted in the preferential formation of the ribbon isomer. The use of short cyclizing linkers has also been reported to drive oxidative folding toward the ribbon connectivity for three other conotoxins: α-conotoxins ImI and AuIB and χ-conotoxin MrIA. Like GeXIVA, these peptides have four Cys residues, with CysI and CysIV being no more than three residues from the N and C termini, respectively. A short cyclizing linker should maintain these two Cys residues in spatial proximity in the reduced form and favor their cross-linking by a disulfide bond during oxidation.
During the oxidative folding of ribbon GeXIVA and ribbon cGeXIVA_GG, a major intermediate was identified. For GeXIVA, the amount of ribbon and globular isomers plateaued after 8 and 12 h of incubation, respectively, whereas the bead form plateaued after 20 h. According to Wedemeyer et al. (54), the rate of formation of disulfide bonds in an unstructured chain decreases with the number of residues between the two cysteines. Because 6, 10, and 6 residues exist between successive cysteines in GeXIVA, we speculate that the major intermediate might involve a disulfide bond between CysI and CysII or between CysIII and CysIV and that this intermediate subsequently translates into the bead isomer. For cGeXIVA_GG, there are only four residues between CysI and CysIV after backbone cyclization, which may favor the formation of either a CysI–CysIV or CysII–CysIII disulfide bond in the intermediate, thus leading to the ribbon isomer. Interestingly, three intermediates with two disulfide connectivities appeared during the course of folding of cGeXIVA_GG. The retention times of these three intermediates were different from the three stable isomers, suggesting that the intermediates have native-like connectivities but have meta-stable conformations.
We hypothesize that the preferential formation of the ribbon isomer probably results from the destabilization of the alternative connectivities rather than through stabilization of the ribbon form (Fig. 8). Backbone cyclization of ribbon GeXIVA possibly affects the folding energy through an entropic effect, mainly impacting the unfolded state. Indeed, the linker has little interaction with the rest of the peptide in molecular models, suggesting that the termini are already maintained in proximity in the linear form, and cyclization does not result in structural perturbation to the core of the peptide. By contrast, the bead and globular isomers probably adopt a different set of conformations after being backbone cyclized because NMR characterization of these isomers indicates that the globular form is disordered, and the N and C termini of the bead isomer are uncoupled (18).
Figure 8.
Schematic representation of the relative energy of the three linear isomers of GeXIVA (black lines) and their cGeXIVA_GG (dotted red lines) analogs. The energy difference between isomers of the same peptide was computed using a Boltzmann distribution of the isomers at equilibrium.
A two-step oxidation strategy is often used to oxidize conotoxins containing two disulfide bonds because the yields are typically improved compared with undirected folding (55). However, the oxidation of GeXIVA using this strategy only produced small amounts of products, hindering high-throughput structure-activity relationship studies and therapeutic studies. The yield of folded ribbon GeXIVA calculated from the final weight of oxidized peptide divided by the weight of reduced peptide was 0.5%. Using the results of Cheneval et al. (56) to extrapolate the yield for synthesis of an unprotected peptide, the yield of cyclic ribbon cGeXIVA_GG through a one-step folding method should be 8.6% using Fmoc-based solid-phase peptide synthesis and considerably higher for a Boc-based approach. Additionally, the HPLC elution peaks of ribbon and bead isomers of GeXIVA overlapped, whereas the ribbon isomer of cGeXIVA_GG is distinct, and ribbon cGeXIVA_GG therefore can be produced in greater purity using one-step oxidation than the ribbon GeXIVA using the same strategy.
An alternative strategy to increase the oxidative folding yield has been used in other cases of disulfide-rich peptides, including the widely used model system bovine pancreatic trypsin inhibitor (BPTI). Undirected folding of this protein results in the formation of two main species, one being native BPTI and the other a native-like two-disulfide intermediate (57). The addition of the N-terminal pro-region increased substantially the native BPTI formation, and it was identified that a cysteine residue in this region was responsible for this increased yield (58). Tethering a cysteine residue to the C terminus with a flexible linker resulted in a similar effect, indicating that this residue acts as an internal catalyst.
Backbone cyclization typically improves the stability of disulfide-rich peptides against enzymatic degradation, as demonstrated for several conotoxins, chlorotoxin, and a spider toxin (32, 35–38, 59, 60). However, it was recently reported that cyclization of κ-conotoxin PVIIA decreased its stability in human serum (61), without significantly perturbing the structure relative to the native form (62). Here, we show that the IC50 for cGeXIVA_GG inhibition of hα9α10 nAChR (37.7 ± 5.0 nm) is comparable with that for ribbon GeXIVA (35.1 ± 2.7 nm). The stability in human serum of ribbon cGeXIVA_GG is only slightly improved compared with linear GeXIVA, suggesting that the main routes of degradation of GeXIVA are by endoproteases or side chain denaturation, which are typically less affected by backbone cyclization. A possible target for such endoproteases are the nine arginine residues in the sequence of GeXIVA (63).
Using short linkers to enhance oxidative folding and other biopharmaceutical properties of bioactive peptides has broader applications than just for toxins, with perhaps a most striking example being for insulin. Insulin is an extremely important hormone but is difficult to synthesize because of its two chain unique structure (64). In one study, the two peptide chains of human insulin were fused, increasing the production yield compared with native human proinsulin (65) while maintaining a native-like structure (66) but with an attendant loss of biological activity (67). In a later study, Hua et al. (68) used a six-residue linker, and the resulting insulin variant maintained biological activity and structure and also had improved folding yield and stability. More recently, the nature of the linker region between the two insulin chains was further investigated, resulting in the identification of an insulin variant with the highest folding efficiency to date (64). In that study, it was shown that the linker region had a direct effect on folding yield by enhancing the formation of the native disulfide bond connectivity. The peptides in the current study have a direct analogy to single-chain insulin analogs: a suitable linker for cGeXIVA_GG led to preferential formation of the ribbon isomer and significantly improved the product yield and stability while retaining the native-like structure and biological activity. The enhanced pharmaceutical properties deriving from a suitable linker provide an opportunity to develop GeXIVA as a potential therapeutic for the treatment of neuropathic pain, a condition with a huge unmet medical need (69). Treatment of neuropathic pain is challenging, and alternative therapeutics are needed because currently available drugs such as morphine, gabapentin, or antidepressants are subject either to the development of tolerance or to significant side effects (70, 71).
In summary, the head-to-tail backbone cyclization via a short linker of ribbon GeXIVA had no impact on the potency of inhibition at hα9α10 nAChR and a rationally designed linker favored the formation of the ribbon disulfide connectivity and improved stability in human serum. The high-yield production of ribbon cGeXIVA_GG compared with linear GeXIVA should facilitate future in vivo studies of the analgesic potential of this compound.
Experimental procedures
Design of linkers
Linkers were designed using molecular modeling. The structure of the ribbon isomer cyclized with linkers from one to five amino acid residues was modeled. Fifty models of the backbone cyclized ribbon GeXIVA were generated for each of the linkers using Modeler 6v13 (72), and the conservation of ribbon GeXIVA native conformation was measured by computing the mean Cα root-mean-square deviation between the models and the 20 lowest-energy NMR structures (18).
Peptide synthesis, cyclization, oxidation, and purification of GeXIVA and variants
The three isomers of GeXIVA and the three cyclic analogs (cGeXIVA_G, cGeXIVA_GG and cGeXIVA_GAG) were synthesized using Fmoc-based solid-phase peptide synthesis on an acid-sensitive resin 2-chlorotrityl chloride. Side chains of Cys residues were protected in pairs with orthogonal protective groups that can be removed selectively under different oxidation conditions. The acid-labile trityl group was used to protect the side chains of Cys2 and Cys27, and the acetamidomethyl group was used to protect Cys9 and Cys20 for the ribbon GeXIVA and its cyclic variants. The backbone cyclization of cyclic variants was achieved using the protocol described recently (56). Briefly, a solution of 1% TFA in dichloromethane (v/v) was used to cleave the linear precursor peptides of the cyclic variants off the resin. The side chain protected precursor peptides were dried, cyclized in O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (5 mm) and N,N′-diisopropylethylamine (10 mm) in N,N-dimethylformamide for 3 h and lyophilized. The protecting group on side chains was removed in TFA with 2% triisopropylsilane and 2% water (v/v) for 2 h. The three isomers of GeXIVA were cleaved and deprotected in TFA with 2% triisopropylsilane and 2% water (v/v) for 2 h.
Two-step oxidative folding was used to form disulfide bonds: the first disulfide bond was formed in 0.1 m ammonium bicarbonate (NH4HCO3, pH 8.3) at room temperature (21–24 °C) for 24 h, and the second disulfide bond was formed in 0.1 m iodine solution for 1 h. All peptides were purified by reversed-phase (RP)-HPLC using Phenomenex C18 columns, and the purification method is 15–25% buffer B within 40 min (increasing 1% buffer B per 4 min). Buffer A is 0.05% TFA in water, and buffer B is 0.045% TFA in 90% acetonitrile.
NMR spectroscopy
Samples of ribbon GeXIVA and its cyclic analogs were prepared by dissolving peptide (∼1 mg) in 90% H2O, 10% D2O at a pH of ∼3. The spectra were recorded on a Bruker Avance III 600 MHz NMR spectrometer at 298 K and included total correlation spectroscopy (mixing time of 80 ms), NOESY (mixing time of 200 ms), and H-N and H-C heteronuclear single quantum coherence. Chemical shifts were referenced to internal 2, 2-dimethyl-2-silapentane-5-sulfonate at 0 ppm. The spectra were processed with Topspin (Bruker Biospin) and analyzed with CcpNMR analysis (version 2.4.1). Secondary αH chemical shifts were calculated as the difference between the observed αH chemical shifts and that of the corresponding residues in a random coil peptide (73). Structure calculations were based upon distance restraints as derived from NOESY spectra and on backbone ϕ and ψ dihedral angle restraints generated using TALOS+ (74). A family of structures consistent with the experimental restraints was calculated using CYANA (75).
Relaxation measurements
NMR relaxation measurements were carried out as described previously (51). Briefly, 13C T1 (spin-lattice) relaxation times were measured using the Bruker pulse program hsqct1etgpsi3d.2. Spectra were acquired with a spectral width of 10 ppm over 2048 complex points in the 1H dimension and 22 ppm over 60 complex points in the 13C dimension. To determine T1 values, nine relaxation delays in the range of 0.01–1.0 s were used. A recycle delay of 3 s was used. 13C NOEs were measured using the pulse program hsqcnoegpsi.2. Peak volumes were measured in CCPNMR. A recycle delay of 5 s was used. All experiments were carried out in triplicate. Order parameters were calculated by a model-free analysis using the same protocol and target function as described previously (51), but here the goodness-of-fit for the NOE values were given a 25% weighting compared with the T1 values to allow for the higher uncertainty in the experimental NOE measurements.
Folding assays
Ribbon GeXIVA and cGeXIVA_GG were reduced with 10 mm dithiothreitol and isolated using RP-HPLC. Reduced peptides were dissolved in 0.1 m NH4HCO3 buffer (pH 8.3) at 20 μm and stirred at room temperature. An aliquot of peptide solution was removed after 0, 1, 4, 8, 12, 16, 20, and 24 h and quenched with 2% TFA. All samples were analyzed with ultra-performance liquid chromatography using a gradient 11–18% buffer B within 28 min (increasing 1% buffer B per 4 min) in an analytical Vydac C18 column.
In vitro cRNA synthesis
Plasmid pT7TS constructs of human α9 and α10 nAChR subunits were linearized with XbaI restriction enzymes (New England Biolabs, Ipswich, MA) for in vitro cRNA transcription using T7 mMessage mMachine® transcription kits (Ambion, Foster City, CA).
Oocyte preparation and microinjection
Stage V–VI oocytes were obtained from X. laevis, defolliculated with 1.5 mg/ml collagenase type II (Worthington Biochemical Corp., Lakewood, NJ) at room temperature (21–23 °C) for 1–2 h in OR-2 solution containing 82.5 mm NaCl, 2 mm KCl, 1 mm MgCl2, and 5 mm HEPES, pH 7.4. The oocytes were injected with 35 ng of cRNA for hα9α10 nAChR (concentration confirmed spectrophotometrically and by gel electrophoresis) using glass pipettes pulled from glass capillaries (3-000-203 GX; Drummond Scientific Co., Broomall, PA). The oocytes were incubated at 18 °C in sterile ND96 solution composed of 96 mm NaCl, 2 mm KCl, 1 mm CaCl2, 1 mm MgCl2, and 5 mm HEPES, pH 7.4, supplemented with 5% fetal bovine serum, 50 mg/liter gentamicin (Gibco) and 10,000 units/ml penicillin-streptomycin (Gibco).
Oocyte two-electrode voltage clamp recording and data analysis
Electrophysiological recordings were carried out 2–5 days after cRNA microinjection. The oocytes expressing hα9α10 nAChRs were incubated in 100 μm BAPTA-AM ∼3 h before recording. Two-electrode voltage clamp recordings were performed at room temperature using a GeneClamp 500B amplifier and pClamp9 software interface (Molecular Devices, Sunnyvale, CA) at a holding potential −80 mV. Voltage-recording and current-injecting electrodes were pulled from GC150T-7.5 borosilicate glass (Harvard Apparatus, Holliston, MA) and filled with 3 m KCl, giving resistances of 0.3–1 MΩ. The oocytes were perfused with ND115 solution containing 115 mm NaCl, 2.5 mm KCl, 1.8 mm CaCl2, and 10 mm HEPES, pH 7.4, using a continuous Legato 270 push/pull syringe pump perfusion system (KD Scientific, Holliston, MA) at a rate of 2 ml/min.
Initially, the oocytes were briefly washed with ND115 solution followed by three applications of ACh at half-maximal effective concentration (EC50) of 6 μm ACh for hα9α10 nAChR. Washout with bath solution was done for 3 min between ACh applications. The oocytes were incubated with peptides for 5 min with the perfusion system turned off, followed by co-application of ACh and peptide with flowing bath solution. All peptide solutions were prepared in ND115 + 0.1% bovine serum albumin. Peak current amplitudes before (ACh alone) and after (ACh + peptide) peptide incubation were measured using Clampfit 10.7 software (Molecular Devices), where the ratio of ACh + peptide-evoked current amplitude to ACh alone-evoked current amplitude was used to assess the activity of the peptides at human nAChRs.
All electrophysiological data were pooled (n = 4 to 12) and represent the means ± S.E. Concentration–response curves were obtained using GraphPad Prism 7 nonlinear regression analysis (GraphPad Software, La Jolla, CA). The half-maximal inhibitory concentration (IC50) was determined from the concentration–response curve and reported with error of the fit.
Stability assays
Serum stability of peptides were carried out in 25% human serum (human male AB plasma; Sigma-Aldrich) over 4 h using a 20 μm final peptide concentration. Serum was prepared by centrifugation at 17,000 × g for 15 min to remove the lipid component, and the supernatant was diluted 1 in 4 in phosphate-buffered saline and incubated at 37 °C for 15 min before the assay. Each peptide was incubated in 25% human serum at 37 °C, and aliquots were taken out at time points 0, 1, 2, 3, and 4 h, respectively. Serum proteins were denatured with 2 m urea and precipitated with 20% TFA. All samples were centrifuged at 17,000 × g for 10 min, and the supernatant was analyzed by analytical RP-HPLC. Remaining peptides were quantified using peak height with absorption at 214 nm.
Author contributions
X. W., Y.-H. H., Q. K., and D. J. C. designed the research. X. W. and Y.-H. H. conducted cyclization, NMR analysis, one-step oxidation, and stability assays. X. W., Y.-H. H., and Q. K. analyzed the results. P. J. H. conducted NMR analysis and structure calculations, and C. K. W. conducted the relaxation analysis. H.-S. T. conducted and analyzed electrophysiological results, and D. J. A. coordinated electrophysiology studies. X. W., Y.-H. H., Q. K., P. J. H., C. K. W., H.-S. T., D. J. A., and D. J. C. wrote the manuscript.
Acknowledgments
We thank Alun Jones and Eivind Undheim (Institute for Molecular Bioscience, The University of Queensland) for mass spectrometry technical support.
This work was supported by Australian Research Council Grant DP150103990. The authors declare that they have no conflicts of interest with the contents of this article.
- nAChR
- nicotinic acetylcholine receptor
- ACh
- acetylcholine
- RMSD
- root-mean-square deviation
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- BPTI
- bovine pancreatic trypsin inhibitor
- RP
- reversed-phase
- BAPTA-AM
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester.
References
- 1. Thal D. M., Sun B., Feng D., Nawaratne V., Leach K., Felder C. C., Bures M. G., Evans D. A., Weis W. I., Bachhawat P., Kobilka T. S., Sexton P. M., Kobilka B. K., and Christopoulos A. (2016) Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 531, 335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taly A., Corringer P. J., Guedin D., Lestage P., and Changeux J. P. (2009) Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug. Discov. 8, 733–750 [DOI] [PubMed] [Google Scholar]
- 3. Albuquerque E. X., Pereira E. F., Alkondon M., and Rogers S. W. (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gotti C., Clementi F., Fornari A., Gaimarri A., Guiducci S., Manfredi I., Moretti M., Pedrazzi P., Pucci L., and Zoli M. (2009) Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 78, 703–711 [DOI] [PubMed] [Google Scholar]
- 5. Terlau H., and Olivera B. M. (2004) Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol. Rev. 84, 41–68 [DOI] [PubMed] [Google Scholar]
- 6. Halai R., and Craik D. J. (2009) Conotoxins: natural product drug leads. Nat. Prod. Rep. 26, 526–536 [DOI] [PubMed] [Google Scholar]
- 7. Kaas Q., Yu R., Jin A. H., Dutertre S., and Craik D. J. (2012) ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 40, D325–D330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lewis R. J., Dutertre S., Vetter I., and Christie M. J. (2012) Conus venom peptide pharmacology. Pharmacol. Rev. 64, 259–298 [DOI] [PubMed] [Google Scholar]
- 9. Akondi K. B., Muttenthaler M., Dutertre S., Kaas Q., Craik D. J., Lewis R. J., and Alewood P. F. (2014) Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 114, 5815–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miljanich G. P. (2004) Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 11, 3029–3040 [DOI] [PubMed] [Google Scholar]
- 11. Jain K. K. (2000) An evaluation of intrathecal ziconotide for the treatment of chronic pain. Expert. Opin. Investig. Drugs 9, 2403–2410 [DOI] [PubMed] [Google Scholar]
- 12. Gorson J., and Holford M. (2016) Small packages, big returns: uncovering the venom diversity of small invertebrate conoidean snails. Integr. Comp. Biol. 56, 962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mohammadi S. A., and Christie M. J. (2015) Conotoxin interactions with α9α10-nAChRs: Is the α9α10-nicotinic acetylcholine receptor an important therapeutic target for pain management? Toxins 7, 3916–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Callaghan B., and Adams D. J. (2010) Analgesic α-conotoxins Vc1.1 and RgIA inhibit N-type calcium channels in sensory neurons of α9 nicotinic receptor knockout mice. Channels (Austin) 4, 51–54 [DOI] [PubMed] [Google Scholar]
- 15. Klimis H., Adams D. J., Callaghan B., Nevin S., Alewood P. F., Vaughan C. W., Mozar C. A., and Christie M. J. (2011) A novel mechanism of inhibition of high-voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 152, 259–266 [DOI] [PubMed] [Google Scholar]
- 16. Nevin S. T., Clark R. J., Klimis H., Christie M. J., Craik D. J., and Adams D. J. (2007) Are α9α10 nicotinic acetylcholine receptors a pain target for α-conotoxins? Mol. Pharmacol. 72, 1406–1410 [DOI] [PubMed] [Google Scholar]
- 17. Callaghan B., Haythornthwaite A., Berecki G., Clark R. J., Craik D. J., and Adams D. J. (2008) Analgesic α-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J. Neurosci. 28, 10943–10951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo S., Zhangsun D., Harvey P. J., Kaas Q., Wu Y., Zhu X., Hu Y., Li X., Tsetlin V. I., Christensen S., Romero H. K., McIntyre M., Dowell C., Baxter J. C., Elmslie K. S., et al. (2015) Cloning, synthesis, and characterization of αO-conotoxin GeXIVA, a potent α9α10 nicotinic acetylcholine receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 112, E4026–E4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu R., Kompella S. N., Adams D. J., Craik D. J., and Kaas Q. (2013) Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J. Med. Chem. 56, 3557–3567 [DOI] [PubMed] [Google Scholar]
- 20. Ellison M., Haberlandt C., Gomez-Casati M. E., Watkins M., Elgoyhen A. B., McIntosh J. M., and Olivera B. M. (2006) α-RgIA: a novel conotoxin that specifically and potently blocks the α9 α10 nAChR. Biochemistry 45, 1511–1517 [DOI] [PubMed] [Google Scholar]
- 21. Li X., Hu Y., Wu Y., Huang Y., Yu S., Ding Q., Zhangsun D., and Luo S. (2016) Anti-hypersensitive effect of intramuscular administration of αO-conotoxin GeXIVA [1,2] and GeXIVA [1,4] in rats of neuropathic pain. Prog. Neuropsychopharmacol. Biol. Psychiatry 66, 112–119 [DOI] [PubMed] [Google Scholar]
- 22. Craik D. J., Fairlie D. P., Liras S., and Price D. (2013) The future of peptide-based drugs. Chem. Biol. Drug. Des. 81, 136–147 [DOI] [PubMed] [Google Scholar]
- 23. King G. F. (2011) Venoms as a platform for human drugs: translating toxins into therapeutics. Expert. Opin. Biol. Ther. 11, 1469–1484 [DOI] [PubMed] [Google Scholar]
- 24. Frokjaer S., and Otzen D. E. (2005) Protein drug stability: a formulation challenge. Nat. Rev. Drug. Discov. 4, 298–306 [DOI] [PubMed] [Google Scholar]
- 25. Craik D. J., Daly N. L., Bond T., and Waine C. (1999) Plant cyclotides: a unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 294, 1327–1336 [DOI] [PubMed] [Google Scholar]
- 26. Conibear A. C., and Craik D. J. (2014) The chemistry and biology of theta defensins. Angew. Chem. Int. Ed. Engl. 53, 10612–10623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klaenhammer T. R. (1993) Genetics of bacteriocins produced by lactic acid bacteria. FEMS Microbiol. Rev. 12, 39–85 [DOI] [PubMed] [Google Scholar]
- 28. Hemu X., Taichi M., Qiu Y., Liu D. X., and Tam J. P. (2013) Biomimetic synthesis of cyclic peptides using novel thioester surrogates. Biopolymers 100, 492–501 [DOI] [PubMed] [Google Scholar]
- 29. Akcan M., and Craik D. (2015) J Engineering venom peptides to improve their stability and bioavailability. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics (King G. F., ed) pp. 275–289, RSC Publishing, Cambridge, UK [Google Scholar]
- 30. Craik D. J. (2006) Seamless proteins tie up their loose ends. Science 311, 1563–1564 [DOI] [PubMed] [Google Scholar]
- 31. Clark R. J., Akcan M., Kaas Q., Daly N. L., and Craik D. J. (2012) Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon 59, 446–455 [DOI] [PubMed] [Google Scholar]
- 32. Wu X., Huang Y. H., Kaas Q., and Craik D. J. (2016) Cyclisation of disulfide-rich conotoxins in drug design applications. Eur. J. Org. Chem. 2016, 3462–3472, 10.1002/ejoc.201600402 [DOI] [Google Scholar]
- 33. Clark R. J., Jensen J., Nevin S. T., Callaghan B. P., Adams D. J., and Craik D. J. (2010) The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 49, 6545–6548 [DOI] [PubMed] [Google Scholar]
- 34. Armishaw C. J., Dutton J. L., Craik D. J., and Alewood P. F. (2010) Establishing regiocontrol of disulfide bond isomers of α-conotoxin ImI via the synthesis of N-to-C cyclic analogs. Biopolymers 94, 307–313 [DOI] [PubMed] [Google Scholar]
- 35. Armishaw C. J., Jensen A. A., Balle L. D., Scott K. C., Sørensen L., and Strømgaard K. (2011) Improving the stability of α-conotoxin AuIB through N-to-C cyclization: the effect of linker length on stability and activity at nicotinic acetylcholine receptors. Antioxid. Redox. Signal. 14, 65–76 [DOI] [PubMed] [Google Scholar]
- 36. Lovelace E. S., Gunasekera S., Alvarmo C., Clark R. J., Nevin S. T., Grishin A. A., Adams D. J., Craik D. J., and Daly N. L. (2011) Stabilization of α-conotoxin AuIB: influences of disulfide connectivity and backbone cyclization. Antioxid. Redox. Signal. 14, 87–95 [DOI] [PubMed] [Google Scholar]
- 37. Dekan Z., Wang C.-I., Andrews R. K., Lewis R. J., and Alewood P. F. (2012) Conotoxin engineering: dual pharmacophoric noradrenaline transport inhibitor/integrin binding peptide with improved stability. Org. Biomol. Chem. 10, 5791–5794 [DOI] [PubMed] [Google Scholar]
- 38. Lovelace E. S., Armishaw C. J., Colgrave M. L., Wahlstrom M. E., Alewood P. F., Daly N. L., and Craik D. J. (2006) Cyclic MrIA: a stable and potent cyclic conotoxin with a novel topological fold that targets the norepinephrine transporter. J. Med. Chem. 49, 6561–6568 [DOI] [PubMed] [Google Scholar]
- 39. Elgoyhen A. B., Vetter D. E., Katz E., Rothlin C. V., Heinemann S. F., and Boulter J. (2001) α10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc. Natl. Acad. Sci. U.S.A. 98, 3501–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koval L., Lykhmus O., Zhmak M., Khruschov A., Tsetlin V., Magrini E., Viola A., Chernyavsky A., Qian J., Grando S., Komisarenko S., and Skok M. (2011) Differential involvement of α4β2, α7 and α9α10 nicotinic acetylcholine receptors in B lymphocyte activation in vitro. Int. J. Biochem. Cell Biol. 43, 516–524 [DOI] [PubMed] [Google Scholar]
- 41. Peng H., Ferris R. L., Matthews T., Hiel H., Lopez-Albaitero A., and Lustig L. R. (2004) Characterization of the human nicotinic acetylcholine receptor subunit α9 (CHRNA9) and α10 (CHRNA10) in lymphocytes. Life. Sci. 76, 263–280 [DOI] [PubMed] [Google Scholar]
- 42. Satkunanathan N., Livett B., Gayler K., Sandall D., Down J., and Khalil Z. (2005) α-Conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain. Res. 1059, 149–158 [DOI] [PubMed] [Google Scholar]
- 43. Wu C. H., Lee C. H., and Ho Y. S. (2011) Nicotinic acetylcholine receptor-based blockade: applications of molecular targets for cancer therapy. Clin. Cancer Res. 17, 3533–3541 [DOI] [PubMed] [Google Scholar]
- 44. Wu R. J., Wang L., and Xiang H. (2015) The structural features of α-conotoxin specifically target different isoforms of nicotinic acetylcholine receptors. Curr. Top. Med. Chem. 16, 156–169 [DOI] [PubMed] [Google Scholar]
- 45. Xu S., Zhang T., Kompella S. N., Yan M., Lu A., Wang Y., Shao X., Chi C., Adams D. J., Ding J., and Wang C. (2015) Conotoxin αD-GeXXA utilizes a novel strategy to antagonize nicotinic acetylcholine receptors. Sci. Rep. 5, 14261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luo S., Christensen S., Zhangsun D., Wu Y., Hu Y., Zhu X., Chhabra S., Norton R. S., and McIntosh J. M. (2013) A novel inhibitor of α9α10 nicotinic acetylcholine receptors from Conus vexillum delineates a new conotoxin superfamily. PLoS One 8, e54648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Halai R., Callaghan B., Daly N. L., Clark R. J., Adams D. J., and Craik D. J. (2011) Effects of cyclization on stability, structure, and activity of α-conotoxin RgIA at the α9α10 nicotinic acetylcholine receptor and GABAB receptor. J. Med. Chem. 54, 6984–6992 [DOI] [PubMed] [Google Scholar]
- 48. Clark R. J., and Craik D. J. (2012) Engineering cyclic peptide toxins. Methods Enzymol. 503, 57–74 [DOI] [PubMed] [Google Scholar]
- 49. Clark R. J., Fischer H., Dempster L., Daly N. L., Rosengren K. J., Nevin S. T., Meunier F. A., Adams D. J., and Craik D. J. (2005) Engineering stable peptide toxins by means of backbone cyclization: stabilization of the α-conotoxin MII. Proc. Natl. Acad. Sci. U.S.A. 102, 13767–13772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carstens B. B., Swedberg J., Berecki G., Adams D. J., Craik D. J., and Clark R. J. (2016) Effects of linker sequence modifications on the structure, stability and biological activity of a cyclic α-conotoxin. Biopolymers 106, 864–875 [DOI] [PubMed] [Google Scholar]
- 51. Wang C. K., Swedberg J. E., Northfield S. E., and Craik D. J. (2015) Effects of cyclization on peptide backbone dynamics. J. Phys. Chem. B 119, 15821–15830 [DOI] [PubMed] [Google Scholar]
- 52. Wu X., Wu Y., Zhu F., Yang Q., Wu Q., Zhangsun D., and Luo S. (2013) Optimal cleavage and oxidative folding of α-conotoxin TxIB as a therapeutic candidate peptide. Mar. Drugs. 11, 3537–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gyanda R., Banerjee J., Chang Y.-P., Phillips A. M., Toll L., and Armishaw C. J. (2013) Oxidative folding and preparation of α-conotoxins for use in high-throughput structure–activity relationship studies. J. Pept. Sci. 19, 16–24 [DOI] [PubMed] [Google Scholar]
- 54. Wedemeyer W. J., Xu X., Welker E., and Scheraga H. A. (2002) Conformational propensities of protein folding intermediates: distribution of species in the 1S, 2S, and 3S ensembles of the [C40A,C95A] mutant of bovine pancreatic ribonuclease A. Biochemistry 41, 1483–1491 [DOI] [PubMed] [Google Scholar]
- 55. Bulaj G., and Olivera B. M. (2008) Folding of conotoxins: formation of the native disulfide bridges during chemical synthesis and biosynthesis of Conus peptides. Antioxid. Redox. Signal. 10, 141–155 [DOI] [PubMed] [Google Scholar]
- 56. Cheneval O., Schroeder C. I., Durek T., Walsh P., Huang Y. H., Liras S., Price D. A., and Craik D. J. (2014) Fmoc-based synthesis of disulfide-rich cyclic peptides. J. Org. Chem. 79, 5538–5544 [DOI] [PubMed] [Google Scholar]
- 57. States D. J., Dobson C. M., Karplus M., and Creighton T. (1984) A new two-disulphide intermediate in the refolding of reduced bovine pancreatic trypsin inhibitor. J. Mol. Biol. 174, 411–418 [DOI] [PubMed] [Google Scholar]
- 58. Weissman J. S., and Kim P. S. (1992) The pro region of BPTI facilitates folding. Cell 71, 841–851 [DOI] [PubMed] [Google Scholar]
- 59. Akcan M., Stroud M. R., Hansen S. J., Clark R. J., Daly N. L., Craik D. J., and Olson J. M. (2011) Chemical re-engineering of chlorotoxin improves bioconjugation properties for tumor imaging and targeted therapy. J. Med. Chem. 54, 782–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jensen J. E., Mobli M., Brust A., Alewood P. F., King G. F., and Rash L. D. (2012) Cyclisation increases the stability of the sea anemone peptide APETx2 but decreases its activity at acid-sensing ion channel 3. Mar. Drugs 10, 1511–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kwon S., Bosmans F., Kaas Q., Cheneval O., Conibear A. C., Rosengren K. J., Wang C. K., Schroeder C. I., and Craik D. J. (2016) Efficient enzymatic cyclization of an inhibitory cystine knot-containing peptide. Biotechnol. Bioeng. 113, 2202–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Scanlon M. J., Naranjo D., Thomas L., Alewood P. F., Lewis R. J., and Craik D. J. (1997) Solution structure and proposed binding mechanism of a novel potassium channel toxin κ-conotoxin PVIIA. Structure 5, 1585–1597 [DOI] [PubMed] [Google Scholar]
- 63. Thomas G. (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zaykov A. N., Mayer J. P., Gelfanov V. M., and DiMarchi R. D. (2014) Chemical synthesis of insulin analogs through a novel precursor. ACS Chem. Biol. 9, 683–691 [DOI] [PubMed] [Google Scholar]
- 65. Markussen J. (1985) Comparative reduction/oxidation studies with single chain des-(B30) insulin and porcine proinsulin. Int. J. Pept. Protein Res. 25, 431–434 [DOI] [PubMed] [Google Scholar]
- 66. Derewenda U., Derewenda Z., Dodson E. J., Dodson G. G., Bing X., and Markussen J. (1991) X-ray analysis of the single chain B29-A1 peptide-linked insulin molecule: a completely inactive analogue. J. Mol. Biol. 220, 425–433 [DOI] [PubMed] [Google Scholar]
- 67. Markussen J., Jørgensen K. H., Sørensen A. R., and Thim L. (1985) Single chain des-(B30) insulin: intramolecular crosslinking of insulin by trypsin catalyzed transpeptidation. Int. J. Pept. Protein Res. 26, 70–77 [PubMed] [Google Scholar]
- 68. Hua Q. X., Nakagawa S. H., Jia W., Huang K., Phillips N. B., Hu S. Q., and Weiss M. A. (2008) Design of an active ultrastable single-chain insulin analog: synthesis, structure, and therapeutic implications. J. Biol. Chem. 283, 14703–14716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Colloca L., Ludman T., Bouhassira D., Baron R., Dickenson A. H., Yarnitsky D., Freeman R., Truini A., Attal N., Finnerup N. B., Eccleston C., Kalso E., Bennett D. L., Dworkin R. H., and Raja S. N. (2017) Neuropathic pain. Nat. Rev. Dis. Primers. 3, 17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dworkin R. H., O'Connor A. B., Backonja M., Farrar J. T., Finnerup N. B., Jensen T. S., Kalso E. A., Loeser J. D., Miaskowski C., Nurmikko T. J., Portenoy R. K., Rice A. S., Stacey B. R., Treede R. D., Turk D. C., et al. (2007) Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 132, 237–251 [DOI] [PubMed] [Google Scholar]
- 71. Cruccu G. (2007) Treatment of painful neuropathy. Curr. Opin. Neurol. 20, 531–535 [DOI] [PubMed] [Google Scholar]
- 72. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 73. Wishart D. S., Sykes B. D., and Richards F. M. (1992) The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31, 1647–1651 [DOI] [PubMed] [Google Scholar]
- 74. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ikeya T., Terauchi T., Güntert P., and Kainosho M. (2006) Evaluation of stereo-array isotope labeling (SAIL) patterns for automated structural analysis of proteins with CYANA. Magn. Reson. Chem. 44, S152–S157 [DOI] [PubMed] [Google Scholar]
- 76. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]