

Abstract
FUS is an RNA‐binding protein (RBP) with a prion‐like domain (PrLD) that condenses into functional liquids, which can aberrantly phase transition into solid aggregates comprised of pathological fibrils connected to neurodegenerative disease. How cells prevent aberrant phase transitions of FUS and other disease‐linked RBPs with PrLDs is poorly understood. In this issue of The EMBO Journal, Monahan et al (2017) establish that phosphorylation of specific serine and threonine residues in the FUS PrLD inhibits aberrant phase separation and toxicity.
Subject Categories: Neuroscience, Protein Biosynthesis & Quality Control
Approximately seventy human RNA‐binding proteins (RBPs) contain a prion‐like domain (PrLD), a type of low‐complexity domain enriched in glycine and uncharged polar amino acids including tyrosine, serine, glutamine, and asparagine (March et al, 2016). This distinctive amino acid composition resembles that of prion domains, which enable yeast proteins such as Sup35 and Swi1 to form prions (March et al, 2016). Whether human RBPs with PrLDs form prions remains an open question. Regardless, the PrLDs of human RBPs render them intrinsically aggregation prone, and an expanding cohort of human RBPs with PrLDs including ataxin 2, TDP‐43, FUS, TAF15, EWSR1, hnRNPA1, hnRNPA2, and TIA1 are linked to several fatal neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (March et al, 2016; Harrison & Shorter, 2017). An archetypal RBP with a PrLD is FUS (Fig 1A), which becomes depleted from the nucleus and mislocalized to cytoplasmic inclusions in the degenerating neurons of a subset of ALS and FTD patients (Harrison & Shorter, 2017). Accumulation of insoluble FUS in the cytoplasm and simultaneous depletion of FUS from the nucleus may elicit neurodegeneration via a gain of toxic function, a loss of function, or both (Sharma et al, 2016; Harrison & Shorter, 2017). Strategies to antagonize the cytoplasmic mislocalization and insolubility of FUS could have therapeutic utility (Harrison & Shorter, 2017; Yasuda et al, 2017). In this issue of The EMBO Journal, elegant studies by Fawzi, Shewmaker, and colleagues reveal that phosphorylation of the FUS PrLD can inhibit pathological FUS aggregation and toxicity (Monahan et al, 2017). Thus, increasing FUS PrLD phosphorylation emerges as a novel therapeutic strategy for ALS and FTD (Monahan et al, 2017).
Figure 1. Phosphorylation of the FUS PrLD inhibits aberrant phase transitions to pathological fibrils.
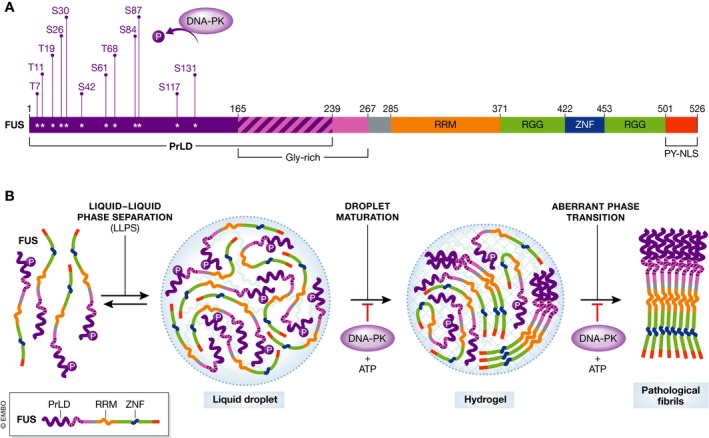
(A) Domain architecture of FUS. FUS harbors an N‐terminal PrLD (residues 1–239), a RNA‐recognition motif (RRM; residues 285–370), two RGG domains (residues 371–421 and 453–500) separated by a zinc‐finger domain (ZNF; residues 422–452), and a C‐terminal PY‐nuclear localization sequence (residues 501–526). (B) The uncharged, polar PrLD enables FUS to undergo liquid–liquid phase separation (LLPS). In these condensed liquid droplets, the FUS PrLD initially remains intrinsically disordered, but the high local concentration of PrLDs promotes their nucleation into cross‐β fibrils. As stable cross‐β fibrils begin to dominate the droplet there is a shift from the liquid state to a hydrogel state and ultimately to a solid aggregate comprised of pathological fibrils. DNA‐PK phosphorylates multiple sites in the FUS PrLD, which permits LLPS of full‐length FUS but prevents the primary nucleation of cross‐β fibrils in the liquid droplet and the aberrant phase transition to pathological fibrils.
FUS shuttles between the nucleus and cytoplasm, but is predominantly localized to the nucleus where it performs critical functions in transcription, pre‐mRNA splicing, RNA processing, and DNA repair (Harrison & Shorter, 2017). For many of these modalities, FUS likely functions in the concentrated and specialized microenvironment of a membraneless organelle or RNP granule (Patel et al, 2015; Harrison & Shorter, 2017). Importantly, the uncharged, polar PrLD enables FUS to undergo liquid–liquid phase separation (LLPS), which contributes to the biogenesis of these membraneless organelles (Fig 1B) (Burke et al, 2015; Murakami et al, 2015; Patel et al, 2015). In these condensed liquid states, the FUS PrLD adopts a molten, disordered conformation just as in the dispersed soluble state, but eventually the high local concentration of PrLDs promotes their nucleation into cross‐β fibrils (Fig 1B) (Burke et al, 2015; Patel et al, 2015; Kato & McKnight, 2017). This aberrant phase transition is accompanied by a shift in the material state of the droplet from liquid to a gel as cross‐β FUS fibrils begin to dominate the droplet (Fig 1B) (Patel et al, 2015; Kato & McKnight, 2017). Initially, short cross‐β FUS fibrils may be labile and reversible (Kato & McKnight, 2017), but ultimately the droplet converts to a deleterious solid state comprised of pathological fibrils (Murakami et al, 2015; Patel et al, 2015). Disease‐linked mutations in the FUS PrLD accelerate this aberrant phase transition from liquid drops to pathological fibrils (Murakami et al, 2015; Patel et al, 2015). In ALS and FTD, this process likely occurs in cytoplasmic stress granules, which may convert into pathological aggregates (March et al, 2016). How cells prevent aberrant phase transitions of FUS and other disease‐linked RBPs with PrLDs is poorly understood. Monahan et al (2017) now establish that phosphorylation of specific serines and threonines in the FUS PrLD inhibits formation of intra‐ and intermolecular contacts that drive aberrant phase separation and toxicity.
Building on prior observations (Han et al, 2012), Monahan et al (2017) establish that the FUS PrLD can be phosphorylated in vitro and in cells by DNA‐dependent protein kinase (DNA‐PK). DNA‐PK phosphorylates the FUS PrLD at multiple consensus S/TQ sites, including T7, T11, T19, S26, S30, S42, S61, T68, S84, S87, S117, and S131. Thus, a complex repertoire of FUS phosphorylation states is likely populated in vivo, which could regulate PrLD behavior. Introduction of negatively charged phosphoserine, phosphothreonine, or both into the predominantly uncharged FUS PrLD would increase electrostatic repulsion between PrLDs and restrict self‐assembly. Indeed, negatively charged residues are likely disfavored in PrLDs for this very reason (March et al, 2016). Importantly, DNA‐PK plus ATP ablated LLPS of a portion of the FUS PrLD (residues 1–163) in vitro, as did substitution of all twelve DNA‐PK consensus S/T residues to phosphomimetic glutamate (termed 12E) (Monahan et al, 2017). Intriguingly, full‐length FUS exhibited more complex phase behavior in vitro indicating the involvement of additional domains. Initial LLPS of full‐length FUS was unaffected by DNA‐PK plus ATP or by the phosphomimetic 12E substitution (Fig 1B) (Monahan et al, 2017). However, LLPS by the 12E variant was more readily inhibited by high salt or high RNA concentrations, indicating increased dependence on electrostatic interactions and an altered molecular grammar of LLPS (Monahan et al, 2017). Importantly, full‐length FUS droplets converted to fibrils, whereas DNA‐PK plus ATP or the 12E substitution prevented this aberrant phase transition and preserved the liquid droplet state (Fig 1B) (Monahan et al, 2017). Not all twelve phosphomimetic substitutions were necessary for inhibition as the 6E variant (with glutamate substitutions at S26, S30, T68, S84, S87, and S117) formed liquid drops but did not fibrillize (Monahan et al, 2017). Thus, partial phosphorylation of the FUS PrLD likely permits LLPS but prevents fibrillization.
The specific serine and threonine residues mutated in 6E likely reside at crucial junctures of the PrLD involved in intermolecular contacts that nucleate or maintain FUS fibrils. NMR experiments revealed that the 12E substitutions did not affect the intrinsically disordered nature the FUS PrLD but that the variant showed reduced transient intra‐ and intermolecular contacts within or between PrLDs (Monahan et al, 2017). Preventing these contacts likely inhibits nucleation events that drive fibrillization (Fig 1B). Importantly, the FUS 6E and 12E variants displayed reduced aggregation and toxicity in yeast and reduced cytoplasmic aggregation in mammalian cells in the context of the ALS‐linked FUSR495X variant (Monahan et al, 2017). Thus, increased FUS PrLD phosphorylation likely mitigates aggregation and toxicity.
Collectively, these advances suggest that strategies to increase FUS PrLD phosphorylation could have therapeutic utility for ALS and FTD, which can now be assessed in more complex disease models including fly, mouse, and patient‐derived neurons. These exciting studies also inspire several questions. For example, the precise role of FUS phosphorylation in physiology and pathology remains unknown. Nevertheless, introduction of multiple negatively charged residues into PrLDs appears to be a powerful mechanism to alter the syntax of self‐assembly and promote liquid states over deleterious solid states (Monahan et al, 2017). However, the FTD‐linked FUS variant, G156E, exhibits accelerated aberrant phase separation (Patel et al, 2015), which raises the possibility that phosphorylation of a single serine or threonine in the FUS PrLD could promote aberrant phase separation. Thus, it is critical that the FUS PrLD gets phosphorylated at multiple sites to mitigate toxicity (Monahan et al, 2017). It also remains unclear whether phosphorylation of the FUS PrLD can drive the dissolution of preformed FUS fibrils and return the aberrant solid aggregates that accumulate in disease to a liquid droplet phase or to soluble monomeric species. It will be important to determine whether key serine and threonine residues that may be buried in the FUS fibril core are accessible to protein kinases. An interesting possibility is that protein kinases might work in conjunction with protein disaggregases to rapidly phosphorylate newly disaggregated FUS and thereby prevent reaggregation (Yasuda et al, 2017).
Another question relates to DNA‐PK, which is localized to the nucleus and likely prevents aberrant FUS phase transitions in this compartment. However, in ALS and FTD, FUS accumulates in the cytoplasm of degenerating neurons. Thus, a goal would be to increase FUS PrLD phosphorylation in the cytoplasm, which may require targeting another kinase or perhaps inhibiting a phosphatase. Precisely how increased FUS PrLD phosphorylation might be elicited pharmacologically is also unclear. Whether phosphorylation of the FUS PrLD negatively regulates FUS functions in transcription, pre‐mRNA splicing, and RNA processing will also be critical to determine as it could be disadvantageous to exacerbate FUS loss of function in disease. Nonetheless, phosphorylation of the FUS PrLD effectively counters toxicity due to cytoplasmic gain of toxic function (Monahan et al, 2017), which is emerging as an important mechanism underpinning neurodegeneration in mouse models (Sharma et al, 2016). Finally, serine, tyrosine, and threonine occur commonly in PrLDs (March et al, 2016). Thus, it will be enlightening to establish whether specific phosphorylation events in PrLDs of other disease‐linked RBPs inhibit aberrant phase separation, which might also be targeted therapeutically. Regardless, the groundbreaking discoveries made by Monahan et al (2017) establish phosphorylation of the FUS PrLD as an intriguing new therapeutic strategy for diverse FUS proteinopathies.
See also: Z Monahan et al (October 2017)
References
- Burke KA, Janke AM, Rhine CL, Fawzi NL (2015) Residue‐by‐residue view of in vitro FUS granules that bind the C‐terminal domain of RNA polymerase II. Mol Cell 60: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL (2012) Cell‐free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149: 768–779 [DOI] [PubMed] [Google Scholar]
- Harrison AF, Shorter J (2017) RNA‐binding proteins with prion‐like domains in health and disease. Biochem J 474: 1417–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, McKnight SL (2017) Cross‐beta polymerization of low complexity sequence domains. Cold Spring Harb Perspect Biol 9: a023598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- March ZM, King OD, Shorter J (2016) Prion‐like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res 1647: 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan Z, Ryan VH, Janke AM, Burke KA, Rhoads SN, Zerze GH, O'Meally R, Dignon GL, Conicella AE, Zheng W, Best RB, Cole RN, Mittal J, Shewmaker F, Fawzi NL (2017) Phosphorylation of the FUS low‐complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J 36: 2951–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, Kronenberg‐Versteeg D, Li Y, Yang SP, Wakutani Y, Meadows W, Ferry RR, Dong L, Tartaglia GG, Favrin G, Lin WL et al (2015) ALS/FTD mutation‐induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88: 678–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162: 1066–1077 [DOI] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA (2016) ALS‐associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7: 10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Clatterbuck‐Soper SF, Jackrel ME, Shorter J, Mili S (2017) FUS inclusions disrupt RNA localization by sequestering kinesin‐1 and inhibiting microtubule detyrosination. J Cell Biol 216: 1015–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]