

Abstract
The intestinal epithelium holds an immense regenerative capacity mobilized by intestinal stem cells (ISCs), much of it supported by Wnt pathway activation. Several unique regulatory mechanisms ensuring optimal levels of Wnt signaling have been recognized in ISCs. Here, we identify another Wnt signaling amplifier, CKIε, which is specifically upregulated in ISCs and is essential for ISC maintenance, especially in the absence of its close isoform CKIδ. Co‐ablation of CKIδ/ε in the mouse gut epithelium results in rapid ISC elimination, with subsequent growth arrest, crypt–villous shrinking, and rapid mouse death. Unexpectedly, Wnt activation is preserved in all CKIδ/ε‐deficient enterocyte populations, with the exception of Lgr5+ ISCs, which exhibit Dvl2‐dependent Wnt signaling attenuation. CKIδ/ε‐depleted gut organoids cease proliferating and die rapidly, yet survive and resume self‐renewal upon reconstitution of Dvl2 expression. Our study underscores a unique regulation mode of the Wnt pathway in ISCs, possibly providing new means of stem cell enrichment for regenerative medicine.
Keywords: adult stem cells, casein kinase I, intestine stem cells, LGR5, Wnt
Subject Categories: Cell Cycle, Signal Transduction, Stem Cells
Introduction
The intestinal epithelium is the most vigorously self‐renewing tissue of adult mammals, replenished every 3–5 days. Proliferation occurs within the crypt compartment and is fueled by Lgr5‐expressing stem cells residing in the crypt (Barker et al, 2007), constantly generating progenitor cells, termed transit amplifying (TA) cells. TA cells undergo a few more divisions before they terminally differentiate into the various intestinal epithelial cell lineages (van der Flier & Clevers, 2009). The proliferation is balanced by apoptosis and cell shedding at the tip of the villus, thus generating a continuous upward movement in the epithelial sheath. The delicate equilibrium of proliferation versus differentiation is governed by a relatively small number of highly evolutionarily conserved signaling pathways, including the canonical Wnt signaling pathway.
Since the discovery of deregulated Wnt signaling as the primary driver of colon cancer (Groden et al, 1991; Kinzler et al, 1991), numerous studies in mouse models have firmly established the significance of Wnt signals for intestinal homeostasis and stem cell maintenance. Blockage of canonical Wnt signaling via Tcf4 deletion (Korinek et al, 1998; van Es et al, 2012), inducible deletion of β‐catenin (Ireland et al, 2004), or overexpression of the Wnt inhibitor Dickkopf 1 (Dkk1) (Pinto et al, 2003; Kuhnert et al, 2004) resulted in impaired proliferation and crypt loss. Conversely, overexpression of Wnt signaling such as knockout for adenomatous polyposis coli (APC) (Sansom et al, 2004; Andreu et al, 2005) resulted in considerable expansion of the proliferative progenitor compartment with increased nuclear localization of β‐catenin and expression of Wnt target genes.
The main target of the canonical Wnt pathway is cytoplasmic β‐catenin, which serves as a transcriptional co‐activator. The transduction of signal depends on the presence or absence of a Wnt ligand. In the absence of Wnt ligand, β‐catenin is constantly phosphorylated at the “destruction complex” composed of APC, Axin, glycogen synthase kinase 3β (GSK3β), and casein kinase I (CKIα), leading to the polyubiquitination and proteasomal degradation of β‐catenin (Clevers & Nusse, 2012). According to a “classical” Wnt signaling model, upon binding of Wnt ligand to the Frizzled receptor, Dishevelled (Dvl) is phosphorylated, subsequently resulting in low‐density lipoprotein receptor‐related proteins 5 and 6 (LRP5/6) co‐receptor phosphorylation by casein kinase 1γ (CK1γ) and sequestration of the “destruction complex” to the Frizzled‐LRP5/6 complex (Mao et al, 2001; Zeng et al, 2005). Other models for Wnt activation proposed inhibition of β‐catenin ubiquitination, rather the disruption of the destruction complex, as the critical event for Wnt response (Li et al, 2012). Nevertheless, both proposed mechanisms ultimately lead to β‐catenin stabilization and translocation to the nucleus, where it binds Tcf/Lef to activate the transcription of numerous target genes.
Further to its regulatory role in intestinal proliferation, Wnt signaling was identified as a critical player in the control of intestinal stem cell (ISC) fate (Beumer & Clevers, 2016), evident by expansion of Lgr5 ISCs in response to R‐spondin‐mediated Wnt upregulation and loss of Lgr5 cells subsequent to expression of Dkk1 Wnt antagonist (Yan et al, 2012). Paneth cells interspersed between ISCs at the bottom of the crypt were specified as a key component of ISC niche (Sato et al, 2011), secreting Wnt3a, thus ensuring a robust and continuous Wnt signal required for ISC maintenance (Farin et al, 2016). Certain regulatory mechanisms for enhancing Wnt activity were found to play an exclusive role in ISCs. Lgr5 and the much broadly expressed Lgr4 were shown to function as receptors for the Wnt agonist family of R‐spondin (Carmon et al, 2011; Glinka et al, 2011; de Lau et al, 2011; Ruffner et al, 2012). Binding of R‐spondin to Lgr5 (Xie et al, 2013; Zebisch et al, 2013) triggers internalization of Znrf3, an E3‐ligase of Frizzled receptors, resulting in Frizzled stabilization and Wnt signaling amplification in intestinal stem cells (Hao et al, 2012; Koo et al, 2012; Xie et al, 2013). Two additional new players in the maintenance of ISC homeostasis, potentiating R‐spondin's action and enhancing Wnt signaling, are the Slit/Robo pathway (Zhou et al, 2013) and Ascl2, a Wnt target by itself, shown to synergize with β‐catenin's transcriptional activity, thus enabling an auto‐regulatory positive feedback loop (Schuijers et al, 2015). On the other hand, several ISC‐specific negative Wnt regulatory mechanisms were also revealed. Yes‐associated protein (YAP) protein was found to control ISC's regeneration through a Wnt inhibitory effect (Gregorieff et al, 2015) and suppression of Dvl nuclear translocation (Barry et al, 2013). Troy, a member of the tumor necrosis factor receptor superfamily expressed in Lgr5‐ISCs, interacts with LGR5 to inhibit LRP6 phosphorylation, thereby suppressing Wnt signaling (Fafilek et al, 2013). Altogether, these data emphasize tight regulation on Wnt signaling in ISCs, facilitating fine‐tuning of the correct Wnt response.
Casein kinase I is a highly conserved serine/threonine kinase family, ubiquitously expressed from yeast to human, and has been demonstrated to be involved in diverse cellular processes such as regulation of Wnt signaling pathway, circadian rhythm regulation, p53 activation, and mRNA translation (Knippschild et al, 2005; Cheong & Virshup, 2011; Elyada et al, 2011; Cruciat, 2014; Shin et al, 2014). Within the CKI isoform group, casein kinase I delta (CKIδ) and casein kinase I epsilon (CKIε) share more than 80% similarity, both in the N‐terminal and in C‐terminal domains, indicating a possible redundancy. Initially, CKI isoforms were identified as positive regulators of the Wnt pathway (Peters et al, 1999; Sakanaka et al, 1999; McKay et al, 2001) by acting upstream of Axin and GSK3 and stabilizing β‐catenin (Peters et al, 1999; McKay et al, 2001). Moreover, CKIε was shown to be activated upon Wnt stimulation, resulting in a positive feedback loop on Wnt signaling (Swiatek et al, 2004; Cruciat et al, 2013). As a primary substrate, CKIε was suggested to phosphorylate and activate the scaffold protein Dvl (Peters et al, 1999; Cong et al, 2004; Klimowski et al, 2006; Del Valle‐Pérez et al, 2011a; Bernatík et al, 2014) resulting in disassociation of the destruction complex and stabilization of β‐catenin. However, the precise function of phosphorylated Dvl is still unknown, as Wnt‐β‐catenin signaling was normally active even upon inhibition of Dvl phosphorylation (Bryja et al, 2007). Additional substrates proposed to mediate CKIδ/ε positive regulation of Wnt signaling included the co‐receptor LRP6 (Zeng et al, 2005), TCF3 (Lee et al, 2001), and p120‐catenin (Casagolda et al, 2010; Del Valle‐Pérez et al, 2011a,b). In addition to β‐catenin stabilization, activation of the canonical Wnt signaling pathway by CKIε was shown to be essential also for suppression of non‐canonical Wnt signaling pathways. CKIε loss‐of‐function mutations in mammary ductal carcinoma (Fuja et al, 2004; Foldynová‐Trantírková et al, 2010; Kim et al, 2010) resulted in non‐canonical Wnt pathway upregulation, important for cellular growth and migration. Notably, CKIε was also implicated as a negative regulator of the canonical Wnt pathway via several mechanisms, including inhibitory phosphorylation of LEF transcription factor (Hämmerlein et al, 2005), the co‐receptor LRP6 (Swiatek et al, 2006), APC (Rubinfeld et al, 2001; Ferrarese et al, 2007), and phosphorylation of the Wnt receptor ROR2 (Witte et al, 2010).
Considering the contrasting roles and suppression effects proposed for the CKI isoforms δ and ε in the Wnt pathway, mostly based on in vitro studies, more information is needed to resolve their Wnt regulatory role in vivo. To this end, we prepared and studied individual and combined ablation of CKI δ and ε in the mouse gut.
Results
Functional redundancy between CKIδ and CKIε in the gut
To investigate the in vivo physiological roles of CKIδ and CKIε, we generated constitutive knockout mice with germline deletion of CKIδ or ε. Consistent with previously published data (Xu et al, 2005), whereas homozygous CKIε−/− mice were viable, born in the expected mendelian ratio and without any notable phenotype, CKIδ−/− mice were born smaller than heterozygous CKIδ+/− mice and died perinatally (data not shown).
To study the role of CKIδ/ε in gut physiology and circumvent the perinatal lethality of constitutive CKIδ KO, we generated mice with floxed‐CKIδ allele and crossed them to Villin‐Cre‐ERT2 mice; in this progeny, CKIδ is deleted upon tamoxifen injection exclusively in the intestinal epithelium (hereafter referred to as CKIδΔgut). Within 5 days of tamoxifen treatment, intestinal epithelial cells (IECs) were completely depleted of CKIδ (Fig 1A). Both CKIδΔgut and CKIε−/− mice were viable and exhibited normal intestinal proliferation and homeostasis as demonstrated by BrdU incorporation (Fig 1B) and normal Wnt activity (Fig 1C). Notably, we observed an increase in CKIε protein and transcript levels in CKIδΔgut mice, and reciprocally, increased CKIδ levels were detected in CKIε−/− enterocytes (Figs 1A and EV1), indicating a compensatory expression mechanism with possible functional redundancy between the two isoforms in the intestinal epithelium.
Figure 1. Functional redundancy between CKIδ and CKIε in the gut.
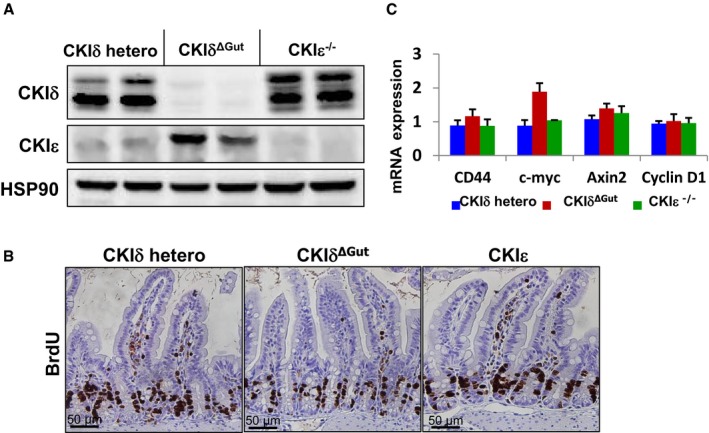
- Western blot (WB) analysis of intestinal epithelial cells (IECs) of CKIδ+/fl ER (CKIδ hetero), CKIδΔGut, and CKIε−/− mice.
- Immunohistochemistry (IHC) of BrdU in intestinal sections of CKIδ hetero, CKIδΔGut, and CKIε−/− mice. Scale bar, 50 μm.
- Quantitative RT–PCR (qRT–PCR) analysis of Wnt target genes in IECs isolated from CKIδ hetero (n = 4), CKIδΔGut (n = 4), and CKIε−/− (n = 4) mice, mean ± SEM.
Figure EV1. Functional redundancy between CKIδ and CKIε in the gut.
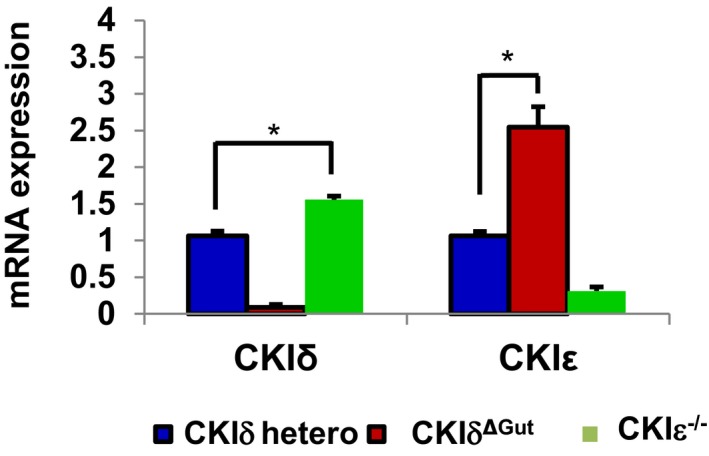
qRT–PCR analysis of CKIδ and CKIε in IECs isolated from CKIδ+/fl ER (CKIδ hetero, n = 3), CKIδΔGut (n = 3), and CKIε−/− mice (n = 3, mean ± SEM); t‐test was performed, *P < 0.05.
Co‐ablation of CKIδ/ε induces intestinal atrophy without compromising Wnt signaling
To assess the functional compensation between CKIδ and ε isoforms in intestinal homeostasis, we crossed CKIδΔgut mice with CKIε−/− mice to generate CKIδ/ε double KO (DKO) mice; in these mice, CKIε is constitutively abolished and CKIδ deletion is inducible upon tamoxifen injection. Remarkably, DKO mice displayed a gradual villi shortening progressing to complete intestinal atrophy within 5 days of the knockout induction (Fig 2A) and died shortly after. Unlike CKIδΔgut or CKIε−/− mice that exhibited normal proliferation rates (Fig 1B), combined ablation of CKIδ and CKIε resulted in a complete cell cycle arrest, as indicated by decreased BrdU and Ki67 staining (Fig 2B), accompanied by a mild increase in intestinal epithelium apoptosis (Fig 2B; cleaved Caspase 3 staining).
Figure 2. Co‐ablation of CKIδ/ε induces intestinal atrophy without compromising Wnt signaling.
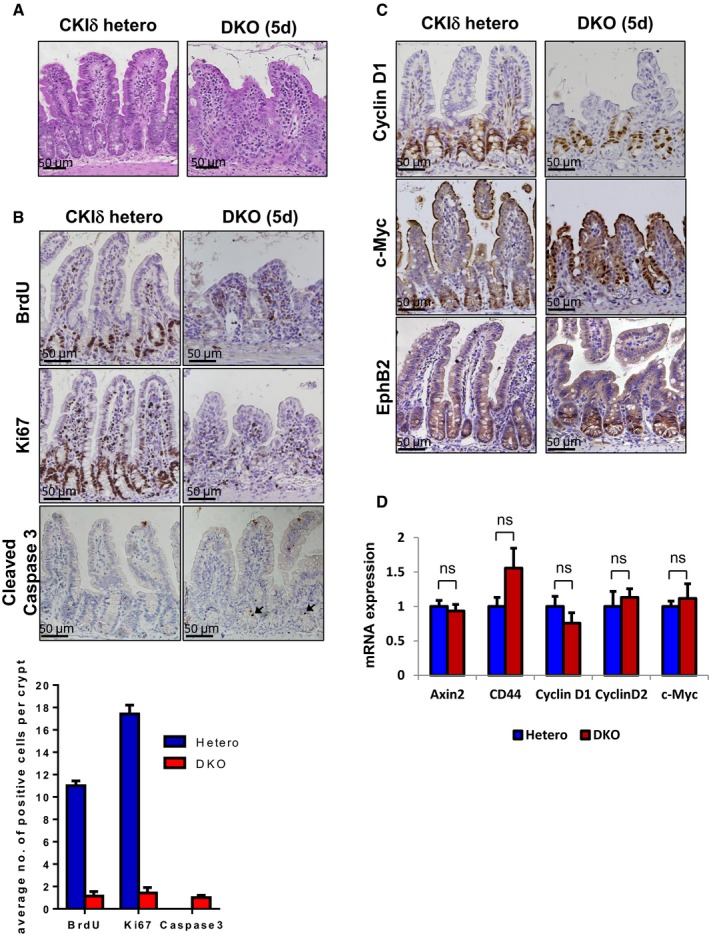
- H&E staining in intestinal sections of CKIδ hetero and CKIδ/ε double KO (DKO) mice, 5 days after KO induction. Scale bar, 50 μm.
- IHC of BrdU, Ki67, and cleaved Caspase 3 in intestinal sections of CKIδ hetero and DKO mice, 5 days after KO induction; black arrowheads point toward apoptotic cleaved Caspase 3+ cells; scale bar, 50 μm. For quantification, 20 crypts per mouse were counted, n = 3, mean ± SEM.
- IHC of cyclin D1, c‐Myc, and EphB2 in intestinal sections of CKIδ hetero and DKO mice, 5 days after KO induction. Scale bar, 50 μm.
- qRT–PCR analysis of Wnt target genes in IECs isolated from CKIδ hetero (n = 3) and DKO mice (n = 3, mean ± SEM), 3 days after KO induction; t‐test was performed, ns = non‐significant.
Considering the major role attributed to Wnt signaling in intestinal proliferation (van der Flier & Clevers, 2009), we sought to determine whether depletion of CKIδ/ε triggers any inhibition of intestinal Wnt activity, explaining the diminished proliferation in DKO mice intestines. Surprisingly, Wnt signaling in the intestinal epithelium was not affected by depletion of CKIδ/ε; Wnt target gene expression, including cyclin D1/D2, c‐Myc, CD44, EphB2, and Axin2, was comparable to control epithelium (Fig 2C and D). Furthermore, Notch signaling, the other signaling pathway crucial for intestinal proliferation, was normally active, per nuclear staining of the major Notch target Hes1 in the crypt regions (Appendix Fig S1).
Several studies have highlighted the contribution of the circadian rhythm to cell cycle progression (Masri et al, 2013). Given the fact that CKIδ/ε was shown to be involved in post‐translational modifications regulating the circadian rhythm (Vielhaber et al, 2000; Shirogane et al, 2005; Xu et al, 2005; Etchegaray et al, 2009, 2010; Lee et al, 2009), we examined the expression levels of several known circadian regulated genes, such as DBP and Rev‐Erbα at two circadian time (CT) points—CT0 and CT12, representing day–night transition time points. Interestingly, while CKIε−/− IECs exhibited a normal circadian rhythm (comparable to CKIδ heterozygous mice), both CKIδΔGut and DKO mice displayed hampered circadian oscillations (Appendix Fig S2), underscoring the specific role of CKIδ in the regulation of the circadian clock, in agreement with previous reports (Etchegaray et al, 2009). Nevertheless, despite the impaired circadian rhythm, intestinal proliferation and homeostasis were preserved in CKIδΔGut mice (Fig 1B), implying that alterations in the circadian clock pathway, observed both in CKIδΔGut and in DKO mice, could not provide a sufficient explanation for the cell cycle arrest phenotype detected specifically in DKO mice.
As we did not detect any change in the activity level of the common signaling pathways known to regulate intestinal proliferation, we sought to determine whether ablation of CKIδ/ε induced the activation of any cell cycle inhibitor that could account for the observed cell cycle arrest phenotype. Indeed, DKO mice showed marked activation of p53, as detected by nuclear p53 and p21 in the crypt regions (Fig EV2A). To assess the contribution of p53 to proliferation arrest in DKO mice, we crossed the CKIδ/ε DKO mice with p53fl/fl (Jonkers et al, 2001) to generate CKIδΔgut/CKIε−/−/p53Δgut triple KO (TKO) mice (Fig EV2B). Unexpectedly, p53 deletion failed to rescue the DKO phenotype, and TKO mice exhibited reduced enterocyte proliferation, developed intestinal atrophy, similarly to DKO mice (Fig EV2C), and died within 2 weeks of TKO induction. These data demonstrate that the cell cycle arrest induced upon CKIδ/ε ablation was p53‐independent.
Figure EV2. CKIδ/ε ablation triggers a p53‐independent cell cycle arrest.
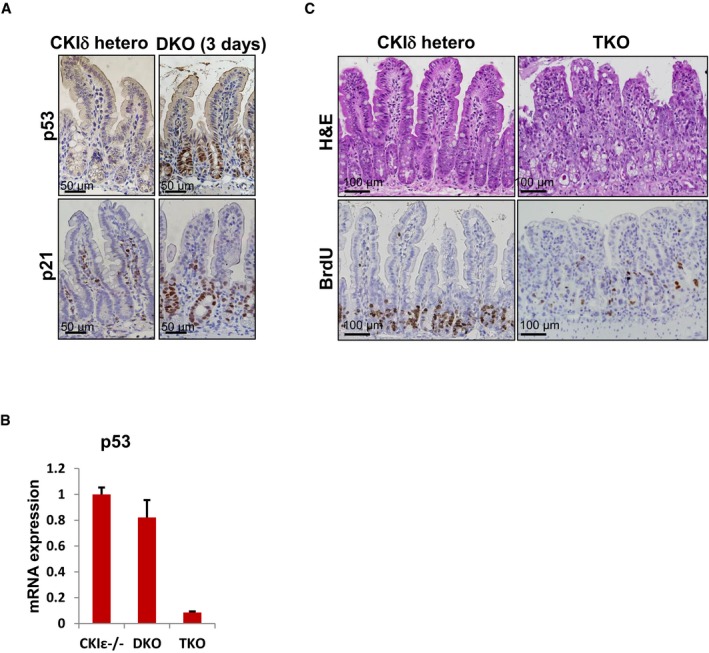
- IHC analysis of p53 and p21 in intestinal sections from CKIδ hetero and DKO mice, 3 days after KO induction. Scale bar, 50 μm.
- qRT–PCR analysis of p53 levels after KO induction in CKIε−/−, DKO, and CKIδ/ CKIε/p53 triple KO mice (TKO) (n = 3, mean ± SEM).
- H&E and IHC of BrdU in intestinal sections from CKIδ hetero and CKIδ/ CKIε/p53 triple KO mice (TKO), 5 days after KO induction. Scale bar, 100 μm.
CKIδ/ε combined deletion compromises intestinal stem cell function
The intestinal epithelium is continuously renewed by an intestinal stem cell (ISC) population residing in the crypt region. To assess the role of CKIδ/ε in ISC self‐renewal, we crossed CKIδfl/fl CKIε−/− mice with Lgr5EGFP‐ires‐CreERT2 (Barker et al, 2007) mice to generate DKO/Lgr5‐CreER mice. These mice express a knock‐in allele of Lgr5, which marks Lgr5‐expressing ISCs with GFP and enables activation of Cre recombinase enzyme only in Lgr5‐expressing cells. Of note, the knock‐in allele in the Lgr5EGFP‐ires‐CreERT2 mouse model is expressed in a variegated manner (Barker et al, 2007), and therefore, tamoxifen injection targets only the crypts in which the knock‐in allele is expressed. The DKO/Lgr5‐CreER mouse model harbors germline‐deleted CKIε alleles, yet enables inducible CKIδ ablation specifically in Lgr5‐GFP+ cells. Since Lgr5 cells function as ISCs, continuously generating progeny that replenishes the intestinal epithelium, all cell lineages derived from the original Lgr5+ cell which underwent CKIδ KO or DKO, will negatively stain for CKIδ, as means of lineage tracing of the Lgr5+ ISCs (Fig 3A). As expected, CKIδ immunostaining in singly‐deleted CKIδ KO/Lgr5‐CreERT2 mice, 7 days after KO induction, revealed regions composed of villi and crypts originated from Cre‐targeted Lgr5EGFP‐ires‐CreERT2 cells, in which CKIδ was completely abolished (Fig 3B). It shows that CKIε expression alone is sufficient to preserve the function of ISC. In contrast, histological analysis of DKO/Lgr5‐CreERT2 mice 7 days after tamoxifen induction revealed no CKIδ‐negative cells (Fig 3B), indicating a strong counter selection against doubly CKIδ/ε‐depleted ISCs; DKO crypts were exclusively repopulated by CKIδ‐positive, CKIε−/− ISCs. These results further demonstrate that while ablation of either CKIδ or CKIε alone is compatible with normal ISC function (self‐renewal and differentiation), loss of both isoforms eliminates functional intestinal stem cells, underscoring the redundancy between CKIδ and ε isoforms for ISC self‐renewal.
Figure 3. Combined CKIδ/ε deletion compromises intestinal stem cell function.
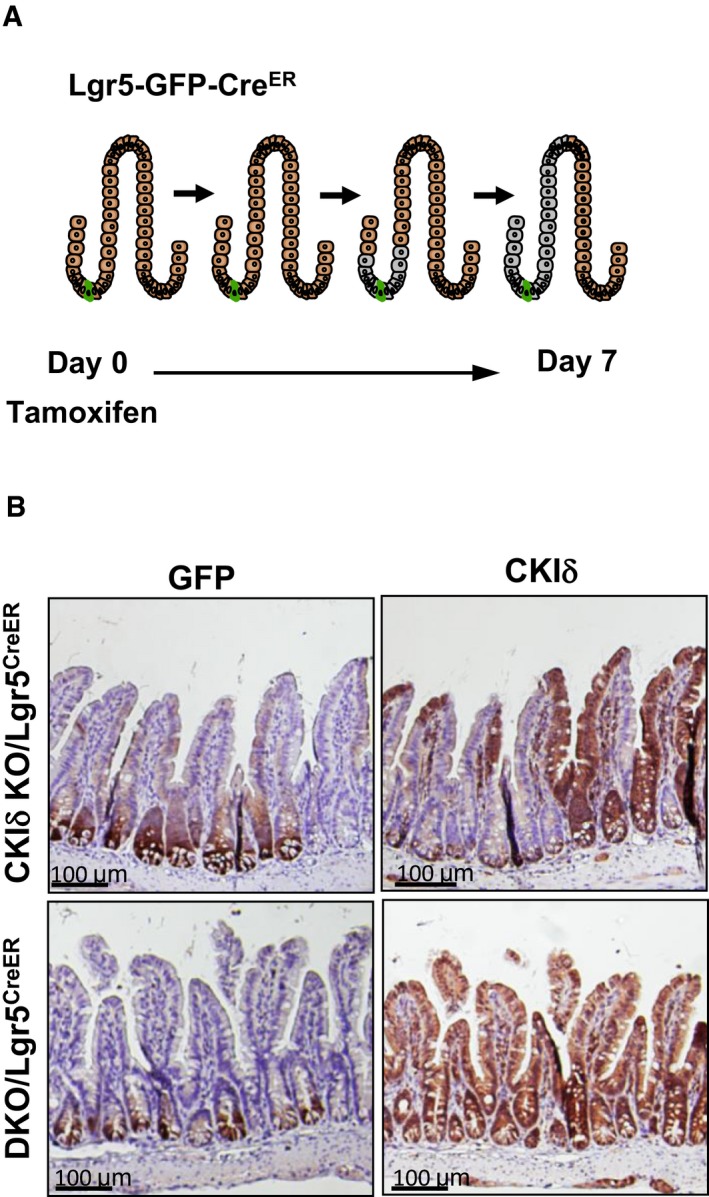
- Schematic representation of lineage tracing experiment illustrated in (B) in Lgr5‐CreER mice, brown staining represent CKIδ in WT or CKIε−/− cells, gray staining represents cells after depletion of CKIδ, and green cell at the bottom of the crypt represent a Lgr5‐EGFP‐CreER cell depleted of CKIδ after tamoxifen injection.
- IHC of GFP used as a marker for Lgr5+ intestinal stem cells (ISCs) and of CKIδ in representative serial intestinal sections of CKIδ KO/Lgr5‐CreER and DKO/Lgr5‐CreER mice, 7 days after KO induction. Scale bar, 100 μm.
Extinction of ISCs upon CKIδ/ε deletion
To determine whether the decreased proliferation in DKO mice (Fig 2B) was associated with loss of the intestinal stem cells, we crossed DKO‐VillinCreERT2 mice with Lgr5EGFP‐ires‐CreERT2 mice to generate DKO‐VillinCreERT2‐Lgr5 mice. This composite mouse model ensures the deletion of CKIδ/ε in the entire intestinal epithelium, including ISCs via the Villin‐Cre recombinase, while using Lgr5‐driven GFP for tracing the fate of ISCs. This model has two advantages over solely using Lgr5EGFP‐ires‐CreERT2 as both deleter and ISC tracing marker: (i) It circumvents the mosaic expression pattern of the Lgr5EGFP‐ires‐CreERT2 knock‐in allele (Barker et al, 2007), allowing efficient targeting of the entire stem cell pool; (ii) it evades de novo generation of Lgr5 cells harboring a WT non‐depleted CKIδ allele due to ISC plasticity (Tian et al, 2011). Notably, expression of a dual Villin‐ and Lgr5‐driven Cre in Lgr5+ ISCs did not disturb itself Lgr5+ ISC homeostasis, evident in the maintenance of Lgr5+ ISC proliferation and stemness function in heterozygous CKIδ+/fl‐VillinCreER‐LGR5CreER control mouse group (Fig 4A–D). In contrast to the heterozygous mice, 5 days after tamoxifen treatment, we could hardly detect any Lgr5‐GFP+ cells in DKO‐VillinCreERT2‐Lgr5 mice (Fig 4A), indicating that the remnant crypts in DKO mice are devoid of stem cells. Reduced Lgr5‐GFP+ cell numbers were accompanied by downregulation of several unique ISC marker expression, previously identified as “the ISC's signature” (Muñoz et al, 2012), including Lgr5, Olfm4, Ascl2, Smoc2, and Clca4 (Fig 4B).
Figure 4. Growth arrest and elimination of ISCs upon CKIδ/ε depletion.
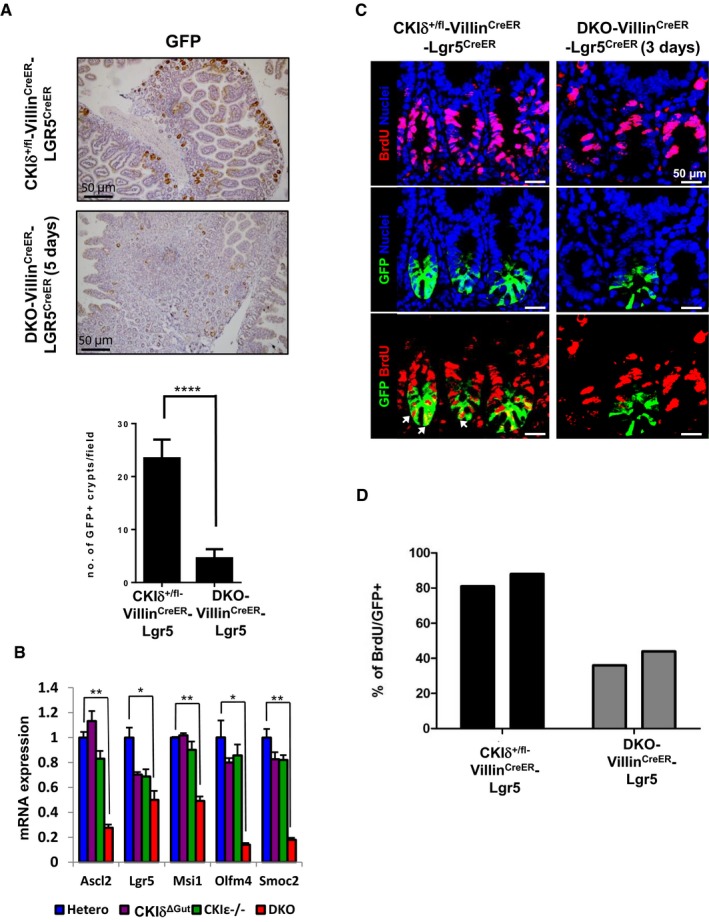
- IHC of GFP used as a marker for Lgr5+ ISCs in intestinal sections of CKIδ+/fl‐VillinCreER‐Lgr5CreER and DKO‐VillinCreER‐Lgr5CreER mice 5 days after KO induction. Quantification was based on number of GFP+ crypts per ×10 field (n = 3, mean ± SEM, t‐test was performed, ****P < 0.0001).
- qRT–PCR analysis of ISC markers in IECs isolated from CKIδ hetero (n = 3), CKIδΔGut (n = 4), CKIε−/− (n = 4), and DKO mice (n = 4, mean ± SEM), 3 days after KO induction, t‐test was performed, *P < 0.05 **P < 0.01.
- Immunofluorescence (IF) of BrdU (red) and GFP (green) in intestinal sections of CKIδ+/fl‐VillinCreER‐Lgr5CreER and DKO‐VillinCreER‐Lgr5CreER mice, 3 days after KO induction; nuclear counterstain Hoechst. Scale bar, 50 μm. Arrows point to cells doubly‐stained with BrdU (red) and GFP (green), representing proliferating Lgr5‐GFP+ cells.
- Quantification of BrdU IF staining, based on %BrdU+ cells out of 300 GFP+ cells in CKIδ+/fl‐VillinCreER‐Lgr5CreER (n = 2) and DKO‐VillinCreER‐Lgr5CreER mice (n = 2).
As proliferation is a mandatory trait for maintaining the “stemness” potential of Lgr5 cells, we examined the proliferation of DKO Lgr5‐expressing ISCs in DKO‐VillinCreERT2‐Lgr5 mice using BrdU labeling. Two and a half days after tamoxifen treatment, DKO‐VillinCreERT2‐Lgr5 mice received two consecutive BrdU injections every 6 h and were sacrificed 12 h later. Given the fact that the average cell cycle length of Lgr5 ISCs is estimated to be ~22 h (Schepers et al, 2011) with an averaged S‐phase duration of 7.5 h (Quastler & Sherman, 1959), we expected BrdU incorporation in approximately 60% of Lgr5‐GFP+ cells. In reality, we observed an average 80% BrdU+ cells in CKIδ+/fl‐VillinCreERT2‐Lgr5 cells. Remarkably, DKO‐VillinCreERT2‐Lgr5 mice showed only 40% BrdU‐positive Lgr5‐GFP+, half the heterozygous control values (Fig 4C and D), indicating attenuated proliferation of Lgr5 ISCs, early upon CKIδ/ε ablation. Furthermore, in addition to limited proliferation, DKO Lgr5‐GFP+ cells exhibited an increase in the apoptosis rate, indicated by cleaved Caspase 3 staining (Fig EV3). Hence, ISC depletion upon CKIδ/ε loss could be due to both apoptosis and proliferation arrest.
Figure EV3. CKIδ/ε depletion induces ISC apoptosis.
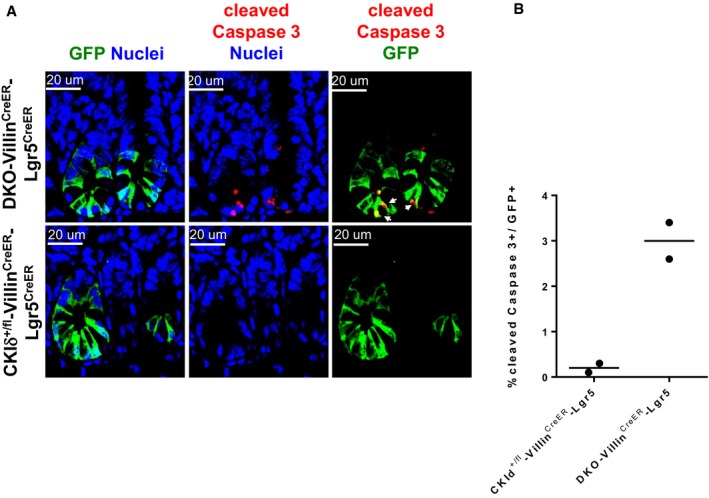
- IF analysis of GFP (green) and cleaved Caspase 3 (red) in intestinal sections of CKIδ+/fl‐VillinCreER‐Lgr5CreER and DKO‐VillinCreER‐Lgr5CreER mice 3 days after KO induction; nuclear counterstain Hoechst. Scale bar, 20 μm. Arrows point to cells doubly stained by GFP and cleaved Caspase 3 representing apoptotic Lgr5‐GFP cells.
- Quantification of cleaved Caspase 3 IF staining, based on %cleaved Caspase 3+ cells out of 300 GFP+ cells in CKId+/fl‐VillinCreER‐Lgr5CreER (n = 2) and DKO‐VillinCreER‐Lgr5CreER mice (n = 2).
Paneth cells are a differentiated secretory epithelial population interspersed between intestinal Lgr5 stem cells at the base of the crypts and have recently been shown as a critical components of the intestinal stem cell niche (Sato et al, 2011). We therefore examined whether ISC depletion could also result from damage to the stem cell niche. However, CKIδ/ε DKO mice displayed normal Paneth cell pattern, at the crypt area per lysozyme staining, even at late time points after KO induction, when ISCs were already absent (Appendix Fig S3). This indicates that the ISC niche in DKO mice is intact and thus cannot account for the decrease in stem cell number.
CKIδ/ε signaling is required for optimal ISC Wnt regulation
To elucidate the role of CKIδ/ε in ISC function, we used the DKO‐VillinCreERT2‐Lgr5 mouse model, in which DKO‐Lgr5 ISCs can be sorted out by flow cytometry according to GFP staining. GFP+ and GFP− cells were collected from DKO‐VillinCreERT2‐Lgr5 mice, CKIε−/−‐VillinCreERT2‐Lgr5, and control CKIδ+/fl‐VillinCreERT2‐Lgr5 mice, 2.5 days after KO induction, an early time point in which ISCs are still present. Interestingly, CKIε expression was markedly upregulated in the control CKIδ+/fl‐VillinCreERT2 Lgr5‐GFP+ ISC population compared to GFP− cells (Fig 5A), consistent with the reported transcriptomic and proteomic analysis of Lgr5 ISCs that showed enrichment of CKIε in these cells (Muñoz et al, 2012). The fact that CKIε was transcriptionally enriched in ISCs and that CKIδ/ε‐depleted ISCs were first non‐functional and ultimately eradicated from the intestine (Figs 3B and 4A) led us to postulate a possible role for CKIδ/ε in intestinal stem cell physiology. Indeed, analysis of the “ISC signature” (Muñoz et al, 2012) in DKO‐Lgr5‐GFP+ cells showed a marked reduction in the expression level of several key ISC markers, such as Lgr5, Olfm4, Ascl2, and Smoc2 (Fig 5B), indicating that reduced expression of these markers observed in total DKO enterocytes (Fig 4B) could have resulted from transcriptional alterations in Lgr5+ cells (Fig 5B), or reduced numbers of total ISCs (Fig 4A). As several of the ISC markers are Wnt‐regulated, we analyzed the Wnt response in the sorted cells. Strikingly, in contrast to the normal expression levels of Wnt target genes in the total population of DKO enterocytes (Fig 2D), isolated DKO‐Lgr5‐GFP+ cells showed reduced expression levels of several “classic” Wnt target genes, such as Axin2, EphB2, EphB3 (Fig 5C), as well as a decrease in Wnt genes restricted to ISCs, such as Lgr5 and Ascl2 (Fig 5B). Wnt signaling suppression in DKO ISCs was further validated using multiple‐color single‐molecule fluorescence in situ hybridization (Itzkovitz et al, 2012) for Lgr5, as a marker for ISCs and Axin2, as a “classical” Wnt target gene. Analysis of 2.5 days DKO ISCs positive for Lgr5 revealed a marked decrease in the number of transcript copies of both Lgr5 and Axin2 compared to CKIδ heterozygous Lgr5 cells (Fig 5D and E), underscoring the role of CKIδ/ε in potentiation of Wnt signaling specifically in ISCs. Notably, whereas CKIε, but not CKIδ expression level, was upregulated in ISCs (Fig 5A), deletion of CKIε alone in Lgr5‐GFP+ cells, unlike of CKIδ/ε co‐ablation, had no effect on the expression levels of Wnt target genes (Fig 5C); most likely explanation is functional compensation for CKIε loss by CKIδ.
Figure 5. CKIδ/ε is indispensable for Wnt signaling in ISCs.
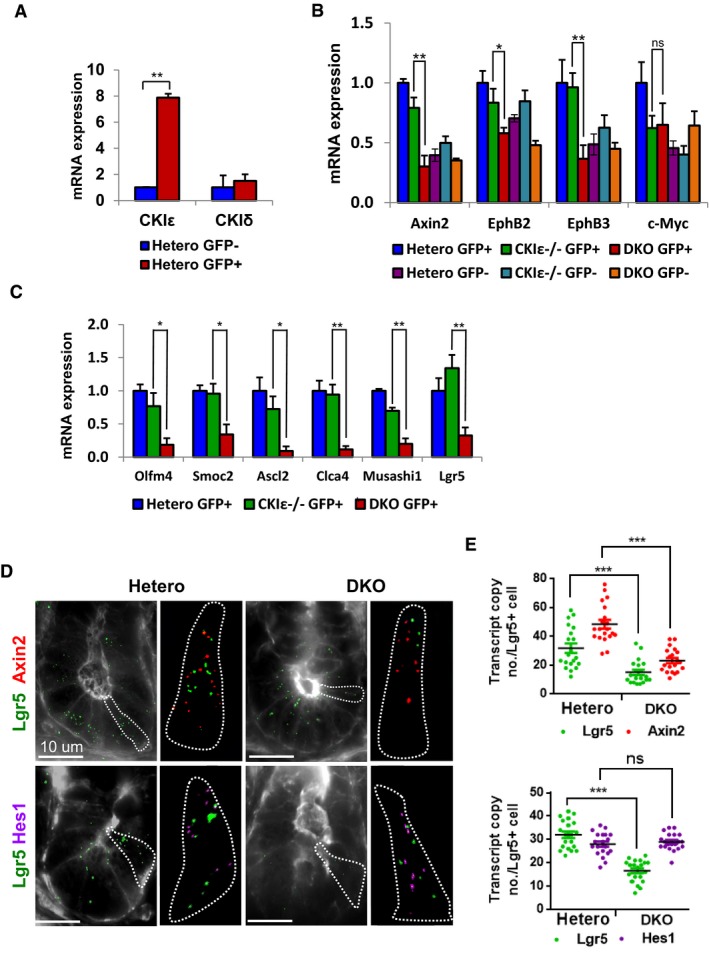
- qRT–PCR analysis of CKIδ and CKIε in sorted GFP+ and GFP− cells isolated from crypts of CKIδ+/fl‐VillinCreER‐Lgr5CreER mice. Data represent the mean of four independent sorting experiments, each with a pool of 6 mice, **P < 0.01 (t‐test). Error bars represent SEM.
- qRT–PCR analysis of Wnt target genes in sorted GFP+ and GFP− cells isolated from crypts of CKIδ hetero (data represent the mean of two independent sorting experiments, each with a pool of 6 mice), CKIε−/−‐VillinCreER‐Lgr5CreER (CKIε−/−), and DKO‐VillinCreER‐Lgr5CreER (DKO), 2.5 days after KO induction. Data represent the mean of four independent sorting experiments, each with a pool of six mice, t‐test was performed, *P < 0.05 **P < 0.01, ns = non‐significant. Error bars represent SEM.
- qRT–PCR analysis of ISC markers in sorted GFP+ and GFP− cells isolated from crypts of CKIδ hetero, CKIε−/−, and DKO, 2.5 days after KO induction. Data represent the mean of three independent sorting experiments, each with a pool of six mice; t‐test was performed, *P < 0.05 **P < 0.01. Error bars represent SEM.
- Three‐color single‐molecule fluorescent in situ hybridization (FISH) analysis of Lgr5 (green), Axin2 (red), and Hes1 (magenta) from representative cells of CKIδ hetero and DKO mice, 2.5 days after KO induction using; cell borders marked by FITC–Phalloidin. Scale bar, 10 μm. Outlines represent borders of a single crypt cell analysis.
- Quantification of Lgr5, Axin2, and Hes1 transcript copy number (mean ± SEM) in Lgr5+ cells based on 25 z‐stacks (0.25 μm apart) of 20 Lgr5+ cells (≥2 Lgr5 transcript spots per cell) from CKIδ hetero and DKO mouse intestinal sections, 2.5 days after KO induction, ***P < 0.001, ns = non‐significant.
Intestinal stem cell activity is controlled by several signaling pathways including Notch, BMP, and Hedgehog. Notch signaling was shown as a critical regulator of intestinal stem cell self‐renewal and proliferation by negative transcriptional regulation of the cyclin‐dependent kinase inhibitors p27KIP1 and p57KIP2 (Riccio et al, 2008). Nevertheless, Notch activity was preserved in DKO‐Lgr5‐GFP+ cells, as indicated by normal expression levels of Hes1, p27, and p57 (Figs 5D and E, and EV4A). Similarly, we did not detect any changes in the expression level of BMP and Hedgehog target genes (Fig EV4A) implicating the reduced Wnt signaling as the primary cause for loss of DKO ISCs.
Figure EV4. Extinction of DKO ISCs is not mediated by Notch signaling downregulation or p53 activation.
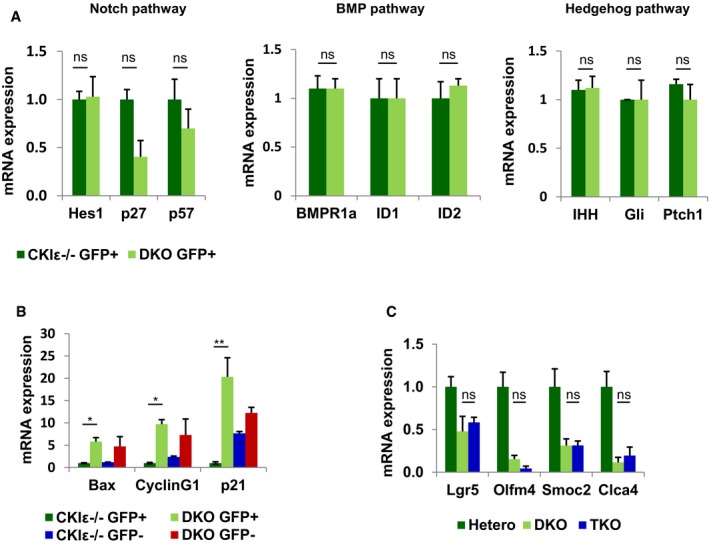
- qRT–PCR analysis of Notch, BMP, and Hedgehog target genes in sorted GFP+ cells isolated from crypts of CKIε−/−‐VillinCreER‐Lgr5CreER (CKIε−/−) and DKO‐VillinCreER‐Lgr5CreER mice (DKO) 2.5 days after KO induction. Data represent mean of three independent sorting experiments, each with a pool of six mice; t‐test was performed, ns = non‐significant. Error bars indicate SEM.
- qRT–PCR analysis of p53 targets in sorted GFP+ and GFP− cells isolated from crypts of CKIε−/−‐VillinCreER‐Lgr5CreER (CKIε−/−) and DKO‐VillinCreER‐Lgr5CreER (DKO), 2.5 days after KO induction. Data represent mean of three independent sorting experiments, each with a pool of six mice; t‐test was performed, *P < 0.05 **P < 0.01. Error bars indicate SEM.
- qRT–PCR analysis of ISC markers in IECs isolated from CKIδ hetero (hetero; n = 3), DKO (n = 3) and TKO (n = 3) mice (mean ± SEM), 5 days after KO induction; t‐test was performed, ns = non‐significant.
Notably, similar to p53 upregulation in the general enterocyte population upon intestinal ablation of CKIδ/ε (Fig EV2A), we observed a robust increase in the expression of p53 target genes in DKO‐Lgr5‐GFP+ cells, such as Bax, cyclin G1, and p21 (Fig EV4B). However, given the fact that TKO mice showed decreased expression levels of ISC markers, similar to DKO mice (Fig EV4C), p53 activation cannot be the major culprit of ISC loss in DKO mice.
To further characterize the contribution of CKIδ/ε to ISC proliferation and crypt homeostasis, CKIδ/ε DKO crypts were studied in the “minigut” organoid culture system (Sato et al, 2009). Epithelial organoids were prepared from CKIε−/−‐VillinCreERT2‐Lgr5CreERT2 and from CKIδfl/fl CKIε−/−‐VillinCreERT2‐Lgr5CreERT2 mice and treated in culture with 4‐hydroxy‐tamoxifen (4OHT), producing CKIε−/− and DKO organoids, respectively. Similar to the effect in gut mucosa, DKO organoids displayed growth arrest and disintegrated within 2–3 days after KO induction (Fig 6A). Considering the observed correlation between suppressed Wnt activity in DKO‐Lgr5‐GFP+ ISCs (Fig 5C–E) and accelerated cell loss (Fig 4A), DKO organoids were treated with valproic acid (V), a histone deacetylase (HDAC) inhibitor, and CHIR99021 (CHIR or C), a glycogen synthase kinase 3β (GSK3β) inhibitor, which is a potent Wnt activator (Sato et al, 2004). This combination was previously shown to increase ISC self‐renewal in organoid culture through Wnt upregulation (Yin et al, 2013). Upon combined VC treatment, both CKIε−/− and DKO organoids acquired enhanced “crypt‐like” structure (“ISC organoids”) and exhibited a remarkable increase in ISC number, evident by extensive Lgr5‐driven GFP expression throughout the organoid (Fig 6A) and a significant increase in Wnt target gene expression (Appendix Fig S4). However, VC‐treated DKO organoids remained smaller than CKIε−/− ones and survived only 12 hours beyond non‐treated DKO organoids (Fig EV5A). In addition, despite the Wnt‐boosting effect of VC treatment, DKO organoids exhibited reduced levels of Dvl2 (Fig 6B), attesting to aberrant Wnt signaling (Bilic et al, 2007). Similar to VC‐treated organoids, decreased levels of Dvl were observed also in vivo in DKO mice; yet, more prominent was a decrease in the total nuclear Dvl levels in DKO crypt enterocytes (phosphorylated and non‐phosphorylated forms) and specifically in DKO ISCs, in which Dvl was almost completely abolished (Fig EV5B and C). Hence, whereas Dvl deficiency due to CKIδ/ε ablation is common to ICS organoids and to crypt enterocytes which are mostly composed of TA cells, Wnt signaling was only compromised in Dvl‐deficient ISCs. Notably, nuclear Dvl deficiency due to aberrant YAP expression was also found to reduce Wnt signaling in ISCs (Barry et al, 2013), underscoring the specific role of nuclear Dvl. To assess the contribution of diminished Dvl levels to the growth arrest phenotype in DKO organoids, we generated organoids expressing Dox‐inducible DVL carrying a nuclear localization signal (DVL‐NLS) (Fig 6B). Remarkably, upon Dvl expression, VC‐treated DKO organoids resumed proliferation, as evident by Ki67 staining (Fig 6C), concomitant with an increase in organoid survival and expression of Lgr5 and Wnt target genes (Fig 6D and E). Interestingly, Dvl expression in control CKIε−/− organoids and DKO organoids was associated with upregulation of Frizzled7 (Fzd7) (Figs 6C and EV5D), which was shown to function as the critical Wnt receptor in Lgr5+ ISCs (Flanagan et al, 2015). Notably, Fzd7 levels increased both in transcript and in protein levels, suggesting a role for nuclear Dvl in Fzd7 transcription. To further establish the role of Dvl and Fzd7 in proliferation arrest of DKO ISCs, organoids were co‐infected with DVL‐NLS and shFzd7 (Fig 6B). As reported previously, Fzd7 deletion is lethal to organoids without external Wnt augmentation (Flanagan et al, 2015). Control CKIε−/− Fzd7 deleted organoids treated with the Wnt activator CHIR99021, survived up to 4 days, yet failed to regenerate upon passage at a later time point (Fig 6D). Strikingly, the effect of Dvl expression in DKO organoids was abolished completely upon Fzd7 deletion, as DKO co‐expressing shFzd7 and Dvl exhibited reduced expression of Wnt target genes and did not survive even at the early time point (Figs 6D and EV5E). These data provide compelling evidence for an essential CKIδ/ε ‐nuclear Dvl–Fzd7 signaling axis critical in ISC Wnt signaling and self‐renewal.
Figure 6. Dvl and Fzd7 expression restores ISC self‐renewal and proliferation in DKO organoids.
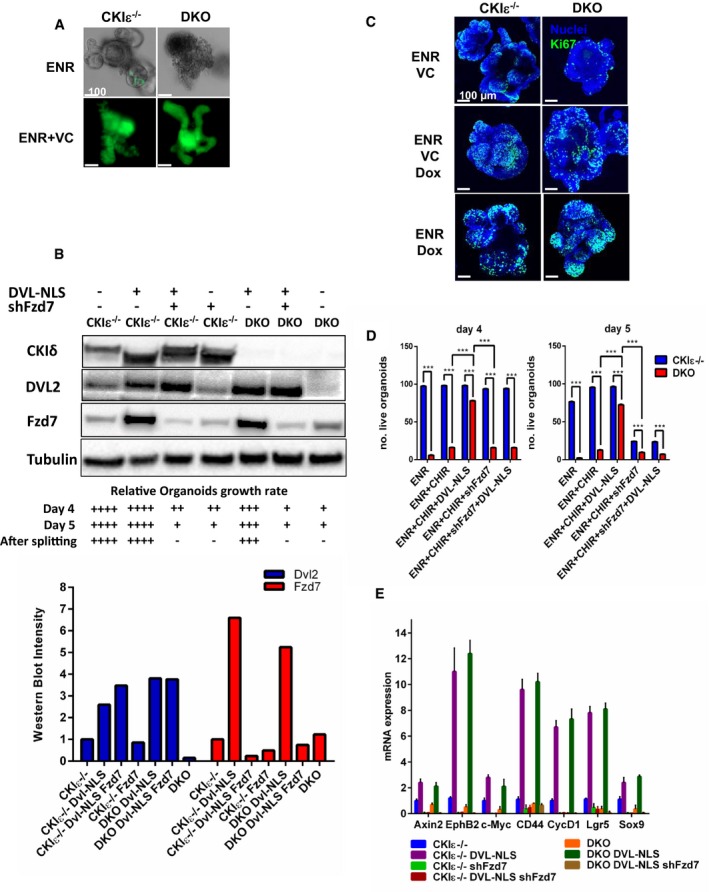
- IF of GFP, marking Lgr5‐GFP+ cells in CKIε−/−‐VillinCreER‐Lgr5CreER (CKIε−/−) and DKO‐VillinCreER‐Lgr5CreER (DKO) organoids treated with regular medium (ENR) or with valproic acid (2 mM) and CHIR99021 (3 μM) added to ENR medium (ENR+VC), 5 days after KO induction. Scale bar, 100 μm.
- WB analysis of CKIε−/− and DKO organoids infected with Dvl‐NLS and shFzd7 treated with ENR or ENR+Doxycycline 4 days after KO induction. The graph shows relative densitometry quantification of Western blot bands using ImageJ software. Relative growth rate of organoids following the different treatments after KO induction (day 0) is indicated at the bottom.
- IF analysis of Ki67 staining (green) in CKIε−/− and DKO organoids treated with ENR, ENR+VC, ENR+Doxycycline, and ENR+VC+Doxycycline medium, 5 days after KO induction; nuclear counterstain Hoechst. Scale bar, 100 μm.
- Survival assessment of CKIε−/− and DKO organoids treated with ENR, ENR+CHIR, ENR+CHIR+Dvl‐NLS, ENR+CHIR+shFzd7, and ENR+CHIR+Dvl‐NLS+shFzd7 was done by scoring 100 organoids for each treatment on day 4 or 5 after tamoxifen treatment and KO induction. Data represent mean of five independent experiments; t‐test was performed, ***P < 0.001.
- qRT–PCR analysis of Lgr5 and Wnt target genes in CKIε−/− and DKO organoids treated ENR, ENR+CHIR, ENR+CHIR+Dvl‐NLS, ENR+CHIR+shFzd7 and ENR+CHIR+Dvl‐NlLS+shFzd7 (n = 3, mean ± SEM), 4 days after KO induction.
Figure EV5. Decreased levels of Dvl2 in crypt DKO IECs.
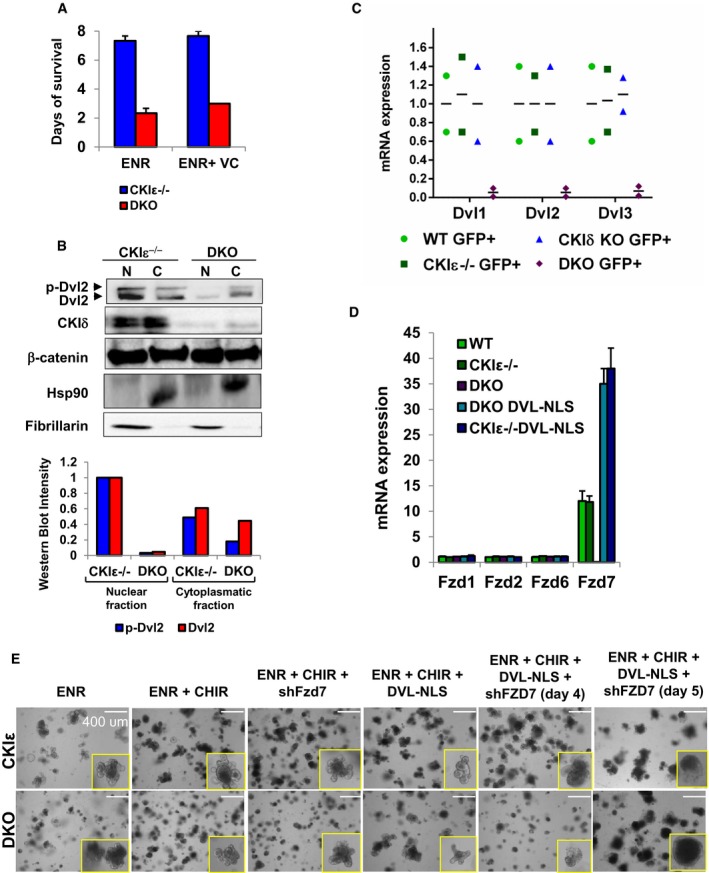
- Survival analysis was done by scoring 100 organoids of CKIε−/− and DKO organoids treated with ENR or ENR+VC. Data represent mean ± SEM of three independent experiments.
- Western blot analysis of nuclear (N) and cytoplasmatic (C) fractions of CKIε−/− and DKO mouse crypt IECs; p‐Dvl2 identified by electrophoretic shift in Dvl mobility. WB quantification shown at the bottom.
- qRT–PCR analysis of Dvl isoforms in sorted GFP+ cells isolated from crypts of WT, CKIε−/−, CKIδ KO, and DKO mice 2.5 days after KO induction. Data represent mean of three independent sorting experiments, each with a pool of six mice.
- qRT–PCR analysis of Fzd isoforms in WT, CKIε−/−, DKO, CKIε−/− Dvl‐NLS, and DKO Dvl‐NLS expressing organoids 4 days after tamoxifen treatment and KO induction (n = 3, mean ± SEM).
- Representative bright field images of CKIε−/− and DKO organoids expressing Dvl‐NLS and shFzd7 at day 4 or 5 after KO and Dvl induction. Insets represent organoids counted as live. Scale bar, 400 μm.
Source data are available online for this figure.
Altogether, our data suggest that ablation of CKIδ/ε in organoids triggers a growth arrest phenotype via nuclear Dvl downregulation, resulting in reduced Fzd7 expression, inhibition of Wnt signaling and ISC loss.
Discussion
The intestinal epithelium sustains an immense regenerative capacity supported by a specialized ISC population dedicated to intestinal self‐renewal (Barker et al, 2007). Multiple lines of evidence have indicated that the proliferative activity of ISCs entails high Wnt activity (Korinek et al, 1998; van Es et al, 2012), secured by several ISC cell‐autonomous Wnt regulatory mechanisms (Carmon et al, 2011; de Lau et al, 2011; Barry et al, 2013; Fafilek et al, 2013; Gregorieff et al, 2015; Schuijers et al, 2015) and an extrinsic Wnt signal from the ISC niche (Sato et al, 2011; Farin et al, 2016), together eliciting a continuous strong Wnt response.
The CKI family has been regarded as a key component in Wnt signaling regulation (Peters et al, 1999; McKay et al, 2001; Cruciat, 2014). Nevertheless, different CKI isoforms have been associated with contradictory affects regarding Wnt activity, both in Wnt potentiation and in suppression (Price, 2006). Notably, of all CKI isoforms, CKIα was shown to function as a clear negative regulator of the Wnt pathway (Amit et al, 2002; Liu et al, 2002; Elyada et al, 2011). This isoform was found to phosphorylate β‐catenin at serine 45, thereby priming GSK3β‐mediated phosphorylation of β‐catenin, linking β‐catenin for targeted proteasomal degradation. However, contrasting evidence, including developmental studies in Xenopus and in vitro experiments using casein kinase I overexpression constructs, has placed casein kinase I‐δ and casein kinase I‐ε (CKIδ/ε) both as activators and as inhibitors of the Wnt signaling pathway (Peters et al, 1999; Sakanaka et al, 1999). Besides, the distinct role of each isoform in vivo and the relationship between the highly similar isoforms δ and ε have not been studied.
In the course of elucidating the role of CKIδ/ε in the gut physiology, we found a striking redundancy with mutual compensatory expression regulation between the two protein isoforms. Whereas intestinal homeostasis was preserved upon ablation of each of the isoforms alone, CKIδ/ε depletion resulted in reduced crypt proliferation till a complete halt. Proliferation in intestinal epithelium is carried out by two distinct cell populations: short‐term TA population and long‐term intestinal stem cell population, the latter residing at the bottom of the crypt, dedicated to intestinal self‐renewal. Both populations exhibit a strong Wnt response, with a gradient along the villus–crypt axis with the highest Wnt activity expressed at the bottom of the crypt. Intriguingly, CKIδ/ε depletion affected Wnt signaling in the TA and ISC populations differentially; whereas Wnt target genes were normally expressed in the TA cells, DKO Lgr5 ISCs had reduced expression levels of both ISC‐specific Wnt targets, such as Lgr5 and Ascl2, and “classical” target genes, such as Axin2. Suppression of Wnt signaling in DKO ISCs was associated with hindered proliferation and increased apoptosis of these cells, subsequently resulting in extinction of DKO ISCs. Further evidence for a critical role of CKIδ/ε in Wnt regulation in ISCs came through observations in DKO intestinal organoids; CKIδ/ε‐depleted organoids exhibited a growth arrest phenotype accompanied by decreased levels of Dvl, Fzd7, and Wnt activities and loss of ISCs, which could be regained upon Wnt signaling enhancement and specifically by nuclear Dvl expression. This rescue effect was accompanied with improved intestinal stem cell self‐renewal, similar to the partial rescue of Lgr4/Lgr5 KO organoids by extrinsic Wnt stimulation (de Lau et al, 2011). Distinctly from the common phenotype reported in mouse models of Wnt inhibition, which manifested mainly as crypt and Paneth cell disappearance (Korinek et al, 1998; van Es et al, 2012), histological analysis of DKO intestines revealed remnant crypts encompassing Paneth cells, even at late stages post‐KO induction, when proliferation was completely abrogated. These findings further substantiate the fact that depletion of CKIδ/ε elicits a Wnt inhibitory mechanism, selectively manifested in ISCs.
CHIR99021, a GSK3β inhibitor, induced upregulation of Wnt activity in DKO organoids as expected, yet was not sufficient to rescue ISC loss and proliferation arrest, suggesting that additional regulatory factors are essential for proper Wnt activity in ISC. Dvl phosphorylation has been highlighted as a central mechanism for Wnt signaling regulation by CKIδ/ε (Peters et al, 1999; Cong et al, 2004; Klimowski et al, 2006), and cytoplasmic Dvl activity was shown to be mandatory for Lrp6 phosphorylation and signalosome formation (Bilic et al, 2007). Recently, however, several studies proposed additional, novel roles for nuclear Dvl, in the regulation of Tcf transcriptional activity (Itoh et al, 2005; Gan et al, 2008; Barry et al, 2013). Our study reinforces the role of nuclear Dvl, yet also positions CKIε as an upstream crucial regulator of nuclear Dvl. This role, which in the absence of CKIε can be replaced by CKIδ, appears to be exclusively essential for ISCs, whereas in TA enterocytes, Dvl regulation may be fulfilled by other regulators. One possible mechanism for the exclusive nuclear role of Dvl, downstream of CKIε in ISCs could be the control of Fzd7 transcription and thereby reinforcing Wnt signaling, specifically in ISCs. Whether CKIδ/ε‐mediated phosphorylation regulates the nuclear translocation of Dvl, or is also necessary to activate nuclear Dvl by phosphorylation, requires further investigation.
In summary, our data provide evidence for a critical role of CKIε specifically in ISCs, aimed at boosting Wnt signaling for securing ISC self‐renewal. This function may be compensated by CKIδ when necessary, as in the CKIε−/− mouse. This may explain why is CKIε expression particularly high in ISCs, contributing to building up optimal levels of Wnt signaling activation required for ISC self‐renewal and maintenance of intestinal homeostasis. Other Wnt supporting factors that are distinctly active in ISC are Lgr4/5, Slit, Znrf3, Ascl2, YAP, and Troy (de Lau et al, 2011; Carmon et al, 2012; Hao et al, 2012; Koo et al, 2012; Barry et al, 2013; Fafilek et al, 2013; Zhou et al, 2013; Gregorieff et al, 2015; Schuijers et al, 2015), possibly synergizing in Wnt activation. CKIε may function independently for the same purpose by activating nuclear Dvl2 via Fzd7 upregulation or may cooperate with one or several other Wnt regulators in ISCs. It would be interesting to look for a similar self‐renewal supportive role of CKIε in other types of stem cells, including embryonal stem cells and iPSs. Further elucidation of CKIε‐associated regulatory mechanisms may deepen our understanding of ISC physiology and self‐renewal in particular, possibly toward developing novel applications for regenerative medicine.
Materials and Methods
Mouse breeding and genotyping
A CKIδ and CKIε expression vector was constructed on the basis of pGEM‐11Zf(+), to which an XbaI‐ and SalI‐digested fragment of a neomycin cassette flanked by two loxP sites was inserted from a pL2‐neo expression vector. Exons 3 of the mouse Csnk1d gene and exons 3 and 4 of the Csnk1e gene were cloned into the vector, flanked by loxP sites using a third loxP site. Short (1‐kilobase) and long (5‐kilobase) homology sequences were cloned upstream and downstream of the targeted exons, respectively. All genomic fragments were amplified by PCR from 129/SvJ‐mouse DNA. The vector was linearized with SalI and purified using phenol–chloroform extraction and ethanol precipitation methods. R1 embryonic stem (ES) cells (129/SvJ‐mouse derived) were electroporated and cultured on a feeder layer of MEFs using DMEM supplemented with 15% ES‐cell‐tested FBS and 1,000 U/ml ESGRO (Chemicon). Neomycin selection was done in 0.2 mg/ml G418 (Sigma). pCA–NLS–Cre was used as a Cre expression vector for transient transfection of Cre into ES cells. Selection was done in 2 μg/ml puromycin.
R1 ES cells were aggregated to CD‐1 mouse morulae. Chimaeric mice were bred with CD‐1 mice to check for germline transmission. Pgk1–Cre transgenic mice (Lallemand et al, 1998) were used as a deleter strain for the generation of germline Csnk1e deletion. For generation of conditional Csnk1d knockout mice, Cre was transiently expressed in ES cells to produce specific deletion of the neomycin cassette and an intact floxed Csnk1d allele. A second aggregation was done, and Vil1–Cre–ERT2 (el Marjou et al, 2004) transgenic mice were used as a gut‐specific deleter to generate a conditional Csnk1d knockout mouse. CKIε−/− mice were crossed with CKIδfl/fl villi‐Cre‐ERT2 mice to generate double KO mice (DKO). Floxed p53 mice (Jonkers et al, 2001) were crossed with DKO mice to generate triple knockout (TKO). Lgr5‐EGFP‐IRES‐CreERT2 (Barker et al, 2007) were crossed with CKIδfl/fl CKIε−/− mice and with CKIδfl/fl CKIε−/−‐Villin‐CreERT2 mice to generate DKO/Lgr5‐EGFP‐IRES‐CreERT2 (DKO/Lgr5‐CreER) and DKO‐Villin‐CreERT2‐Lgr5‐EGFP‐IRES‐CreERT2 (DKO‐VillinCreERT2‐Lgr5) mice, respectively.
Mice were kept under specific pathogen‐free conditions at the animal house in Hadassah Medical School of the Hebrew University and were maintained in a 12‐h light/12‐h dark lighting cycle with lights on at 07:00. All mouse experiments were performed in accordance with guidelines of the relevant institution's ethics committee.
For mouse genotyping, DNA from the tail or ear of 4‐week‐old pups was extracted by means of standard protocols. For embryo genotyping, a small section of the embryo was taken for DNA preparation. The following primers were used for genotyping: 5′‐TTGCTACCAGTCAAATGC (forward primer for wild‐type and floxed‐CKIδ); 5′‐GCTAGAGTAAATTACTGGGTT (reverse primer for wild‐type and floxed‐CKIδ ); 5′‐GGGGCTGAGGGAGACTATAACGTG (forward primer for WT‐CKIε); 5′‐TCCACCAGACCTTCCCCACTATTA (reverse primer for WT‐CKIε); 5′‐GGGCGAATTCTGCAGATATC (forward primer for null‐CKIε); 5′‐CATCCTCGAACCCCCACA (reverse primer for null‐CKIε); 5′‐CACAAAAAACAGGTTAAACCCAG (forward primer for wild‐type and floxed p53); 5′‐AGCACATAGGAGGCAGAGAC (reverse primer for wild‐type and floxed p53); 5′‐ATGTCCAATTTACTGACCGTACACC (forward primer for cre); and 5′‐CGCCTGAAGATATAGAAGATAATCG (reverse primer for cre); 5′‐GTGTGGGACAGAGAACAAACC (forward primer for Villin‐CreER T2); 5′‐ACATCTTCAGGTTCTGCGGG (reverse primer for Villin‐CreER T2); 5′‐CTGCTCTCTGCTCCCAGTCT (forward primer for WT and mutant‐Lgr5); 5′‐ATACCCCATCCCTTTTGAGC (reverse primer for WT‐Lgr5); 5′‐GAACTTCAGGGTCAGCTTGC (reverse primer for mutant‐Lgr5).
Tamoxifen administration and BrdU labeling
Tamoxifen (Sigma) was dissolved in corn oil (Sigma), and mice were injected intraperitoneally (120 mg/kg), three injections on two consecutive days. At 3–5 days after the last injection, each mouse was injected intraperitoneally with 10 μl/g of 5‐bromo‐2‐deoxyuridine (BrdU; GE Healthcare) and sacrificed 2 h later.
Tissue preparation
Jejunum, ileum, and the entire large intestine were flushed with ice‐cold PBS, cut open longitudinally, and subjected to fixation in 4% formaldehyde and paraffin embedding (FFPE). Intestinal epithelial cells (IECs) were isolated from the middle part of the small intestine, as described previously (Greten et al, 2004) with slight modifications: Intestinal cells were separated into single cells in HBSS containing 10 mM HEPES, 5 mM EDTA, and 0.5 mM DTT, at 37°C for 30 min. Crypt–villi fractionations were performed by 3 serial 10‐min incubations in HBSS containing 10 mM HEPES, 5 mM EDTA, and 0.5 mM DTT, at 37°C with fractions 1 and 2 representing villus enterocytes and fraction 3 representing crypt cells.
Histology, immunohistochemistry, and immunofluorescence
5‐μm sections were cut for hematoxylin/eosin (H&E) staining and immunohistochemistry (IHC). For IHC and IF, sections were dewaxed, by placing into xylene for 15 min, rehydrated through downgraded organic solutions, and put into distilled water. Antigen retrieval (AR) was performed as per the pressure cooker method, in appropriate buffer.
Slides were washed 3 × 5 min in PBS, then incubated for 30 min with 3% BSA (blocking), and briefly washed and incubated with primary antibodies. Primary antibodies were CKIδ (AR Citrate; 128A ICOS kindly provided by Eli Lilly and Co; 1:8,000), Ki67 (AR EDTA; SP6 Neomarkers; 1:250), BrdU (AR Citrate; Ab3; 1:150, Neomarkers), cyclin D1 (AR EDTA; SP4 Lab Vision; 1:125), p53 (AR Citrate; CM5 Novocastra; 1:200), p21 (AR Citrate; F‐5 Santa Cruz; 1:50), c‐Myc (AR Citrate; N‐262 Santa Cruz; 1:200), cleaved Caspase‐3 (AR Citrate; Cell Signaling; 1:100), GFP (AR Citrate; ab6673 Abcam; 1:1,000), EphB2 (AR Citrate; AF‐467 R&D Systems; 1:200), Hes1 (AR Citrate; 11988 Cell Signaling; 1:500), lysozyme (AR Citrate; A0099 DAKO; 1:5,000). Secondary antibodies were HRP‐polymer anti‐mouse, anti‐rabbit, anti‐goat, and anti‐rat (Nichirei, Dako, and Biocare), and detection was done using DAB chromogen (LabVision).
For double immunofluorescence of GFP and BrdU or GFP and cleaved Caspase‐3, sections were incubated with primary antibodies, followed by Alexa‐488 donkey anti‐goat (1:1,000; Molecular Probes) and Alexa‐647 donkey anti‐mouse (1:1,000; Molecular Probes) or Alexa‐647 donkey anti‐rabbit (1:1,000; Molecular Probes). Hoechst (1 μg/ml; Molecular Probes) was used as nuclear counterstain. % BrdU+/GFP+ and % cleaved Caspase 3+/GFP+ quantification was based on 300 GFP+ cells in each mice group.
Protein extraction and western blotting
Protein was extracted (whole‐cell extract protocol) from cell pellets using protein lysis buffer containing 50 mM Tris pH 7.6, 150 mM NaCl, 5 mM EDTA, and 0.5% Nonidet P‐40 (NP‐40), with protease and phosphatase inhibitors. Western blot (WB) analysis was performed using standard techniques. Briefly, membranes were first checked by Ponceau staining, to confirm transfer efficiency. Then, according to the expected size (MW) of the proteins of interest, the membrane was cut into few strips. For example, in Fig EV5B, we were interested primarily for DVL2 (90–95 kDa), β‐catenin (90 kDa), HSP90 (95 kDa), Fibrillarin (39 kDa), and CKIδ (45 kDa). We therefore marked the membrane at approximately 62 kDa and 27 kDa and cut it into three portions. The strip above 62 kDa was then used for incubating with DVL2 antibody first and then restriped twice and incubate with β‐catenin and HSP90 antibodies, respectively. The middle strip (below 62 kDa and above 27 kDa) was incubated with Fibrillarin antibody, restriped, once and then incubated with CKIδ.
Blots were incubated with primary antibodies: CKIε (BD), CKIδ (ICOS A128), p‐Lrp6 (Cell Signaling), Dvl2 (Cell Signaling), β‐catenin (BD), Fzd7 (Abcam), Tubulin (Sigma), PP2AC, and HSP90 (Calbiocem). HRP‐linked goat anti‐mouse, goat anti‐rabbit, and rabbit anti‐goat were used as secondary antibodies. Blots were developed using ECL (GE Healthcare).
RNA analysis
Total RNA was extracted from cell pellets using TRI reagent (Sigma) and phenol–chloroform methods. 2 μg RNA was subjected to reverse transcription using M‐MLV‐RT (Invitrogen), and mRNA representative expression levels were measured by qRT–PCR using SYBR‐Green (Invitrogen) in QuantStudio™ 12K Flex Real‐Time PCR System (ABI). qRT–PCR primers were designed to detect an exon–exon boundary. RT–PCR primers are listed in Table 1. Relative quantities of gene transcripts were analyzed in qBase 1.3.5 software and normalized to UBC, HPRT, and GAPDH transcripts.
Table 1.
Primers for quantitative real‐time PCR
| Detector | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Ascl2 | CTACTCGTCGGAGGAAAG | ACTAGACAGCATGGGTAAG |
| Axin2 | TAGGCGGAATGAAGATGGAC | CTGGTCACCCAACAAGGAGT |
| Bax | ATGCGTCCACCAAGAAGCTGA | AGCAATCATCCTCTGCAGCTCC |
| CD44 | CAGTATCTCCCGGACTGAGG | GCCAACTTCATTTGGTCCAT |
| CKIδ | AACCAAACATCCTCAGCTCCAC | GTCCCAGTAGCTCCATCACCAT |
| CKIε | AGCCTGGAGGACCTCTTCAACT | CGGTGGATGAAGTTCTTGGAGT |
| Clca4 | TCACTGCTCAAATCCAGACAG | GCTGCTTGGTTCATTAGGGTC |
| c‐Myc | TGAGCCCCTAGTGCTGCAT | AGCCCGACTCCGACCTCTT |
| cyclin D1 | TTGACTGCCGAGAAGTTGTG | CCACTTGAGCTTGTTCACCA |
| cyclin D2 | CACCGACAACTCTGTGAAGC | TGCTCAATGAAGTCGTGAGG |
| cyclin G1 | GCTGGCGCTATCTATCCTTG | GGTCAAATCTCGGCCACTTA |
| EphB2 | CCAACGGTGTGATCCTGGAC | CCCGCACCTGGAAGACATAG |
| EphB3 | CCTGCACCTGCGCTACTG | CCCTTCTCCTTGCTTTGCTT |
| GAPDH | ACAACTTTGGCATTGTGGAA | GATGCAGGGATGATGTTCTG |
| GFP | AAGTTCATCTGCACCACCG | TCCTTGAAGAAGATGGTGCG |
| Hes1 | AAACCAAAGACGGCCTCT | GCTTAGACTTTCATTTATTCTTGCC |
| HPRT | GTTAAGCAGTACAGCCCCAAA | AGGGCATATCCAACAACAAACTT |
| Lgr5 | GACAATGCTCTCACAGAC | GGAGTGGATTCTATTATTATGG |
| Musashi1 | ATGCCATGCTGATGTTCGAC | TGGATCTCACAAACTTTCTCTACGA |
| Olfm4 | CACAGCTCACATCCTTTCTCAG | ACTCGGACCGTCAGGTTCAG |
| p21 | TCCACAGCGATATCCAGACA | AGACAACGGCACACTTTGCT |
| p27 | GTTAGCGGAGCAGTGTCCAG | TTCTGTTCTGTTGGCCCTTT |
| p57 | TGAAGGACCAGCCTCTCTCG | TTCGACGCCTTGTTCTCCTG |
| Smoc2 | CGGATACTGCTGGTGTGTGCT | CGGGCTGTGTTGTCACACTTA |
| UBC | CAGCCGTATATCTTCCCAGACT | CTCAGAGGGATGCCAGTAATCTA |
Multiple‐color single‐molecule fluorescence in situ hybridization (smFISH)
Multiple‐color smFISH was done as described previously (Itzkovitz et al, 2012). Briefly, mouse ileum samples were placed into pre‐chilled cryoprotecting solution (4% formaldehyde–paraformaldehyde—30% sucrose–PBS) and incubated overnight at 4°C with gentle agitation. After the overnight incubation, the tissue was placed into the molds filled with cold OCT and stored at −80°C. Hybridizations were done overnight with two differentially labeled probes using Axin2‐Alexa594 Hes1‐Alexa594 and Lgr5‐tetramethylrhodamine (TMR) fluorophores. An additional fluorescein isothiocyanate‐conjugated antibody for Phalloidin was added to the hybridization mix and used for protein immunofluorescence to detect cell borders. DAPI (4′,6‐diamidino‐2‐phenylindole) dye for nuclear staining was added during the washes. Images were taken with a Nikon TE2000 inverted fluorescence microscope equipped with a 100× oil‐immersion objective and a Princeton Instruments camera using MetaMorph software (Molecular Devices, Downingtown, PA). All images are filtered with a Laplacian or Gaussian filter and are maximal projections of 25 stacks spaced 0.25 μm apart in the z direction. Transcription rate quantification of gene of interest was measured using TransQuant (Bahar Halpern & Itzkovitz, 2016).
Intestinal crypt culture
Intestinal crypts from CKIδfl/fl ε−/−‐Villin‐CreERT2 mice were isolated on the basis of the previously published method (Sato et al, 2009) with several modifications: Small intestines were flushed with cold PBS, cut into 5‐cm pieces and opened longitudinally, washed with 70% ethanol, and agitated in PBS containing 2 mM EDTA for 30 min at 4°C. Crypts were collected as described (Sato et al, 2009) and embedded onto Matrigel (BD Biosciences) at 50 μl/well in 24‐well plates. Culture medium was DMEM/F12 medium (Gibco), containing Glutamax (Gibco) and penicillin/streptomycin. Medium was supplemented with B27 (Gibco, 1:50), 100 ng/ml murine Noggin (Peprotech), 20 ng/ml murine EGF (Peprotech), 10 ng/ml human basic‐FGF (Peprotech), and 500 ng/ml human R‐Spondin1 (Peprotech). The crypts were split 1:3–1:4 and embedded onto new Matrigel every 5–7 days. Induction of knockout in the crypt cultures was carried out by incubation with 300 nM 4‐hydroxy‐tamoxifen (4OHT; Sigma) for 48 h. Organoids were treated with valproic acid (2 mM) and CHIR99021 (3 μm) diluted in the organoid regular media. For whole‐mount IF staining, organoids were cultured on round cover slips in a 24‐well plate. Incubation with 10 μM bromodeoxyuridine (BrdU) was performed for 16 h in the culture medium. Fixation was performed with 4% PFA for 30 min. For BrdU staining, fixed organoids were treated with 2M HCl for 25 min and then neutralized with 0.1 M sodium borate (pH = 8.5) for 5 min. Then, organoids were permeabilized in blocking solution (1% BSA, 5% normal goat serum, 0.3% Triton X‐100 in PBS) and incubated with anti‐BrdU antibody (Ab3; 1:200; Neomarkers) for overnight at 4°. Secondary antibody was applied for overnight at 4°C. Hoechst was used as nuclear counterstain. Stained organoids were mounted in mounting medium and imaged with the Olympus confocal microscope.
Lentiviral infection of organoids: The DVL‐NLS plasmid was a kind gift from Dr. Ferdinand Camargo, Harvard Stem Cell Institute. DVL‐NLS and shFzd7 (Sigma) were transduced with lentiviral particles according to previously published protocols (Van Lidth de Jeude et al, 2015). Infected organoids were treated with 3 μg of Doxycycline, 4 days prior to tamoxifen treatment for DVL‐NLS induction.
Lgr5‐GFP+ cells sorting
Lgr5‐GFP+ cells were sorted as described previously (Muñoz et al, 2012) with slight modifications: For each experiment, cells sorted from six mice were pooled together. Freshly isolated mouse small intestines were incised along their length and villi were removed by scraping. The tissue was then incubated in PBS/EDTA (5 mM) for 5 min. Gentle shaking removed remaining villi, and intestinal tissue was subsequently incubated in PBS/EDTA for 30 min at 4°C. Vigorous shaking yielded a supernatant containing isolated crypts. Remaining intestinal tissue was incubated in PBS/EDTA for 30 min at 4°C once more, followed by vigorous shaking and supernatant collection. Combined collected crypt supernatant was centrifuged 5 min at 400 × g, and supernatant was dismissed. Crypt pellet was incubated in TrypLE Express (Invitrogen) with DNase I (0.5 μg/μl) for 5 min at 37°C followed by pippetation with SMEM and filtration through a 40‐μm mesh. Subsequently, cells were spun down, resuspended in SMEM (Invitrogen), and filtered again through a 40‐μm mesh. GFPhigh and GFPlow expressing cells were isolated using ARIA‐II sorter (BD). Single viable epithelial cells were gated by forward scatter, side scatter, and by negative staining for propidium iodide. Sorted cells were spun down, and cell pellets were used for RNA extraction using miRNA mini kit (QIAGEN). Experiments were repeated in biological triplicates.
Author contributions
YM and UDA designed and performed experiments, analyzed data, and wrote the manuscript; EE generated the CKIδ/ε KO mice and conceived the project; MA performed part of the experimental work; BT, AM, and SI performed the multiple‐color smFISH analysis; YB‐N supervised the research and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Acknowledgements
We thank Dr. Ferdinand Camargo for providing the DVL‐NLS expression plasmid. This work was supported by grants from Israel Science Foundation (ISF)‐Centers of Excellence, the European Research Council within the FP‐7 (294390 PICHO), the Dr. Miriam and Sheldon G, Adelson Medical Research Foundation (AMRF), and the Israel Cancer Research Fund.
The EMBO Journal (2017) 36: 3046–3061
References
- Amit S, Hatzubai A, Birman Y, Andersen JS, Ben‐Shushan E, Mann M, Ben‐Neriah Y, Alkalay I (2002) Axin‐mediated CKI phosphorylation of beta‐catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 16: 1066–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu P, Colnot S, Godard C, Gad S, Chafey P, Niwa‐Kawakita M, Laurent‐Puig P, Kahn A, Robine S, Perret C, Romagnolo B (2005) Crypt‐restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development 132: 1443–1451 [DOI] [PubMed] [Google Scholar]
- Bahar Halpern K, Itzkovitz S (2016) Single molecule approaches for quantifying transcription and degradation rates in intact mammalian tissues. Methods 98: 134–142 [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007 [DOI] [PubMed] [Google Scholar]
- Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, Kuo CJ, Camargo FD (2013) Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493: 106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatík O, Šedová K, Schille C, Ganji RS, Červenka I, Trantírek L, Schambony A, Zdráhal Z, Bryja V (2014) Functional analysis of dishevelled‐3 phosphorylation identifies distinct mechanisms driven by casein kinase 1 and frizzled5. J Biol Chem 289: 23520–23533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beumer J, Clevers H (2016) Regulation and plasticity of intestinal stem cells during homeostasis and regeneration. Development 143: 3639–3649 [DOI] [PubMed] [Google Scholar]
- Bilic J, Huang Y‐L, Davidson G, Zimmermann T, Cruciat C‐M, Bienz M, Niehrs C (2007) Wnt induces LRP6 signalosomes and promotes dishevelled‐dependent LRP6 phosphorylation. Science 316: 1619–1622 [DOI] [PubMed] [Google Scholar]
- Bryja V, Schulte G, Arenas E (2007) Wnt‐3a utilizes a novel low dose and rapid pathway that does not require casein kinase 1‐mediated phosphorylation of Dvl to activate beta‐catenin. Cell Signal 19: 610–616 [DOI] [PubMed] [Google Scholar]
- Carmon KS, Gong X, Lin Q, Thomas A, Liu Q (2011) R‐spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta‐catenin signaling. Proc Natl Acad Sci USA 108: 11452–11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmon KS, Lin Q, Gong X, Thomas A, Liu Q (2012) LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/β‐catenin signaling. Mol Cell Biol 32: 2054–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casagolda D, Del Valle‐Pérez B, Valls G, Lugilde E, Vinyoles M, Casado‐Vela J, Solanas G, Batlle E, Reynolds AB, Casal JI, de Herreros AG, Duñach M (2010) A p120‐catenin‐CK1epsilon complex regulates Wnt signaling. J Cell Sci 123: 2621–2631 [DOI] [PubMed] [Google Scholar]
- Cheong JK, Virshup DM (2011) Casein kinase 1: complexity in the family. Int J Biochem Cell Biol 43: 465–469 [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R (2012) Wnt/β‐catenin signaling and disease. Cell 149: 1192–1205 [DOI] [PubMed] [Google Scholar]
- Cong F, Schweizer L, Varmus H (2004) Casein kinase I modulates the signaling specificities of dishevelled. Mol Cell Biol 24: 2000–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciat C‐M, Dolde C, de Groot REA, Ohkawara B, Reinhard C, Korswagen HC, Niehrs C (2013) RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt‐β‐catenin signaling. Science 339: 1436–1441 [DOI] [PubMed] [Google Scholar]
- Cruciat C‐M (2014) Casein kinase 1 and Wnt/β‐catenin signaling. Curr Opin Cell Biol 31: 46–55 [DOI] [PubMed] [Google Scholar]
- Del Valle‐Pérez B, Arqués O, Vinyoles M, de Herreros AG, Duñach M (2011a) Coordinated action of CK1 isoforms in canonical Wnt signaling. Mol Cell Biol 31: 2877–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle‐Pérez B, Casagolda D, Lugilde E, Valls G, Codina M, Dave N, de Herreros AG, Duñach M (2011b) Wnt controls the transcriptional activity of Kaiso through CK1ε‐dependent phosphorylation of p120‐catenin. J Cell Sci 124: 2298–2309 [DOI] [PubMed] [Google Scholar]
- Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, Snir‐Alkalay I, Burstain I, Haffner‐Krausz R, Jung S, Wiener Z, Alitalo K, Oren M, Pikarsky E, Ben‐Neriah Y (2011) CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 470: 409–413 [DOI] [PubMed] [Google Scholar]
- van Es JH, Haegebarth A, Kujala P, Itzkovitz S, Koo B‐K, Boj SF, Korving J, van den Born M, van Oudenaarden A, Robine S, Clevers H (2012) A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self‐renewal. Mol Cell Biol 32: 1918–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray J‐P, Machida KK, Noton E, Constance CM, Dallmann R, Di Napoli MN, DeBruyne JP, Lambert CM, Yu EA, Reppert SM, Weaver DR (2009) Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol Cell Biol 29: 3853–3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray J‐P, Yu EA, Indic P, Dallmann R, Weaver DR (2010) Casein kinase 1 delta (CK1delta) regulates period length of the mouse suprachiasmatic circadian clock in vitro . PLoS ONE 5: e10303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fafilek B, Krausova M, Vojtechova M, Pospichalova V, Tumova L, Sloncova E, Huranova M, Stancikova J, Hlavata A, Svec J, Sedlacek R, Luksan O, Oliverius M, Voska L, Jirsa M, Paces J, Kolar M, Krivjanska M, Klimesova K, Tlaskalova‐Hogenova H et al (2013) Troy, a tumor necrosis factor receptor family member, interacts with lgr5 to inhibit wnt signaling in intestinal stem cells. Gastroenterology 144: 381–391 [DOI] [PubMed] [Google Scholar]
- Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DVF, de Punder K, Angers S, Peters PJ, Maurice MM, Clevers H (2016) Visualization of a short‐range Wnt gradient in the intestinal stem‐cell niche. Nature 530: 340–343 [DOI] [PubMed] [Google Scholar]
- Ferrarese A, Marin O, Bustos VH, Venerando A, Antonelli M, Allende JE, Pinna LA (2007) Chemical dissection of the APC Repeat 3 multistep phosphorylation by the concerted action of protein kinases CK1 and GSK3. Biochemistry 46: 11902–11910 [DOI] [PubMed] [Google Scholar]
- Flanagan DJ, Phesse TJ, Barker N, Schwab RHM, Amin N, Malaterre J, Stange DE, Nowell CJ, Currie SA, Saw JTS, Beuchert E, Ramsay RG, Sansom OJ, Ernst M, Clevers H, Vincan E, Ashton GH, Morton JP, Myant K, Phesse TJ et al (2015) Frizzled7 functions as a Wnt receptor in intestinal epithelial Lgr5(+) stem cells. Stem Cell Reports 4: 759–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, Clevers H (2009) Stem cells, self‐renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 71: 241–260 [DOI] [PubMed] [Google Scholar]
- Foldynová‐Trantírková S, Sekyrová P, Tmejová K, Brumovská E, Bernatík O, Blankenfeldt W, Krejcí P, Kozubík A, Dolezal T, Trantírek L, Bryja V (2010) Breast cancer‐specific mutations in CK1epsilon inhibit Wnt/beta‐catenin and activate the Wnt/Rac1/JNK and NFAT pathways to decrease cell adhesion and promote cell migration. Breast Cancer Res 12: R30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuja TJ, Lin F, Osann KE, Bryant PJ (2004) Somatic mutations and altered expression of the candidate tumor suppressors CSNK1 epsilon, DLG1, and EDD/hHYD in mammary ductal carcinoma. Cancer Res 64: 942–951 [DOI] [PubMed] [Google Scholar]
- Gan X, Wang J, Xi Y, Wu Z, Li Y, Li L (2008) Nuclear Dvl, c‐Jun, beta‐catenin, and TCF form a complex leading to stabilization of beta‐catenin‐TCF interaction. J Cell Biol 180: 1087–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka A, Dolde C, Kirsch N, Huang Y‐L, Kazanskaya O, Ingelfinger D, Boutros M, Cruciat C‐M, Niehrs C (2011) LGR4 and LGR5 are R‐spondin receptors mediating Wnt/β‐catenin and Wnt/PCP signalling. EMBO Rep 12: 1055–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL (2015) Yap‐dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature 526: 715–718 [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li Z‐W, Egan LJ, Kagnoff MF, Karin M (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis‐associated cancer. Cell 118: 285–296 [DOI] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M (1991) Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66: 589–600 [DOI] [PubMed] [Google Scholar]
- Hämmerlein A, Weiske J, Huber O (2005) A second protein kinase CK1‐mediated step negatively regulates Wnt signalling by disrupting the lymphocyte enhancer factor‐1/beta‐catenin complex. Cell Mol Life Sci 62: 606–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H‐X, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H, Mao X, Ma Q, Zamponi R, Bouwmeester T, Finan PM, Kirschner MW, Porter JA, Serluca FC, Cong F (2012) ZNRF3 promotes Wnt receptor turnover in an R‐spondin‐sensitive manner. Nature 485: 195–200 [DOI] [PubMed] [Google Scholar]
- Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ (2004) Inducible Cre‐mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta‐catenin. Gastroenterology 126: 1236–1246 [DOI] [PubMed] [Google Scholar]
- Itoh K, Brott BK, Bae G‐U, Ratcliffe MJ, Sokol SY (2005) Nuclear localization is required for Dishevelled function in Wnt/beta‐catenin signaling. J Biol 4: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzkovitz S, Lyubimova A, Blat IC, Maynard M, van Es J, Lees J, Jacks T, Clevers H, van Oudenaarden A (2012) Single‐molecule transcript counting of stem‐cell markers in the mouse intestine. Nat Cell Biol 14: 106–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A (2001) Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 29: 418–425 [DOI] [PubMed] [Google Scholar]
- Kim SY, Dunn IF, Firestein R, Gupta P, Wardwell L, Repich K, Schinzel AC, Wittner B, Silver SJ, Root DE, Boehm JS, Ramaswamy S, Lander ES, Hahn WC (2010) CK1epsilon is required for breast cancers dependent on beta‐catenin activity. PLoS ONE 5: e8979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D (1991) Identification of FAP locus genes from chromosome 5q21. Science 253: 661–665 [DOI] [PubMed] [Google Scholar]
- Klimowski LK, Garcia BA, Shabanowitz J, Hunt DF, Virshup DM (2006) Site‐specific casein kinase 1epsilon‐dependent phosphorylation of Dishevelled modulates beta‐catenin signaling. FEBS J 273: 4594–4602 [DOI] [PubMed] [Google Scholar]
- Knippschild U, Wolff S, Giamas G, Brockschmidt C, Wittau M, Würl PU, Eismann T, Stöter M (2005) The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Onkologie 28: 508–514 [DOI] [PubMed] [Google Scholar]
- Koo B‐K, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJR, Maurice MM, Clevers H (2012) Tumour suppressor RNF43 is a stem‐cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488: 665–669 [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H (1998) Depletion of epithelial stem‐cell compartments in the small intestine of mice lacking Tcf‐4. Nat Genet 19: 379–383 [DOI] [PubMed] [Google Scholar]
- Kuhnert F, Davis CR, Wang H‐T, Chu P, Lee M, Yuan J, Nusse R, Kuo CJ (2004) Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf‐1. Proc Natl Acad Sci USA 101: 266–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand Y, Luria V, Haffner‐Krausz R, Lonai P (1998) Maternally expressed PGK‐Cre transgene as a tool for early and uniform activation of the Cre site‐specific recombinase. Transgenic Res 7: 105–112 [DOI] [PubMed] [Google Scholar]
- de Lau W, Barker N, Low TY, Koo B‐K, Li VSW, Teunissen H, Kujala P, Haegebarth A, Peters PJ, van de Wetering M, Stange DE, van Es JE, Guardavaccaro D, Schasfoort RBM, Mohri Y, Nishimori K, Mohammed S, Heck AJR, Clevers H (2011) Lgr5 homologues associate with Wnt receptors and mediate R‐spondin signalling. Nature 476: 293–297 [DOI] [PubMed] [Google Scholar]
- Lee E, Salic A, Kirschner MW (2001) Physiological regulation of [beta]‐catenin stability by Tcf3 and CK1epsilon. J Cell Biol 154: 983–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Chen R, Lee Y, Yoo S, Lee C (2009) Essential roles of CKIdelta and CKIepsilon in the mammalian circadian clock. Proc Natl Acad Sci USA 106: 21359–21364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li VSW, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJR, Maurice MM, Mahmoudi T, Clevers H (2012) Wnt signaling through inhibition of β‐catenin degradation in an intact Axin1 complex. Cell 149: 1245–1256 [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X (2002) Control of beta‐catenin phosphorylation/degradation by a dual‐kinase mechanism. Cell 108: 837–847 [DOI] [PubMed] [Google Scholar]
- Mao J, Wang J, Liu B, Pan W, Farr GH, Flynn C, Yuan H, Takada S, Kimelman D, Li L, Wu D (2001) Low‐density lipoprotein receptor‐related protein‐5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell 7: 801–809 [DOI] [PubMed] [Google Scholar]
- el Marjou F, Janssen K‐P, Chang BH‐J, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S (2004) Tissue‐specific and inducible Cre‐mediated recombination in the gut epithelium. Genesis 39: 186–193 [DOI] [PubMed] [Google Scholar]
- Masri S, Cervantes M, Sassone‐Corsi P (2013) The circadian clock and cell cycle: interconnected biological circuits. Curr Opin Cell Biol 25: 730–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay RM, Peters JM, Graff JM (2001) The casein kinase I family in Wnt signaling. Dev Biol 235: 388–396 [DOI] [PubMed] [Google Scholar]
- Muñoz J, Stange DE, Schepers AG, van de Wetering M, Koo B‐K, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S, Myant K, Versteeg R, Sansom OJ, van Es JH, Barker N, van Oudenaarden A, Mohammed S, Heck AJR, Clevers H (2012) The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J 31: 3079–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, McKay RM, McKay JP, Graff JM (1999) Casein kinase I transduces Wnt signals. Nature 401: 345–350 [DOI] [PubMed] [Google Scholar]
- Pinto D, Gregorieff A, Begthel H, Clevers H (2003) Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev 17: 1709–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MA (2006) CKI, there's more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev 20: 399–410 [DOI] [PubMed] [Google Scholar]
- Quastler H, Sherman FG (1959) Cell population kinetics in the intestinal epithelium of the mouse. Exp Cell Res 17: 420–438 [DOI] [PubMed] [Google Scholar]
- Riccio O, van Gijn ME, Bezdek AC, Pellegrinet L, van Es JH, Zimber‐Strobl U, Strobl LJ, Honjo T, Clevers H, Radtke F (2008) Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep 9: 377–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Tice DA, Polakis P (2001) Axin‐dependent phosphorylation of the adenomatous polyposis coli protein mediated by casein kinase 1epsilon. J Biol Chem 276: 39037–39045 [DOI] [PubMed] [Google Scholar]
- Ruffner H, Sprunger J, Charlat O, Leighton‐Davies J, Grosshans B, Salathe A, Zietzling S, Beck V, Therier M, Isken A, Xie Y, Zhang Y, Hao H, Shi X, Liu D, Song Q, Clay I, Hintzen G, Tchorz J, Bouchez LC et al (2012) R‐spondin potentiates Wnt/β‐catenin signaling through orphan receptors LGR4 and LGR5. PLoS ONE 7: e40976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakanaka C, Leong P, Xu L, Harrison SD, Williams LT (1999) Casein kinase Ivarepsilon in the Wnt pathway: regulation of beta ‐catenin function. Proc Natl Acad Sci 96: 12548–12552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon‐Assmann P, Clevers H, Nathke IS, Clarke AR, Winton DJ (2004) Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev 18: 1385–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH (2004) Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK‐3‐specific inhibitor. Nat Med 10: 55–63 [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H (2009) Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 459: 262–265 [DOI] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H (2011) Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers AG, Vries R, van den Born M, van de Wetering M, Clevers H (2011) Lgr5 intestinal stem cells have high telomerase activity and randomly segregate their chromosomes. EMBO J 30: 1104–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijers J, Junker JP, Mokry M, Hatzis P, Koo B‐K, Sasselli V, van der Flier LG, Cuppen E, van Oudenaarden A, Clevers H, Acar M, Mettetal JT, van Oudenaarden A, Andreu P, Colnot S, Godard C, Gad S, Chafey P, Niwa‐Kawakita M, Laurent‐Puig P et al (2015) Ascl2 acts as an R‐spondin/Wnt‐responsive switch to control stemness in intestinal crypts. Cell Stem Cell 16: 158–170 [DOI] [PubMed] [Google Scholar]
- Shin S, Wolgamott L, Roux PP, Yoon S‐O (2014) Casein Kinase 1{varepsilon} Promotes Cell Proliferation by Regulating mRNA Translation. Cancer Res 74: 201–211 [DOI] [PubMed] [Google Scholar]
- Shirogane T, Jin J, Ang XL, Harper JW (2005) SCFbeta‐TRCP controls clock‐dependent transcription via casein kinase 1‐dependent degradation of the mammalian period‐1 (Per1) protein. J Biol Chem 280: 26863–26872 [DOI] [PubMed] [Google Scholar]
- Swiatek W, Tsai I‐C, Klimowski L, Pepler A, Barnette J, Yost HJ, Virshup DM (2004) Regulation of casein kinase I epsilon activity by Wnt signaling. J Biol Chem 279: 13011–13017 [DOI] [PubMed] [Google Scholar]
- Swiatek W, Kang H, Garcia BA, Shabanowitz J, Coombs GS, Hunt DF, Virshup DM (2006) Negative regulation of LRP6 function by casein kinase I epsilon phosphorylation. J Biol Chem 281: 12233–12241 [DOI] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ (2011) A reserve stem cell population in small intestine renders Lgr5‐positive cells dispensable. Nature 478: 255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lidth de Jeude JF, Vermeulen JLM, Montenegro‐Miranda PS, Van den Brink GR, Heijmans J (2015) A protocol for lentiviral transduction and downstream analysis of intestinal organoids. J Vis Exp 98: e52531 https://doi.org/10.3791/52531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielhaber E, Eide E, Rivers A, Gao ZH, Virshup DM (2000) Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase I epsilon. Mol Cell Biol 20: 4888–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte F, Bernatik O, Kirchner K, Masek J, Mahl A, Krejci P, Mundlos S, Schambony A, Bryja V, Stricker S (2010) Negative regulation of Wnt signaling mediated by CK1‐phosphorylated Dishevelled via Ror2. FASEB J 24: 2417–2426 [DOI] [PubMed] [Google Scholar]
- Xie Y, Zamponi R, Charlat O, Ramones M, Swalley S, Jiang X, Rivera D, Tschantz W, Lu B, Quinn L, Dimitri C, Parker J, Jeffery D, Wilcox SK, Watrobka M, Lemotte P, Granda B, Porter JA, Myer VE, Loew A et al (2013) Interaction with both ZNRF3 and LGR4 is required for the signalling activity of R‐spondin. EMBO Rep 14: 1120–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptácek LJ, Fu Y‐H (2005) Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature 434: 640–644 [DOI] [PubMed] [Google Scholar]
- Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR, Sangiorgi E, Capecchi MR, Kuo CJ (2012) The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA 109: 466–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Farin HF, van Es JH, Clevers H, Langer R, Karp JM (2013) Niche‐independent high‐purity cultures of Lgr5(+) intestinal stem cells and their progeny. Nat Methods 11: 106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebisch M, Xu Y, Krastev C, Macdonald BT, Chen M, Gilbert RJC, He X, Jones EY (2013) Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R‐spondin. Nat Commun 4: 2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X (2005) A dual‐kinase mechanism for Wnt co‐receptor phosphorylation and activation. Nature 438: 873–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W‐J, Geng ZH, Spence JR, Geng J‐G (2013) Induction of intestinal stem cells by R‐spondin 1 and Slit2 augments chemoradioprotection. Nature 501: 107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File