

Abstract
A mild and convenient one‐step preparation of 4H‐1,3‐benzoxazin‐4‐ones by a domino carbonylation–cyclization process is developed. Readily available ortho‐iodophenols are subjected to palladium‐catalyzed carbonylative coupling with Mo(CO)6 and cyanamide, followed by a spontaneous, intramolecular cyclization to afford 4H‐1,3‐benzoxazin‐4‐ones in moderate to excellent yields. Furthermore, the scope of the reaction is extended to include challenging ortho‐bromophenols. Finally, to highlight the versatility of the developed method, Mo(CO)6 is successfully replaced with a wide array of CO‐releasing reagents, such as oxalyl chloride, phenyl formate, 9‐methylfluorene‐9‐carbonyl chloride, and formic acid, making this an appealing strategy for the synthesis of 4H‐benzo[e][1,3]oxazin‐4‐ones.
Keywords: 4H-benzo[e][1,3]oxazin-4-ones; carbonylation; domino reactions; halophenols; heterocycles
1. Introduction
Heterocycles are arguably one of the most important classes of organic compounds due to their wide range of biological activities,1 and there is a constant need for new synthetic methods for their preparation. Benzoxazines are of particular interest and have been reported to possess antimicrobial,2 anticancer,3, 4 and antiplatelet5, 6 activities. However, preparation of this valuable heterocycle is limited to a handful of synthetic routes which often require hazardous reagents, cumbersome preparation of starting materials and reagents or, alternatively, are restricted to electron‐withdrawing substituents in the benzene ring (Scheme 1 a–e).7, 8, 9, 10, 11, 12 Palladium‐catalyzed carbonylation reactions such as aminocarbonylation and cross‐coupling reactions have become essential tools for synthesizing heterocycles and immense effort has been invested in developing safer methods for handling the toxic carbon monoxide gas.13, 14, 15 Over the last decade, several non‐gaseous CO sources have been reported both for in situ and ex situ use, for example, formates,16, 17 formamides,18 aldehydes,19 formic acid20 and metal carbonyls such as Mo(CO)6.21, 22, 23 In addition, Pd‐catalyzed decomposition of 9‐methylfluorene‐9‐carbonyl chloride (COgen)24 and fluoride‐mediated CO release from silacarboxylic acid25 enable the use of substoichiometric amounts of CO as well as the possibility to isotopically label the carbonyl carbon. Furthermore, base‐mediated decomposition of oxalyl chloride26 and chloroform27, 28 have been reported as effective CO‐generating strategies for carbonylation chemistry. Notably, the latter facilitates the preparation of 13C‐ and 14C‐labeled carbonyl derivatives.
Scheme 1.
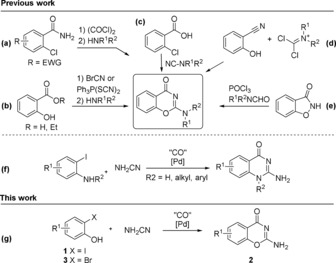
Synthetic procedures for the preparation of benzoxazinones, carbonylation–cyclization domino reactions used in the synthesis of 2‐aminoquinazolinones, and the work presented herein.
We recently reported a novel Mo(CO)6‐mediated carbonylation–cyclization sequence to prepare 2‐amino‐quinazolin‐4(3H)‐ones and 2‐aminoquinazolin‐4(1H)‐ones from ortho‐iodoanilines and cyanamide (Scheme 1 f).29 As a continuation of our previous work, we sought to develop an analogous approach for ortho‐halophenols (1 or 3) to give 2‐amino‐4H‐benzo[e][1,3]oxazin‐4‐ones (2, Scheme 1 g). This approach offers a number of advantages including 1) the use of readily available starting materials, 2) facile synthesis under low CO pressures without the use of CO gas, and 3) access to the unsubstituted exocyclic amine.
2. Results and Discussion
Initially, 2‐iodophenol (1 a) and cyanamide were reacted in chamber A with Pd(PPh3)4 and triethylamine in 1,4‐dioxane using a bridged two‐chamber system,22 where CO was generated ex situ from Mo(CO)6 in chamber B at 65 °C for 20 h. The reaction afforded full conversion of 1 a to 2‐amino‐4H‐benzo[e][1,3]oxazin‐4‐one (2 a). Notably, the product was conveniently isolated by precipitation, affording 2 a in 76 % yield (Scheme 2). The structure of 2 a was confirmed by NMR spectroscopy and X‐ray crystallography (see the Supporting Information). Furthermore, the phenol nucleophile afforded ring closure readily at 65 °C, whereas the aniline required higher temperatures for complete cyclization.
Scheme 2.
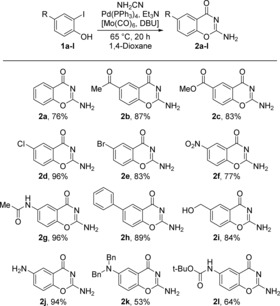
Synthesis of 2‐amino‐4H‐benzo[e][1,3]oxazin‐4‐ones 2 a–l from ortho‐iodophenols 1 a–l (isolated yields).
The method was then evaluated for a set of ortho‐iodophenols to test the scope and limitations of the reaction (Scheme 2). Overall, the products were obtained in moderate to excellent yields and good chemoselectivity was observed under the mild reaction conditions. The reaction worked well for electron‐deficient ortho‐iodophenols and acetyl‐substituted ortho‐iodophenol provided the corresponding 6‐acetyl‐2‐amino‐4H‐benzo[e][1,3]oxazin‐4‐one (2 b) in 87 % isolated yield. Methyl‐ester‐substituted ortho‐iodophenol furnished the corresponding methyl 2‐amino‐4‐oxo‐4H‐benzo[e][1,3]oxazine‐6‐carboxylate (2 c) without cleavage of the ester functionality in 83 % isolated yield. Chloro‐ and bromo‐substituted ortho‐iodophenols gave the corresponding 6‐chloro‐ and 6‐bromo‐substituted cyclic structures (2 d and 2 e, respectively) in 96 % and 83 % yields, respectively. The bromide was intact and fully orthogonal to the iodide under the mild reaction conditions. The introduction of the strongly electron‐withdrawing nitro functionality para to the nucleophilic phenol, resulted only in a slight reduction in yield (2 f, 77 %) compared to other electron‐deficient substrates. This suggests that the phenoxide anion present under the reaction conditions is a sufficiently strong nucleophile to counteract the electron‐withdrawing properties of substituents on the aryl ring. Mo(CO)6 has been reported as a reducing agent of aryl nitro groups at elevated temperatures.30 However, given that Mo(CO)6 is separated from the carbonylative reaction mixture and the mild conditions used in this procedure, nitro substituents can successfully be incorporated without reduction of the nitro group. 2‐Amino‐6‐(hydroxymethyl)‐4H‐benzo[e][1,3]oxazin‐4‐one (2 i) was successfully obtained in 84 % isolated yield without side reactions taking place. Electron‐rich amino‐substituted ortho‐iodophenol furnished 2,6‐diamino‐4H‐benzo[e][1,3]oxazin‐4‐one (2 j) and dibenzyl‐protected 2,6‐diamino‐4H‐benzo[e][1,3]oxazin‐4‐one (2 k) in 94 % and 53 % isolated yields, respectively. In contrast, if 2‐iodo‐5‐methoxyphenol was used, a complex reaction mixture was obtained from which the pure compound could not be isolated. We attribute this result to reduced susceptibility of the halide towards oxidative addition due to an electron‐donating effect from the methoxy group. Finally, Boc‐protected amino‐substituted ortho‐iodophenol furnished the desired tert‐butyl (2‐amino‐4‐oxo‐4H‐benzo[e][1,3]oxazin‐6‐yl)carbamate (2 l) in 64 % isolated yield without apparent cleavage of the carbamate functionality. Overall, both electron‐deficient and electron‐rich substrates performed well in the reaction. It is worth noting that the outcome of the reaction is not exclusively dependent on electronic properties but also on physical properties such as solubility; this was illustrated by the difference in yield of the electronically equivalent 2 d (96 %) and 2 e (83 %).
To further broaden the generality of the reaction we decided to develop a viable procedure for reacting ortho‐bromophenols. There are few examples in the literature of carbonylative coupling reactions of ortho‐bromophenols, and these reactions often require harsh conditions and suffer from low yields.31 There are, however, reports of palladium‐catalyzed aminocarbonylations of electron‐rich aryl bromides using monodentate and bidentate phosphine ligands, for example, di(1‐adamantyl)‐n‐butylphosphine (cataCXium A),32 1,1′‐bis(diphenylphosphino)ferrocene (dppf),22 and bis[(2‐diphenylphosphino)phenyl] ether (DPEphos).33 Initial optimization studies were performed using 2‐bromophenol (3 a) as a standard substrate. However, it was quickly discovered that the desired product 2 a reacted further with cyanamide resulting in the guanidine‐substituted side product 4 a along with the desired benzoxazinone. We have recently shown by X‐ray crystallography that the corresponding quinazolinone‐guanidine side product is formed in the carbonylation reaction of ortho‐iodoanilines with an excess of cyanamide.29 Compound 4 a was isolated and subsequently fully characterized; spectral data were in agreement with the proposed structure. We postulated that the generation of the desired product is slow from the electron‐rich unsubstituted 2‐bromophenol and the resulting large excess of cyanamide present in the reaction mixture enables the formation of the guanidine side product. Therefore, 2‐bromo‐4‐chlorophenol (3 b), which is more activated towards oxidative addition, was used as the standard substrate in the optimization studies (Table 1). All ligands were found to activate the C−Br bond and facilitated consumption of starting material (3 b) albeit with varying efficiency. Use of neither the monodentate phosphine ligand cataCXium A and precatalyst Pd(dppf)Cl2 nor the bidentate phosphine ligand 1,3‐bis(dicyclohexylphosphino)propane (dcpp) provided full conversion of the starting bromide and the guanidine side‐product (4 b) was predominantly formed over the desired 2 d (Table 1, entries 1–3). However, if 4,5‐bis(diphenylphosphino)‐9,9‐dimethylxanthene (Xantphos) was used as a ligand, 2 d could be isolated in 10 % yield, although 4 b was formed as the major product (Table 1, entry 4). By changing the ligand to DPEphos the starting material was fully consumed and 2 d was formed as the major product and was isolated in 68 % yield (Table 1, entry 5). Next, we attempted to circumvent the formation of the guanidine side product by altering the reaction temperature to 45 and 85 °C. By doing so, we hoped to either 1) suppress the nucleophilic attack leading to 4 b at the lower temperature, or 2) increase the reaction rate for the oxidative addition step at elevated temperature and thereby reduce the concentration of cyanamide present in the reaction mixture after the product had formed. Unfortunately, the formation of 4 b was observed despite the reduction in temperature and the starting material was not fully consumed in contrast to the reaction at 65 °C (Table 1, entry 6). At 85 °C the starting material was fully consumed, nonetheless 4 b was the major product (Table 1, entry 7). Next, LiCl34, 35 was added to stabilize the Pd0 species, however, this favored the formation of 4 b, and 2 d was isolated in 11 % yield (Table 1, entry 8). We next decided to evaluate if sodium phenoxide36 or DMAP37, 38 nucleophiles could accelerate the reaction and thereby lower the amount of 4 b. However, addition of sodium phenoxide reduced the conversion of starting bromide and 4 b was obtained as the major product. In contrast, addition of DMAP maintained a high conversion of starting material and the desired 2 d could be isolated in 58 % yield leading us to continue with the conditions given in Table 1, entry 5.
Table 1.
Optimization of synthesis of 2 d from 2‐bromo‐4‐chlorophenol (3 b).[a]
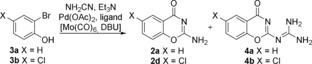
| Entry | Ligand | Additive | T [°C] | Yield 2 d [b] [%] |
|---|---|---|---|---|
| 1 | cataCXium A | – | 65 | trace |
| 2 | –[c] | – | 65 | trace |
| 3 | dcpp⋅BF4 | – | 65 | trace |
| 4 | Xantphos | – | 65 | 10 |
| 5 | DPEphos | – | 65 | 68 |
| 6 | DPEphos | – | 45 | 8 |
| 7 | DPEphos | – | 85 | 36 |
| 8 | DPEphos | LiCl | 65 | 11 |
| 9 | DPEphos | NaOPh | 65 | – |
| 10 | DPEphos | DMAP | 65 | 58 |
[a] Reaction conditions: chamber A: 2‐bromo‐4‐chlorophenol (1 mmol), cyanamide (1 equiv), Pd(OAc)2 (5 mol %), ligand, Et3N (2 equiv), 1,4‐dioxane (3 mL), 65 °C, 20 h; chamber B: Mo(CO)6 (0.8 equiv), DBU (2.4 equiv), 1,4‐dioxane (3 mL), 65 °C, 20 h. [b] Isolated yield. [c] Pd(dppf)Cl2 (5 mol %).
With suitable reaction conditions determined (Table 1, entry 5), we extended the scope of the reaction to a set of commercially available ortho‐bromophenols (Scheme 3). 2‐Bromophenol (1 a) was fully consumed, however the side reactions took precedence, leading to the formation of the guanidine adduct as the major product and the desired 2 a was isolated in 20 % yield. In contrast, the electron‐poor chloro‐substituted ortho‐bromophenol gave the desired 2 d as major product which could be isolated in 68 % yield. This correlation became even more clear with a comparison of the reactions of fluoro‐substituted analogues (3 c–e). Among these substrates, 3 c has almost no electron‐withdrawing effect on the bromo position, and thus the corresponding 2 m was obtained in 21 % yield. In contrast, 3 d is activated for oxidative addition, providing 2 n in 34 % isolated yield. The difluoro‐substituted 3 e gave the corresponding 2‐amino‐6,7‐difluoro‐4H‐benzo[e][1,3]oxazin‐4‐one (2 o) in 45 % isolated yield. The strongly electron‐withdrawing nitrile‐substituted ortho‐bromophenol 3 f returned the desired product 2 p in 90 % isolated yield and 2‐bromo‐4‐methylphenol (3 g) afforded 2 q in 46 % yield.
Scheme 3.
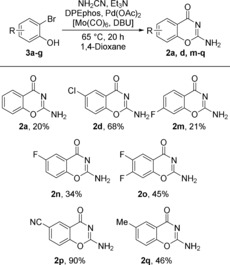
Synthesis of 2‐amino‐4H‐benzo[e][1,3]oxazin‐4‐ones 2 a, 2 d, and 2 m ‐q from ortho‐bromophenols 3 a–g (isolated yields).
Overall, the ortho‐bromophenols afforded lower yields than the analogous iodides and the reaction towards electron‐poor halides was clearly favored. Nonetheless, this is one of the first examples of palladium‐catalyzed carbonylation of ortho‐bromophenols that typically prove challenging.
To verify that the reaction proceeds via the proposed N‐cyanobenzamide intermediate 5, phenol 1 a was protected with a para‐methoxybenzyl (PMB) group according to standard procedures, yielding PMB‐protected compound 6 in 62 % isolated yield (Scheme 4). Compound 6 was then treated with cyanamide under the carbonylative conditions described in Scheme 2. Pleasingly, this afforded the corresponding PMB‐protected cyanobenzamide 7 in 45 % isolated yield. It is worth noting that the deprotected and cyclized benzoxazinone product 2 a was also generated and isolated in 14 % yield after the carbonylation step. The benzyl ether 7 was cleaved using standard acidic conditions, which gave a mixture of O‐debenzylated cyanobenzamide 5 and cyclized 2 a. The crude mixture was carried forward without further purification and was heated with Et3N in 1,4‐dioxane at 65 °C to complete the cyclization in 80 % isolated yield over two steps.
Scheme 4.
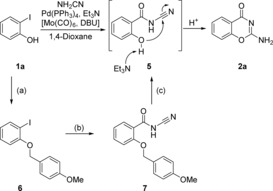
Mechanistic studies for the formation of 2 a from 1 a. Reaction conditions: a) 4‐methoxybenzylbromide, K2CO3, DMF, 50 °C, 5 h; b) NH2CN, Pd(PPh3)4, Et3N, Mo(CO)6, DBU, 1,4‐dioxane, 65 °C, 20 h; c) TFA, CH2Cl2, 0 °C to RT, 1 h.
The field of non‐gaseous carbonylation reactions has flourished over the last decade with the development of several novel CO sources that have eradicated the need to use cumbersome CO gas.39, 40 The main focus has been to identify and develop cheap, easy to handle and convenient reagents that can be used for carbonylations, including isotopic labeling. To assess if the method developed herein was viable with previously reported CO surrogates we replaced Mo(CO)6 as the CO source in both ex situ (two‐chamber system22) and in situ (single vial) reactions (Table 2). For each CO surrogate, the internal pressure was monitored over the course of the CO‐releasing reaction as a means to follow CO generation (see the Supporting Information for details). Ex situ use of Mo(CO)6 at 65 and 85 °C generated a yield of 76 % and 69 % respectively (Table 2, entries 1 and 2) with moderately rapid release of CO to relatively high internal pressures. Secondly, CO was delivered from a balloon following ex situ generation from oxalyl chloride,26 returning 2 a in 68 % yield (Table 2, entry 3). At atmospheric pressure a slight decrease in yield was observed and the same outcome was noted for prolonged CO‐release times (Table 2, entries 3, 5 and 8). CO generated from phenyl formate17 ex situ (Table 2, entry 5) and alkaline hydrolysis of chloroform27, 28 (Table 2, entry 8) resulted in the slow release of CO (T Pmax=22 h and 108 min, respectively), which caused increased formation of guanidine side‐product and thus a decrease in yield of 2 a. Unfortunately, neither phenyl formate nor hydrolysis of chloroform were successful in situ and no product was detected (Table 2, entries 5 and 9). Both palladium‐catalyzed release of CO from COgen24 and fluoride‐mediated decarbonylation of silacarboxylic acid25 proved efficient and compatible for ex situ use (Table 2, entries 6 and 7). Finally, ex situ introduction of CO by dehydration of formic acid by sulfuric acid20 gave 2 a in equivalent yield (Table 2, entry 10).
Table 2.
Evaluation of various CO sources.[a–c]

| Entry | CO source | T [°C] | P max [bar] | T Pmax [min] | Yield [%] |
|---|---|---|---|---|---|
| 1 | Mo(CO)6 [d] | 65 | 2.3 | 40 | 76 |
| 2 | Mo(CO)6 [d] | 85 | 2.4 | 20 | 69 |
| 3 | Oxalyl chloride[d] | 65 | atm[f] | – | 68 |
| 4 | Phenyl formate[e] | 65 | 2.5 | 20 h | – |
| 5 | Phenyl formate[d] | 65 | 3.1 | 22 h | 67 |
| 6 | COgen[d] | 80 | 3.1 | 21 | 75 |
| 7 | MePh2SiCOOH[d] | 65 | 2.9 | 3 | 79 |
| 8 | CHCl3 [d] | 80 | 2.7 | 108 | 61 |
| 9 | CHCl3 [e] | 80 | 6.3 | 39 | – |
| 10 | Formic acid[d] | 80 | 3.5 | 15 | 75 |
[a] P max: maximum pressure achieved; T Pmax: time required to reach P max. [b] See the Supporting Information for full experimental conditions. [c] Isolated yields. [d] Ex situ. [e] In situ. [f] CO collected in and distributed using a balloon.
The results in Table 2 show that the method developed herein could be easily paired with a variety of ex situ CO surrogates. Depending on the aim of the chemistry, for example, cheap and readily available precursors, solid shelf‐stable CO sources, or isotopic labeling, the reported method provides an attractive synthetic route to a variety of 4H‐benzo[e][1,3]oxazin‐4‐ones (2 a–q).
3. Conclusions
We have developed a low‐pressure, gas‐free, one‐step carbonylation method for the synthesis of 4H‐benzo[e][1,3]oxazin‐4‐ones from readily available ortho‐halophenols and cyanamide that involves a palladium‐catalyzed carbonylation–cyclization sequence. The reaction products are readily isolated by precipitation and the reaction occurs under mild conditions with good substrate compatibility. Additionally, the protocol was further extended to include ortho‐bromophenols as substrates. Finally, the versatility of the reaction was demonstrated by replacing Mo(CO)6 with a variety of ex situ CO‐releasing reagents, including introduction of CO with a balloon at atmospheric pressure, with retained product yields.
Experimental Section
General Information
Analytical thin‐layer chromatography (TLC) was performed on silica gel 60 F254 plates and visualized with UV light. Flash column chromatography was performed on silica gel 60 (40–63 μm). 1H, 13C, and 19F NMR spectra were recorded at 400, 100 and 377 MHz, respectively. The chemical shifts for 1H, 13C and 19F NMR spectra are referenced to tetramethylsilane according to residual solvent signals, a sealed capillary filled with CFCl3 was used as an internal reference in 19F NMR (1H: CD3OD at 3.31 ppm, CDCl3 at 7.26 ppm and [D6]DMSO at 2.50 ppm; 13C: CD3OD at 49.0 ppm, CDCl3 at 77.0 ppm and [D6]DMSO at 39.5 ppm; 19F: CFCl3 at 0.0 ppm). The data are reported as (s=singlet, br s=broadened singlet, d=doublet, t=triplet, q=quartet, or m=multiplet, coupling constant(s), integral). Analytical HPLC–MS was performed using electrospray ionization (ESI) and a C18 column (50×3.0 mm, 2.6 μm particle size, 100 Å pore size) with CH3CN/H2O in 0.05 % aqueous HCOOH as mobile phase at a flow rate of 1.5 mL min−1. Analysis of purity by LC was performed using a gradient of 5–100 % CH3CN/H2O in 0.05 % aqueous HCOOH as mobile phase at a flow rate of 1.5 mL min−1 for 5 min, unless otherwise stated, on a C18 column. High‐resolution molecular masses were determined on a mass spectrometer equipped with an ESI source and time‐of‐flight (TOF) mass analyzer.
Synthesis of ortho‐Iodophenol Precursors 1
1. ‐Chloro‐2‐iodophenol (1 d)
Prepared following a literature procedure41 to give a white solid (618 mg, 47 %). Compound 1 d is known,41 and spectral data were in agreement with the proposed structure and matched those reported in the literature.
2. ‐Bromo‐2‐iodophenol (1 e)
Prepared following a modified literature procedure41 to give a white solid (1.05 g, 66 %). Compound 1 e is known,42 and spectral data were in agreement with the proposed structure and matched those reported in the literature.
N‐(4‐Hydroxy‐3‐iodophenyl)acetamide (1 g)
Acetic anhydride (0.10 mL, 1.1 mmol) was added to a solution of 4‐amino‐2‐iodophenol (250 mg, 1.06 mmol) in absolute ethanol (5 mL) and the reaction mixture was stirred at RT for 1 h. The reaction mixture was concentrated to give a brown residue, which was purified using silica gel column chromatography pentane/EtOAc (4:1) to give 1 g as a brown solid (184 mg, 62 %). 1H NMR (400 MHz, CD3OD): δ=7.90 (d, J=2.5 Hz, 1 H), 7.31 (dd, J=8.7, 2.5 Hz, 1 H), 6.76 (d, J=8.7 Hz, 1 H), 2.08 ppm (s, 3 H); 13C NMR (400 MHz, CD3OD): δ=171.3, 154.9, 132.9, 132.4, 123.0, 115.3, 83.9, 23.5 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H9INO2: 277.9678 [M+H]+; found: 277.9671; LC purity (254 nm): 95 %.
3. ‐(Hydroxymethyl)‐2‐iodophenol (1 i)
Methyl 4‐hydroxy‐3‐iodobenzoate (562 mg, 2.02 mmol) was dissolved in anhydrous CH2Cl2 (50 mL) under inert conditions and cooled to −78 °C. Then, diisobutylaluminum hydride (7.0 mL, 7.0 mmol, 1 m in heptane) was added dropwise over 15 min. The reaction mixture was stirred at −78 °C for 30 min and then the cold bath was removed and the reaction mixture was stirred at RT for 2 h. The reaction was quenched by the addition of water (50 mL) and acetic acid (10 mL). The aqueous layer was extracted with CH2Cl2 (2×50 mL) and the combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The crude mixture was purified by silica gel column chromatography using pentane/EtOAc (1:1) to give 1 i as a white solid (304 mg, 60 %). 1H NMR (400 MHz, [D6]DMSO): δ=10.14 (s, OH), 7.60 (d, J=2.1 Hz, 1 H), 7.11 (dd, J=8.2, 2.1 Hz, 1 H), 6.82 (d, J=8.2 Hz, 1 H), 5.06 (t, J=5.8 Hz; OH), 4.34 ppm (d, J=5.2 Hz, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=155.3, 137.0, 135.2, 128.0, 114.6, 84.1, 61.8 ppm; HRMS (ESI‐TOF): m/z: calcd for C7H6IO2: 248.9413 [M−H]−; found: 248.9420; LC purity (254 nm): >98 %.
4. ‐(Dibenzylamino)‐2‐iodophenol (1 k)
4‐Amino‐2‐iodoaniline (250 mg, 1.06 mmol) and benzaldehyde (0.32 mL, 3.2 mmol) were dissolved in MeOH (5 mL) and acetic acid (0.18 mL, 3.1 mmol) was added. The reaction mixture was stirred at RT for 30 min, cooled to 0 °C and NaBH3CN (174 mg, 2.77 mmol) was added. The reaction was allowed to warm from 0 °C to RT and stirred for 48 h. The reaction was quenched with saturated aq NaHCO3 (30 mL) and then extracted with EtOAc (3×50 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The crude mixture was purified by silica gel column chromatography using pentane/EtOAc (20:1) to yield 1 k as a pale purple solid (239 mg, 54 %). 1H NMR (400 MHz, CDCl3): δ=7.36–7.30 (m, 4 H), 7.29–7.21 (m, 6 H), 7.06 (s, 1 H), 6.81 (d, J=8.6 Hz, 1 H), 6.68 (m, 1 H), 4.75 (s; OH), 4.52 ppm (s, 4 H); 13C NMR (100 MHz, CDCl3): δ=147.0, 144.9, 138.4, 128.8, 127.2, 127.0, 122.5, 116.0, 115.3, 86.6, 55.1 ppm; HRMS (ESI‐TOF): m/z: calcd for C20H19INO: 416.0511 [M+H]+; found: 416.0506; LC purity (254 nm): 95 %
tert‐Butyl (4‐Hydroxy‐3‐iodophenyl)carbamate (1 l)
4‐Amino‐2‐iodophenol (250 mg, 1.06 mmol) and di‐tert‐butyl carbonate (232 mg, 1.06 mmol) were dissolved in anhydrous THF and stirred at RT for 4 h. The reaction mixture was concentrated and the crude material was purified by silica gel column chromatography using pentane/EtOAc (5:1) to yield 1 l as an off‐white solid (317 mg, 89 %). 1H NMR (400 MHz, CDCl3): δ=7.77 (br s, 1 H), 7.10 (dd, J=8.8, 2.7 Hz, 1 H), 6.86 (m, 1 H), 6.39 (br s, 1 H), 5.43 (br s, 1 H), 1.50 ppm (s, 9 H); 13C NMR (100 MHz, CDCl3): δ=153.2, 151.4, 132.3, 129.1, 121.7, 114.9, 85.3, 80.9, 28.5 ppm; HRMS (ESI‐TOF): m/z: calcd for C11H13INO3: 333.9940 [M−H]−; found: 333.9948; LC purity (254 nm): 98 %.
Method A: General Procedure for the Synthesis of 2‐Amino‐4H‐benzo[e][1,3]oxazin‐4‐ones 2 a–l from ortho‐Iodophenols 1 a–l
To the reaction chamber (chamber A) of an oven‐dried, bridged two‐chamber system22 was added ortho‐iodophenol (1.0 mmol), cyanamide (50 mg, 1.2 mmol) and Pd(PPh3)4 (58 mg, 5 mol %). Mo(CO)6 (211 mg, 0.8 mmol) was added to the CO‐generation chamber (chamber B).43 The two chambers were capped with gastight caps, and the atmosphere was exchanged for nitrogen. 1,4‐Dioxane (3 mL) was added through the septa into each chamber, followed by the addition of Et3N (0.28 mL, 2 mmol) into chamber A and DBU (0.36 mL, 2.4 mmol) into chamber B. The two‐chamber system was immediately heated with vigorous stirring in a heating block at 65 °C for 20 h. After 20 h, the reaction mixture in chamber A was poured into iso‐hexane (30 mL) and collected by filtration. The solid was washed with water (10 mL) and EtOAc (10 mL) to give the pure compound.
Method B: General Procedure for the Synthesis of 2‐Amino‐4H‐benzo[e][1,3]oxazin‐4‐ones 2 a, 2 d, and 2 m–q from ortho‐Bromophenols 3 a–g
To the reaction chamber (chamber A) of an oven‐dried, bridged two‐chamber system22 was added ortho‐bromophenol (1.0 mmol), cyanamide (42 mg, 1.0 mmol), Pd(OAc)2 (11 mg, 5 mol %) and DPEphos (32 mg, 6 mol %). Mo(CO)6 (211 mg, 0.8 mmol) was added into the CO‐generation (chamber B). The two chambers were capped with gastight caps, and the atmosphere was exchanged for nitrogen. 1,4‐Dioxane (3 mL) was added through the septa into each chamber, followed by the addition of Et3N (0.28 mL, 2 mmol) into chamber A and DBU (0.36 mL, 2.4 mmol) into chamber B. The two‐chamber system was immediately heated and vigorously stirred in a heating block at 65 °C for 20 h. After 20 h, the reaction mixture in chamber A was poured into iso‐hexane (30 mL) and collected by filtration. The solid was washed with water (10 mL) and EtOAc (10 mL) to give the pure compound.
1. ‐Amino‐4H‐benzo[e][1,3]oxazin‐4‐one (2 a)
Method A: Tan solid (128 mg, 76 %). Method B: Tan solid (34 mg, 20 %). R f=0.29 (SiO2, 5 % MeOH/CH2Cl2+Et3N); m.p. 256–257 °C; 1H NMR (400 MHz, CD3OD): δ=8.00 (dd, J=7.8, 1.7 Hz, 1 H), 7.71 (ddd, J=8.7, 7.3, 1.7 Hz, 1 H), 7.41 (m, 1 H), 7.34 ppm (dd, J=8.3, 1.0 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.0, 159.8, 153.4, 134.1, 126.6, 125.2, 117.1, 115.7 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H7N2O2: 163.0508 [M+H]+; found: 163.0503; LC purity (254 nm): >98 %.
2. ‐Acetyl‐2‐amino‐4H‐benzo[e][1,3]oxazin‐4‐one (2 b)
Method A: Tan solid (178 mg, 87 %). R f=0.26 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO/CD3OD 3:1) δ=8.43 (d, J=2.2 Hz, 1 H), 8.20 (dd, J=8.7, 2.3 Hz, 1 H), 7.39 (d, J=8.6 Hz, 1 H), 2.59 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=196.4, 165.5, 159.6, 156.3, 133.6, 133.4, 127.2, 117.0, 116.5, 26.7 ppm; HRMS (ESI‐TOF): m/z: calcd for C10H9N2O3: 205.0613 [M+H]+; found: 205.0615; LC purity (254 nm): >98 %.
Methyl 2‐Amino‐4‐oxo‐4H‐benzo[e][1,3]oxazine‐6‐carboxylate (2 c)
Method A: Off‐white solid (185 mg, 83 %). R f=0.26 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO): δ=8.41 (d, J=2.2 Hz, 1 H), 8.36 (br s, 1 H; NH), 8.28 (br s, 1 H; NH), 8.20 (dd, J=8.6, 2.3 Hz, 1 H), 7.45 (d, J=8.6 Hz, 1 H), 3.89 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=165.3, 165.1, 159.6, 156.4, 134.4, 128.0, 126.4, 117.2, 116.7, 52.4 ppm; HRMS (ESI‐TOF): m/z: calcd for C10H9N2O4: 221.0562 [M+H]+; found: 221.0572; LC purity (254 nm): 95 %.
3. ‐Amino‐6‐chloro‐4H‐benzo[e][1,3]oxazin‐4‐one (2 d)
Method A: Tan solid (189 mg, 96 %). Method B: Gray solid (110 mg, 68 %). R f=0.23 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, CD3OD): δ=7.95 (d, J=2.6 Hz, 1 H), 7.70 (dd, J=8.8, 2.6 Hz, 1 H), 7.35 ppm (d, J=8.8 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO) δ=165.0, 159.7, 152.1, 133.9, 129.2, 125.7, 118.5, 118.2 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H6ClN2O2: 197.0118 [M+H]+; found: 197.0111; LC purity (254 nm): >98 %.
4. ‐Amino‐6‐bromo‐4H‐benzo[e][1,3]oxazin‐4‐one (2 e)
Method A: Tan solid (197 mg, 83 %). R f=0.23 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO/CD3OD 3:1) δ=7.94 (d, J=2.6 Hz, 1 H), 7.79 (dd, J=8.8, 2.5 Hz, 1 H), 7.26 ppm (d, J=8.8 Hz, 1 H); 13C NMR ([D6]DMSO): δ=164.8, 159.6, 152.5, 136.7, 128.7, 118.9, 118.5, 116.9 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H6BrN2O2: 240.9613 [M+H]+; found: 240.9612; LC purity (254 nm): 97 %.
5. ‐Amino‐6‐nitro‐4H‐benzo[e][1,3]oxazin‐4‐one (2 f)
Method A: Pale yellow solid (166 mg, 77 %). R f=0.20 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO) δ=8.55 (d, J=2.8 Hz, 1 H), 8.53 (br s, 1 H; NH), 8.48 (dd, J=9.0, 2.9 Hz, 1 H), 8.44 (br s, 1 H; NH), 7.57 ppm (d, J=9.1 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=164.7, 159.6, 157.2, 144.1, 128.8, 122.2, 117.9, 117.6 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H6N3O4: 208.0358 [M+H]+; found: 208.0357; LC purity (254 nm): >98 %.
N‐(2‐Amino‐4‐oxo‐4H‐benzo[e][1,3]oxazin‐6‐yl)acetamide (2 g)
Method A: Brown solid (111 mg, 96 %). R f=0.17 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, CD3OD): δ=8.12 (d, J=2.6 Hz, 1 H), 7.96 (dd, J=9.0, 2.6 Hz, 1 H), 7.30 (d, J=9.0 Hz, 1 H), 2.15 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=168.8, 166.5, 160.2, 149.4, 136.8, 125.3, 117.6, 116.5, 116.2, 24.3 ppm; HRMS (ESI‐TOF): m/z: calcd for C10H10N3O3: 220.0722 [M+H]+; found: 220.0729; LC purity (254 nm): >98 %.
6. ‐Amino‐6‐phenyl‐4H‐benzo[e][1,3]oxazin‐4‐one (2 h)
Method A: Off‐white solid (209 mg, 89 %). R f=0.46 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO): δ=8.23 (br s, 1 H; NH, exchanges with deuterium), 8.16 (br s, 1 H; NH, exchanges with deuterium), 8.08 (d, J=2.4 Hz, 1 H), 7.98 (dd, J=8.6, 2.4 Hz, 1 H), 7.74–7.64 (m, 2 H), 7.54–7.45 (m, 2 H), 7.46–7.37 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.0, 159.8, 153.0, 138.7, 137.2, 132.4, 129.2, 127.8, 126.7, 124.1, 117.5, 116.5 ppm; HRMS (ESI‐TOF): m/z: calcd for C14H11N2O2: 239.0821 [M+H]+; found: 239.0814; LC purity (254 nm): 98 %.
7. ‐Amino‐6‐(hydroxymethyl)‐4H‐benzo[e][1,3]oxazin‐4‐one (2 i)
Method A: Tan solid (163 mg, 84 %). R f=0.14 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, CD3OD): δ=7.98 (d, J=2.1 Hz, 1 H), 7.71 (dd, J=8.5, 2.2 Hz, 1 H), 7.32 (d, J=8.5 Hz, 1 H), 4.67 ppm (s, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.6, 160.2, 152.7, 140.0, 132.6, 124.5, 117.1, 115.8, 62.5 ppm; HRMS (ESI‐TOF): m/z: calcd for C9H9N2O3: 193.0613 [M+H]+; found: 193.0607; LC purity (254 nm): >98 %.
8. ,6‐Diamino‐4H‐benzo[e][1,3]oxazin‐4‐one (2 j)
Method A: Brown solid (175 mg, 94 %). R f=0.23 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, CD3OD): δ=7.20 (dd, J=2.8, 0.5 Hz, 1 H), 7.09 (dd, J=8.8, 0.5 Hz, 1 H), 7.04 ppm (dd, J=8.8, 2.7 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.7, 159.8, 146.2, 144.9, 120.2, 117.5, 115.9, 108.6 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H8N3O2: 178.0617 [M+H]+; found: 178.0609; LC purity (254 nm): 95 %.
9. ‐Amino‐6‐(dibenzylamino)‐4H‐benzo[e][1,3]oxazin‐4‐one (2 k)
Method A: Pale yellow solid (92 mg, 53 %). 1H NMR (400 MHz, [D6]DMSO): δ=7.86 (br s, 2 H, NH), 7.36–7.31 (m, 4 H), 7.29–7.21 (m, 6 H), 7.11 (m, 1 H), 7.03–6.98 (m, 2 H), 4.75 ppm (s, 4 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.4, 159.8, 145.6, 145.1, 138.4, 128.6, 126.8, 126.5, 118.7, 117.4, 116.2, 107.4, 54.6 ppm; HRMS (ESI‐TOF): m/z: calcd for C22H20N3O2: 358.1556 [M+H]+; found: 358.1559; LC purity (254 nm): 97 %.
tert‐Butyl (2‐amino‐4‐oxo‐4H‐benzo[e][1,3]oxazin‐6‐yl)carbamate (2 l)
Method A: Off‐white solid (126 mg, 64 %). R f=0.32 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, CD3OD) δ=7.99 (d, J=2.7 Hz, 1 H), 7.79 (dd, J=9.1, 2.7 Hz, 1 H), 7.26 (d, J=9.0 Hz, 1 H), 1.53 ppm (s, 9 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.1, 159.8, 152.8, 148.5, 136.6, 124.1, 117.2, 116.0, 114.7, 79.4, 28.1 ppm; HRMS (ESI‐TOF): m/z: calcd for C13H16N3O4: 278.1141 [M+H]+; found: 278.1151; LC purity (254 nm): >98 %.
10. ‐Amino‐7‐fluoro‐4H‐benzo[e][1,3]oxazin‐4‐one (2 m)
Method B: Gray solid (38 mg, 21 %). R f=0.35 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO): δ=8.21 (br s, 1 H; NH, exchanges with deuterium), 8.16 (br s, 1 H; NH, exchanges with deuterium), 7.93 (m, 1 H), 7.36–7.15 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=165.3, 164.9 (d, 1 J CF=251.0 Hz), 159.7, 154.5 (d, 3 J CF=14.0 Hz), 129.2 (d, 3 J CF=10.7 Hz), 114.1 (d, 4 J CF=2.8 Hz), 113.0 (d, 2 J CF=22.5 Hz), 103.3 ppm (d, 2 J CF=26.2 Hz); 19F NMR (377 MHz, CD3OD): δ=−103.6 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H6FN2O2: 181.0413 [M+H]+; found: 181.0407; LC purity (254 nm): 95 %.
11. ‐Amino‐6‐fluoro‐4H‐benzo[e][1,3]oxazin‐4‐one (2 n)
Method B: Gray solid (56 mg, 34 %). R f=0.32 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO): δ=8.22 (br s, 1 H; NH exchanges with deuterium), 8.16 (br s, 1 H; NH exchanges with deuterium), 7.64–7.49 (m, 2 H), 7.41 ppm (m, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=165.5, 160.0, 158.7 (d, 1 J CF=242.9 Hz), 149.8 (d, 4 J CF=1.8 Hz), 121.7 (d, 2 J CF=24.8 Hz), 118.4 (d, 3 J CF=7.1 Hz), 118.3 (d, 3 J CF=8.0 Hz), 111.9 ppm (d, 2 J CF=23.8 Hz); 19F NMR (377 MHz, CD3OD): δ=−117.0 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H6FN2O2: 181.0413 [M+H]+; found: 181.0410; LC purity (254 nm): >98 %.
12. ‐Amino‐6,7‐difluoro‐4H‐benzo[e][1,3]oxazin‐4‐one (2 o)
Method B: Gray solid (91 mg, 45 %). R f=0.35 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO): δ=8.15 (br s, 2 H; NH2, exchanges with deuterium), 7.79 (m, 1 H), 7.57 ppm (m, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=164.6, 159.8, 152.8 (dd, 1 J CF=253.5 Hz, 2 J CF=14.8 Hz), 150.0–149.8 (m, 1 C), 147.0 (dd, 1 J CF=246.3 Hz, 2 J CF=13.3 Hz), 114.8–113.8 (m, 2 C), 106.1 ppm (d, J=21.8 Hz); 19F NMR (377 MHz, CD3OD): δ=−128.2, −141.7 ppm; HRMS (ESI‐TOF): m/z: calcd for C8H5F2N2O2: 199.0319 [M+H]+; found: 199.0317; LC purity (254 nm): >98 %.
13. ‐Amino‐4‐oxo‐4H‐benzo[e][1,3]oxazine‐6‐carbonitrile (2 p)
Method B: Gray solid (161 mg, 90 %). R f=0.27 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO): δ=8.37–8.16 (m, 3 H), 8.09 (m, 1 H), 7.50 ppm (m, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=164.1, 159.3, 155.8, 137.1, 131.3, 117.9, 117.50, 117.47, 107.9 ppm; HRMS (ESI‐TOF): m/z: calcd for C9H6N3O2: 188.0460 [M+H]+; found: 188.0463; LC purity (254 nm): 97 %.
14. ‐Amino‐6‐methyl‐4H‐benzo[e][1,3]oxazin‐4‐one (2 q)
Method B: Gray solid (84 mg, 46 %). R f=0.41 (SiO2, 5 % MeOH/CH2Cl2+Et3N); 1H NMR (400 MHz, [D6]DMSO): δ=8.07 (br s, 1 H; NH, exchanges with deuterium), 8.03 (br s, 1 H; NH, exchanges with deuterium), 7.67 (m, 1 H), 7.49 (m, 1 H), 7.21 (d, J=8.4 Hz, 1 H), 2.36 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.2, 159.8, 151.5, 134.8, 134.5, 126.3, 116.8, 115.5, 20.3 ppm; HRMS (ESI‐TOF): m/z: calcd for C9H9N2O2: 177.0664 [M+H]+; found: 177.0661; LC purity (254 nm): >98 %.
Characterization of Side Product 2‐(4‐Oxo‐4H‐benzo[e][1,3]oxazin‐2‐yl)guanidine (4 a)
To the reaction chamber (chamber A) of an oven‐dried, bridged two‐chamber system22 was added ortho‐bromophenol (0.12 mL, 0.99 mmol), cyanamide (44 mg, 1.1 mmol), Pd(OAc)2 (11 mg, 5 mol %) and DPEphos (38 mg, 7 mol %). Mo(CO)6 (215 mg, 0.8 mmol) was added into the CO‐generation chamber (chamber B). The two chambers were capped with gastight caps, and the atmosphere was exchanged for nitrogen. 1,4‐Dioxane (3 mL) was added through the septa into each chamber, followed by the addition of Et3N (0.28 mL, 2.0 mmol) into chamber A and DBU (0.36 mL, 2.4 mmol) into chamber B. The two‐chamber system was immediately heated and vigorously stirred in a heating block at 65 °C for 20 h. After 20 h, the reaction mixture in chamber A was poured into iso‐hexane (30 mL) and the resulting precipitate was filtered off. The filtrate was concentrated under reduced pressure and purified by silica gel column chromatography using a gradient of 1–5 % MeOH in CH2Cl2 containing 1 % Et3N. The combined pure fractions were concentrated under reduced pressure, dissolved in CH2Cl2 (10 mL) and washed with 0.1 % aq HCl (2×10 mL) and water (10 mL) to remove excess Et3N. The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure to yield 4 a as a white solid (19 mg, 9 %). 1H NMR (400 MHz, CDCl3/CD3OD 1:3): δ=8.03 (dd, J=8.0, 1.8 Hz, 1 H), 7.45 (m, 1 H), 7.02–6.93 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.6, 159.4, 157.4, 135.2, 130.9, 119.7, 117.2, 116.8, 116.0 ppm; HRMS (ESI‐TOF): m/z: calcd for C9H7N4O2: 203.0569 [M−H]−; found: 203.0574; LC purity (254 nm): 97 %.
Characterization of Side Product 2‐(6‐Chloro‐4‐oxo‐4H‐benzo[e][1,3]oxazin‐2‐yl)guanidine (4 b)
To the reaction chamber (chamber A) of an oven‐dried, bridged two‐chamber system22 was added 2‐bromo‐4‐chlorophenol (208 mg, 1.00 mmol), cyanamide (43 mg, 1.0 mmol), Pd(OAc)2 (11 mg, 5 mol %) and CataCXium A (55 mg, 15 mol %). Mo(CO)6 (215 mg, 0.8 mmol) was added into the CO‐generation chamber (chamber B). The two chambers were capped with gastight caps, and the atmosphere was exchanged for nitrogen. 1,4‐Dioxane (3 mL) was added through the septa into each chamber, followed by the addition of Et3N (0.28 mL, 2.0 mmol) into chamber A and DBU (0.36 mL, 2.4 mmol) into chamber B. The two‐chamber system was immediately heated and vigorously stirred in a heating block at 65 °C for 20 h. After 20 h, the reaction mixture in chamber A was poured into iso‐hexane (30 mL) and the resulting precipitate was filtered off. The filtrate was concentrated under reduced pressure and purified by silica gel column chromatography using a gradient of 1–5 % MeOH in CH2Cl2 containing 1 % Et3N. The combined pure fractions were concentrated under reduced pressure, dissolved in CH2Cl2 (10 mL) and extracted with 0.1 % aq HCl (2×10 mL) to remove excess Et3N. Compound 4 b precipitated out in the acidic aqueous phase and was filtered off and dissolved in EtOAc. The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure to yield 4 b as a yellow solid (20 mg, 8 %). 1H NMR (400 MHz, [D6]DMSO): δ=8.92 (br s, 1 H), 7.68 (d, J=2.9 Hz, 1 H), 7.46 (br s, 1 H), 7.20 (dd, J=8.8, 2.9 Hz, 1 H), 6.68 (d, J=8.8 Hz, 1 H), 4.11 ppm (br s, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=171.6, 166.1, 163.0 (detected by HMBC), 132.8, 128.7, 124.7, 120.5 (two signals overlapping, confirmed by HMBC), 119.2 ppm; HRMS (ESI‐TOF): m/z: calcd for C9H6ClN4O2: 237.0179 [M−H]−; found: 237.0184; LC purity (254 nm): >98 %.
Synthesis of 4‐Methoxybenzyl 2′‐Iodophenyl Ether (6)
1‐(Bromomethyl)‐4‐methoxybenzene (0.41 mL, 2.8 mmol) was added to a stirred suspension of 2‐iodophenol (565 mg, 2.57 mmol) and potassium carbonate (1.80 g, 13 mmol) in DMF. The reaction mixture was stirred at 50 °C for 5 h. The crude mixture was separated between diethyl ether and water and the organic phase was washed with water (2×15 mL) and brine (3×15 mL) consecutively to remove DMF. The organic phase was dried over MgSO4, filtered and concentrated to yield 6 as a tan solid (543 mg, 62 %). Compound 6 is known44 and spectral data were in agreement with the proposed structure and matched those reported in the literature.
Synthesis of N‐Cyano‐2‐[(4‐methoxybenzyl)oxy]benzamide (7)
To the reaction chamber (chamber A) of an oven‐dried, bridged two‐chamber system22 was added 6 (107 mg, 0.31 mmol), cyanamide (19 mg, 0.45 mmol) and Pd(PPh3)4 (20 mg, 6 mol %). Mo(CO)6 (147 mg, 0.56 mmol) was added to the CO‐generation chamber (chamber B). The two chambers were capped with gastight caps, and the atmosphere was exchanged for nitrogen. 1,4‐Dioxane (3 mL) was added through the septa into each chamber, followed by the addition of Et3N (0.08 mL, 0.6 mmol) into chamber A and DBU (0.18 mL, 1.2 mmol) into chamber B. The two‐chamber system was immediately heated and vigorously stirred in a heating block at 65 °C for 20 h. After 20 h, the reaction mixture in chamber A was concentrated and 0.1 % HCl (10 mL) was added. The residue was extracted with EtOAc (3×10 mL) and the combined organic phases were dried over MgSO4, filtered and concentrated. The crude residue was purified by silica gel column chromatography using a gradient 0–5 % MeOH in CH2Cl2 to yield 7 as a yellow oil (40 mg, 45 %). R f=0.67 (SiO2, 5 % MeOH/CH2Cl2); 1H NMR (400 MHz, CDCl3): δ=9.74 (br s, 1 H; NH), 8.19 (m, 1 H), 7.58 (ddd, J=8.4, 7.3, 1.8 Hz, 1 H), 7.39–7.32 (m, 2 H), 7.19–7.11 (m, 2 H), 7.01–6.92 (m, 2 H), 5.17 (s, 2 H), 3.84 ppm (s, 2 H); 13C NMR (100 MHz, CDCl3, 50 °C): δ=164.0, 160.8, 157.5, 135.7, 133.2, 129.9, 126.5, 122.4, 118.4, 115.0, 113.7, 106.9, 72.4, 55.5 ppm; HRMS (ESI‐TOF): m/z: calcd for C16H13N2O3: 281.0926 [M−H]−; found: 281.0918; LC purity (254 nm): >98 %.
Deprotection of Compound 7 and Cyclization to Benzoxazinone 2 a
TFA (0.10 mL, 1.3 mmol) was added to a solution of compound 7 (37 mg, 0.13 mmol) in dichloromethane (3 mL) on an ice bath. The reaction mixture was stirred at RT for 1 h and then concentrated and co‐evaporated with toluene (3×5 mL) to give a brown residue. The crude residue was dissolved in 1,4‐dioxane (3 mL) followed by the addition of Et3N (0.02 mL, 0.1 mmol) and the resulting solution was stirred at 65 °C for 3 h. The crude mixture was purified by silica gel column chromatography using a gradient of 0–5 % MeOH/CH2Cl2 to yield 2 a as a white solid (17 mg, 80 %).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Magnus Fagrell (Fagrell Produktutveckling AB, Sweden) for technical support with the pressure measurements and Dr. Lisa D. Haigh (Imperial College London, UK) and Alfred Svan (Uppsala University) for assistance with accurate mass determination. The research was supported by Uppsala University.
L. Åkerbladh, S. Y. Chow, L. R. Odell, M. Larhed, ChemistryOpen 2017, 6, 620.
Contributor Information
Dr. Luke R. Odell, Email: luke.odell@orgfarm.uu.se
Prof. Mats Larhed, Email: mats.larhed@orgfarm.uu.se
References
- 1. Vitaku E., Smith D. T., Njardarson J. T., J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 2. Alper-Hayta S., Aki-Sener E., Tekiner-Gulbas B., Yildiz I., Temiz-Arpaci O., Yalcin I., Altanlar N., Eur. J. Med. Chem. 2006, 41, 1398–1404. [DOI] [PubMed] [Google Scholar]
- 3. Morrison R., Al-Rawi J. M. A., Jennings I. G., Thompson P. E., Angove M. J., Eur. J. Med. Chem. 2016, 110, 326–339. [DOI] [PubMed] [Google Scholar]
- 4. Ihmaid S. K., Al-Rawi J. M. A., Bradley C. J., Angove M. J., Robertson M. N., Eur. J. Med. Chem. 2012, 57, 85–101. [DOI] [PubMed] [Google Scholar]
- 5. Morrison R., Zheng Z., Jennings I. G., Thompson P. E., Al-Rawi J. M. A., Bioorg. Med. Chem. Lett. 2016, 26, 5534–5538. [DOI] [PubMed] [Google Scholar]
- 6. Pritchard K. M., Al-Rawi J., Bradley C., Eur. J. Med. Chem. 2007, 42, 1200–1210. [DOI] [PubMed] [Google Scholar]
- 7. Hedayatullah M., Lion C., Pailler J., J. Heterocycl. Chem. 1991, 28, 133–137. [Google Scholar]
- 8. Ried W., Oremek G., Guryn R., Erle H.-E., Chem. Ber. 1980, 113, 2818–2822. [Google Scholar]
- 9. Li P., Zhang X., Wu Y., Cheng M., Zhang D., Li G., Lin Z., Zhang X., Huang H., Tetrahedron Lett. 2015, 56, 4683–4685. [Google Scholar]
- 10. Pritchard K. M., Al‐Rawi J. M., Hughes A. B., Synth. Commun. 2005, 35, 1601–1611. [Google Scholar]
- 11. Raw S. A., O'Kearney-Mcmullan A. M., Graham M. A., Tetrahedron Lett. 2011, 52, 6775–6778. [Google Scholar]
- 12. Kokel B., Menichi G., Hubert-Habart M., Tetrahedron Lett. 1984, 25, 3837–3840. [Google Scholar]
- 13. Grigg R., Mutton S. P., Tetrahedron 2010, 66, 5515–5548. [Google Scholar]
- 14. Brennführer A., Neumann H., Beller M., Angew. Chem. Int. Ed. 2009, 48, 4114–4133; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4176–4196. [Google Scholar]
- 15. Morimoto T., Kakiuchi K., Angew. Chem. Int. Ed. 2004, 43, 5580–5588; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 5698–5706. [Google Scholar]
- 16. Schareina T., Zapf A., Cotté A., Gotta M., Beller M., Adv. Synth. Catal. 2010, 352, 1205–1209. [Google Scholar]
- 17. Ueda T., Konishi H., Manabe K., Org. Lett. 2012, 14, 3100–3103. [DOI] [PubMed] [Google Scholar]
- 18. Kondo T., Okada T., Mitsudo T., Organometallics 1999, 18, 4123–4127. [Google Scholar]
- 19. Morimoto T., Fuji K., Tsutsumi K., Kakiuchi K., J. Am. Chem. Soc. 2002, 124, 3806–3807. [DOI] [PubMed] [Google Scholar]
- 20. Brancour C., Fukuyama T., Mukai Y., Skrydstrup T., Ryu I., Org. Lett. 2013, 15, 2794–2797. [DOI] [PubMed] [Google Scholar]
- 21. Odell L. R., Russo F., Larhed M., Synlett 2012, 5, 685–698. [Google Scholar]
- 22. Nordeman P., Odell L. R., Larhed M., J. Org. Chem. 2012, 77, 11393–11398. [DOI] [PubMed] [Google Scholar]
- 23. Kaiser N. K., Hallberg A., Larhed M., J. Comb. Chem. 2002, 4, 109–111. [DOI] [PubMed] [Google Scholar]
- 24. Hermange P., Lindhardt A. T., Taaning R. H., Bjerglund K., Lupp D., Skrydstrup T., J. Am. Chem. Soc. 2011, 133, 6061–6071. [DOI] [PubMed] [Google Scholar]
- 25. Friis S. D., Taaning R. H., Lindhardt A. T., Skrydstrup T., J. Am. Chem. Soc. 2011, 133, 18114–18117. [DOI] [PubMed] [Google Scholar]
- 26. Hansen S. V. F., Ulven T., Org. Lett. 2015, 17, 2832–2835. [DOI] [PubMed] [Google Scholar]
- 27. Gockel S. N., Hull K. L., Org. Lett. 2015, 17, 3236–3239. [DOI] [PubMed] [Google Scholar]
- 28. Grushin V. V., Alper H., Organometallics 1993, 12, 3846–3850. [Google Scholar]
- 29. Åkerbladh L., Odell L. R., J. Org. Chem. 2016, 81, 2966–2973. [DOI] [PubMed] [Google Scholar]
- 30. Spencer J., Anjum N., Patel H., Rathnam R. P., Verma J., Synlett 2007, 16, 2557–2558. [Google Scholar]
- 31. Pri-bar I., Buchman O., J. Org. Chem. 1988, 53, 624–626. [Google Scholar]
- 32. Wu X.-F., Neumann H., Beller M., Chem. Eur. J. 2010, 16, 9750–9753. [DOI] [PubMed] [Google Scholar]
- 33. Dang T. T., Zhu Y., Ngiam J. S. Y., Ghosh S. C., Chen A., Seayad A. M., ACS Catal. 2013, 3, 1406–1410. [Google Scholar]
- 34. Amatore C., Azzabi M., Jutand A., J. Am. Chem. Soc. 1991, 113, 8375–8384. [Google Scholar]
- 35. Amatore C., Jutand A., Suarez A., J. Am. Chem. Soc. 1993, 115, 9531–9541. [Google Scholar]
- 36. Martinelli J. R., Clark T. P., Watson D. A., Munday R. H., Buchwald S. L., Angew. Chem. Int. Ed. 2007, 46, 8460–8463; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8612–8615. [Google Scholar]
- 37. Odell L. R., Sävmarker J., Larhed M., Tetrahedron Lett. 2008, 49, 6115–6118. [Google Scholar]
- 38. Wiéckowska A., Fransson R., Odell L. R., Larhed M., J. Org. Chem. 2011, 76, 978–981. [DOI] [PubMed] [Google Scholar]
- 39. Gautam P., Bhanage B. M., Catal. Sci. Technol. 2015, 5, 4663–4702. [Google Scholar]
- 40. Friis S. D., Lindhardt A. T., Skrydstrup T., Acc. Chem. Res. 2016, 49, 594–605. [DOI] [PubMed] [Google Scholar]
- 41. Bovonsombat P., Leykajarakul J., Khan C., Pla-on K., Krause M. M., Khanthapura P., Ali R., Doowa N., Tetrahedron Lett. 2009, 50, 2664–2667. [Google Scholar]
- 42. Schmidt B., Riemer M., Karras M., J. Org. Chem. 2013, 78, 8680–8688. [DOI] [PubMed] [Google Scholar]
- 43.In a related aminocarbonylation process it was shown that two CO molecules were released from the Mo(CO)6 complex; see Wannberg J., Larhed M., J. Org. Chem. 2003, 68, 5750–5753. [DOI] [PubMed] [Google Scholar]
- 44. Lane B. S., Sames D., Org. Lett. 2004, 6, 2897–2900. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary