

Abstract
The chemoenzymatic flow synthesis of enantiomerically pure captopril, a widely used antihypertensive drug, is accomplished starting from simple, inexpensive, and readily available reagents. The first step is a heterogeneous biocatalyzed regio‐ and stereoselective oxidation of cheap prochiral 2‐methyl‐1,3‐propandiol, performed in flow using immobilized whole cells of Acetobacter aceti MIM 2000/28, thus avoiding the use of aggressive and environmentally harmful chemical oxidants. The isolation of the highly hydrophilic intermediate (R)‐3‐hydroxy‐2‐methylpropanoic acid is achieved in‐line by using a catch‐and‐release strategy. Then, three sequential high‐throughput chemical steps lead to the isolation of captopril in only 75 min. In‐line quenching and liquid–liquid separation enable breaks in the workflow and other manipulations to be avoided.
Keywords: biocatalysis, captopril, flow chemistry, oxidation, reactor design
1. Introduction
The design and development of reproducible, environmentally benign and economical synthetic routes to drugs is of paramount importance in the pharmaceutical industry, and, in this context, continuous‐flow synthesis is receiving increasing attention.1, 2, 3 As a result, in recent years, many new flow‐based routes have been designed towards commercially available active pharmaceutical ingredients.4, 5
We herein report an efficient chemoenzymatic flow synthesis of captopril [(S)‐1‐[(S)‐3‐mercapto‐2‐methylpropanoyl]pyrrolidine‐2‐carboxylic acid, 5, Scheme 1]. Captopril is an angiotensin‐converting enzyme (ACE) inhibitor and was discovered and developed at E. R. Squibb & Sons Pharmaceuticals in the 1970s, and is still a widely prescribed drug for the treatment of hypertension.
Scheme 1.
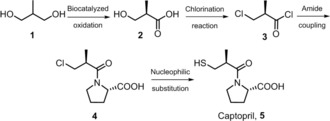
Four‐step chemoenzymatic synthesis of enantiomerically pure captopril.
Our flow‐based synthetic route to obtain captopril includes a first biocatalyzed heterogeneous regio‐ and stereoselective oxidation reaction starting from a cheap prochiral diol (2‐methyl‐1,3‐propandiol, 1)6, 7 followed, after isolation of carboxylic acid 2, by three sequential chemical steps. The biocatalyzed flow oxidation offers an efficient alternative for environmentally benign oxidations in answer to the critical economic and environmental issues that urgently demand greener, more atom‐efficient and scalable oxidation methods.8, 9 The oxidation was performed using Acetobacter aceti MIM 2000/28;10, 11, 12 this strain is able to completely convert the substrate into (R)‐3‐hydroxy‐2‐methylpropanoic acid (2) with an excellent ee (96–97 %) using a conventional batch‐mode biotransformation. To exploit all the advantages of performing (bio)catalyzed reactions in flow, in particular the high local concentration of the biocatalyst,13, 14, 15, 16 A. aceti has been immobilized as dried alginate‐entrapped cells, that are economical, easy to prepare and can be efficiently used in a packed bed column.17 Also, importantly, the use of whole cells in biocatalytic oxidation has the great advantage of not requiring an additional cofactor regeneration system.
The following three chemical steps that yield captopril under continuous flow conditions are depicted in Scheme 1. To achieve this continuous‐flow synthesis, the reactions have been designed so that the excess of reagents and reaction by‐products from each reaction were compatible with the downstream reactions, in order to perform steps in sequence without breaks in the workflow and manipulation. The risks associated with exothermic reactions and quenches (i.e., the use and neutralization of thionyl chloride) were mitigated owing to two of the advantages of continuous flow over batch synthesis: 1) only a small amount of material relative to the overall output of the system is utilized at any given time, and 2) the large surface‐area‐to‐volume ratios that allow precise reaction control through rapid heat transfer and mixing. Moreover, in‐line liquid–liquid extractions safely neutralized and removed problematic reagents and by‐products.
2. Results and Discussion
First, commercially available diol 1 (1 g L−1 in 20 mm acetate buffer, pH 6) was successfully oxidized with dried alginate beads of Acetobacter aceti MIM 2000/28 with high enantio‐ and regioselectivity. The immobilized cells were prepared according to a protocol recently reported by us,17 packed into a glass column (inner diameter 15 mm) and swelled by flowing buffer through the column until their volume tripled. No release of catalyst in the exiting flow stream was observed. The oxygen supply, essential for cofactor recycling, was ensured by means of a segmented air–liquid flow stream (for details, see the Experimental Section) formed before contact with the immobilized biocatalyst (Scheme 2).
Scheme 2.
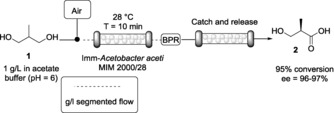
Biocatalyzed heterogeneous oxidation of prochiral 2‐methyl‐1,3‐propandiol (1) and in‐line purification of the product through a catch‐and‐release protocol.
The reaction reached 95 % conversion in only 10 min with an excellent ee (96–97 %), as determined by chiral gas chromatography. The stability of the biocatalyst under continuous work was assessed by performing the biotransformation under the conditions reported in the Experimental Section. Using 400 mg of immobilized biocatalyst, a total volume of approximately 50 mL was collected. Samples collected at different times were analyzed by HPLC and the results obtained in terms of conversion were comparable over time, indicating good stability under continuous work conditions. The maximum conversion (≈95 %) was observed for the first 35 mL of collected solution. Then, the conversion slowly decreased to 80 % in the following fractions. Therefore, the use of dried alginate beads in flow offers good stability over the time, giving 95 % conversion for approximately 10 h of continuous work. However, the buffer flow stream, even if characterized by low ionic strength (20 mm), might interfere with the alginate bead structure, which could slowly release the biocatalyst and consequently decrease the conversion.
Attempts to perform in‐line acidification and extraction with an organic solvent (e.g., EtOAc) using a liquid–liquid separator were not successful due to the high hydrophilicity of the obtained carboxylic acid 2. Consequently, we exploited a catch‐and‐release strategy using a column packed with Ambersep 900 OH resin that trapped the acid, leaving unreacted starting material in the exiting flow stream. Compound 2 was then released from the resin by using 1 n HCl and lyophilized.
Then, starting from the isolated compound 2, we studied the chlorination reaction with thionyl chloride with heating in a pressurized system to effect both the formation of the acid chloride and the direct chlorination of the primary alcohol (Scheme 3). A 200 psi backpressure regulator (BPR) was used to prevent outgassing. The possibility of conducting such an exothermic reaction under heating conditions demonstrates one of the operational advantages and capabilities of continuous flow synthesis over analogous batch processes that require cooling during the addition of thionyl chloride.
Scheme 3.
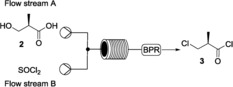
The chlorination reaction using a 10 mL reactor coil. BPR: 200 psi. Solvent, temperature and residence time were optimized as summarized in Table 1.
The reaction was tested in anhydrous toluene, THF, and CH2Cl2; the reaction proceeded similarly with all the selected solvents. Therefore, toluene was selected and a 1 m solution of compound 2 was used. An excess of thionyl chloride (3 equiv, 3 m solution in toluene) was necessary to drive the reaction to completion. A catalytic amount of imidazole and 10 % DMF (v/v) were added to the solution of 2. The use of DMF has a double effect: first, it favored the reaction due to the formation of the Vilsmeier reagent and, second, it increased the solubility of the starting material in toluene. Different reaction conditions were then tested and they are summarized in Table 1. The outcome was monitored by 1H NMR spectroscopy after evaporation of the solvent. If no complete conversion was observed, the starting material 2 and/or the acid chloride (without substitution of the hydroxy group) were identified as the only impurities. We also tried the reaction using only DMF to prepare the solution of 2 (Table 1, entries 6 and 7) and observed complete conversion at 100 °C in 30 min.
Table 1.
Conditions tested for the chlorination reaction.[a]
| Entry | T [°C] | t r [min] | Yield 3 [b] [%] |
|---|---|---|---|
| 1[c] | 85 | 60 | 80 |
| 2[c] | 100 | 60 | 100 |
| 3[c] | 110 | 30 | 100 |
| 4[c] | 125 | 15 | 70 |
| 5[c] | 150 | 15 | 70 |
| 6[d] | 100 | 30 | 100 |
| 7[d] | 100 | 15 | 85 |
[a] See the Experimental Section for the reaction conditions. [b] Conversions were determined by 1H NMR, after evaporation of the solvent. [c] Flow stream A: 2 (1 m) in toluene/DMF (9:1) containing 0.1 equiv imidazole; flow stream B: SOCl2 in toluene (3 m). [d] Flow stream A: 2 in DMF containing 1 % imidazole; flow stream B: neat SOCl2.
Intermediate 3 was then submitted to amide coupling with l‐proline (Scheme 4). Because the final goal was to have all the synthetic steps in‐line, without a break in the workflow, we decided to use for this reaction the same solvent as in the previous step.
Scheme 4.
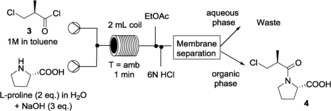
The coupling reaction using a 2 mL reactor coil. Pressure: 200 psi.
Thus, 3 was dissolved in toluene (1 m solution), mixed with an aqueous basic (NaOH, 3 equiv) solution of l‐proline (2 m solution, 2 equiv) and flowed into a 2 mL perfluoroalkoxy alkane (PFA) reactor coil. The reaction was performed at room temperature, without cooling, and was rapid, giving full conversion within only 1 min of residence time. In‐line acidification to pH 2 followed by a liquid–liquid separation using a Zaiput separator were performed, affording the desired product 4.
The final step was the nucleophilic substitution of the chlorine with the thiol group (Scheme 5). Again, we used toluene as an organic solvent and the reaction was studied in a biphasic system toluene/degassed water, used to reduce the possible formation of oxidized byproducts.
Scheme 5.
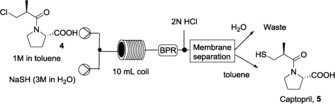
The nucleophilic substitution using a 10 mL reactor coil. Pressure: 200 psi.
To perform the substitution, an excess of NaSH (3 equiv) was efficiently used. An in‐line acidification followed by a liquid–liquid separation allowed captopril to be obtained from the organic phase that was evaporated and analyzed by 1H NMR spectroscopy. Temperature and residence time were optimized and the results are summarized in Table 2. The reaction was complete in 30 min at 125 °C (Table 2, entry 4).
Table 2.
Conditions tested for the nucleophilic substitution.[a]
| Entry | T [°C] | t r [min] | Yield 5 [b] [%] |
|---|---|---|---|
| 1 | 50 | 60 | 70 |
| 2 | 80 | 60 | 90 |
| 3 | 100 | 60 | 93 |
| 4 | 125 | 30 | 100 |
| 5 | 125 | 15 | 75 |
[a] See the Experimental Section for the reaction conditions. [b] Conversions were determined by 1H NMR spectroscopy after evaporation of the solvent.
Once the reactions were separately optimized, we next developed a continuous multistep production of captopril that would be sufficiently robust for multiple hours of continuous work (Scheme 6). We assembled the system such that the product stream from the chlorination reaction, obtained using the conditions reported in Table 1 (entry 3), was immediately submitted to the coupling reaction in a toluene/water biphasic system, as described above. To this end, first we evaluated the amount of NaOH necessary to neutralize the excess acid from the chlorination step. The T‐junction was cooled to 0 °C for the neutralization and, in this way, the uncontrolled exothermic neutralization and the release of large quantities of HCl vapors were avoided. In this set‐up, the residence time for the coupling reaction in the 2 mL reactor coil was approximately 3 min (Scheme 6). After separation of the two phases, the exiting organic solution of compound 4 was collected in a vial, acting as a reservoir, and then directly used for the substitution reaction. The solution was pumped into a 10 mL reactor coil after mixing with the solution of NaSH in degassed water and heated at 125 °C.
Scheme 6.
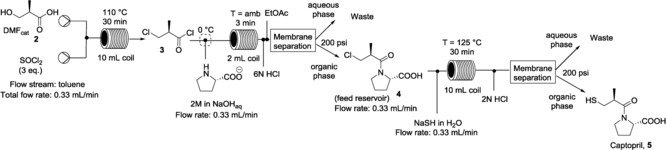
Three‐step flow chemical synthesis of captopril. BPR: 200 psi.
After in‐line acidification, extraction and evaporation of the solvent, the resulting crude material was purified by column chromatography to yield pure captopril as a white solid. The 1H NMR spectrum and the optical rotation matched those reported in the literature.18
3. Conclusions
The multistep flow synthesis presented here enabled captopril to be obtained in an overall yield of 50 % after crystallization. Biocatalytic oxidation facilitated the conversion of a prochiral substrate into a chiral intermediate with high enantiomeric excess. Three chemical transformations were performed without isolation of the intermediates. The separation of co‐products, by‐products, and excess reagents was achieved in‐line. These results clearly highlight the benefits of performing multistep chemical synthesis in a flow environment. As a further advantage, our synthetic protocol benefits from the use of an environmentally friendly biocatalytic oxidation, avoiding the use of toxic chemical oxidants. The bioprocess might be further linked to the chemical transformations by optimizing the extraction of acid (2) with organic solvents, for example, using specific organic‐acid‐complexing carriers.19 In light of a future scale‐up, whereas none of the chemical steps have foreseen limitations, further efforts should be directed toward increasing the productivity of the first biocatalyzed step, which at present represents a bottleneck in the process. The new proposed protocol for the synthesis of captopril represents a powerful integration of biocatalysis and flow‐chemistry technology.
Experimental Section
All reagents and solvents were purchased from Sigma–Aldrich. The continuous flow reactions were performed using a commercial R2C/R4 flow reactor (Vapourtec, Bury St. Edmunds, Suffolk, UK) equipped with Omnifit glass columns (15 mm i.d. × 100 mm length) and PFA reactor coils (2 and 10 mL, respectively). The R2C unit is the pumping unit that contains two adapted Knauer pumps, which are able to pump highly concentrated and corrosive acids. R4 is the heating unit with four heating positions. The additional HPLC pumps necessary to perform the overall synthesis were provided by another R2+/R4 flow reactor (Vapourtec) and by two external pumps (ThalesNano). The temperature sensor sits on the wall of the PFA tubing. The pressure was controlled by using two 100 psi BPRs. In‐line liquid–liquid extractions were performed using a Zaiput separator. 1H NMR and 13C NMR spectra were recorded with a Varian Mercury 300 (300 MHz) spectrometer. Chemical shifts (δ) are expressed in ppm, and coupling constants (J) are expressed in Hz. The molar conversion of the biotransformation was determined by HPLC analysis using a Luna NH2 100 Å column (250 mm×4.6 mm, particle size 5 μm, Phenomenex, Aschaffenburg, Germany) and a Biorad refractor index detector with an acidic aqueous KH2PO4 buffer (20 mm, pH 2.7) as the mobile phase (flow rate 0.2 mL min−1). The samples (40 μL) were injected as soon as collected and without further treatment. The enantiomeric composition of 2 was determined by gas chromatographic analysis of the corresponding methyl ester [methyl (R)‐3‐hydroxy‐2‐methylpropionate: t r=3.6 min], obtained after treatment with diazomethane, using a chiral capillary column (diameter 0.25 mm, length 25 m, DMePeBeta‐CDX‐PS086, MEGA, Legnano, Italy). Optical rotation determinations were performed using a Jasco P‐1010 spectropolarimeter coupled with a Haake N3‐B thermostat. MS analyses were performed on a Varian 320‐MS triple quadrupole mass spectrometer with an electrospray ionization (ESI) source. Microanalyses (C, H, N) were within ±0.4 % of theoretical values.
Strain Preparation
The A. aceti MIM 2000/28 strain was routinely maintained on GYC agar plates [glucose (50 g L−1), yeast extract (10 g L−1), CaCO3 (30 g L−1), agar (15 g L−1), pH 6.3] at 28 °C. Strain was inoculated into 100 mL Erlenmeyer baffled flask containing GLY medium [yeast extract (10 g L−1) and glycerol (25 g L−1), pH 5; 20 mL]. After growth for 24 h (shaking at 150 rpm, 28 °C), the liquid culture was entirely used to inoculate a 1 L Erlenmeyer baffled flask containing GLY medium (150 mL). Flasks were grown for 24 h at 28 °C, with shaking at 150 rpm.
Preparation of Dry Alginate Beads
Gel beads were prepared by ionotropic gelation, by following a protocol previously developed by us:17 a 4 % (w/v) sodium alginate solution was prepared in distilled water and stirred until a homogeneous clear solution was formed. The solution was allowed to settle for 2 h in order to eliminate the air bubbles. The alginate solution was then gently mixed in a 1:1 (w/w) ratio with a suspension of A. aceti cells (40 OD mL−1) in sodium acetate buffer (20 mm, pH 6). The resulting mixture was then pumped dropwise into a slightly agitated CaCl2 solution (0.2 m). Calcium alginate beads were agitated for 20 min, then filtered, washed with deionized water and dried at 25 °C for 16 h.
Synthesis of (R)‐3‐Hydroxy‐2‐methylpropanoic Acid (2)
Dry alginate beads (400 mg) were packed into a glass column (i.d. 15 mm) and swelled until their volume tripled by flowing acetate buffer (20 mm, pH 6) through the column (flow rate: 400 μL min−1, 60 min). The final volume of the bed was 5.1 mL. Air was delivered at 17 psi; its flow was measured using the method described in Ref. 20. To ensure a constant flow, a BRP (40 psi) was applied before the air tank. An aqueous solution of 1 (1 g L−1, 40 mL) was pumped at 60 μL min−1, joining the airflow at the T‐junction, before entering the column in which the oxidation occurs in approximately 10 min. The exiting flow stream was directed into a decompression column. A BPR (5 psi) ensured a constant and controlled flow of aqueous phase leaving the column. The aqueous stream was directed into a column filled with Ambersep 900 OH resin (2 g) and, after washing the column with water (20 mL, 0.5 mL min−1), the trapped acid was released by flowing HCl (1 n, 5 mL). After lyophilization, compound 2 was isolated as pale yellow oil (42 mg).
To obtain a larger amount of 2, 1.6 g of alginate beads were used, with a reactor volume of 20 mL. At this scale, 170 mg of compound 2 was isolated (91 % yield). [α] =−11.55 (c=1.00 in EtOH); 1H NMR (300 MHz, CDCl3): δ = 1.22 (d, J=7.4 Hz, 3 H), 2.10 (s, 1 H), 2.68–2.80 (m, 1 H), 3.75 (d, J=6.0 Hz, 2 H), 5.70 ppm (br s, 1 H); 13C NMR (75 MHz, CDCl3): δ = 13.2, 41.6, 64.0, 180.1 ppm; MS (ESI): m/z: 102.9 [M–H]−;21 HPLC analysis: 1, t r=22 min; 2, t r=18 min.
Synthesis of (R)‐3‐Chloro‐2‐methylpropanoyl Chloride (3)
A solution of 2 (52 mg, 0.5 mmol) was prepared in anhydrous toluene (410 μL). Imidazole (3.5 mg, 0.1 equiv) and DMF (50 μL) were added to the solution. A second solution of thionyl chloride (125 μL, 3.5 equiv) was prepared in anhydrous toluene (375 μL). The two solutions were mixed into a T‐piece and flowed through a 10 mL reactor coil according to the conditions reported in Table 1. A 200 psi backpressure regulator was applied to the system. The exiting solution was collected, the solvent was evaporated under reduced pressure to yield 3 as a crude oil, which was analyzed by 1H NMR spectroscopy. 1H NMR (300 MHz, CDCl3): δ=1.42 (d, 3 H), 3.20–3.31 (m, 1 H), 3.66–3.80 ppm (m, 2 H).
Synthesis of (S)‐1‐[(S)‐3‐Chloro‐2‐methylpropanoyl]pyrrolidine‐2‐carboxylic Acid (4)
A solution of compound 3 (70 mg, 0.5 mmol) in toluene (0.5 mL) was prepared. A second solution of l‐proline (115 mg, 2 equiv) and NaOH (60 mg, 3 equiv) was prepared in water (0.5 mL). The two solutions were flowed at a total rate of 2 mL min−1 through a 2 mL reactor coil, that had been washed with a mixture of toluene and water, at room temperature. The aqueous phase was acidified to pH 2 by introducing a flow of HCl (6 n, 1 mL min−1) and the resulting biphasic system was mixed using a T‐junction into an EtOAc stream (1 mL min−1) to perform in‐line extraction. The two phases were separated in‐line using a Zaiput liquid–liquid separator. A 200 psi backpressure was applied to the system. The organic solvent was dried over anhydrous Na2SO4 and evaporated under reduced pressure to yield 4 as a crude solid, which was analyzed by 1H NMR spectroscopy. [α] =−103.6 (c=1 in EtOH); 1H NMR (300 MHz, CDCl3): δ=1.24 (d, 3 H, J=7.0), 2.02–2.18 (m, 4 H), 2.96–3.08 (m, 1 H), 3.44–3.51 (m, 1 H), 3.57–3.72 (m, 2 H), 3.81 (t, 1 H, J=10.4), 4.62–4.69 (m, 1 H), 11.30 (br s, 1 H); 13C NMR (75 MHz, CDCl3): δ=15.8, 24.9, 27.7, 41.3, 45.8, 47.7, 59.6, 173, 175 ppm; MS (ESI): m/z: 217.9 [M–H]−; elemental analysis calcd (%) for C9H14ClNO3: C 49.21, H 6.42, N 6.38; found: C 49.00, H 6.31, N 6.50.
Synthesis of (S)‐1‐[(S)‐3‐Chloro‐2‐methylpropanoyl]pyrrolidine‐2‐carboxylic Acid (Captopril, 5)
A solution of compound 4 (22 mg, 0.1 mmol) in toluene (0.1 mL) was prepared. A second solution of NaSH (21 mg, 3 equiv) was prepared using degassed water (0.1 mL). The two solutions were flowed through a 10 mL reactor coil. A 200 psi backpressure was applied to the system. Reaction time and temperature were optimized, as reported in Table 2. The aqueous phase was acidified at pH 2 by introducing a flow of HCl (2 n) and the resulting biphasic system was mixed using a T‐junction into an EtOAc stream to perform in‐line extraction. The two phases were separated in‐line using a Zaiput liquid–liquid separator. The organic solvent was dried over anhydrous Na2SO4 and evaporated under reduced pressure to yield 5 as a pale yellow crude solid, which was analyzed by 1H NMR spectroscopy. [α] =−128.5 (c=1 in EtOH); lit=−129.4 (c=1.35 in EtOH at 22 °C);18 1H NMR (300 MHz, CDCl3): δ=1.24 (d, J=7.0 Hz, 3 H), 1.58 (t, J=9.2 Hz, 1 H), 2.02–2.18 (m, 4 H), 2.43–2.51 (m, 1 H), 2.79–2.90 (m, 2 H), 3.55–3.71 (m, 2 H), 4.62–4.69 (m, 1 H), 11.30 ppm (br s, 1 H); 13C NMR (75 MHz, CDCl3): δ=17.0, 20.8, 24.7, 27.4, 42.5, 47.4, 59.2, 173.0, 175.0 ppm; MS (ESI): m/z: 215.9 [M–H]−; elemental analysis calcd (%) for C9H15NO3S: C 49.75, H 6.96, N 6.45; found: C 49.60, H 6.89, N 6.52.
Three‐Step Continuous‐Flow Synthesis of Captopril (5)
A solution of 2 (156 mg, 1.5 mmol) in anhydrous toluene (1.23 mL) was prepared. Imidazole (10.5 mg, 0.1 equiv) and DMF (150 μL) were added to the solution. A second solution of thionyl chloride (375 μL, 3.5 equiv) was prepared using anhydrous toluene (1.12 mL). The two solutions were flowed through a 10 mL coil reactor that was maintained at 110 °C, with a residence time of 30 min (total flow rate 0.33 mL min−1). Then, the exiting flow was merged at a T‐junction with an aqueous solution (1.5 mL) of l‐proline (2 equiv) and NaOH (7 equiv). This then entered a 2 mL reactor coil, maintained at room temperature with a residence time of 3 min (total flow rate 0.66 mL min−1). The aqueous phase was acidified to pH 2 by adding a flow stream of HCl (6 n, 0.33 mL min−1) and the resulting biphasic system was mixed using a T‐junction into an EtOAc stream (0.33 mL min−1) to perform an in‐line extraction. The two phases were separated in‐line using a Zaiput liquid–liquid separator. The organic phase was collected in a vial acting as a substrate reservoir for the final nucleophilic substitution reaction. The organic phase was pumped and mixed with a solution of NaSH (c=1.5 m, 3 equiv) in degassed water, and entered a 10 mL coil reactor maintained at 125 °C for 30 min (total flow rate 0.66 mL min−1). A 200 psi backpressure was added to the whole system. The exiting flow was then acidified to pH 1 with HCl (2 n) and a continuous extraction with the Zaiput liquid–liquid separator was performed. The organic solvent was dried over anhydrous Na2SO4 and evaporated under reduced pressure to yield a crude material that was purified by column chromatography (dichloromethane/methanol 98:2) to yield captopril, which was crystallized from ethyl acetate/hexane (1:1, total volume 1 mL; 160 mg, 50 %).
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank the Cariplo Foundation (Project 2014‐0568: INBOX, Innovative Biocatalytic Oxidations) for funding.
V. De Vitis, F. Dall'Oglio, A. Pinto, C. De Micheli, F. Molinari, P. Conti, D. Romano, L. Tamborini, ChemistryOpen 2017, 6, 668.
Contributor Information
Dr. Diego Romano, Email: diego.romano@unimi.it
Dr. Lucia Tamborini, Email: lucia.tamborini@unimi.it
References
- 1. Tsubogo T., Oyamada H., Kobayashi S., Nature 2015, 520, 329–332. [DOI] [PubMed] [Google Scholar]
- 2. Webb D., Jamison T. F., Chem. Sci. 2010, 1, 675–680. [Google Scholar]
- 3. Hartman R. L., McMullen J. P., Jensen K. F., Angew. Chem. Int. Ed. 2011, 50, 7502–7519; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7642–7661. [Google Scholar]
- 4. Baumann M., Baxendale I. R., Beilstein J. Org. Chem. 2015, 11, 1194–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Porta R., Benaglia M., Puglisi A., Org. Process Res. Dev. 2016, 20, 2–25. [Google Scholar]
- 6. Molinari F., Gandolfi R., Villa R., Urban E., Kiener A., Tetrahedron: Asymmetry 2003, 14, 2041–2043. [Google Scholar]
- 7. Brenna E., Cannavale F., Crotti M., De Vitis V., Gatti F. G., Migliazza G., Molinari F., Parmeggiani F., Romano D., Santangelo S., ChemCatChem 2016, 8, 3796–3803. [Google Scholar]
- 8. Hollmann F., Arends I. W. C. E., Buehler K., Schallmey A., Bühler B., Green Chem. 2011, 13, 226–265. [Google Scholar]
- 9. Romano D., Villa R., Molinari F., ChemCatChem 2012, 4, 739–749. [Google Scholar]
- 10. Borrometi A., Romano A., Gandolfi R., Sinisterra J. V., Molinari F., Tetrahedron: Asymmetry 2002, 13, 2345–2349. [Google Scholar]
- 11. Romano A., Gandolfi R., Nitti P., Rollini M., Molinari F., J. Mol. Catal. B 2002, 17, 235–240. [Google Scholar]
- 12. Romano D., Contente M., Granato T., Remelli W., Zambelli P., Molinari F., Monatsh. Chem. 2013, 144, 735–737. [Google Scholar]
- 13. Newman S. G., Jensen K. F., Green Chem. 2013, 15, 1456–1472. [Google Scholar]
- 14. Tamborini L., Romano D., Pinto A., Contente M., Iannuzzi M. C., Conti P., Molinari F., Tetrahedron Lett. 2013, 54, 6090–6093. [Google Scholar]
- 15. Tamborini L., Romano D., Pinto A., Bertolani A., Conti P., Molinari F., J. Mol. Catal. B 2012, 84, 78–82. [Google Scholar]
- 16. Planchestainer M., Contente M. L., Cassidy J., Molinari F., Tamborini L., Paradisi F., Green Chem. 2017, 19, 372–375. [Google Scholar]
- 17. Zambelli P., Tamborini L., Cazzamalli S., Pinto A., Arioli S., Balzaretti S., Plou F. J., Fernandez-Arrojo L., Molinari F., Conti P., Romano D., Food Chem. 2016, 190, 607–613. [DOI] [PubMed] [Google Scholar]
- 18. Bashiardes G., Davies S. G., Tetrahedron Lett. 1987, 28, 5563–5564. [Google Scholar]
- 19. León R., Molinari F., Prazeres D. M. F., Cabral J. M. S., J. Chem. Technol. Biotechnol. 2000, 75, 617–624. [Google Scholar]
- 20. Wang S., Wang Z., Yang L., Dong J., Chi C., Sui D., Wang Y., Ren J., Hung M., Jiang Y., J. Mol. Catal. A 2007, 264, 60–65. [Google Scholar]
- 21. Tomaszewski B., Schmid A., Buehler K., Org. Process Res. Dev. 2014, 18, 1516–1526. [Google Scholar]