

Abstract
Cells lining the kidney proximal tubule (PT) respond to acute changes in glomerular filtration rate and the accompanying fluid shear stress (FSS) to regulate reabsorption of ions, glucose, and other filtered molecules and maintain glomerulotubular balance. Recently, we discovered that exposure of PT cells to FSS also stimulates an increase in apical endocytic capacity (Raghavan et al. PNAS, 111:8506–8511, 2014). We found that FSS triggered an increase in intracellular Ca2+ concentration ([Ca2+]i) that required release of extracellular ATP and the presence of primary cilia. In this study, we elucidate steps downstream of the increase in [Ca2+]i that link FSS‐induced calcium increase to increased apical endocytic capacity. Using an intramolecular FRET probe, we show that activation of Cdc42 is a necessary step in the FSS‐stimulated apical endocytosis cascade. Cdc42 activation requires the primary cilia and the FSS‐mediated increase in [Ca2+]i. Moreover, Cdc42 activity and FSS‐stimulated endocytosis are coordinately modulated by activators and inhibitors of calmodulin. Together, these data suggest a mechanism by which PT cell exposure to FSS is translated into enhanced endocytic uptake of filtered molecules.
Keywords: Calcium, calmodulin, cilia, cubilin, endocytosis, fluid shear stress, megalin, proximal tubule
Introduction
Renal epithelial cells that line the kidney nephron encounter wide fluctuations in the magnitude of FSS due to frequent changes in glomerular filtration rate (GFR). FSS produced by the movement of the glomerular ultrafiltrate through the renal tubule acutely regulates PT ion transport. This mechanism prevents loss of solute after increases in GFR and maintains the adequate distal delivery of sodium and fluid when GFR is reduced. PT cells respond to mechanical forces, including FSS, with immediate downstream signal transduction events as well as long‐term adaptation mechanisms that enable them to fine‐tune their cytoskeleton structure, intercellular junctions, and cell‐substratum interactions to adjust to the mechanical challenge (Weinbaum et al. 2010; Raghavan and Weisz 2015).
High tubular flow rates are common complications of kidney diseases, and FSS is instrumental in coordinating biochemical signaling pathways that regulate cell morphology, migration and growth (Rohatgi and Flores 2010). It is well recognized that the primary cilia present on renal epithelial cells acts as a flow sensor that couples upstream flow stimuli to rapid increases in [Ca2+]i (Praetorius 2015). Mutations in ciliary proteins are associated with deregulated FSS‐mediated [Ca2+]i responses and with the development of polycystic kidney disease, suggesting that failure to properly sense biomechanical signals may contribute to cyst development (Yoder 2007; Kotsis et al. 2013). However, despite considerable evidence of the crucial role of FSS in regulating renal epithelial integrity and function, the molecular mechanisms by which FSS is transduced into downstream signals remain unclear (Delling et al. 2016).
An essential function of the PT is the clathrin‐dependent apical uptake of filtered low molecular weight proteins and albumin by the multiligand receptors megalin and cubilin (Eshbach and Weisz 2017). We previously reported that exposure of kidney PT cells to FSS causes a rapid and sustained increase in endocytic capacity (Raghavan et al. 2014). FSS also triggered an increase in [Ca2+]i in these cells that required the presence of primary cilia and extracellular release of ATP. Our findings led us to hypothesize that flow‐mediated Ca2+ signaling initiates a downstream cascade that coordinates reorganization of the cytoskeleton to stimulate endocytosis. However, how increased [Ca2+]i ultimately stimulates endocytic uptake in PT cells is not known.
Cdc42 is a member of the Rho family of GTPases that function to regulate numerous cellular activities, including cytoskeletal dynamics and endocytosis. Cdc42 is known to be activated in response to increased [Ca2+]i and has been implicated in endothelial cell responses to FSS (Li et al. 1999; Tzima et al. 2003). The subapical domain of PT cells is actin‐rich and we previously hypothesized that changes in actin dynamics might regulate apical endocytosis in response to FSS (Raghavan et al. 2014). In this study, we use two complementary approaches to investigate the underlying intracellular signaling pathway that couples FSS to apical endocytosis. In the first, we used sensitive fluorescence resonance energy transfer (FRET) probes to monitor activation of Cdc42 in PT cells upon exposure to laminar FSS or pharmacologic modulators of [Ca2+]i‐dependent signaling pathways. In a parallel approach, we measured the effects of these maneuvers on apical endocytosis. Our data suggest a mechanism by which Cdc42 alters actin dynamics to enhance PT endocytic uptake in response to FSS.
Materials and Methods
Cell Culture
Opossum kidney OK cells (Didelphis virginiana, adult female, kidney cortex) were originally provided by Moshe Levi (University of Colorado) and cultured in DMEM/F12 Hamm with 10% FBS. For most experiments, 4.5 × 105 cells were plated in Ibidi μ‐slide I 0.4 or 0.8 collagen‐coated chambers (Ibidi). For confocal imaging studies, 3 × 105 cells/well were plated in Ibidi μ‐slide VI 0.4 chambers. Medium was replaced twice daily for 3 days before experiments. For FRET and endocytosis experiments, cells were pretreated with drugs [ML141 (Tocris‐4266, 10 μmol/L); Calp3 (Tocris‐2321, 5 μmol/L; ryanodine (Tocris‐1329, 25 μmol/L), or W13 (Tocris‐0361, 60 μmol/L), all prepared at ≥1000× stocks in DMSO or water as recommended] for 1 h in culture medium prior to initiation of FSS. Cells were transfected with Raichu plasmids immediately prior to plating using Lipofectamine 3000 reagent (Invitrogen) according to manufacturer guidelines. Where indicated, cells were deciliated by incubation for 3 h in 30 mmol/L ammonium sulfate in FBS‐supplemented medium as previously described (Raghavan et al. 2014) and allowed to recover overnight where indicated.
Measurement of Cdc42 activation using FRET
The Raichu‐Cdc42 intramolecular FRET probe has been described previously (Itoh et al. 2002; Nakamura et al. 2006). In brief, Raichu‐Cdc42 consists of YFP, the CRIB domain of the Cdc42 effector protein PAK1, Cdc42, CFP, and the CAAX box of K i‐Ras to enable membrane tethering, all linked via spacer sequences. The probe is designed so that the signal‐induced conformational change in the probe brings CFP into close proximity with YFP, which results in a decrease in CFP fluorescence and a concomitant increase in YFP fluorescence.
Cells expressing the Raichu‐Cdc42 probe were imaged using a Nikon Ti‐E microscope (Nikon) equipped with a Z‐driven piezo motor. Images were acquired using the Nikon A1 confocal unit through a 60 × 1.45 N/A objective (Nikon). Data were acquired using Nikon Elements Software (Nikon Corporation). Fluorescent proteins were excited with 440‐nm (CFP) and 514‐nm (YFP) lasers (Melles Griot). Six to twenty cells [regions of interest (ROIs)] that had comparable starting ratios of YFP/CFP were selected prior to initiating the experiment. For FRET, an excitation wavelength of 440 ± 10 nm was used to excite CFP, and YFP and CFP emissions were collected using 535 ± 12 nm (YFP) and 480 ± 15 nm (CFP) filters. A syringe pump was connected to the cell‐seeded Ibidi μ‐slide I 0.4 microchannel slides and a flow rate of 111 μL/min was used to generate unidirectional laminar shear stress of ~0.1 dyne/cm2 according to the manufacturer's instructions. Typically, the experimental timeline was as follows: baseline images (static condition images) were acquired every minute for 10 min, after which the cells were exposed to FSS for 30 min. Subsequently, the flow was stopped and the cells were imaged for an additional 20 min under static conditions. NIS software was used to determine the fluorescence intensity for each ROI as well as the background. Further analysis was performed with Excel software (Microsoft). YFP divided by CFP fluorescence ratios were obtained by taking the fluorescence ratio of background‐normalized YFP and background‐normalized CFP signal intensities. These values were normalized against basal signal intensities (before initiation of flow). The average and standard deviation were calculated from the YFP/CFP ratios for at least three independent experiments for each condition and statistical significance assessed by two‐way ANOVA.
Quantitation of endocytic uptake in response to FSS
OK cells cultured in Ibidi μ‐slide I 0.4 chambers were exposed to 0.1 dyne/cm2 laminar FSS for 1 h at 37°C in the presence of 40 μg/mL AlexaFluor 647‐albumin (Molecular Probes) in DMEM/F12 Hamm medium supplemented with 25 mmol/L HEPES, pH 7.5, then washed five times with ice cold PBS supplemented with 1 mmol/L Ca2+ and 1 mmol/L Mg2+ (PBS‐CM). Cells were solubilized in 200 μL 50 mmol/L Tris‐HCl, 1% (v/v) IGEPAL, 0.4% deoxycholate, 62.5 mmol/L EDTA, pH 8.0 at ambient temperature for 30 min and fluorescence quantified using a Glomax‐Multi Detection spectrofluorimeter (Promega). Background values (lysate buffer) were <10% of the lowest experimental values and were subtracted prior to data analysis.
Confocal imaging of albumin uptake
For experiments measuring effects of fluid shear stress (FSS) on endocytosis, cells in Ibidi μ‐slide VI 0.4 six‐well chambers were perfused or maintained under static conditions for 1 h at 37°C. After washing, cells were fixed with 4% paraformaldehyde for 20 min at ambient temperature, rinsed twice with PBS‐CM, and 50 μL of ProlongGold (Invitrogen) added to each well. The chambers were imaged using a Leica TCS SP5 confocal microscope and maximum intensity projections of stacks were processed identically using Fiji and Adobe Photoshop.
[Ca2+]i measurements
OK cells cultured on collagen‐coated Ibidi μ‐slide I (0.8‐mm clearance) chambers were loaded with Fura‐2 AM (2.5 μmol/L) in HEPES‐buffered saline (HBS: 135 mmol/L NaCl, 4.5 mmol/L KCl, 2.5 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L HEPES, 10 mmol/L glucose, pH 7.4) for 20 min at 37°C, rinsed with HBS, and incubated for 10 min to de‐esterify the dye at ambient temperature. Ibidi μ‐slides with Fura‐2 loaded cells were placed in a stage top incubator (Tokai Hit) set at 37°C for 10 min. Cells were imaged with a Nikon Eclipse Ti upright live cell microscope using an ORCA‐Flash 2.8 camera (Hamamatsu). Cells were excited with a PhotoFluor II metal halide light source (89 North) connected to a Lambda 10‐3 filter wheel system (Sutter Instruments) with 340‐ and 380‐nm filters (Chroma Technology). To generate a FSS of 0.1 dyne/cm2, Ibidi μ‐slides were perfused with an infusion pump (Harvard Apparatus) at a flow rate of 400 μL/min. Temperature was maintained at 37°C throughout the experiment using an inline heater (SH27B, Warner Instruments) controlled by a dual channel bipolar temperature controller (TC‐344B, Warner Instruments).
Baseline measurements were acquired every 10 sec for 1 min before initiation of FSS and then every 5 sec, 10 sec, or 30 sec. In situ calibration with ionomycin (Alomone) was conducted to define the Ca2+ calibration parameters under zero and saturating [Ca2+]i concentrations. Cells were perfused with Ca2+‐free buffer (135 mmol/L NaCl, 4.5 mmol/L KCl, 2 mmol/L EGTA, 1 mmol/L MgCl2, 10 mmol/L HEPES, 10 mmol/L glucose, pH 7.4) containing 2.5 μmol/L ionomycin to estimate ratios at zero concentration of [Ca2+]i. Subsequently, cells were perfused with HBS with 2.5 μmol/L ionomycin to estimate Fura‐2 ratios at saturating levels of [Ca2+]i. NIS Elements (Nikon) was used to select individual cells (6–10 per experiment) and the ratio of 340:380‐nm emission intensity was calculated at each time point. The [Ca2+]i values of cells at each time point was normalized to the initial [Ca2+]i value (at t = 0 sec) to estimate the fold changes in [Ca2+]i over time. [Ca2+]i changes across experimental and control conditions were averaged across 4–6 experiments (34–43 cells per condition).
Results
Cdc42 is activated by FSS and is required for the endocytic response to FSS
To determine whether exposure to FSS activates Cdc42, we transfected OK cells with the “Ras and interacting protein chimeric unit” Raichu‐Cdc42 construct and measured time courses of intramolecular FRET changes as readout for activation of this small GTPase in live cells (Itoh et al. 2002). Activation of Cdc42 in this membrane‐tethered construct facilitates binding of the attached CRIB domain of the Cdc42 effector PAK1. This change brings CFP into close proximity with YFP, causing a decrease in CFP fluorescence and a concomitant increase in YFP fluorescence due to FRET. The high affinity of the intramolecular PAK1 interaction with Cdc42 in the Raichu‐Cdc42 construct is essentially irreversible upon activation of Cdc42 (Thompson et al. 1998; Itoh et al. 2002). Despite this limitation, this probe is a useful tool to confirm Cdc42 activation in response to acute stimuli.
FRET ratios (YFP/CFP) were stable (or sometimes decreased slightly due to unequal bleaching of CFP and YFP) when cells were maintained under static conditions over a 1 h period (Fig. 1A and B). In contrast, when cells were imaged for 10 min and then exposed to 0.1 dyne/cm2 FSS (arrowhead in Fig. 1A), Cdc42 became gradually activated. Moreover, cells exposed to FSS developed protrusions, changed shape, and migrated more rapidly compared with cells maintained under static conditions, consistent with known effects of activated Cdc42 on cellular dynamics (Fig. 1B). The FRET signal remained steady or even increased slightly even after FSS was stopped (arrow in Fig. 1A), consistent with the irreversibility of this FRET probe.
Figure 1.
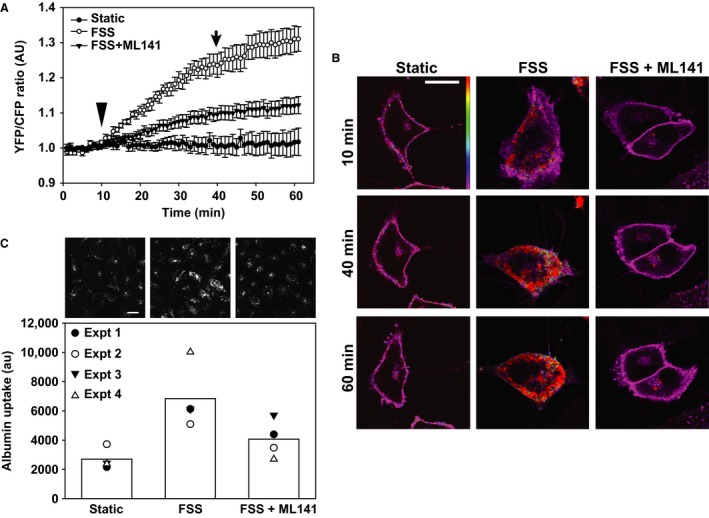
Cdc42 is activated by fluid shear stress and is required for the endocytic response to flow. (A) FRET measurements of OK cells transfected with the Raichu‐Cdc42 FRET probe and cultured in Ibidi chambers. Cells were maintained under static conditions throughout the 1 h imaging period or were exposed to 0.1 dyne/cm2 FSS starting at 10 min (arrowhead) until 40 min (arrow). The Cdc42 inhibitor ML141 (10 μmol/L) was included where indicated. Data (mean ± SEM) from five independent experiments (6–20 regions of interest analyzed per experiment) are plotted. All curves are significantly different from each other (P < 0.001) by two‐way ANOVA. (B) YFP/CFP merged images of Raichu‐Cdc42 taken at the indicated times reveal selective activation of Cdc42 in cells exposed to FSS. Note the increase in protrusions and change in cell shape upon exposure to FSS in control but not ML141‐treated cells. Scale bar: 25 μmol/L. (C) OK cells cultured on Ibidi chambers were incubated for 1 h at 37°C with AlexaFluor 647‐albumin under static conditions or at 0.1 dyne/cm2 FSS, and cell associated albumin quantified as described in Methods. ML141 was included where indicated. Data from four individual experiments, each shown using a different symbol, are plotted, and the bar shows the mean uptake for each condition. Representative images from a separate experiment in which cells were fixed and albumin uptake imaged using confocal microscopy are shown above each bar. Scale bar: 25 μm.
ML141 is a high affinity and selective inhibitor of Cdc42 [EC50~1.4 μmol/L (Hong et al. 2013)] that exhibits minimal inhibition of other related GTPases including Rac or Rho at concentrations up to 100 μmol/L (Surviladze et al. 2010). Incubation of cells with 10 μmol/L ML141 (Surviladze et al. 2010; Hong et al. 2013), starting 1 h prior to the experiment, reduced the FSS‐induced increase in Cdc42 FRET, and also prevented morphological and migratory changes in cells (Fig. 1A and B).
To test whether Cdc42 activation is necessary for the stimulation of endocytosis that we previously observed in response to FSS, we incubated OK cells under static conditions or at 0.1 dyne/cm2 for 1 h in the presence of 40 μg/mL AlexaFluor 647‐albumin, then washed and solubilized the cells and quantified cell‐associated fluorescence using spectrofluorimetry (Fig. 1C, lower panel). Exposure to acute FSS increased albumin uptake in cells by ~50–100%. Parallel studies in which cells were treated similarly, then fixed and imaged by confocal microscopy, confirmed an increase in intracellular albumin in cell exposed to FSS compared with cells maintained under static conditions (Fig. 1C, upper panel). This increase was markedly inhibited when cells were preincubated with ML141 for 1 h prior to addition of AlexaFluor 647‐albumin (Fig. 1C). Consistent with the well‐established role of Cdc42 in modulating actin dynamics, we confirmed that endocytosis in OK cells is actin dependent, as albumin uptake was profoundly inhibited by cytochalasin D (S.B. and V.R., unpublished observations). We conclude that activation of Cdc42 is an important step in the cascade leading from FSS to increased endocytosis.
Cilia and calcium are required for Cdc42 activation by FSS
Estimates of FSS experienced by cells lining the PT differ widely, from ~0.1 to ≥1 dyne/cm2 (Essig and Friedlander 2003; Du et al. 2004; Duan et al. 2008; Jang et al. 2013). We previously demonstrated that exposure of OK cells to ≥2 dyne/cm2 FSS causes a transient spike in [Ca2+]i that requires the primary cilium, extracellular Ca2+, and Ca2+ release from intracellular stores (Raghavan et al. 2014). Because Ca2+ signaling may vary with FSS (Xu et al. 2009; Liu et al. 2011), we modified our imaging setup to enable us to monitor changes in [Ca2+]i at the lower FSS used in our Cdc42 activation and endocytosis experiments. OK cells were loaded with Fura‐2 and imaged prior to and during exposure to 0.1 dyne/cm2 FSS. Initial [Ca2+]i concentrations were somewhat variable when measured using Fura‐2, however, fold‐dependent increases in [Ca2+]i were very consistent across cells, similar to our previous observations (Raghavan et al. 2014). As shown in Figure 2A, exposure to acute FSS caused a transient 4–5‐fold increase in [Ca2+]i that declined within ~2 min to a new higher baseline (~3‐fold higher than starting levels) and remained stable for at least 10 min. This profile is similar to that observed upon stimulation of Ca2+ signaling in vivo and in studies in MDCK cells using FSS <1 dyne/cm2 (Rydholm et al. 2010; Szebenyi et al. 2015). In contrast, in previous studies where OK and other cells were exposed to higher shear stress, [Ca2+]i levels rapidly increased 2–4‐fold but rapidly (<5 min) returned to near baseline levels (Nauli et al. 2003; Raghavan et al. 2014).
Figure 2.
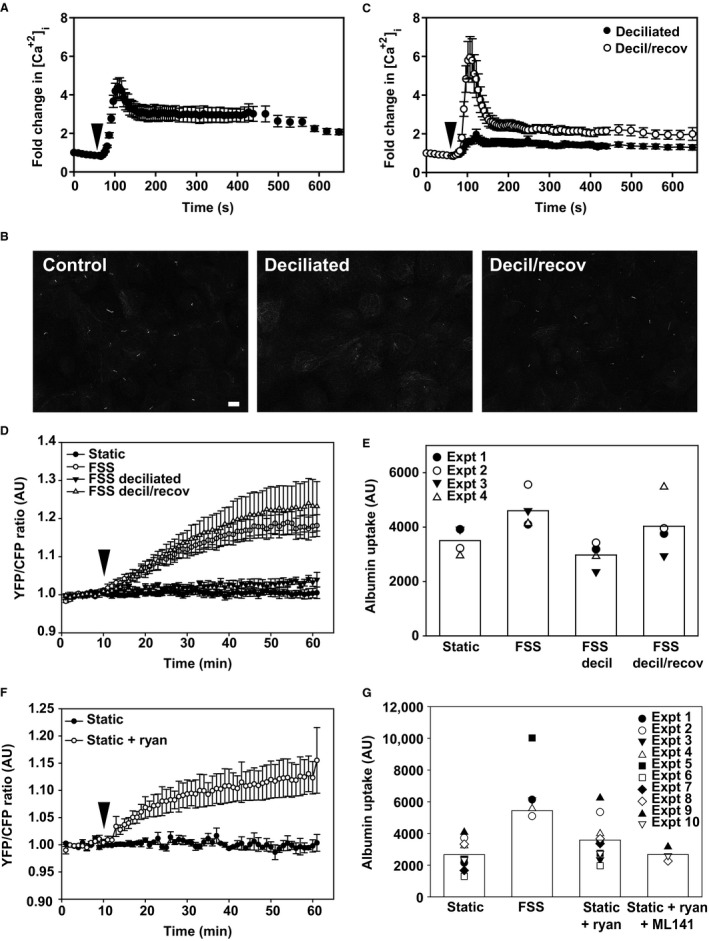
Primary cilia and calcium are required for the activation of Cdc42 by FSS. (A) OK cells cultured in Ibidi chambers were loaded with Fura‐2 and [Ca2+]i levels were monitored before and upon exposure to 0.1 dyne/cm2 FSS (arrowhead). (B) Indirect immunofluorescence of acetylated tubulin staining to visualize primary cilia in control cells, cells deciliated with ammonium sulfate (decil), and deciliated cells after overnight recovery to enable cilia regrowth (decil/recov). Scale bar: 10 μm. (C) [Ca2+]i levels were monitored as above in deciliated cells immediately after deciliation and after overnight recovery. Kruskal–Wallis test (nonparametric) followed by Dunn's multiple comparisons test confirmed statistically significant differences in peak [Ca2+]i levels between control and deciliated cells (P < 0.001) and between deciliated and recovered cells (P < 0.001). (D) Cdc42 is not activated in deciliated cells exposed to FSS. Cells were deciliated for 3 h prior to exposure to 0.1 dyne/cm2 FSS for 30 min starting at 10 min (arrowhead) or incubated overnight to recover cilia prior to measuring the Raichu‐Cdc42 FRET response to FSS. Mean ± SEM is plotted (static, n = 6; FSS, n = 7; deciliation, n = 8, decil/recov, n = 3). Profiles for FSS versus rescue and static versus deciliated conditions were not significantly different from each other; P values for all other comparisons were <0.001 by two‐way ANOVA. (E) Endocytosis is not stimulated by FSS in deciliated cells. OK cells cultured on Ibidi chambers were deciliated immediately prior to quantitation of AlexaFluor 647‐albumin uptake or incubated overnight to recover cilia. Data from four individual experiments, each shown using a different symbol, are plotted, and the bar shows the mean uptake for each condition. Representative images from a separate experiment in which cells were fixed and albumin uptake imaged using confocal microscopy are shown above each bar. Scale bar: 25 μm. (F) Ryanodine activates Cdc42 in the absence of FSS. FRET ratios were monitored in OK cells transfected with the Raichu‐Cdc42 under static conditions. Ryanodine (25 μmol/L) was added at 10 min (arrowhead) and the incubation continued for an additional 50 min. The mean of from three experiments is plotted. Profiles for static versus static + ryanodine are significantly different from each other (P < 0.001) by two‐way ANOVA. (G) Ryanodine‐stimulated albumin uptake in the absence of FSS is inhibited by ML‐141. Data from 10 experiments are plotted, and the bar shows the mean uptake for each condition.
We previously demonstrated that deciliation of OK cells compromised the Ca response to ≥2 dyne/cm2 FSS. To determine whether this also occurs at 0.1 dyne/cm2, we incubated cells with 30 mmol/L ammonium sulfate for 3 h to remove cilia, and loaded them with Fura‐2 immediately afterward or after overnight incubation to recover cilia (Raghavan et al. 2014). Indirect immunofluorescence of cells with antiacetylated tubulin to visualize primary cilia confirmed that ammonium sulfate treatment effectively removed primary cilia, and that deciliated cells regenerated primary cilia after overnight incubation (Fig. 2B). Deciliated cells loaded readily with Fura‐2, suggesting that membrane permeability was not compromised by this maneuver. Similar to our previous results, the [Ca2+]i response to FSS was markedly blunted in deciliated cells (Fig. 2C). When cells were allowed to recover overnight after deciliation the response to FSS was similar to that in control cells (Fig. 2C).
Next, we monitored FRET in cells expressing Raichu‐Cdc42 to determine whether intact primary cilia are required for FSS‐dependent activation of Cdc42. No increase in FRET was observed in deciliated cells upon exposure to FSS, consistent with a requirement for ciliary‐mediated increase in [Ca2+]i to activate Cdc42 (Fig. 2D). FSS‐dependent activation of Cdc42 was fully restored when cells were allowed to recover overnight (Fig. 2D). Additionally, we confirmed our previously published observation that deciliation impairs albumin uptake in response to FSS, and that this response is restored after cell recovery [(Fig. 2E) and (Raghavan et al. 2014)].
To assess whether the increase in [Ca2+]i upon exposure to FSS is upstream of Cdc42 activation, we asked whether increasing [Ca2+]i by addition of ryanodine is sufficient to activate Cdc42 in the absence of FSS. As shown in Figure 2F, addition of ryanodine increased Cdc42 FRET, albeit to a lesser degree compared with FSS. Similarly, ryanodine enhanced endocytic uptake of albumin in OK cells maintained under static conditions (Fig. 2G). Importantly, the ryanodine‐mediated increase in albumin uptake was not observed in the presence of ML141 (Fig. 2G). Together, these data suggest that the primary cilium is required for FSS‐dependent mobilization of [Ca2+]i that activates Cdc42 and stimulates FSS‐dependent endocytosis.
CaM and CaMKII mediate the activation of Cdc42 and the endocytic response to FSS
Activation of Ca2+‐binding protein calmodulin (CaM) is a central mediator of downstream responses to increases in [Ca2+]i. To test whether CaM is required for the activation of Cdc42 in response to FSS, we measured Cdc42 activation by FRET upon addition of the CaM activator Calp3 (Villain et al. 2000). Addition of Calp3 to cells maintained under static conditions increased Cdc42 FRET (Fig. 3A), similar to our results using ryanodine. Moreover, Calp3 enhanced constitutive uptake of AlexaFluor 647‐albumin under static conditions (Fig. 3B). In contrast, when cells were exposed to FSS in the presence of the CaM inhibitor W13 (Wei et al. 1983), Cdc42 activation was not observed (Fig. 3C). Moreover, W13 blunted the endocytic response to FSS (Fig. 3D).
Figure 3.
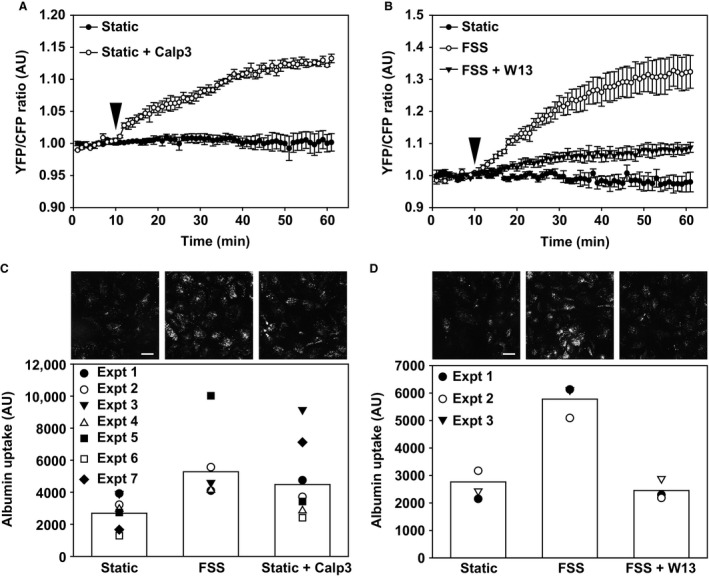
CaM activation is required for Cdc42 activation and FSS‐stimulated endocytosis. (A) FRET ratios were monitored in OK cells transfected with the Raichu‐Cdc42 under static conditions. The calmodulin activator Calp3 (5 μmol/L) was added at 10 min (arrowhead). The mean ± SEM of three independent experiments is plotted. Profiles for static versus static + Calp3 are significantly different from each other (P < 0.001) by two‐way ANOVA. (B) FRET measurements of OK cells transfected with the Raichu‐Cdc42‐ transfected OK cells were maintained under static conditions or exposed to 0.1 dyne/cm2 FSS for 30 min starting at 10 min (arrowhead). The calmodulin inhibitor W13 (60 μmol/L) was included where indicated. Data (mean ± SEM) from three independent experiments are plotted. Profiles for all conditions are significantly different from each other (P < 0.001) by two‐way ANOVA. (C) Calp3 increases albumin uptake under static conditions. Data from seven experiments are plotted, and the bar shows the mean uptake for each condition. Representative images from a separate experiment in which cells were fixed and albumin uptake imaged using confocal microscopy are shown above each bar. Scale bar: 25 μm. (D) W13 inhibits albumin uptake in response to FSS. Data from three experiments are plotted, and the bar shows the mean uptake for each condition. Representative images from a separate experiment in which cells were fixed and albumin uptake imaged using confocal microscopy are shown above each bar. Scale bar: 25 μm.
Discussion
Studies in many cell types have focused on the potent role of flow in regulating cell functions. Aberrant signaling responses to mechanical stress are implicated in the progression of various diseases, including vascular disease, osteoporosis, and cancer (Iolascon et al. 2013; Baeyens et al. 2016; Brown et al. 2016; Tarbell and Cancel 2016). Similarly, tubular flow should be considered as a powerful regulator of proximal cell function. FSS resulting from fluid movement in renal tubules affects the organization of the cytoskeleton and the brush border, with consequences to many cellular functions including ion transport and endocytosis (Duan et al. 2008; Ferrell et al. 2012; Maggiorani et al. 2015).
Our prior studies demonstrate that exposure of PT cells to FSS results in mobilization of [Ca2+]i via a pathway dependent on the primary cilia. We have now investigated the downstream events triggered by the increase in [Ca2+]i that lead to increased endocytosis. We found that activation of Cdc42 is a necessary step in the PT cell signaling cascade activated by the low levels of FSS (0.1 dyne/cm2) used in our studies. Mobilization of [Ca2+]i and activation of CaM in the absence of FSS were sufficient to activate Cdc42 and enhance endocytosis. Our current model for how acute changes in FSS are coupled to endocytic capacity in PT cells is shown in Figure 4.
Figure 4.
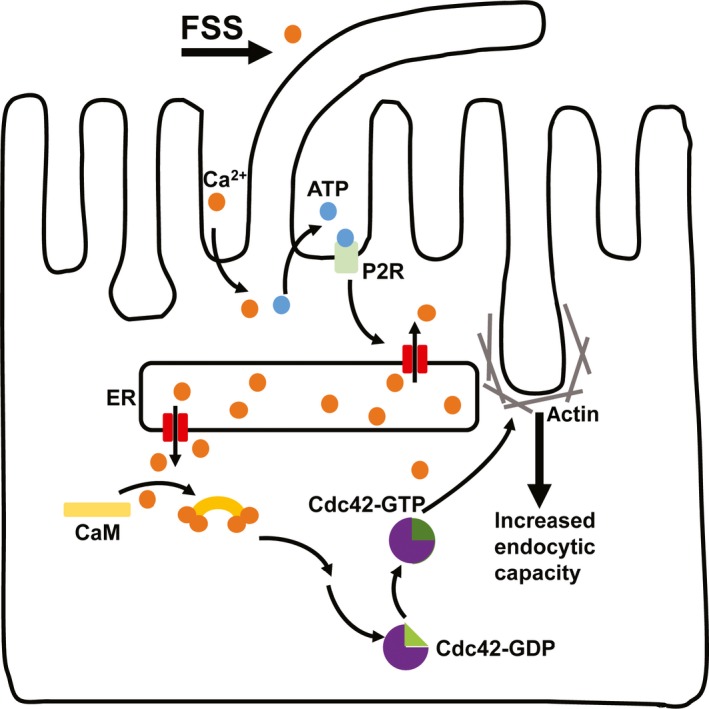
Updated model for the translation of FSS to stimulated endocytosis. Our data support a model in which exposure to FSS increases apical endocytic capacity in PT cells via a pathway that requires mechanosensation by the primary cilium. Our previous studies demonstrated that this response requires extracellular Ca2+ and is mediated by release of ATP and purinergic receptor (P2R) signaling that leads to release of Ca2+ from endoplasmic reticulum (ER) stores. Here we show that FSS‐stimulated endocytosis requires calmodulin (CaM) mediated activation of Cdc42. We hypothesize that Cdc42 modulates cytoskeletal dynamics that lead to increased endocytic capacity.
Cdc42 is a known master regulator of actin dynamics, and disruption of actin inhibited albumin uptake in cells maintained under static conditions as well as in response to FSS. A role for actin in apical clathrin‐dependent endocytosis has been previously demonstrated in MDCK cells (Gottlieb et al. 1993; Boulant et al. 2011). Additionally, actin is known to play a role in the uptake of large virus particles through elongated clathrin‐coated structures (Cureton et al. 2009). In this regard, it is interesting that clathrin‐coated invaginations in PT cells are highly irregular in shape (Rodman et al. 1986; Birn et al. 1993). Of note, Cdc42 is a potent activator of Toca‐1 and N‐WASP‐Arp2/3, which regulate the formation of branched actin filaments and which have previously been implicated in endocytosis (Bu et al. 2010). We hypothesize that activation of N‐WASP/Arp2/3 leads to structural remodeling of the actin cytoskeleton to enable the formation of clathrin‐coated structures.
Our data also suggest that the [Ca2+]i response to flow varies with shear stress. We previously found that renal cell exposure to higher FSS (≥2 dyne/cm2) results in a rapid and transient increase in [Ca2+]i. The lower FSS used here also led to an initial spike in [Ca2+]i but this quickly reset to a higher baseline [Ca2+]i that remained steady for at least 10 min. This difference in Ca2+ responses could reflect variable sensitivity to FSS of the machinery needed to re‐equilibrate [Ca2+]i. Estimates of FSS experienced by PT cells in vivo vary widely, and cellular responses to higher levels of FSS may differ from those we observed at the lower end of the reported range. Interestingly, a recent study using a genetically encoded Ca2+ sensor in mice demonstrated that the effect of ATP on PT [Ca2+]i levels in vivo is considerably longer than in vitro (Szebenyi et al. 2015). Thus, Ca2+ responses to physiologic stimuli may be longer lived in the PT than previously thought.
Our studies point to a critical role for primary cilia in the activation of Cdc42 by FSS, however, we cannot rule out the possibility that our method of deciliation has nonciliary effects on cell function. That said, our deciliated cells are clearly viable, have intact plasma membranes, and are able to regrow cilia. It is generally accepted that primary cilia are required for flow‐dependent mechanosensation; however, their specific role in signaling remains unknown. Flow‐sensitive Ca2+ responses in distal tubules of mice with impaired ciliary biogenesis are blunted, and deciliation has also been shown to impair the flow‐dependent Ca2+ response in MDCK cells (Liu et al. 2005; Praetorius and Leipziger 2009). However, other studies have reported cilium‐independent increases in [Ca2+]i in response to FSS (Liu et al. 2003; Rodat‐Despoix et al. 2013). Moreover, changes in Ca2+ within the cilium occur subsequent to cytoplasmic changes when cells are stimulated with ATP or FSS (Su et al. 2013; Delling et al. 2016).
In summary, the studies described here deepen our understanding of how acute changes in FSS modulate endocytic capacity. Activation of Cdc42 in response to cilia‐initiated changes in [Ca2+]i is a necessary step in this cascade, and is regulated upstream by CaM. Activation of Cdc42 may also contribute to other effects of FSS on PT function, including the modulation of tight junction dynamics (Rojas et al. 2001; Duan et al. 2008). Future studies will be aimed at identifying the downstream targets of Cdc42 in response to FSS and the mechanism by which endocytic capacity is enhanced in PT cells in culture and in vivo.
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank Michiyuki Matsuda for the Raichu Cdc42 FRET construct.
Bhattacharyya S., Jean‐Alphonse F. G., Raghavan V., McGarvey J. C., Rbaibi Y., Vilardaga J.‐P., Carattino M. D., Weisz O. A.. Cdc42 activation couples fluid shear stress to apical endocytosis in proximal tubule cells. Physiol Rep, 5 (19), 2017, e13460, https://doi.org/10.14814/phy2.13460
Funding Information
This project was supported by R01‐DK101484 to O. A. Weisz and by R01‐DK102495 and R01‐DK087688 to J. P. Vilardaga. We are grateful for support from the morphology and physiology cores of the Pittsburgh Center for Kidney Research (P30 DK079307).
References
- Baeyens, N. , Bandyopadhyay C., Coon B. G., Yun S., and Schwartz M. A.. 2016. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Invest. 126:821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birn, H. , Christensen E. I., and Nielsen S.. 1993. Kinetics of endocytosis in renal proximal tubule studied with ruthenium red as membrane marker. Am. J. Physiol. 264:F239–F250. [DOI] [PubMed] [Google Scholar]
- Boulant, S. , Kural C., Zeeh J. C., Ubelmann F., and Kirchhausen T.. 2011. Actin dynamics counteract membrane tension during clathrin‐mediated endocytosis. Nat. Cell Biol. 13:1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, A. J. , Teng Z., Evans P. C., Gillard J. H., Samady H., and Bennett M. R.. 2016. Role of biomechanical forces in the natural history of coronary atherosclerosis. Nat. Rev. Cardiol. 13:210–220. [DOI] [PubMed] [Google Scholar]
- Bu, W. , Lim K. B., Yu Y. H., Chou A. M., Sudhaharan T., and Ahmed S.. 2010. Cdc42 interaction with N‐WASP and Toca‐1 regulates membrane tubulation, vesicle formation and vesicle motility: implications for endocytosis. PLoS ONE 5:e12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton, D. K. , Massol R. H., Saffarian S., Kirchhausen T. L., and Whelan S. P.. 2009. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 5:e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delling, M. , Indzhykulian A. A., Liu X., Li Y., Xie T., Corey D. P., et al. 2016. Primary cilia are not calcium‐responsive mechanosensors. Nature 531:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, Z. , Duan Y., Yan Q., Weinstein A. M., Weinbaum S., and Wang T.. 2004. Mechanosensory function of microvilli of the kidney proximal tubule. Proc. Natl Acad. Sci. USA 101:13068–13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, Y. , Gotoh N., Yan Q., Du Z., Weinstein A. M., Wang T., et al. 2008. Shear‐induced reorganization of renal proximal tubule cell actin cytoskeleton and apical junctional complexes. Proc. Natl Acad. Sci. USA 105:11418–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshbach, M. L. , and Weisz O. A.. 2017. Receptor‐mediated endocytosis in the proximal tubule. Annu. Rev. Physiol. 79:425–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essig, M. , and Friedlander G.. 2003. Tubular shear stress and phenotype of renal proximal tubular cells. J. Am. Soc. Nephrol. 14(Suppl 1):S33–S35. [DOI] [PubMed] [Google Scholar]
- Ferrell, N. , Ricci K. B., Groszek J., Marmerstein J. T., and Fissell W. H.. 2012. Albumin handling by renal tubular epithelial cells in a microfluidic bioreactor. Biotechnol. Bioeng. 109:797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb, T. A. , Ivanov I. E., Adesnik M., and Sabatini D. D.. 1993. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J. Cell Biol. 120:695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, L. , Kenney S. R., Phillips G. K., Simpson D., Schroeder C. E., Noth J., et al. 2013. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J. Biol. Chem. 288:8531–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iolascon, G. , Resmini G., and Tarantino U.. 2013. Mechanobiology of bone. Aging Clin. Exp. Res. 25(Suppl 1):S3–S7. [DOI] [PubMed] [Google Scholar]
- Itoh, R. E. , Kurokawa K., Ohba Y., Yoshizaki H., Mochizuki N., and Matsuda M.. 2002. Activation of rac and cdc42 video imaged by fluorescent resonance energy transfer‐based single‐molecule probes in the membrane of living cells. Mol. Cell. Biol. 22:6582–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, K. J. , Mehr A. P., Hamilton G. A., McPartlin L. A., Chung S., Suh K. Y., et al. 2013. Human kidney proximal tubule‐on‐a‐chip for drug transport and nephrotoxicity assessment. Integr. Biol. (Camb) 5:1119–1129. [DOI] [PubMed] [Google Scholar]
- Kotsis, F. , Boehlke C., and Kuehn E. W.. 2013. The ciliary flow sensor and polycystic kidney disease. Nephrol. Dial. Transplant. 28:518–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Chen B. P., Azuma N., Hu Y. L., Wu S. Z., Sumpio B. E., et al. 1999. Distinct roles for the small GTPases Cdc42 and Rho in endothelial responses to shear stress. J. Clin. Invest. 103:1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. , Xu S., Woda C., Kim P., Weinbaum S., and Satlin L. M.. 2003. Effect of flow and stretch on the [Ca2 + ]i response of principal and intercalated cells in cortical collecting duct. Am. J. Physiol. Renal Physiol. 285:F998–F1012. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Murcia N. S., Duan Y., Weinbaum S., Yoder B. K., Schwiebert E., et al. 2005. Mechanoregulation of intracellular Ca2 + concentration is attenuated in collecting duct of monocilium‐impaired orpk mice. Am. J. Physiol. Renal Physiol. 289:F978–F988. [DOI] [PubMed] [Google Scholar]
- Liu, B. , Lu S., Zheng S., Jiang Z., and Wang Y.. 2011. Two distinct phases of calcium signalling under flow. Cardiovasc. Res. 91:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggiorani, D. , Dissard R., Belloy M., Saulnier‐Blache J. S., Casemayou A., Ducasse L., et al. 2015. Shear stress‐induced alteration of epithelial organization in human renal tubular cells. PLoS ONE 10:e0131416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T. , Kurokawa K., Kiyokawa E., and Matsuda M.. 2006. Analysis of the spatiotemporal activation of rho GTPases using Raichu probes. Methods Enzymol. 406:315–332. [DOI] [PubMed] [Google Scholar]
- Nauli, S. M. , Alenghat F. J., Luo Y., Williams E., Vassilev P., Li X., et al. 2003. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33:129–137. [DOI] [PubMed] [Google Scholar]
- Praetorius, H. A. 2015. The primary cilium as sensor of fluid flow: new building blocks to the model. A review in the theme: cell signaling: proteins, pathways and mechanisms. Am. J. Physiol. Cell Physiol. 308:C198–C208. [DOI] [PubMed] [Google Scholar]
- Praetorius, H. A. , and Leipziger J.. 2009. Released nucleotides amplify the cilium‐dependent, flow‐induced [Ca2 + ]i response in MDCK cells. Acta Physiol. (Oxf) 197:241–251. [DOI] [PubMed] [Google Scholar]
- Raghavan, V. , and Weisz O. A.. 2015. Flow stimulated endocytosis in the proximal tubule. Curr. Opin. Nephrol. Hypertens. 24:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan, V. , Rbaibi Y., Pastor‐Soler N. M., Carattino M. D., and Weisz O. A.. 2014. Shear stress‐dependent regulation of apical endocytosis in renal proximal tubule cells mediated by primary cilia. Proc. Natl Acad. Sci. USA 111:8506–8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodat‐Despoix, L. , Hao J., Dandonneau M., and Delmas P.. 2013. Shear stress‐induced Ca(2)(+) mobilization in MDCK cells is ATP dependent, no matter the primary cilium. Cell Calcium 53:327–337. [DOI] [PubMed] [Google Scholar]
- Rodman, J. S. , Seidman L., and Farquhar M. G.. 1986. The membrane composition of coated pits, microvilli, endosomes, and lysosomes is distinctive in the rat kidney proximal tubule cell. J. Cell Biol. 102:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi, R. , and Flores D.. 2010. Intratubular hydrodynamic forces influence tubulointerstitial fibrosis in the kidney. Curr. Opin. Nephrol. Hypertens. 19:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas, R. , Ruiz W. G., Leung S. M., Jou T. S., and Apodaca G.. 2001. Cdc42‐dependent modulation of tight junctions and membrane protein traffic in polarized Madin‐Darby canine kidney cells. Mol. Biol. Cell 12:2257–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydholm, S. , Zwartz G., Kowalewski J. M., Kamali‐Zare P., Frisk T., and Brismar H.. 2010. Mechanical properties of primary cilia regulate the response to fluid flow. Am. J. Physiol. Renal Physiol. 298:F1096–F1102. [DOI] [PubMed] [Google Scholar]
- Su, S. , Phua S. C., DeRose R., Chiba S., Narita K., Kalugin P. N., et al. 2013. Genetically encoded calcium indicator illuminates calcium dynamics in primary cilia. Nat. Methods 10:1105–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surviladze, Z. , Waller A., Strouse J. J., Bologa C., Ursu O., Salas V., et al. 2010. A potent and selective inhibitor of Cdc42 GTPase Probe reports from the NIH Molecular Libraries Program. Bethesda (MD). [Google Scholar]
- Szebenyi, K. , Furedi A., Kolacsek O., Csohany R., Prokai A., Kis‐Petik K., et al. 2015. Visualization of calcium dynamics in kidney proximal tubules. J. Am. Soc. Nephrol. 26:2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbell, J. M. , and Cancel L. M.. 2016. The glycocalyx and its significance in human medicine. J. Intern. Med. 280:97–113. [DOI] [PubMed] [Google Scholar]
- Thompson, G. , Owen D., Chalk P. A., and Lowe P. N.. 1998. Delineation of the Cdc42/Rac‐binding domain of p21‐activated kinase. Biochemistry 37:7885–7891. [DOI] [PubMed] [Google Scholar]
- Tzima, E. , Kiosses W. B., del Pozo M. A., and Schwartz M. A.. 2003. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J. Biol. Chem. 278:31020–31023. [DOI] [PubMed] [Google Scholar]
- Villain, M. , Jackson P. L., Manion M. K., Dong W. J., Su Z., Fassina G., et al. 2000. De novo design of peptides targeted to the EF hands of calmodulin. J. Biol. Chem. 275:2676–2685. [DOI] [PubMed] [Google Scholar]
- Wei, J. W. , Hickie R. A., and Klaassen D. J.. 1983. Inhibition of human breast cancer colony formation by anticalmodulin agents: trifluoperazine, W‐7, and W‐13. Cancer Chemother. Pharmacol. 11:86–90. [DOI] [PubMed] [Google Scholar]
- Weinbaum, S. , Duan Y., Satlin L. M., Wang T., and Weinstein A. M.. 2010. Mechanotransduction in the renal tubule. Am. J. Physiol. Renal Physiol. 299:F1220–F1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C. , Shmukler B. E., Nishimura K., Kaczmarek E., Rossetti S., Harris P. C., et al. 2009. Attenuated, flow‐induced ATP release contributes to absence of flow‐sensitive, purinergic Cai2 + signaling in human ADPKD cyst epithelial cells. Am. J. Physiol. Renal Physiol. 296:F1464–F1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder, B. K. 2007. Role of primary cilia in the pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 18:1381–1388. [DOI] [PubMed] [Google Scholar]