

Abstract
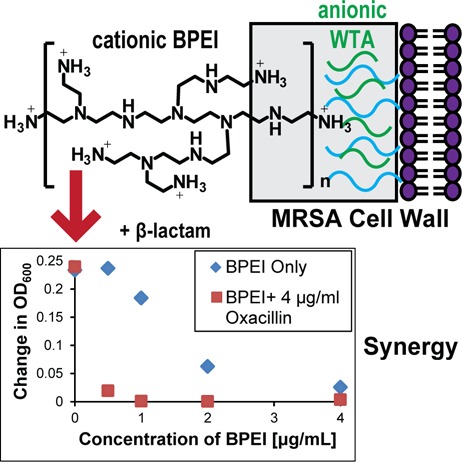
Methicillin-resistant Staphylococcus aureus (MRSA) is a medical concern. Here, we show that branched polyethylenimine (BPEI), a nontoxic, cationic polymer, restores MRSA’s susceptibility to β-lactam antibiotics. Checkerboard assays with MRSA demonstrated synergy between BPEI and β-lactam antibiotics. A time-killing curve showed BPEI to be bactericidal in combination with oxacillin. BPEI did not potentiate efficacy with vancomycin, chloramphenicol, or linezolid. When exposed to BPEI, MRSA increased in size and had difficulty forming septa. BPEI electrostatically binds to wall teichoic acid (WTA), a cell wall anionic polymer of Gram-positive bacteria that is important for localization of certain cell wall proteins. Lack of potentiation in a WTA knockout mutant supports the WTA-based mechanism. These data suggest that BPEI may prevent proper localization of cell wall machinery by binding to WTA; leading to cell death when administered in combination with β-lactam antibiotics. Negligible in vitro toxicity suggests the combination could be a viable treatment option.
Keywords: Branched polyethylenimine, MRSA, wall teichoic acid, bacteria, antibiotic resistance, β-lactams
Clinicians prescribed 118 million courses of β-lactam antibiotics in 2011 to treat the majority of patients presenting bacterial infection symptoms.1 Patient outcomes are generally positive unless β-lactam resistant bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA), are present. In cases where symptoms are attributed to MRSA, β-lactams are avoided and effective antibiotics are given without delay.2 However, most MRSA infections are not diagnosed immediately.3 After initial infection symptoms arise, patients suffer while ineffective antibiotics, usually β-lactams, are given.3 MRSA colonies survive and invade host tissue to release toxins that cause tissue injury. Therefore, the need exists to decrease morbidity and mortality while improving the quality of life and medical outcomes of patients infected by MRSA. These needs can be met by strengthening the arsenal of clinical first-line antibiotics and using these new weapons to fight MRSA. A new antibiotic treatment option with efficacy against MRSA and MSSA could be based on branched polyethylenimine (BPEI) and β-lactams.
β-Lactam antibiotics, such as oxacillin and ampicillin, prevent bacterial cell wall synthesis by irreversibly binding to the active site of penicillin-binding proteins (PBPs) that are responsible for cross-linking peptidoglycan. MRSA has the mecA gene that codes for PBP2a, an extra PBP with a low binding affinity for β-lactam antibiotics, thereby reducing their effectiveness in treatment.4 PBP4 has also been shown to aid community-acquired MRSA in β-lactam resistance.5 Wall teichoic acid (WTA) is important for proper function and localization of PBP2a and PBP4.6−8 As an anionic polymer found in the cell wall of Gram-positive bacteria, WTA is also important for metal binding, cell adhesion, virulence, biofilm formation, and localization of autolysins.9−14 As inhibition of WTA synthesis makes MRSA susceptible to β-lactam antibiotics,7,8 similar potentiation effects could arise by disrupting WTA function with BPEI.
An initial study published by our lab found that BPEI and ampicillin had synergy against MRSA, which we proposed was due to electrostatic binding of BPEI to WTA.15 Expanding on our initial study, we investigated the anti-MRSA properties of BPEI and discovered that small amounts (<1 μg/mL, <1.6 μM) of BPEI potentiate β-lactam antibiotics against MRSA. We found that BPEI has synergy with oxacillin, ampicillin, and amoxicillin against MRSA. BPEIs of different molecular weights were tested, but due to toxicity concerns of the higher Dalton BPEIs, the lowest MW (600 Da BPEI) was considered to be the lead potentiator. Additionally, BPEI increased cell size and reduced autolytic rates. Microscopy images show perturbation of the cell wall while time–kill curves demonstrate that the combination of oxacillin and BPEI is bactericidal. Also, the minimum bactericidal concentration (MBC) for the combination is less than four-fold higher than the minimum inhibitory concentration (MIC), which indicates a bactericidal mechanism. We believe that BPEI works by causing steric hindrance of WTA to prevent proper localization of PBP2a and PBP4, which effectively disables this β-lactam resistance factor.
BPEI’s value in drug discovery is supported by mammalian cell cytotoxicity assays that demonstrate that 600 Da BPEI does not impact viability until concentrations are much higher than those required for in vitro efficacy. BPEI may have oral bioavailability because BPEI is a hydrophilic weak base with high water solubility; whose absorption by the gut epithelium was demonstrated by Cheng et al. in vitro using Caco-2 epithelial cells and an in vivo rat model.16 Future animal studies will test this hypothesis for BPEI + antibiotic combinations. Enormous benefits to patient health are possible by providing clinicians with improved first-line antibiotics to lower health care costs, decrease morbidity, and reduce the need for MRSA screening. We recognize that a large amount of work remains in order to demonstrate in vivo efficacy, safety, bioavailability, and optimal drug formulation. Yet, the biophysical properties of BPEI and the data generated in our laboratory provide guarded optimism that we can successfully address these concerns and overcome obstacles within the drug discovery pipeline.
MRSA is a concern for human health due to its ability to infect humans and its resistance to commonly used β-lactam antibiotics. The CDC considers MRSA to be a serious threat to public health. It is estimated that 1 in 7 severe cases of MRSA resulted in death in 2011.17 Additionally, many patients with MRSA undergo surgery to remove infected tissue. Drugs of last resort, such as vancomycin, require hospitalization. Meanwhile, strains of MRSA resistant to vancomycin, daptomycin, and linezolid have been found.18−21 Orally available β-lactams, the most common class of first-line antibiotics,1 could be used in a new anti-MRSA drug combination by overcoming a leading cause of β-lactam antibiotic resistance in MRSA: PBP2a and PBP4.5,22 Disabling PBP2a and PBP4 with oral β-lactams has not reached clinical use, but success may be possible with a discovery made in our laboratory.15 β-Lactam antibiotics that kill MSSA are potentiated to stop MRSA growth if coadministered with 600 Da BPEI, which is noncytotoxic.
BPEI creates synergy with β-lactam antibiotics to stop MRSA growth. Our previously published work showed that BPEI potentiated ampicillin susceptibility in MRSA.15 Additional checkerboard assays demonstrate anti-MRSA properties of 600 Da BPEI mixed with β-lactam antibiotics (Table 1) against a MRSA strain containing PBP2a (ATCC 700787) that is also moderately resistant to vancomycin (vancomycin-intermediate S. aureus, VISA). BPEI has synergy (FIC < 0.5) with oxacillin, ampicillin, and amoxicillin (Figure 1A–C). Adding 600 Da BPEI (0.5 μg/mL, 0.8 μM) reduced the MIC of oxacillin from 64 μg/mL (149 μM) to 4 μg/mL (9 μM) against MRSA 700787. Increasing the concentration of 600 Da BPEI to 1 μg/mL (1.6 μM) resulted in a 128-fold decrease of the MIC of oxacillin to 0.5 μg/mL (1.1 μM). Likewise, increasing the 600 Da BPEI concentration increases potentiation and decreases MIC values of amoxicillin and ampicillin (Table 1, Figure 1B,C). BPEI did not have synergy with non-β-lactam antibiotics, such as vancomycin, linezolid, and chloramphenicol (Table 1, Figure 1D). Additionally, BPEI only had modest additivity with oxacillin (64 ng/mL, 140 nM, Figure 1E), ampicillin (64 ng/mL, 170 nM, Figure S1A), and amoxicillin (64 ng/mL, 160 nM, Figure S1B) against MSSA (ATCC 25923) (Table 1). The different BPEI potentiation effects against MRSA versus MSSA support a mechanism involving PBP2a, which is present in MRSA 700787 but not in MSSA. We also report that the combination of BPEI and β-lactam antibiotics does not have synergy against Gram-negative E. coli 11775 (Figure 1F) or Gram-positive B. subtilis 6051 (Figure S1F). These data lead us to believe that the mechanism by which BPEI potentiates β-lactams is specific for MRSA.
Table 1. Synergy of 600 Da BPEI and β-Lactams against MRSA 700787 and MSSA 25923a.
| MICA (μg/mL)/μM |
MICB (μg/mL)/μM |
||||||
|---|---|---|---|---|---|---|---|
| strain | antibiotic | alone | comb. | alone | comb. | FICI | outcome |
| MRSA 700787 | oxacillin | 64/149 | 4/9 | 4/6.6 | 0.5/0.8 | 0.188 | synergy |
| 0.5/1.1 | 1/1.6 | 0.258 | |||||
| ampicillin | 128/345 | 8/22 | 4/6.6 | 0.5/0.8 | 0.188 | synergy | |
| 2/5 | 1/1.6 | 0.266 | |||||
| amoxicillin | 128/305 | 8/22 | 4/6.6 | 1/1.6 | 0.313 | synergy | |
| methicillin | 64/160 | 4/10 | 4/6.6 | 2/3.3 | 0.563 | additivity | |
| meropenem | 2/4.6 | 0.5/1.1 | 4/6.6 | 0.5/0.8 | 0.375 | synergy | |
| chloramphenicol | 8/25 | 2/6 | 4/6.6 | 1/1.6 | 0.500 | additivity | |
| linezolid | 2/5.9 | 0.5/1.5 | 4/6.6 | 1/1.6 | 0.500 | additivity | |
| vancomycin | 2/1.3 | 2/1.3 | 4/6.6 | 4/6.7 | 1.0 | no synergy | |
| MSSA 25923 | oxacillin | 0.064/0.14 | 0.032/0.072 | 32/53 | 8/13 | 0.75 | additivity |
| ampicillin | 0.064/0.17 | 0.032/0.086 | 32/53 | 8/13 | 0.75 | additivity | |
| amoxicillin | 0.064/0.16 | 0.016/0.038 | 32/53 | 16/27 | 0.75 | additivity | |
MICA is minimum inhibitory concentration of the antibiotic; MICB is minimum inhibitory concentration of BPEI; FICI is fractional inhibitory concentration index.
Figure 1.
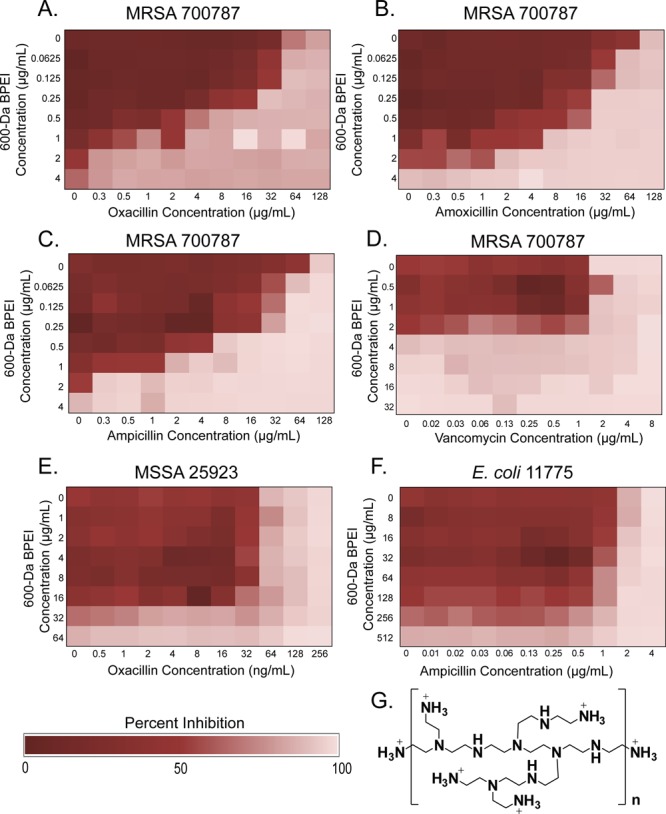
BPEI potentiates β-lactam activity against MRSA 700787, but not MSSA 25923 or E. coli 11775. Checkerboard assays show that BPEI potentiates oxacillin (A), amoxicillin (B), and ampicillin (C) activity against MRSA 700787. BPEI does not potentiate vancomycin (D) activity. No synergy is seen between β-lactams and BPEI against MSSA 25923 (E) or E. coli 11775 (F). Each assay was performed as three separate trials, and the presented data is shown as the average change in OD600. The structure of BPEI (G) is shown (n = 1, MW = 600 Da; n = 2, MW = 1200 Da; n = 3, MW = 1800 Da).
BPEI is commercially available in a wide range of sizes (600 to 1 000 000 Da). We were motivated to determine if increasing molecular weight of BPEI increases efficacy against MRSA. The MICs of 1200 and 1800 Da BPEI alone (0.5 μg/mL, 0.42 μM, and 0.28 μM, respectively) were lower compared to that of 600 Da BPEI alone (4 μg/mL, 6.6 μM). The 1200 and 1800 Da BPEI had synergy with oxacillin (Figure S1C,D). Only 0.125 μg/mL (0.10 μM) 1200 Da BPEI was required to reduce the MIC of oxacillin from 64 μg/mL (149 μM) to 4 μg/mL (9 μM) against MRSA 700787. The 10 000 Da BPEI had an MIC of 4 μg/mL (0.4 μM) but did not demonstrate synergy with oxacillin (Figure S1E). Compared to the 600 Da BPEI+ β-lactam combination, the 1200 and 1800 Da + β-lactam combinations have superior in vitro efficacy. However, as shown below, 600 Da BPEI shows negligible in vitro cytotoxicity against mammalian cells, whereas toxicity increases for 1200, 1800, and 10 000 Da BPEI.
The 600 Da BPEI has low cytotoxicity. A possible critique of utilizing BPEI in drug discovery is that cationic BPEIs are cytotoxic and thus unlikely to be useful as clinical antibacterial treatments. Reports show that cationic compounds, such as aminoglycosides and polymyxins, lead to nephrotoxicity.23,24 However, presumptions that all BPEIs are toxic overlook the stipulation that toxicity depends on molecular weight and concentration. High molecular weight BPEIs (over 25 000 Da) are cytotoxic, whereas the lower molecular weight BPEIs are nontoxic unless their concentrations are orders of magnitude higher than the amount required for potentiation (Table 1).25,26 Low cytotoxicity was confirmed in our lab with in vitro cytotoxicity data using mouse fibroblasts,15 human HeLa, humancolon, and human kidney cell lines (Table 2). The IC50 values for 600 Da BPEI (100–1000 μg/mL, 166–1670 μM) are orders of magnitude higher than the amount required for potentiation (∼1 μg/mL, 1.6 μM). An in vitro hemolysis assay, published by Gibney et al., showed that 600 Da BPEI had no hemolytic activity and that 10 000 Da BPEI had minimal hemolysis (>5%) up to 2000 μg/mL.25 An in vitro nephrotoxicity assay was performed using primary human renal proximal tubule epithelial cells (hRPTECs). The assay detects the release of the metabolic enzyme, lactate dehydrogenase (LDH). A lack of LDH release suggests that the membrane is not damaged and that the test agent does not cause in vitro nephrotoxicity. Exposure to 600 Da BPEI caused minimal release of LDH (∼1% at 8 μg/mL [13 μM], 16 μg/mL [27 μM], and 31 μg/mL [52 μM]; 3.5% at 62 μg/mL [103 μM]; and 8% at 125 μg/mL [208 μM], Figure 2). As per the literature, these values are indicative of low toxicity.27 These values are much lower than the LDH release values for cationic and nephrotoxic colistin (1% at 8 μg/mL [7 μM]; 2.3% at 16 μg/mL [14 μM]; 18% at 31 μg/mL [27 μM]; 26% at 62 μg/mL [54 μM]; and 28% at 125 μg/mL [108 μM], Figure 2). The 1200, 1800, and 10 000 Da BPEIs were more toxic than the 600 Da BPEI as seen with the CellTiter Blue cell viability (Table 2) and LDH assays (Figure 2). These data suggest 600 Da BPEI is preferred as a lead potentiator in drug discovery due to its low toxicity and low nephrotoxicity.
Table 2. In Vitro Mammalian Cytotoxic Activity of BPEIa.
| mean
IC50 ± SD (μM) |
||||
|---|---|---|---|---|
| human cell line | 600 Da | 1200 Da | 1800 Da | 10 kDa |
| cervical cancer (HeLa) | 1820 ± 250 | 692 ± 13 | 519 ± 34 | 0.66 ± 0.089 |
| colon cancer (HCT116) | 487 ± 75 | 457 ± 48 | 100 ± 42 | 1.11 ± 0.22 |
| kidney cancer (HEK293) | 1150 ± 130 | 155 ± 23 | 33 ± 13 | 0.19 ± 0.05 |
Values are reported as the average of three trials ± standard deviation. Cells were treated for 48 h and assayed with the CellTiter Blue method.
Figure 2.
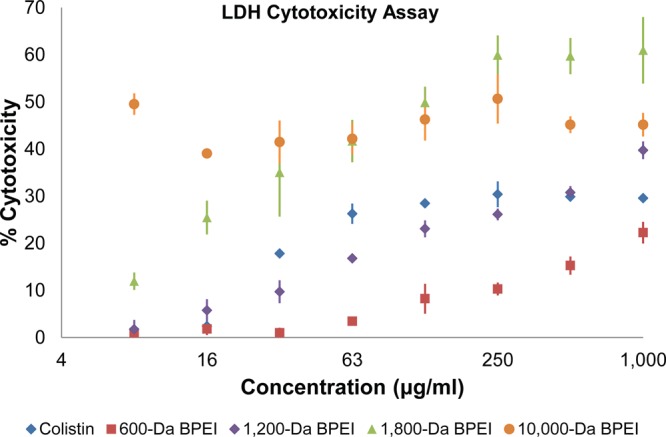
The 600 Da BPEI had minimal LDH release in a primary kidney proximal tubule cell line (PCS-400-010). The 1800 and 10 000 Da BPEIs had higher LDH release than the nephrotoxic drug colistin. Error bars denote standard deviation (n = 3).
The low toxicity of our lead potentiator is the result of its hydrophilic nature. The 600 Da BPEI is very hydrophilic and completely miscible with water. Also, 600 Da BPEI molecules are very small and do not contain regions of hydrophobic character, as seen with cationic peptides, aminoglycosides, and polymyxins. Thus, 600 Da BPEI lacks the energetic force that drives hydrophobic compounds into lipid membranes. Conversely, the higher MW BPEIs possess hydrophobic interiors that increase their lipophilicity and lead to membrane penetration and damage and, ultimately, higher toxicity.
The 600 Da BPEI:oxacillin combination is bactericidal. Monitoring the growth of MRSA 700787 reveals that bacteria exposed to subinhibitory concentrations of BPEI and oxacillin fail to reach the exponential phase when the two compounds are combined (Figure 3A). Aliquots were transferred to agar plates, and CFUs were counted from the 0, 4, 8, and 24 h time points to determine if the lack of growth observed for the BPEI and oxacillin combination was bacteriostatic or bactericidal. After 8 h, the control sample had the highest cell density (1 × 109 cells/μL). The samples that contained BPEI or oxacillin had lower viable cell numbers in comparison to the control sample, but these values were still higher than the initial cell density (>1 × 105 cells/μL). The viable cell count decreased for growth in both BPEI and oxacillin (47 CFUs/μL at 8 h growth) and did not increase after 24 h (Figure 3B). This data demonstrates that the mechanism by which BPEI and oxacillin prevents growth of MRSA is bactericidal. To support this conclusion, MBCs were determined from a checkerboard assay of MRSA containing BPEI and oxacillin. The MBC of BPEI was the same as the MIC (4 μg/mL, 6.7 μM), while the MBC for oxacillin was at least twice as large as the MIC (MIC = 64 μg/mL, 149 μM; MBC > 128 μg/mL, 290 μM). The MBCs for the combination (1 μg/mL BPEI, 2 μg/mL oxacillin; Figure S2) were two-fold greater than the combination MICs (1 μg/mL BPEI, 1 μg/mL oxacillin). If the MBC is less than four-fold of the MIC, the drug is considered bactericidal and does not require the assistance of host defenses.28 Since the MBC for BPEI and its oxacillin combination is not greater than four times the MIC, both BPEI alone and the BPEI–oxacillin combination are bactericidal.
Figure 3.
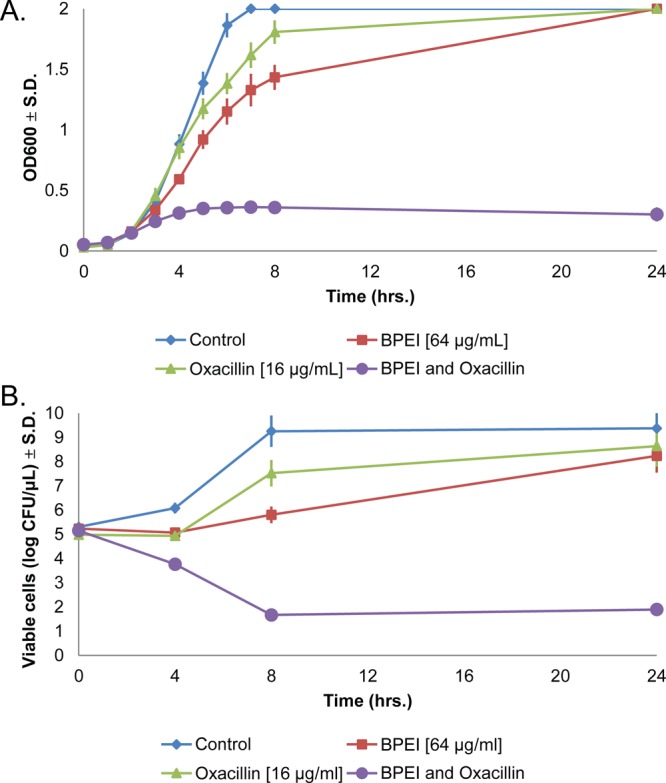
The 600 Da BPEI and oxacillin comprise a bactericidal combination in MRSA 700787. Cells treated individually with BPEI or oxacillin were still able to grow, but growth was inhibited when both BPEI and oxacillin were used (A). A time–kill curve shows that the combination has a bactericidal mechanism of action as seen with the drop in viable cell counts after 4 h (B). Error bars denote standard deviation (n = 3).
WTA is important for BPEI potentiation of β-lactam antibiotics. Studies have shown that WTA is important for β-lactam resistance in MRSA and aids the localization of PBP4 to the division septum.7,8,29,30 WTA is dispensable for cell growth; however, WTA deficient mutants of MRSA have an increased susceptibility to β-lactam antibiotics.7 Also, by inhibiting WTA synthesis by chemical inhibition of TarO, the protein responsible for the first step of WTA synthesis results in a decreased resistance. Recent work has been done to develop new WTA inhibitors to fight MRSA with reduced protein binding effects.30−32 Removal of WTA through genetic or chemical means reduced CFUs in mice treated with imipenem.29 Disabling mature WTA in the cell wall should have a similar effect.
BPEI, with its polycationic properties, has the potential for very strong electrostatic interactions with the polyanionic WTA molecules. Localization of BPEI to the Gram-positive cell wall results from interactions between the primary amines of BPEI and the phosphate groups of WTA. Previously, using laser scanning confocal microscopy, we showed that BPEI tagged with Alexa Fluor 488 localizes on the perimeter of Gram-positive MRSA cells but not on Gram-negative E. coli cells.15 Using NMR, we further found that phosphate–amine binding from the WTA–BPEI interactions likely occurs through electrostatic attraction between the numerous cationic primary amines of BPEI and anionic phosphate groups of WTA.15 BPEI and ampicillin have synergy against MRSA MW2 (Figure S3A). However, BPEI did not exhibit synergy with ampicillin against MRSA MW2 ΔtarO (strain referenced in ref (7)), which lacks WTA (Figure S3B). This, combined with the data in Figure 1, suggests that BPEI potentiation arises through binding to WTA, preventing proper localization of PBP2a/4, and thereby allowing cell death via β-lactam inhibition of other PBPs.
BPEI prevents proper cell division in MRSA. WTA deficient mutants have altered morphologies. SEM images at midexponential phase showed that a WTA-deficient mutant was significantly larger and had a rougher surface than the wild-type cells.33 We were motivated to determine if BPEI caused any morphological changes in MRSA. MRSA cells were grown in sublethal concentrations of BPEI, isolated at late-lag phase, and imaged. SEM images show that BPEI does not dramatically alter the shape of MRSA 700787. However, the size of the cells was significantly increased (p-value < 0.001) from 0.86 ± 0.11 to 0.98 ± 0.14 μm when grown in BPEI, and the numerous MRSA aggregates suggest the inability to properly form septa and complete the cell division process (Figure 4). The cell morphologies do not appear to be significantly altered; however, the cleavage furrow does not form properly for the cells treated with BPEI. The phenotypic similarity between cells grown in BPEI and knockout mutant cells suggests that the altered morphologies may arise from an interaction between BPEI and WTA. If so, autolysin activity, which is controlled by WTA, should also be affected.
Figure 4.

BPEI caused a significant increase in MRSA 700787 cell size. The average cell size (largest diameter) for untreated cells is 0.86 ± 0.11 μm (A) but increases to 0.98 ± 0.14 μm when grown in 64 μg/mL (107 μM) BPEI (B). Normal cleavage furrows are shown with black arrows. Enlarged cells that do not have cleavage furrows present are shown with white arrows. Cells were fixed and imaged at late-lag phase. Scale bar equals 1 μm. A size analysis graph (C) shows the average cell size (center line), the standard deviation (outside of box), and minimum and maximum values (error bars) of 100 measured cells (p-value < 0.001).
Autolysins are used by bacterial cells to lyse peptidoglycan strands during cell division, allowing separation of daughter cells. The importance of WTA and lipoteichoic acid (LTA) in the regulation of autolysin activity is well-known. Schlag et al. found that autolysis increased in WTA knockout mutants and proposed that mature WTA prevents binding of Atl, an autolytic protein in S. aureus, to the mature cell wall.33 WTA knockout mutants were unable to correctly localize Atl to the division septum.33 Campbell et al. suggests that, when WTA is absent, inefficient septal formation occurs due to the degradation of the autolysins track that allows for cell separation.7 Our SEM images show that MRSA aggregates arise after BPEI exposure, which could have resulted if autolysis was hindered (Figure 4). Triton X-100 induced autolytic rate measurements were performed to determine if BPEI affects autolysis in MRSA 700787. The autolytic rate of MRSA cells grown in 16 μg/mL (27 μM) of BPEI was slowed in comparison to control samples (Figure S4). Autolysis was completely stopped for MRSA cells grown in 64 μg/mL (107 μM) of BPEI. Due to a larger initial cell density (∼1 × 108 CFUs/mL), the concentration of BPEI used was higher than the MIC found in the checkerboard assays (∼5 × 105 CFUs/mL). Triton X-100 induced autolysis is believed to occur via the release of LTA from the cell wall.34 The origin of BPEI-induced slowing of autolysis is unknown, but a possible explanation is the prevention of LTA release by the electrostatic binding of BPEI to LTA, which causes steric restraint and prevents Atl localization. Further work is necessary to test this hypothesis.
Diagnosed or suspected MRSA infections require treatment with vancomycin, linezolid, or daptomycin;2 and to date, no MRSA strain is resistant to more than one of them.20,35 Other drugs such as ceftaroline, teflaro, and telavancin have been approved for patient use in severe cases, but all must be given intravenously.2 Yet, people suffer from MRSA infections because MRSA is either misdiagnosed or not suspected, and ineffective first-line antibiotics, usually β-lactams,1 are given. After MRSA diagnosis, clinicians turn to drugs of last resort, but treatment delays can result in mortality or increased morbidity due to release of MRSA toxins into tissue.35,36 In 2011, MRSA infected 80 500 people; nearly 1 in 7 cases resulted in death (11 300; 14%).17 Our expected research contribution is to address these drawbacks by blocking the PBP2a and PBP4 resistance pathways with BPEI, thereby potentiating β-lactam antibiotics to kill MRSA. The typical route is inhibiting WTA synthesis to prevent PBP2a/4 function. Here, we have described a different approach that, in our opinion, departs from the status quo by deactivating mature WTA within the cell wall through electrostatic interactions with BPEI.15 Nontoxic 600 Da BPEI was able to potentiate β-lactam efficacy against MRSA. The mechanism of action is electrostatically binding to WTA and thereby preventing proper localization of PBP2a/PBP4 (Figures S5 and S6). Future studies are planned to probe the structure–activity relationship, test in vivo efficacy, and further elucidate the mechanism of action. The impact of our contribution is meeting the need for new first-line antibiotics that can be given promptly at the first sign of any S. aureus infection to kill MSSA and/or MRSA without the need to identify MRSA as the infectious agent.
Acknowledgments
This work was possible due to the kindness and contributions of Prof. Daniel Glatzhofer, Prof. Robert Cichewicz, Jarrod King, Dr. Rama Kothapalli, Prof. Anthony Burgett, Dr. Preston Larson, and Greg Strout. We also acknowledge Dr. Suzanne Walker (Harvard) for providing MRSA MW2 and the ΔtarO mutant.
Glossary
ABBREVIATIONS
- MRSA
methicillin-resistant Staphylococcus aureus
- MSSA
methicillin-susceptible Staphylococcus aureus
- BPEI
branched polyethylenimine
- NMR
nuclear magnetic resonance
- SEM
scanning electron microscopy
- WTA
wall teichoic acid
- LTA
lipoteichoic acid
- PBP
penicillin-binding protein
- FIC
fractional inhibitory concentration
- MIC
minimum inhibitory concentration
- MBC
minimum bactericidal concentration
- OD600
optical density at 600 nm
- Da
Dalton
- MW
molecular weight
- LDH
lactate dehydrogenase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00285.
Additional figures and experimental section (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have approved the final version of the manuscript.
Funding was provided by the Oklahoma Center of Advancement of Science and Technology (HR16-084-1), National Institutes of Health (1R01GM090064-01), and the University of Oklahoma.
The authors declare no competing financial interest.
Supplementary Material
References
- Hicks L. A.; Bartoces M. G.; Roberts R. M.; Suda K. J.; Hunkler R. J.; Taylor T. H. Jr.; Schrag S. J. US outpatient antibiotic prescribing variation according to geography, patient population, and provider specialty in 2011. Clin. Infect. Dis. 2015, 60 (9), 1308–1316. 10.1093/cid/civ076. [DOI] [PubMed] [Google Scholar]
- Liu C.; Bayer A.; Cosgrove S. E.; Daum R. S.; Fridkin S. K.; Gorwitz R. J.; Kaplan S. L.; Karchmer A. W.; Levine D. P.; Murray B. E.; Rybak M. J.; Talan D. A.; Chambers H. F. Clinical Practice Guidelines by the Infectious Diseases Society of America for the Treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin. Infect. Dis. 2011, 52 (3), 18–55. 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- Gandra S.; Barter D. M.; Laxminarayan R. Economic burden of antibiotic resistance: how much do we really know?. Clin. Microbiol. Infect. 2014, 20 (10), 973–979. 10.1111/1469-0691.12798. [DOI] [PubMed] [Google Scholar]
- Waxman D. J.; Strominger J. L. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annu. Rev. Biochem. 1983, 52 (1), 825–869. 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- Memmi G.; Filipe S. R.; Pinho M. G.; Fu Z.; Cheung A. Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob. Agents Chemother. 2008, 52 (11), 3955–3966. 10.1128/AAC.00049-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atilano M. L.; Pereira P. M.; Yates J.; Reed P.; Veiga H.; Pinho M. G.; Filipe S. R. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (44), 18991–18996. 10.1073/pnas.1004304107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J.; Singh A. K.; Santa Maria J. P.; Kim Y. Jr.; Brown S.; Swoboda J. G.; Mylonakis E.; Wilkinson B. J.; Walker S. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 2011, 6 (1), 106–116. 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farha M. A.; Leung A.; Sewell E. W.; D’Elia M. A.; Allison S. E.; Ejim L.; Pereira P. M.; Pinho M. G.; Wright G. D.; Brown E. D. Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to beta-lactams. ACS Chem. Biol. 2012, 8 (1), 226–233. 10.1021/cb300413m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidenmaier C.; Peschel A.; Xiong Y.-Q.; Kristian S. A.; Dietz K.; Yeaman M. R.; Bayer A. S. Lack of wall teichoic acids in Staphylococcus aureus leads to reduced interactions with endothelial cells and to attenuated virulence in a rabbit model of endocarditis. J. Infect. Dis. 2005, 191 (10), 1771–1777. 10.1086/429692. [DOI] [PubMed] [Google Scholar]
- Vergara-Irigaray M.; Maira-Litrán T.; Merino N.; Pier G. B.; Penadés J. R.; Lasa I. Wall teichoic acids are dispensable for anchoring the PNAG exopolysaccharide to the Staphylococcus aureus cell surface. Microbiology 2008, 154 (3), 865–877. 10.1099/mic.0.2007/013292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland L. M.; Conlon B.; O’Gara J. P. Mutation of tagO reveals an essential role for wall teichoic acids in Staphylococcus epidermidis biofilm development. Microbiology 2011, 157 (2), 408–418. 10.1099/mic.0.042234-0. [DOI] [PubMed] [Google Scholar]
- Weidenmaier C.; Kokai-Kun J. F.; Kristian S. A.; Chanturiya T.; Kalbacher H.; Gross M.; Nicholson G.; Neumeister B.; Mond J. J.; Peschel A. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat. Med. 2004, 10 (3), 243–245. 10.1038/nm991. [DOI] [PubMed] [Google Scholar]
- Thomas K. J. 3rd; Rice C. V. Revised model of calcium and magnesium binding to the bacterial cell wall. BioMetals 2014, 27 (6), 1361–1370. 10.1007/s10534-014-9797-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar A.; Golemi-Kotra D. Dual roles of FmtA in Staphylococcus aureus cell wall biosynthesis and autolysis. Antimicrob. Agents Chemother. 2012, 56 (7), 3797–3805. 10.1128/AAC.00187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxley M. A.; Friedline A. W.; Jensen J. M.; Nimmo S. L.; Scull E. M.; King J. B.; Strange S.; Xiao M. T.; Smith B. E.; Thomas Iii K. J.; Glatzhofer D. T.; Cichewicz R. H.; Rice C. V. Efficacy of ampicillin against methicillin-resistant Staphylococcus aureus restored through synergy with branched poly(ethylenimine). J. Antibiot. 2016, 69 (12), 871–878. 10.1038/ja.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W. P.; Gray A. I.; Tetley L.; Hang T. L. B.; Schatzlein A. G.; Uchegbu I. F. Polyelectrolyte nanoparticles with high drug loading enhance the oral uptake of hydrophobic compounds. Biomacromolecules 2006, 7 (5), 1509–1520. 10.1021/bm060130l. [DOI] [PubMed] [Google Scholar]
- Dantes R.; Mu Y.; Belflower R.; Aragon D.; Dumyati G.; Harrison L. H.; Lessa F. C.; Lynfield R.; Nadle J.; Petit S.; Ray S. M.; Schaffner W.; Townes J.; Fridkin S. Emerging infections program-active bacterial core surveillance, M. S. I., National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Int. Med. 2013, 173 (21), 1970–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.; Sievert D. M.; Hageman J. C.; Boulton M. L.; Tenover F. C.; Downes F. P.; Shah S.; Rudrik J. T.; Pupp G. R.; Brown W. J.; Cardo D.; Fridkin S. K. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 2003, 348 (14), 1342–1347. 10.1056/NEJMoa025025. [DOI] [PubMed] [Google Scholar]
- Sievert D. M.; Rudrik J. T.; Patel J. B.; McDonald L. C.; Wilkins M. J.; Hageman J. C. Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin. Infect. Dis. 2008, 46 (5), 668–674. 10.1086/527392. [DOI] [PubMed] [Google Scholar]
- Mangili A.; Bica I.; Snydman D. R.; Hamer D. H. Daptomycin-resistant, methicillin-resistant Staphylococcus aureus bacteremia. Clin. Infect. Dis. 2005, 40 (7), 1058–1060. 10.1086/428616. [DOI] [PubMed] [Google Scholar]
- Tsiodras S.; Gold H. S.; Sakoulas G.; Eliopoulos G. M.; Wennersten C.; Venkataraman L.; Moellering R. C. Jr; Ferraro M. J. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 2001, 358 (9277), 207–208. 10.1016/S0140-6736(01)05410-1. [DOI] [PubMed] [Google Scholar]
- Leski T. A.; Tomasz A. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J. Bacteriol. 2005, 187 (5), 1815–1824. 10.1128/JB.187.5.1815-1824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo-Godoy A.; Muldoon C.; Becker B.; Elliott A. G.; Lash L. H.; Huang J. X.; Butler M. S.; Pelingon R.; Kavanagh A. M.; Ramu S.; Phetsang W.; Blaskovich M. A.; Cooper M. A. Activity and predicted nephrotoxicity of synthetic antibiotics based on polymyxin B. J. Med. Chem. 2016, 59 (3), 1068–1077. 10.1021/acs.jmedchem.5b01593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. X.; Kaeslin G.; Ranall M. V.; Blaskovich M. A.; Becker B.; Butler M. S.; Little M. H.; Lash L. H.; Cooper M. A. Evaluation of biomarkers for in vitro prediction of drug-induced nephrotoxicity: comparison of HK-2, immortalized human proximal tubule epithelial, and primary cultures of human proximal tubular cells. Pharmacol. Res. Perspect. 2015, 3 (3), e00148. 10.1002/prp2.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney K.; Sovadinova I.; Lopez A. I.; Urban M.; Ridgway Z.; Caputo G. A.; Kuroda K. Poly(ethylene imine)s as antimicrobial agents with selective activity. Macromol. Biosci. 2012, 12 (9), 1279–1289. 10.1002/mabi.201200052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G. P.; Guo H. Y.; Alexis F.; Wang X.; Zeng S.; Lim T. M.; Ding J.; Yang Y. Y.; Wang S. Low molecular weight polyethylenimines linked by β-cyclodextrin for gene transfer into the nervous system. J. Gene Med. 2006, 8 (6), 736–744. 10.1002/jgm.874. [DOI] [PubMed] [Google Scholar]
- Fischer D.; Li Y.; Ahlemeyer B.; Krieglstein J.; Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24 (7), 1121–1131. 10.1016/S0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- Levison M. E.; Levison J. H. Pharmacokinetics and pharmacodynamics of antibacterial agents. Infectious disease clinics of North America 2009, 23 (4), 791–815. 10.1016/j.idc.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Wang H.; Labroli M.; Koseoglu S.; Zuck P.; Mayhood T.; Gill C.; Mann P.; Sher X.; Ha S.; Yang S.-W.; Mandal M.; Yang C.; Liang L.; Tan Z.; Tawa P.; Hou Y.; Kuvelkar R.; DeVito K.; Wen X.; Xiao J.; Batchlett M.; Balibar C. J.; Liu J.; Xiao J.; Murgolo N.; Garlisi C. G.; Sheth P. R.; Flattery A.; Su J.; Tan C.; Roemer T. TarO-specific inhibitors of wall teichoic acid biosynthesis restore β-lactam efficacy against methicillin-resistant staphylococci. Sci. Transl. Med. 2016, 8 (329), 329ra32. 10.1126/scitranslmed.aad7364. [DOI] [PubMed] [Google Scholar]
- Mandal M.; Tan Z.; Madsen-Duggan C.; Buevich A. V.; Caldwell J. P.; Dejesus R.; Flattery A.; Garlisi C. G.; Gill C.; Ha S. N.; Ho G.; Koseoglu S.; Labroli M.; Basu K.; Lee S. H.; Liang L.; Liu J.; Mayhood T.; McGuinness D.; McLaren D. G.; Wen X.; Parmee E.; Rindgen D.; Roemer T.; Sheth P.; Tawa P.; Tata J.; Yang C.; Yang S.-W.; Xiao L.; Wang H.; Tan C.; Tang H.; Walsh P.; Walsh E.; Wu J.; Su J. Can we make small molecules lean? Optimization of a highly lipophilic TarO inhibitor. J. Med. Chem. 2017, 60 (9), 3851–3865. 10.1021/acs.jmedchem.7b00113. [DOI] [PubMed] [Google Scholar]
- Farha M. A.; Koteva K.; Gale R. T.; Sewell E. W.; Wright G. D.; Brown E. D. Designing analogs of ticlopidine, a wall teichoic acid inhibitor, to avoid formation of its oxidative metabolites. Bioorg. Med. Chem. Lett. 2014, 24 (3), 905–910. 10.1016/j.bmcl.2013.12.069. [DOI] [PubMed] [Google Scholar]
- Yang S.-W.; Pan J.; Yang C.; Labroli M.; Pan W.; Caldwell J.; Ha S.; Koseoglu S.; Xiao J. C.; Mayhood T.; Sheth P. R.; Garlisi C. G.; Wu J.; Lee S. H.; Wang H.; Tan C. M.; Roemer T.; Su J. Benzimidazole analogs as WTA biosynthesis inhibitors targeting methicillin resistant Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2016, 26 (19), 4743–4747. 10.1016/j.bmcl.2016.08.036. [DOI] [PubMed] [Google Scholar]
- Schlag M.; Biswas R.; Krismer B.; Kohler T.; Zoll S.; Yu W.; Schwarz H.; Peschel A.; Gotz F. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 2010, 75 (4), 864–873. 10.1111/j.1365-2958.2009.07007.x. [DOI] [PubMed] [Google Scholar]
- Suzuki J.; Komatsuzawa H.; Sugai M.; Ohta K.; Kozai K.; Nagasaka N.; Suginaka H. Effects of various types of Triton X on the susceptibilities of methicillin-resistant staphylococci to oxacillin. FEMS Microbiol. Lett. 1997, 153 (2), 327–331. 10.1111/j.1574-6968.1997.tb12592.x. [DOI] [PubMed] [Google Scholar]
- Pastagia M.; Kleinman L. C.; de la Cruz E. G. L.; Jenkins S. G. Predicting risk for death from MRSA bacteremia. Emerging Infect. Dis. 2012, 18 (7), 1072–1080. 10.3201/eid1807.101371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove S. E.; Qi Y. L.; Kaye K. S.; Harbarth S.; Karchmer A. W.; Carmeli Y. The impact of methicillin-resistance in Staphylococcus aureus bacteremia on patient outcomes: Mortality, length of stay, and hospital charges. Infect. Cont. Hosp. Ep. 2005, 26 (2), 166–174. 10.1086/502522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.