

Abstract
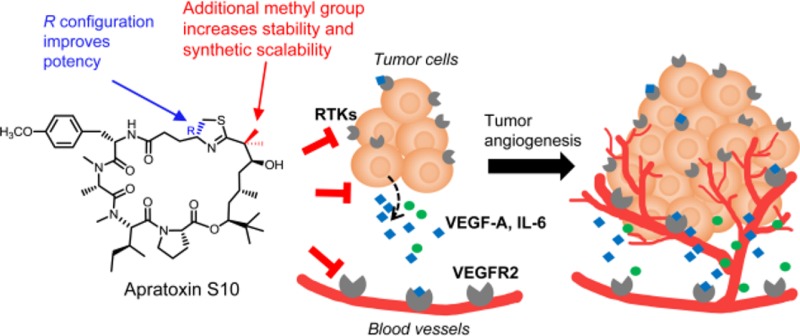
Renal, hepatocellular, and neuroendocrine carcinomas are known as highly vascularized tumors. Although vascular endothelial growth factor A (VEGF-A)-targeted therapies have shown efficacy in the treatment of these cancers, drug resistance is a major concern and might be mediated by interleukin 6 (IL-6). Furthermore, upon antiangiogenic drug exposure, tumor cells may adapt to survive in a vascular-independent manner. Apratoxins are potent marine-derived cytotoxic in vivo-active agents, preventing cotranslational translocation in the secretory pathway, and show promise to overcome resistance by targeting angiogenesis and tumor growth simultaneously. We designed and synthesized a novel apratoxin analogue, apratoxin S10, with a balanced potency and stability as well as synthetic accessibility and scalability. We showed that apratoxin S10 potently inhibits both angiogenesis in vitro and growth of cancer cells from vascularized tumors. Apratoxin S10 down-regulated vascular endothelial growth factor receptor 2 (VEGFR2) on endothelial cells and blocked the secretion of VEGF-A and IL-6 from cancer cells. It inhibited cancer cell growth through down-regulation of multiple receptor tyrosine kinases (RTKs) and compares favorably to currently approved RTK inhibitors in both angiogenesis and cancer cell growth.
Keywords: Antiangiogenic agents, antiproliferative agents, vascular endothelial growth factor, receptor tyrosine kinases, total synthesis, cotranslational translocation inhibitor
The concept of treating cancer by inhibiting new blood vessel formation in tumors was established by Judah Folkman about 50 years ago.1 Tumor angiogenesis, or the formation of blood vessels, is a vital step for tumor development and metastasis. Without angiogenesis, a tumor will not grow beyond 2–3 mm in size.2 The tumor cells secreted growth factors that caused new capillaries to sprout, grow toward, and then infiltrate into the tumor mass, supplying nutrients and oxygen and supporting the growth of a hyper-proliferating tumor. It is widely accepted that vascular endothelial growth factor A (VEGF-A) is the key angiogenic regulator contributing to tumor angiogenesis. VEGF-A is highly secreted from tumor mass and diffused into the tumor microenvironment and binds to its cognate receptors on endothelial cells, with vascular endothelial growth factor receptor 2 (VEGFR2) as the major one, leading to activation of signaling pathways involved in mediating proliferation, migration, and survival of endothelial cells and promoting vascular permeability.3,4
Tumor angiogenesis is a complex mechanism that depends on the tumor type. Renal cell carcinoma (RCC) is a highly vascularized tumor (Figure 1), which is often due to a hyper-activated proangiogenic signaling pathway triggered by von Hippel Lindau (VHL) gene mutations.5,6 Loss-of-function of the VHL protein causes unregulated activation of hypoxia inducible factor (HIF) and overexpression of VEGF-A. In addition to RCC, other indications including hepatocellular carcinoma and neuroendocrine carcinoma (Figure 1) are also considered to be highly vascularized tumors, which have been the focus of development for antiangiogenic agents.
Figure 1.
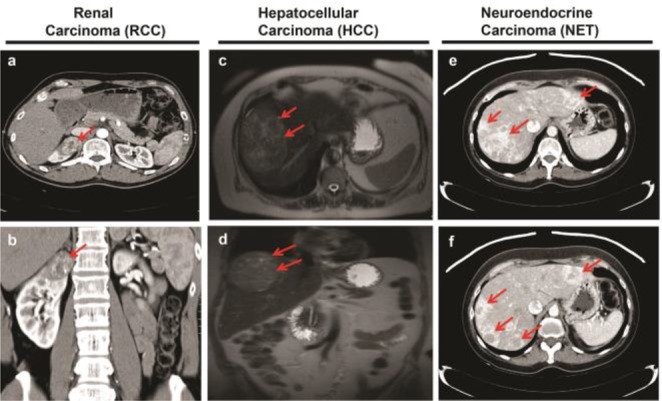
Images of highly vascularized tumors from deidentified cancer patients. Red arrows show the tumors. Tumor vascularization is indicated by white contrast enhancement either infiltrating (a–d) or surrounding (e,f) the tumors. Contrast enhanced computed tomography (CT) images showing highly vascularized primary renal cell cancer of the right kidney (a,b) and highly vascularized neuroendocrine tumor metastatic to the liver (e,f). Contrast enhanced magnetic resonance images showing highly vascularized primary hepatocellular carcinoma (c,d).
Due to the crucial role of VEGF-A/VEGFR signaling in tumor angiogenesis, VEGF-targeted therapies have been developed to inhibit blood vessel growth and thus starve tumors of necessary oxygen and nutrients. Currently, the first and second line treatment for these vascularized cancers include VEGF-A monoclonal antibody, bevacizumab, and several antiangiogenic receptor tyrosine kinase (RTK) inhibitors, including sunitinib and sorafenib. Despite the efficacy of antiangiogenic therapy, the development of drug resistance leading to transient clinical benefits and failure of antiangiogenic drugs is still a major concern. Individual responses are variable, with some patients never responding to the drugs (intrinsic resistance) and others going on to develop resistance following a brief period of treatment (acquired resistance).
There is increasing evidence that reveals significant relationships between interleukin 6 (IL-6) and both tumor angiogenesis and resistance against antiangiogenic therapy.7−13 IL-6 is a multifunctional cytokine, secreted by a number of different types of cells including tumor cells. In renal cell carcinoma cells, IL-6 functions as an autocrine growth factor and induces cell growth in vitro.14,15 It has been shown that high serum IL-6 levels in cancer patients were associated with a poor outcome and bevacizumab and sunitinib resistance.7,16 Siltuximab, an anti-IL-6 chimeric monoclonal antibody, is under investigation in clinical trials for multiple types of cancers with favorable results observed in a phase I/II study.17,18 With such compelling evidence highlighting the roles of IL-6 in angiogenesis, it would appear to be an attractive target for combination therapy in numerous tumor types with VEGF-A-targeting drugs.
Furthermore, in some scenario, upon antiangiogenic drug treatment, despite a strong vascular response, shrinkage of tumor tissue is negligible.19−21 Given that the development of intrinsic or acquired resistance to antiangiogenic therapy is mediated through a complex mechanism,22−24 the molecular mechanism attributed to the stabilized tumor growth despite a diminished vascular supply is still under investigation, but it is possible that tumor cells may be adapted to survive in a vascular-independent manner.2 Currently, most antiangiogenic drugs are primarily targeting the tumor endothelium rather than tumor cells, aiming to reduce vascular density and starving the nutrient for cancer cells.25 The above information has raised the possibility that acquired resistance might be circumvented if an antitumor effect could be achieved simultaneously with antiangiogenic treatment; in other words, a combination of antitumor and antiangiogenic therapy would be more beneficial. We have identified a class of marine natural products, apratoxins, which have the necessary attributes to be a dual inhibitor.
Apratoxins are potent cytotoxic agents derived from marine cyanobacteria.26−31 Apratoxin A possesses broad-spectrum differential in vitro activities,32 and its cytotoxicity is due to potent inhibition of cotranslational translocation33 at the level of the Sec61 translocon,34 leading to both down-regulation of various receptor tyrosine kinases and reduced growth factor secretion.33 This dual effect on RTKs and their ligands, including VEGF and its receptor VEGFR2, give a one–two punch to cancer cells, particularly those cancers that rely on autocrine loops.35 Having this striking feature, we aimed to further explore this effect on other cancer-related cell types, specifically, endothelial cells. As mentioned above, endothelial cells are enriched in VEGF receptors that recognize VEGF secreted from tumor cells leading to formation of blood vessels. We proposed that apratoxins could also down-regulate VEGFR2 on endothelial cells and through which an additional antiangiogenic effect is achieved. Provided that apratoxins exert both antiangiogenic and antitumor effects, this structure class could be a promising scaffold to develop inhibitors overcoming drug resistance of antiangiogenic therapy.
Our group has spent considerable efforts on improving therapeutic index of apratoxin A through a medicinal chemistry campaign, which led to apratoxins S4, S8, and S9 (Figure 2), possessing potent in vitro as well as in vivo anticancer activities and enhanced in vitro stability.35,36 Recognizing potential liabilities that might be responsible for the irreversible toxicity and low tolerability of apratoxin A in vivo, we previously replaced the α,β-unsaturated system at C27–C31 in apratoxin A with a corresponding saturated unit in apratoxin E, which resulted in a synthetic apratoxin A/E hybrid, apratoxin S4, possessing further improved potency, in vivo selectivity, and antitumor activity.35 An extended SAR study of apratoxins S4 (30S) and S9 (30R)36 coupled with our recent work on the total synthesis of (30R)-apratoxin E (natural product) and its epimer (30S)-apratoxin E37 demonstrated the positive impact of the modified cysteine (moCys) moiety on activity with R configuration at C30 more potent than S. Although apratoxins S4 and S9 possess excellent potency, we observed a high tendency of dehydration at C34–35 during the scale-up synthesis. Knowing that this issue may result in (1) pharmacological deactivation and (2) side products during the synthesis that would lower the synthetic yield and require HPLC purification of the intermediate, we introduced an additional methyl group at C34, which prevented the dehydration in apratoxin S8 and led to an enhanced three-step yield (70%) during macrocyclization and a total yield (3%) without sacrificing potency in vivo.36 With apratoxin S8 having the highest yield during macrocyclization and S9 as the most potent apratoxin analogue against human colon cancer cells, we logically proposed a new hybrid apratoxin S8/S9 compound, apratoxin S10 (1), aiming to achieve a balance between potency, stability, and synthetic yield.
Figure 2.
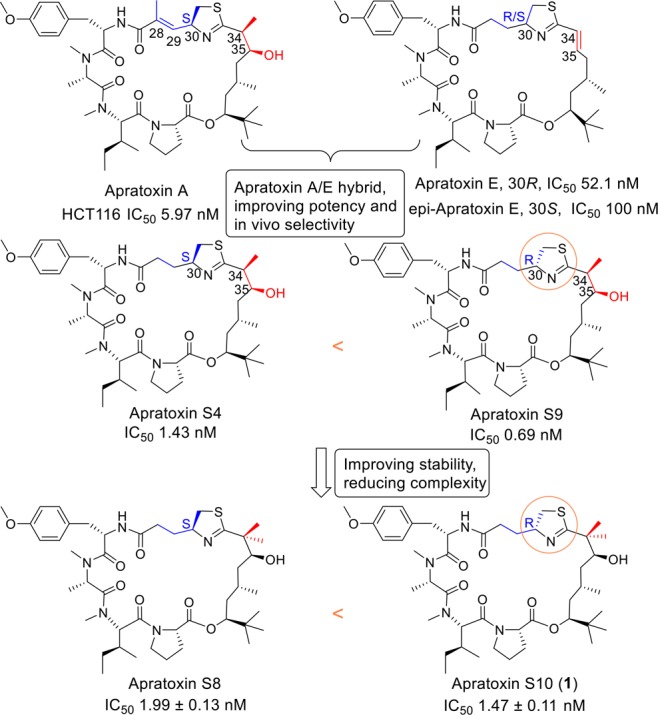
SAR study: Apratoxin A and E (natural products) and evolution of synthetic analogues with corresponding IC50 values for HCT116 colorectal cancer cells. C30R configuration (apratoxins S9 and S10) leads to a higher potency than C30S (apratoxins S4 and S8).
In our current study, we aimed to (1) conduct total synthesis of 1 and (2) evaluate its effect on both angiogenesis and tumor growth in highly vascularized cancer cell models.
The synthetic route of 1 is depicted in Scheme 1. We applied a similar synthetic strategy that we previously developed for the synthesis of apratoxins S4–S9,35,36 which is a modification of other published methods.38−44 Recently, some other papers were published on total syntheses of apratoxins.45−51 The known compounds 2, 3, and 7 were synthesized as we established previously.36 The N-Boc group in 3 was selectively removed with TMSOTf in the presence of 2,6-lutidine, which was subsequently followed by a coupling reaction of deprotected 3 with 2 to afford compound 4 in 92% yield. Using a modified Kelly’s method, the thiazoline ring was formed smoothly catalyzed by TiCl4. Subsequent removal of Troc using Zn-NH4OAc gave compound 5 with a good two-step yield of 60%. The allyl group in 5 was removed through a standard deprotection reaction using Pd(PPh3)4/N-methylaniline to obtain acid 6 in 95% yield. The Fmoc-protected tripeptide 7 was treated with Et2NH in MeCN, and the liberated free amine in 7 was then coupled with carboxylic acid in 6 to afford a linear compound 8 in 92% yield (two steps) using PyAOP as coupling reagent. Finally, the target compound 1 was synthesized from 8 through three sequential reaction steps: cleavage of allyl ester by Pd(PPh3)4/N-methylaniline, removal of Fmoc by Et2NH/MeCN, and macrocyclization reaction mediated by PyAOP, with an excellent yield of 80% in three steps (95% HPLC purity and 100% chiral purity). About 5% epimerization happened during macrocyclization, and the epimer was able to be separated by HPLC.
Scheme 1. Total Synthesis of Apratoxin S10.

It is important to note that the synthetic yield of apratoxin S10 (1) is higher than that of apratoxin S8 in terms of both total yield and three-step macrocyclization yield (Table S1). The high cyclization efficiency of the precursor of 1 was reproducible in parallel reactions. The intermediates 4, 5, and 6 were also obtained in a higher yield than the corresponding intermediates in the synthesis of apratoxin S8, which suggests that the structural conformation with C30R is more favorable than C30S. Furthermore, the antiproliferative activity of 1 was directly compared to the two parent compounds apratoxin S8 and S9 using HCT116, a human colon cancer cell model that we used previously for the characterization of apratoxins S8 and S9. As expected, the hybridization of S8 and S9 led to an improved potency of S10 (IC50 = 1.47 ± 0.11 nM) compared with S8 (IC50 = 1.99 ± 0.13 nM) (Figure 2). Additionally, apratoxins S4, S8, S9, and S10 were stable (t1/2 > 24 h) under acidic aqueous condition, pH 2.32, a pH representing gastric condition, with apratoxin S7 (a nonmethyl-C34 analogue)36 being an exception (Figure S1). These findings suggest that an increased C30 methylation status increases stability and that we have successfully achieved a balance between synthetic yield and potency by hybridizing the two lead compounds in terms of yields (S8) and potency (S9).
Following the successful design and synthesis of 1 with an improvement in compound stability and synthesis yield, we then aimed to evaluate its effect as an antiangiogenic agent. We tested 1 in a human endothelial cell (HUVEC)-based in vitro angiogenesis model, mimicking the process of blood vessel formations. Compared with solvent control, the presence of 1 significantly inhibited the formation of tube-like structures (Figure 3a). Concentrations as low as 10 pM caused detectable diminishing of branch points and number of junctions (Figure 3b,c). Increasing concentrations of 1 further decreased tube-like structures by both visual inspection and automatic quantifications. We have also tested known RTKs inhibitors in parallel (Figure 3). Sunitinib, a first line antiangiogenic drug for renal cancer, inhibited tube formation at 100 nM and 1 μM (Figure 3a). However, its effect under concentrations lower than 100 nM was not observed. As expected, erlotinib, an EGFR inhibitor, did not show any antiangiogenic effect within the tested concentration range (Figure 3a). Cabozantinib, a multi-RTK inhibitor, although showing better effect than both sunitinib and erlotinib, still showed a slightly weaker effect than 1 (Figure 3a). These results indicated a potent antiangiogenic effect of 1 in vitro. To rule out the possibility that apratoxins disrupted angiogenesis in vitro due to cytotoxicity on HUVEC cells, we monitored its effect on HUVEC cell viability using MTT assay, indicating a negligible effect on cell viability (Figure 3d). Knowing that VEGFR2 is highly expressed in endothelial cells, we next tested 1’s effect on VEGFR2 expression. Consistent with the proposed mode of action,33,351 down-regulated the expression level of functional (glycosylated) VEGFR2 on endothelial cells at 10 nM and above (Figure 3e), which potentially explained its antiangiogenic effect in this assay.
Figure 3.
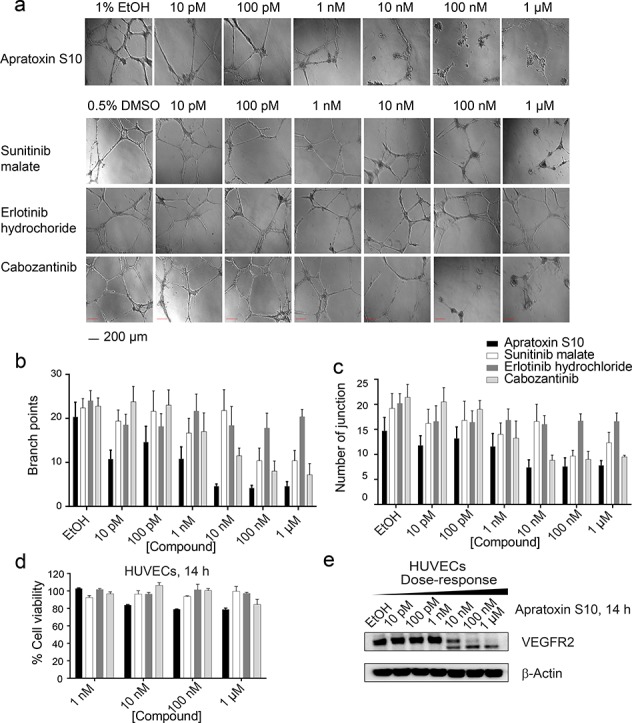
(a) Apratoxin S10 inhibited angiogenesis in vitro in a dose-dependent manner, determined by matrigel assay using HUVECs (scale bar 200 μm), 14 h. Three known RTK inhibitors were tested in parallel. (b) Branch point counting was used as quantification method. Five random microscope view-fields were counted, and the number of branch points was averaged. (c) Number of junctions analyzed by the Angiogenesis Analyzer plug-in for ImageJ (n = 5 per group). Error bars in (c) and (d) indicate mean ± SEM of five fields. (d) Antiproliferative effect of apratoxin S10 and known RTK inhibitors on HUVECs. Error bars indicate mean ± SD of three replicates. (e) Immunoblot analysis using lysates from apratoxin S10-treated HUVECs, 14 h. The higher bands are functional (glycosylated) VEGFR2. The lower bands correspond to the unprocessed (non-glycosylated) form of VEGFR2.
Our previous studies indicated that apratoxins effectively blocked VEGF-A secretion from human colon cancer cells (HCT116).35,36 Here, we evaluated the effect of 1 on VEGF-A secretion in highly vascularized cancer cell models: renal cancer (A498), hepatocellular carcinoma (Huh7), and neuroendocrine cancer (NCI-H727). Indeed, VEGF-A secretion in all three cell lines was blocked by 1 (Figure 4). Since IL-6 has also been implicated in angiogenesis, we evaluated the effect of 1 on IL-6 secretion in these three cell lines. Except for NCI-H727 cells, which do not produce a detectable amount of IL-6, the other two cell lines (A498 and Huh7) produced high and detectable levels of IL-6, respectively, which were all effectively inhibited by 1 (Figure 4).
Figure 4.

Activity of apratoxin S10 on VEGF-A and IL-6 secretion, 24 h. VEGF-A secretion from (a) A498, (b) Huh7, and (c) NCI-H727 cells detected using AlphaLISA Human VEGF-A Immunoassay Kit (PerkinElmer). IL-6 secretion from (d) A498 and (e) Huh7 cells detected using AlphaLISA Human IL-6 Immunoassay Kit (PerkinElmer). Error bars indicate mean ± SD of three replicates.
In addition to its antiangiogenic effects, we also evaluated 1 for its effect on cancer cell growth using the three representative cell lines above. Apratoxin S10 exerted potent antiproliferative effects against all three cancer cell lines with IC50 values in the low-nanomolar range (Table 1). In contrast, the three known RTK inhibitors that we tested are 2000–5000 times less potent than 1, with IC50 values in micromolar range. Possible explanations for the tremendous difference in potency between 1 and known RTKs inhibitors are that (1) apratoxin S10 (1) blocks both RTKs and secretive factors (VEGF-A and IL-6), leading to disruption of positive feedback autocrine loops necessary for cancer cell growth15,52,53 and (2) apratoxin S10 inhibits a broader spectrum of RTKs, which prevents resistance through activation of alternative RTKs, and (3) efficacy in cell types with mutated (oncogenic) KRAS confers to intrinsic resistance to RTK inhibitors. In agreement with our previous study on human colon cancer cells,35,361 exerts its potent antiproliferative effect against these three cancer cell types through down-regulation of multiple RTKs including VEGFR2, EGFR, MET, IGF1Rβ, and FGFR4 (Figure 5). We also observed differential potency of 1 against different RTKs within the same cell line as well as VEGF-A and IL-6; among them, VEGF-A was the most sensitive protein in all cell lines (Figure 4). These data suggested that the apratoxin scaffold can be used to potentially modulate substrate selectivity, as we previously proposed,35 and differential effects also appear to be cell-type dependent.
Table 1. Activities of Apratoxin S10 and Known RTKs Inhibitors on a Range of Cancer Cells, IC50 (nM)a.
| renal | hepatocellular | neuroendocrine | |
|---|---|---|---|
| A498 | Huh7 | NCI-H727 | |
| apratoxin S10 (1) | 3.35 ± 0.34 | 0.83 ± 0.06 | 2.55 ± 0.19 |
| sunitinib | 7981 ± 1194 | 4707 ± 492 | 10120 ± 1179 |
| erlotinib | ∼12500 | ∼25000 | >6240 |
| cabozantinib | 8456 ± 2046 | 4348 ± 708 | 14010 ± 2813 |
Determined after 48 h (n = 3).
Figure 5.

Immunoblot analysis of apratoxin S10-treated (a) A498, (b) NCI-H727, and (c) Huh7 cells, 24 h. Apratoxin S10 down-regulated multiple RTKs.
In conclusion, we reported a novel apratoxin analogue, apratoxin S10 (1), which is considered one of the lead candidates of the apratoxin family in terms of potency, stability, and synthetic accessibility. We conducted the total synthesis of 1 and evaluated its dual inhibitory effects against both angiogenesis and cancer cell growth. Apratoxin S10 (1) inhibited angiogenesis in vitro, mediated through down-regulation of VEGFR2 expression of endothelial cells, and blocked secretions of VEGF-A and IL-6 from cancer cells, which are considered triggers for endothelial cell proliferation, migration, and blood vessel formation. In addition to its antiangiogenic effects, 1 possesses potent inhibitory effects against cancer cells from highly vascularized tumor through down-regulations of multiple RTKs. RTKs are differentially susceptible to 1’s inhibitory effect, suggesting apratoxins selectively inhibit different substrates in the process of cotranslational translocation through a novel mechanism that warrants further investigation.
Acknowledgments
We thank Dr. Chen Liu for providing Huh7 cells.
Glossary
ABBREVIATIONS
- RTK
receptor tyrosine kinase
- SAR
structure–activity relationship
- VEGF-A
vascular endothelial growth factor A
- VEGFR
VEGF receptor
- MET
met proto-oncogene (hepatocyte growth factor receptor)
- EGFR
epidermal growth factor receptor
- IGF1Rβ
insulin-like growth factor 1 receptor β
- FGFR4
fibroblast growth factor receptor 4
- IL-6
interleukin 6
- Cys
cysteine
- IC50
half-maximum inhibitory concentration
- moCys
modified cysteine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PyAOP
(7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- TMS
trimethylsilyl
- TMSOTf
trimethylsilyl trifluoromethanesulfonate
- Troc
2,2,2-trichloroethoxycarbonyl
- Trt
trityl/triphenylmethyl
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00192.
Experimental details, supplementary data, and copies of NMR spectra for synthetic compounds (PDF)
Author Contributions
§ Weijing Cai (biology) and Qi-Yin Chen (chemistry) contributed equally.
This research was supported by the National Institutes of Health grant R01CA172310 and Ocala Royal Dames for Cancer Research, Inc.
The authors declare the following competing financial interest(s): H. Luesch is co-founder of Oceanyx Pharmaceuticals, Inc., which has licensed patents and patent applications related to the subject matter.
Supplementary Material
References
- Folkman J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285 (21), 1182–1186. 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Vasudev N. S.; Reynolds A. R. Anti-Angiogenic Therapy for Cancer: Current Progress, Unresolved Questions and Future Directions. Angiogenesis 2014, 17 (3), 471–494. 10.1007/s10456-014-9420-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis L. M.; Hicklin D. J. VEGF-Targeted Therapy: Mechanisms of Anti-Tumour Activity. Nat. Rev. Cancer 2008, 8 (8), 579–591. 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- Ferrara N.; Adamis A. P. Ten Years of Anti-Vascular Endothelial Growth Factor Therapy. Nat. Rev. Drug Discovery 2016, 15 (6), 385–403. 10.1038/nrd.2015.17. [DOI] [PubMed] [Google Scholar]
- Escudier B.; Szczylik C.; Porta C.; Gore M. Treatment Selection in Metastatic Renal Cell Carcinoma: Expert Consensus. Nat. Rev. Clin. Oncol. 2012, 9 (6), 327–337. 10.1038/nrclinonc.2012.59. [DOI] [PubMed] [Google Scholar]
- Rini B. I.; Campbell S. C.; Escudier B. Renal Cell Carcinoma. Lancet 2009, 373 (9669), 1119–1132. 10.1016/S0140-6736(09)60229-4. [DOI] [PubMed] [Google Scholar]
- Hara M.; Nagasaki T.; Shiga K.; Takahashi H.; Takeyama H. High Serum Levels of Interleukin-6 in Patients with Advanced or Metastatic Colorectal Cancer: The Effect on the Outcome and the Response to Chemotherapy plus Bevacizumab. Surg. Today 2017, 47 (4), 483–489. 10.1007/s00595-016-1404-7. [DOI] [PubMed] [Google Scholar]
- Porta C.; Paglino C.; Imarisio I.; Ganini C.; Sacchi L.; Quaglini S.; Giunta V.; De Amici M. Changes in Circulating pro-Angiogenic Cytokines, Other than VEGF, before Progression to Sunitinib Therapy in Advanced Renal Cell Carcinoma Patients. Oncology 2012, 84 (2), 115–122. 10.1159/000342099. [DOI] [PubMed] [Google Scholar]
- Wei L.-H.; Kuo M.-L.; Chen C.-A.; Chou C.-H.; Lai K.-B.; Lee C.-N.; Hsieh C.-Y. Interleukin-6 Promotes Cervical Tumor Growth by VEGF-Dependent Angiogenesis via a STAT3 Pathway. Oncogene 2003, 22 (10), 1517–1527. 10.1038/sj.onc.1206226. [DOI] [PubMed] [Google Scholar]
- Negrier S.; Perol D.; Menetrier-Caux C.; Escudier B.; Pallardy M.; Ravaud A.; Douillard J. Y.; Chevreau C.; Lasset C.; Blay J. Y. Interleukin-6, Interleukin-10, and Vascular Endothelial Growth Factor in Metastatic Renal Cell Carcinoma: Prognostic Value of Interleukin-6–from the Groupe Francais d’Immunotherapie. J. Clin. Oncol. 2004, 22 (12), 2371–2378. 10.1200/JCO.2004.06.121. [DOI] [PubMed] [Google Scholar]
- Songur N.; Kuru B.; Kalkan F.; Ozdilekcan C.; Cakmak H.; Hizel N. Serum Interleukin-6 Levels Correlate with Malnutrition and Survival in Patients with Advanced Non-Small Cell Lung Cancer. Tumori 2004, 90 (2), 196–200. [DOI] [PubMed] [Google Scholar]
- Eichten A.; Su J.; Adler A. P.; Zhang L.; Ioffe E.; Parveen A. A.; Yancopoulos G. D.; Rudge J.; Lowy I.; Lin H. C.; MacDonald D.; Daly C.; Duan X.; Thurston G. Resistance to Anti-VEGF Therapy Mediated by Autocrine IL6/STAT3 Signaling and Overcome by IL6 Blockade. Cancer Res. 2016, 76 (8), 2327–2339. 10.1158/0008-5472.CAN-15-1443. [DOI] [PubMed] [Google Scholar]
- Fulciniti M.; Hideshima T.; Vermot-Desroches C.; Pozzi S.; Nanjappa P.; Shen Z.; Patel N.; Smith E. S.; Wang W.; Prabhala R.; Tai Y. T.; Tassone P.; Anderson K. C.; Munshi N. C. A High-Affinity Fully Human Anti-IL-6 mAb, 1339, for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2009, 15 (23), 7144–7152. 10.1158/1078-0432.CCR-09-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T. IL-6: From Its Discovery to Clinical Applications. Int. Immunol. 2010, 22 (5), 347–352. 10.1093/intimm/dxq030. [DOI] [PubMed] [Google Scholar]
- Miki S.; Iwano M.; Miki Y.; Yamamoto M.; Tang B.; Yokokawa K.; Sonoda T.; Hirano T.; Kishimoto T. Interleukin-6 (IL-6) Functions as an in Vitro Autocrine Growth Factor in Renal Cell Carcinomas. FEBS Lett. 1989, 250 (2), 607–610. 10.1016/0014-5793(89)80805-1. [DOI] [PubMed] [Google Scholar]
- Zhu A. X.; Sahani D. V.; Duda D. G.; di Tomaso E.; Ancukiewicz M.; Catalano O. A.; Sindhwani V.; Blaszkowsky L. S.; Yoon S. S.; Lahdenranta J.; Bhargava P.; Meyerhardt J.; Clark J. W.; Kwak E. L.; Hezel A. F.; Miksad R.; Abrams T. A.; Enzinger P. C.; Fuchs C. S.; Ryan D. P.; Jain R. K. Efficacy, Safety, and Potential Biomarkers of Sunitinib Monotherapy in Advanced Hepatocellular Carcinoma: A Phase II Study. J. Clin. Oncol. 2009, 27 (18), 3027–3035. 10.1200/JCO.2008.20.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J.-F.; Négrier S.; James N. D.; Kocak I.; Hawkins R.; Davis H.; Prabhakar U.; Qin X.; Mulders P.; Berns B. A Phase I/II Study of Siltuximab (CNTO 328), an Anti-Interleukin-6 Monoclonal Antibody, in Metastatic Renal Cell Cancer. Br. J. Cancer 2010, 103 (8), 1154–1162. 10.1038/sj.bjc.6605872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angevin E.; Tabernero J.; Elez E.; Cohen S. J.; Bahleda R.; van Laethem J.-L.; Ottensmeier C.; Lopez-Martin J. A.; Clive S.; Joly F.; Ray-Coquard I.; Dirix L.; Machiels J.-P.; Steven N.; Reddy M.; Hall B.; Puchalski T. A.; Bandekar R.; van de Velde H.; Tromp B.; Vermeulen J.; Kurzrock R. A Phase I/II, Multiple-Dose, Dose-Escalation Study of Siltuximab, an Anti-Interleukin-6 Monoclonal Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20 (8), 2192–2204. 10.1158/1078-0432.CCR-13-2200. [DOI] [PubMed] [Google Scholar]
- Smith A. D.; Lieber M. L.; Shah S. N. Assessing Tumor Response and Detecting Recurrence in Metastatic Renal Cell Carcinoma on Targeted Therapy: Importance of Size and Attenuation on Contrast-Enhanced CT. AJR, Am. J. Roentgenol. 2010, 194 (1), 157–165. 10.2214/AJR.09.2941. [DOI] [PubMed] [Google Scholar]
- Smith A. D.; Shah S. N.; Rini B. I.; Lieber M. L.; Remer E. M. Morphology, Attenuation, Size, and Structure (MASS) Criteria: Assessing Response and Predicting Clinical Outcome in Metastatic Renal Cell Carcinoma on Antiangiogenic Targeted Therapy. AJR, Am. J. Roentgenol. 2010, 194 (6), 1470–1478. 10.2214/AJR.09.3456. [DOI] [PubMed] [Google Scholar]
- Vasudev N. S.; Goh V.; Juttla J. K.; Thompson V. L.; Larkin J. M. G.; Gore M.; Nathan P. D.; Reynolds A. R. Changes in Tumour Vessel Density upon Treatment with Anti-Angiogenic Agents: Relationship with Response and Resistance to Therapy. Br. J. Cancer 2013, 109 (5), 1230–1242. 10.1038/bjc.2013.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G.; Hanahan D. Modes of Resistance to Anti-Angiogenic Therapy. Nat. Rev. Cancer 2008, 8 (8), 592–603. 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loges S.; Schmidt T.; Carmeliet P. Mechanisms of Resistance to Anti-Angiogenic Therapy and Development of Third-Generation Anti-Angiogenic Drug Candidates. Genes Cancer 2010, 1 (1), 12–25. 10.1177/1947601909356574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N. R.; Baker D.; Farren M.; Pommier A.; Swann R.; Wang X.; Mistry S.; McDaid K.; Kendrew J.; Womack C.; Wedge S. R.; Barry S. T. Tumor Stromal Architecture Can Define the Intrinsic Tumor Response to VEGF-Targeted Therapy. Clin. Cancer Res. 2013, 19 (24), 6943–6956. 10.1158/1078-0432.CCR-13-1637. [DOI] [PubMed] [Google Scholar]
- Huang D.; Ding Y.; Li Y.; Luo W. M.; Zhang Z. F.; Snider J.; VandenBeldt K.; Qian C. N.; Teh B. T. Sunitinib Acts Primarily on Tumor Endothelium rather than Tumor Cells to Inhibit the Growth of Renal Cell Carcinoma. Cancer Res. 2010, 70 (3), 1053–1062. 10.1158/0008-5472.CAN-09-3722. [DOI] [PubMed] [Google Scholar]
- Luesch H.; Yoshida W. Y.; Moore R. E.; Paul V. J.; Corbett T. H. Total Structure Determination of Apratoxin A, a Potent Novel Cytotoxin from the Marine Cyanobacterium Lyngbya Majuscula. J. Am. Chem. Soc. 2001, 123 (23), 5418–5423. 10.1021/ja010453j. [DOI] [PubMed] [Google Scholar]
- Luesch H.; Yoshida W. Y.; Moore R. E.; Paul V. J. New Apratoxins of Marine Cyanobacterial Origin from Guam and Palau. Bioorg. Med. Chem. 2002, 10 (6), 1973–1978. 10.1016/S0968-0896(02)00014-7. [DOI] [PubMed] [Google Scholar]
- Matthew S.; Schupp P. J.; Luesch H. Apratoxin E, a Cytotoxic Peptolide from a Guamanian Collection of the Marine Cyanobacterium Lyngbya Bouillonii. J. Nat. Prod. 2008, 71 (6), 1113–1116. 10.1021/np700717s. [DOI] [PubMed] [Google Scholar]
- Gutiérrez M.; Suyama T. L.; Engene N.; Wingerd J. S.; Matainaho T.; Gerwick W. H. Apratoxin D, a Potent Cytotoxic Cyclodepsipeptide from Papua New Guinea Collections of the Marine Cyanobacteria Lyngbya Majuscula and Lyngbya Sordida. J. Nat. Prod. 2008, 71 (6), 1099–1103. 10.1021/np800121a. [DOI] [PubMed] [Google Scholar]
- Tidgewell K.; Engene N.; Byrum T.; Media J.; Doi T.; Valeriote F. A.; Gerwick W. H. Evolved Diversification of a Modular Natural Product Pathway: Apratoxins F and G, Two Cytotoxic Cyclic Depsipeptides from a Palmyra Collection of Lyngbya Bouillonii. ChemBioChem 2010, 11 (10), 1458–1466. 10.1002/cbic.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg C. C.; Cowley E. S.; Sikorska J.; Shaala L. A.; Ishmael J. E.; Youssef D. T. A.; McPhail K. L. Apratoxin H and Apratoxin A Sulfoxide from the Red Sea Cyanobacterium Moorea Producens. J. Nat. Prod. 2013, 76 (9), 1781–1788. 10.1021/np4004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luesch H.; Chanda S. K.; Raya R. M.; DeJesus P. D.; Orth A. P.; Walker J. R.; Izpisúa Belmonte J. C.; Schultz P. G. A Functional Genomics Approach to the Mode of Action of Apratoxin A. Nat. Chem. Biol. 2006, 2 (3), 158–167. 10.1038/nchembio769. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Law B. K.; Luesch H. Apratoxin a Reversibly Inhibits the Secretory Pathway by Preventing Cotranslational Translocation. Mol. Pharmacol. 2009, 76 (1), 91–104. 10.1124/mol.109.056085. [DOI] [PubMed] [Google Scholar]
- Paatero A. O.; Kellosalo J.; Dunyak B. M.; Almaliti J.; Gestwicki J. E.; Gerwick W. H.; Taunton J.; Paavilainen V. O. Apratoxin Kills Cells by Direct Blockade of the Sec61 Protein Translocation Channel. Cell Chem. Biol. 2016, 23 (5), 561–566. 10.1016/j.chembiol.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Chen Q. Y.; Liu Y.; Luesch H. Systematic Chemical Mutagenesis Identifies a Potent Novel Apratoxin A/E Hybrid with Improved in Vivo Antitumor Activity. ACS Med. Chem. Lett. 2011, 2 (11), 861–865. 10.1021/ml200176m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q. Y.; Liu Y.; Cai W.; Luesch H. Improved Total Synthesis and Biological Evaluation of Potent Apratoxin S4 Based Anticancer Agents with Differential Stability and Further Enhanced Activity. J. Med. Chem. 2014, 57 (7), 3011–3029. 10.1021/jm4019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.; Cai W.; Chen Q. Y.; Xu S.; Yin R.; Li Y.; Zhang W.; Luesch H. Total Synthesis and Biological Evaluation of Apratoxin E and Its C30 Epimer: Configurational Reassignment of the Natural Product. Org. Lett. 2016, 18 (20), 5400–5403. 10.1021/acs.orglett.6b02780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Forsyth C. J. Total Synthesis of Apratoxin A. J. Am. Chem. Soc. 2003, 125 (29), 8734–8735. 10.1021/ja036050w. [DOI] [PubMed] [Google Scholar]
- Ma D.; Zou B.; Cai G.; Hu X.; Liu J. O. Total Synthesis of the Cyclodepsipeptide Apratoxin A and Its Analogues and Assessment of Their Biological Activities. Chem. - Eur. J. 2006, 12 (29), 7615–7626. 10.1002/chem.200600599. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Chen Z.; Ye T. Synthesis of the Polyketide Segment of Apratoxin A. Tetrahedron: Asymmetry 2004, 15 (2), 355–363. 10.1016/j.tetasy.2003.11.026. [DOI] [Google Scholar]
- Zou B.; Wei J.; Cai G.; Ma D. Synthesis of an Oxazoline Analogue of Apratoxin A. Org. Lett. 2003, 5 (19), 3503–3506. 10.1021/ol035332y. [DOI] [PubMed] [Google Scholar]
- Doi T.; Numajiri Y.; Munakata A.; Takahashi T. Total Synthesis of Apratoxin A. Org. Lett. 2006, 8 (3), 531–534. 10.1021/ol052907d. [DOI] [PubMed] [Google Scholar]
- Numajiri Y.; Takahashi T.; Doi T. Total Synthesis of (−)-Apratoxin A, 34-Epimer, and Its Oxazoline Analogue. Chem. - Asian J. 2009, 4 (1), 111–125. 10.1002/asia.200800365. [DOI] [PubMed] [Google Scholar]
- Doi T.; Numajiri Y.; Takahashi T.; Takagi M.; Shin-Ya K. Solid-Phase Total Synthesis of (−)-Apratoxin a and Its Analogues and Their Biological Evaluation. Chem. - Asian J. 2011, 6 (1), 180–188. 10.1002/asia.201000549. [DOI] [PubMed] [Google Scholar]
- Gilles A.; Martinez J.; Cavelier F. Supported Synthesis of Oxoapratoxin A. J. Org. Chem. 2009, 74 (11), 4298–4304. 10.1021/jo900583j. [DOI] [PubMed] [Google Scholar]
- Robertson B. D.; Wengryniuk S. E.; Coltart D. M. Asymmetric Total Synthesis of Apratoxin D. Org. Lett. 2012, 14 (20), 5192–5195. 10.1021/ol302309c. [DOI] [PubMed] [Google Scholar]
- Masuda Y.; Suzuki J.; Onda Y.; Fujino Y.; Yoshida M.; Doi T. Total Synthesis and Conformational Analysis of Apratoxin C. J. Org. Chem. 2014, 79 (17), 8000–8009. 10.1021/jo501130b. [DOI] [PubMed] [Google Scholar]
- Tarsis E. M.; Rastelli E. J.; Wengryniuk S. E.; Coltart D. M. The Apratoxin Marine Natural Products: Isolation, Structure Determination, and Asymmetric Total Synthesis. Tetrahedron 2015, 71, 5029–5044. 10.1016/j.tet.2015.05.047. [DOI] [Google Scholar]
- Mao Z. Y.; Si C. M.; Liu Y. W.; Dong H. Q.; Wei B. G.; Lin G. Q. Asymmetric Synthesis of Apratoxin E. J. Org. Chem. 2016, 81 (20), 9903–9911. 10.1021/acs.joc.6b02086. [DOI] [PubMed] [Google Scholar]
- Yin R.; Zhang W.; Liu G.; Wu P.; Lau C.; Li Y. Synthesis, Conformational Analysis and Biological Evaluation of the Lactam Analogue of the Cyclodepsipeptide Apratoxin A. Tetrahedron 2016, 72 (27–28), 3823–3831. 10.1016/j.tet.2016.04.050. [DOI] [Google Scholar]
- Yoshida M.; Onda Y.; Masuda Y.; Doi T. Potent Oxazoline Analog of Apratoxin C: Synthesis, Biological Evaluation, and Conformational Analysis. Biopolymers 2016, 106, 404–414. 10.1002/bip.22781. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Wang E.; Pal K.; Dutta S. K.; Bar-Sagi D.; Mukhopadhyay D. VEGF Exerts an Angiogenesis-Independent Function in Cancer Cells to Promote Their Malignant Progression. Cancer Res. 2012, 72 (16), 3912–3918. 10.1158/0008-5472.CAN-11-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti L.; Thomachot M. C.; Bachelot T.; Menetrier-Caux C.; Puisieux I.; Blay J. Y. IL-6 as an Intracrine Growth Factor for Renal Carcinoma Cell Lines. Int. J. Cancer 2004, 111 (5), 653–661. 10.1002/ijc.20287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.